



Introduction
Lung cancer is the most common histological subtype of non-small cell lung cancer (NSCLC), accounting for over 80% of all lung cancer cases [1]. NSCLC is first diagnosed at an advanced stage in China due to the lack of widespread screening and limited diagnostic tools in most areas [2]. As a result, advanced NSCLC has a poor prognosis, with a 5-year overall survival rate as low as 15% [3]. Classification of NSCLC considers mutations at the molecular level that may affect treatment. However, most NSCLC patients (close to 85–90%) do not have an identified drug-targeted driver mutations, and thus chemotherapy remains the standard of care [4]. Surgical treatment is not preferred in advanced NSCLC. Some conventional chemotherapeutic agents are also often used after targeted drug therapy. Cisplatin (Cis/DDP), mitomycin C (MMC), paclitaxel (PTX), and doxorubicin (DOX) are commonly used as conventional chemotherapeutic agents. Cisplatin is often used clinically in a small number of patients with drug-targeted driver mutations in NSCLC. The main drawback of conventional chemotherapy is the limited tumor suppression and toxic side effects, including nausea, vomiting, hair loss, fatigue, and diarrhea. In addition, residual tumor cells and systemic toxicity associated with chemotherapy can exacerbate host immunosuppression, thus increase tumor progression [5, 6]. Effective chemotherapy improves the quality of life and overall survival rate in NSCLC. The development of chemoresistance during treatment greatly limits chemotherapy efficacy and clinical utility [7]. Therefore, the resistance mechanisms in platinum-based therapies in NSCLC should be explored. In addition, appropriate measures should be taken to reduce the development of resistance in NSCLC and increase the efficacy of first-line chemotherapy.
Ferroptosis is a form of cell death distinct from apoptosis, necrosis, and autophagy. It is characterized by iron-dependent lipid peroxidation. The mitochondrial morphology is characterized by consolidation, loss of cristae, and an increase in bilayer density. Under normal physiological conditions, iron is involved in oxygen transport, DNA biosynthesis, and coenzymes in the tricarboxylic acid cycle and electron transfer. Excess intracellular iron ions catalyze the production of large amounts of reactive oxygen species (ROS), leading to cellular damage or death and inhibiting tumor cell proliferation [8, 9]. Current research suggests that ferroptosis is induced by the following: (1) elevation of intracellular divalent ferrous ions: extracellular trivalent ferric ions are bound to transferrin and then reduced to divalent ferrous ions via transferrin receptor on the cell membrane. Divalent ferrous ions are stored in a bound form to ferritin (FT). The excess divalent iron ions are oxidized to trivalent iron for extracellular transport, and this process requires Ferroprotein. An increase in transferrin receptor (TFR) and a decrease in FT and FTP associated with the transport and binding of iron, respectively, will lead to an increase in divalent iron ions inducing ferroptosis. (2) Excessive production of lipid peroxides: Through the Fenton reaction, divalent iron ions can produce precursors of lipid peroxides with hydrogen peroxide. (3) Inhibition of lipid peroxidation damage repair: Glutathione peroxidase 4 (GPX4) and xCT (reverse cystine/glutamate transporter) play an important role in this process. The xCT transporter transports extracellular cystine into the cell for the synthesis of glutathione (GSH). GPX4 uses the reduced glutathione to detoxify lipid peroxides to the corresponding alcohols or water [10]. Studies on the mechanism of cisplatin resistance have shown a high expression of GSH in the resistant cells. Ferroptosis can be induced by inhibition of GSH. Thus, ferroptosis is an area of interest in the study of cisplatin resistance. It has been shown that cisplatin resistance in head and neck cancer cells is associated with increased xCT and GPX4 [11]. In contrast, ferroptosis and cisplatin resistance in NSCLC has not been extensively studied, and the mechanism of action is unclear.
Nigella sativa is an herb that has been used in folk medicine for the treatment of various conditions [12–14]. It possesses anti-inflammatory, hypoglycemic, hypotensive, antioxidant, and antitumor effects [15]. α-Hederin (α-Hed) is one of its main extracts. Various studies have reported that α-Hederin possesses anti-inflammatory activity [16], relaxes airway and gastrointestinal smooth muscle [16, 17], and increases cell permeability [18]. However, studies have not reported any significant adverse effects or side effects associated with α-Hederin. The potential antitumor effects of α-Hederin have been demonstrated in breast cancer, laryngeal cancer, lung cancer, melanoma, and leukemia [19–21]. Studies have shown that α-Hederin can block the JAK2/STAT3 signaling pathway. Moreover, α-Hederin was shown to inhibit the proliferation, invasion, and migration of colon cancer cells SW620 by regulating the JAK2/STAT3 signaling pathway. α-Hederin was also shown to block the expression of JAK2 or activated STAT3, significantly inhibiting IL-6-induced epithelial-mesenchymal transition in colon cancer cells [22]. Another study also reported that α-Hederin inhibited tumor cell proliferation and induced tumor cell apoptosis-related to an imbalance of antioxidants in the cell [23]. In contrast, excessive ROS has been reported to cause downstream signaling, decrease mitochondrial Apaf-1 and Cyt C expression, increase caspase-3 and caspase-9 activity, thus activating oxidative damage of mitochondrial membrane and triggering apoptosis in breast cancer cells. Wang et al. found that α-Hederin could activate the mitochondrial apoptotic pathway by depleting intracellular GSH, thus inducing ROS accumulation and antitumor effects in hepatocellular carcinoma [24]. However, the role of α-Hederin in cisplatin resistance in NSCLC has not been reported.
The present study aimed to explore the potential effects and underlying mechanisms of α-Hederin and ferroptosis in cisplatin resistance in NSCLC.
Methods
Cell treatment
A549/DDP and PC-9/DDP cells (DDP-resistant human lung cancer cells) were purchased from MeiXuan Biological Science and Technology, Inc., (Shanghai, China). The cells were cultured in F12K medium supplemented with 100 ml/l fetal bovine serum (FBS), 100 kU/l penicillin, and 100 mg/l chloramphenicol in a cell incubator at 37°C and 5% CO2. The drugs α-Hederin (98%; Shanghai Yuanye Bio-Technology Co., Ltd., Shanghai, China) and cisplatin (DDP; Macklin Inc., Shanghai, China) were dissolved in dimethyl sulfoxide (DMSO) and stored at −80°C.
Cell transfection
Two siRNAs against EGR1 and DDIT3 were synthesized by GeneChem (Shanghai, China). The expression efficiency was determined using qPCR in cells transfected with vector or siRNA. miR-96-5p inhibitor and miR-96-5p mimic were constructed by Hanbio (Shanghai, China). Cells were transfected with the appropriate amount of vector using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA). The cells were then cultured for 24 hours.
Animal studies
For orthotopic assay, a total of 50 μl cell suspension of A549/DPP (1 × 107) was mixed with 50 ml of Matrigel. Subsequently, the mixture was injected into the left lung of mice through the chest wall to a depth of 3 mm. Magnetic resonance imaging (MRI) was used to exam tumor formation after one week. For one treatment cycle in a week (starting from week 1 to week 6), cisplatin (5 mg/kg, intraperitoneal injection, 1 time) and α-Hederin (40 mg/kg, oral administration, 2 times) were given. Total three treatment cycles were conducted in this experiment. Each group contained five mice.
All animals’ experiments were conducted in accordance with the recommendations of the Animal Care Committee of Huzhou Central Hospital.
Statistical analysis
Statistical analysis was performed using R (R version 3.6.3). T-test was used to analyze data between two independent groups, while one-way ANOVA was used to analyze differences among various groups. p < 0.05 was considered statistically significant.
Detailed materials and methods are available in the Supplementary Materials and Methods.
Availability of data and materials
The data generated or analyzed during this study are included in this article, or if absent are available from the corresponding author upon reasonable request.
Results
α-Hederin inhibited in vitro and in vivo cisplatin resistance in NSCLC
To evaluate the effect of α-Hederin on cisplatin resistance in NSCLC, we first cultured cisplatin resistance NSCLC A549/DPP and PC-9/DPP cell lines. The cells were then treated with α-Hederin for 24 h, to suppress the viability of the cell lines in a dose-dependent manner (Figure 1A). A CCK8 assay determined at 0, 24, 48, and 72 hours showed that α-Hederin inhibited cell proliferation (Figure 1B). In addition, α-Hederin sensitized A549/DPP and PC-9/DPP cells to cisplatin in a dose-dependent manner (Figure 1C). Colony formation assay showed that A549/DPP and PC-9/DPP treated with 25 and 50 μM of α-Hederin significantly inhibited cell proliferation (Figure 1D). Cell apoptosis was determined via flow cytometry and Propidium Iodide (PI) staining after treatment with α-Hederin for one day. As shown in Figure 1E, 1F, α-Hederin significantly promoted cell apoptosis. To assess the effect of α-Hederin on cell metastatic ability, Transwell assay was used to detect cell migration and invasion, while Western blot was used to determine protein levels of E-cadherin, N-cadherin, and snail. Figure 2A–2C shows that α-Hederin abrogates cell migration and invasion. The results showed that N-cadherin and snail protein expression were suppressed while E-cadherin protein expression was enhanced. α-Hederin at a concentration of 25 and 50 μM was also shown to weaken the sphere-forming ability of NSCLC cells and decrease the protein levels of stemness markers CD133 and CD44 (Figure 2D, 2E).
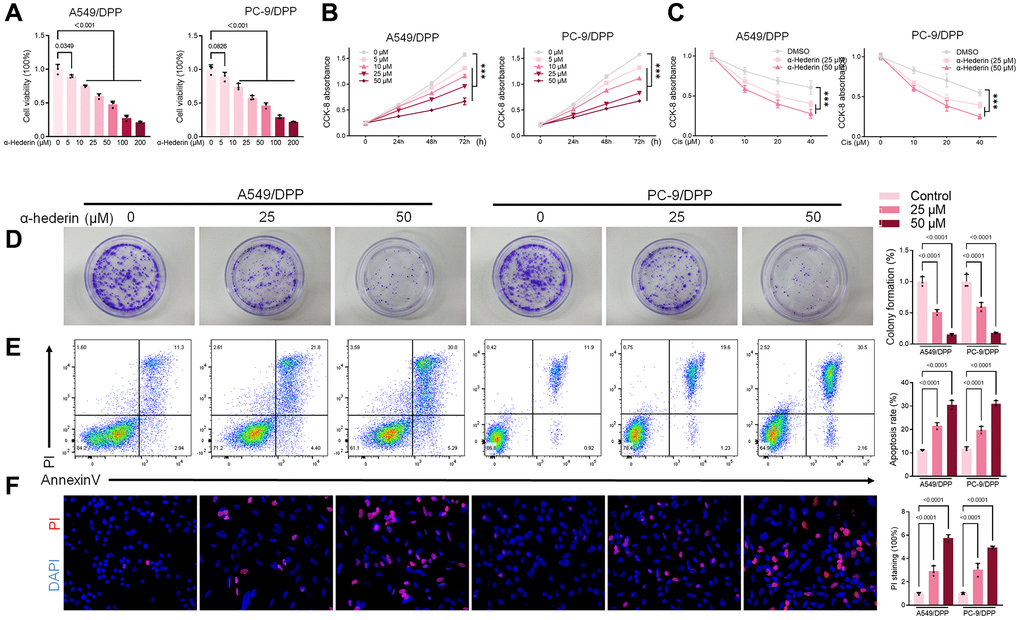
Figure 1. α-Hederin reduces cisplatin resistance in cisplatin-resistant NSCLC cell lines. (A) CCK8 results showing the viability of cisplatin-resistant A549/DPP and PC-9/DPP cell lines after treatment with different concentrations of α-Hederin for 24 hours. (B) CCK8 results showing the viability of A549/DPP and PC-9/DPP cell lines after treatment with different concentrations of α-Hederin for 0, 24, 48, and 72 hours. (C) CCK8 results showing the viability of cell after treatment with different concentrations of cisplatin for 24 hours. (D) Colony formation assay showing proliferation in NSCLC cells after treatment with α-Hederin for 24 hours. (E) Cell apoptosis detected by flow cytometry. (F) Propidium iodide (PI) staining (red) indicates apoptotic/necrotic cells. (n = 3). Data are shown as mean ± SD, One-way ANOVA, ***P < 0.001.
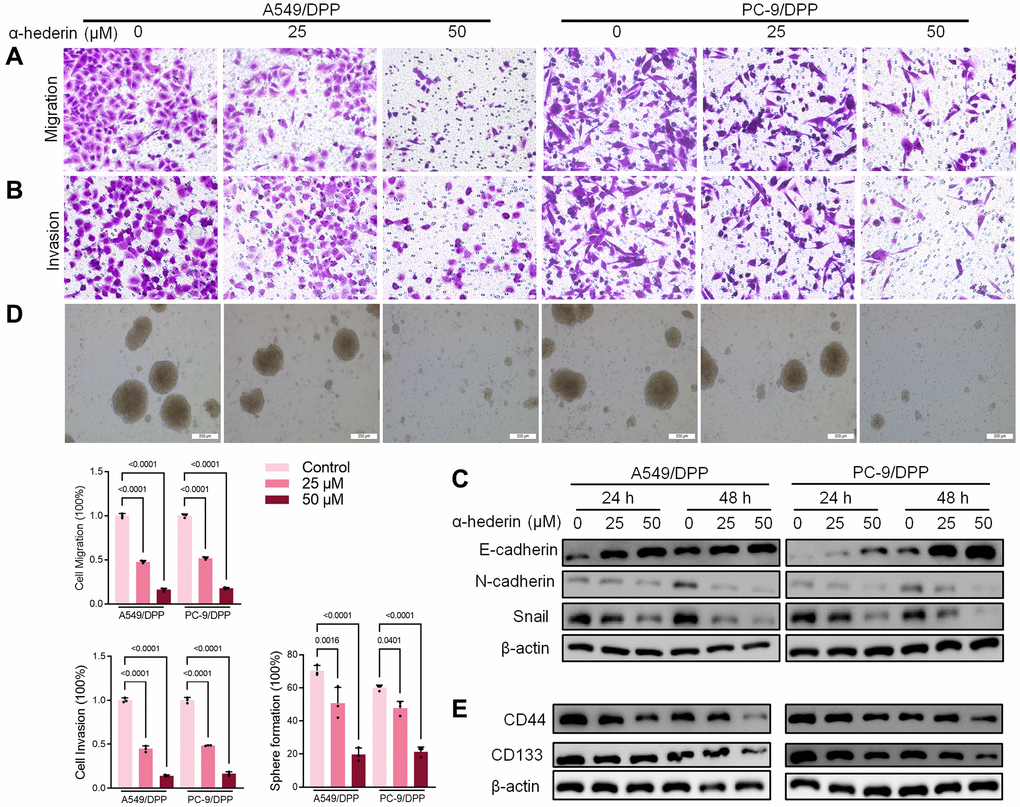
Figure 2. α-Hederin decreases metastasis and stemness in cisplatin-resistant NSCLC cells. (A, B) Cell migration and invasion were detected by Transwell assay. (C) Western blot demonstrating E-cadherin, N-cadherin, and Snail levels in NSCLC cells after treatment with α-Hederin for 24 hours. (D) The spheroid formation ability assessed in NSCLC cells. (E) Western blot demonstrating CD44 and CD133 levels in NSCLC cells. (n = 3). Data are shown as mean ± SD, One-way ANOVA.
The mice xenograft model of A549/DPP tumor construction confirmed α-Hederin (40 mg/kg) treatment repressed tumor volume compared with the control group. Moreover, α-Hederin (40 mg/kg) combined with cisplatin (5 mg/kg) further inhibited xenograft tumor formation compared with cisplatin treatment (Figure 3A, 3B). Immunohistochemical detection indicated that α-Hederin (40 mg/kg) treatment increased Tunel and E-cad and suppressed Ki-67 and CD44 expression in xenograft tumor tissues (Figure 3C). Live imaging showed that α-Hederin (40 mg/kg) combined with cisplatin (5 mg/kg) abrogated NSCLC metastasis compared with cisplatin treatment (Figure 3D). Orthotopic models of lung tumor tissues were also reproduced in nude mice in the different treatment groups (Figure 3E).
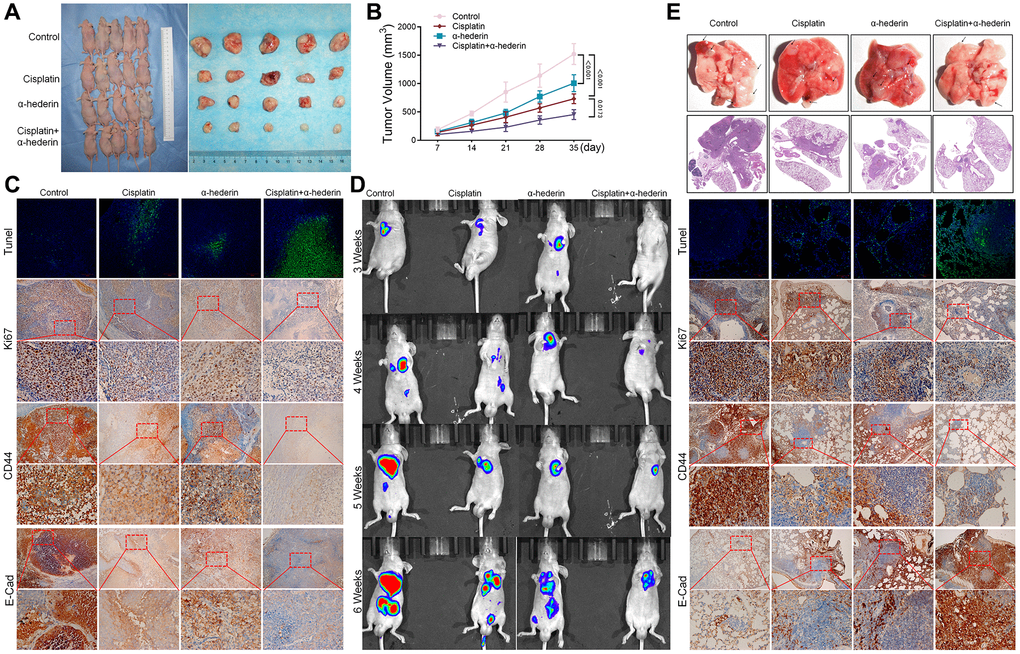
Figure 3. α-Hederin decreases A549/DPP cell growth in vivo. (A) Representative images of xenograft tumors isolated from nude mice in the different groups. (B) Tumor sizes in the different groups. (C) Representative images of Tunel, Ki-67, CD44, E-Cad staining isolated from nude mice xenograft tumor tissues in the different groups. (D) Live imaging showing metastasis of A549/DPP cells of an intravenous tail injection with α-Hederin in different weeks. (E) Representative images of orthotopic tumor model isolated from nude mice in the different groups and HE, Tunel, Ki-67, CD44, E-Cad staining in orthotopic lung tissue. (n = 5). Data are shown as mean ± SD, One-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
α-Hederin treatment promoted ferroptosis in NSCLC cells
MetaboAnalyst software was used to explore underlying alterations in metabolic pathways. As shown in Figure 4A, 4B, α-Hederin significantly altered the glutathione pathway. GSH depletion and lipid peroxidation are significant hallmarks of ferroptosis related cell death. Thus, we evaluated ferroptosis and lipid peroxidation-related indicators such as GSH, NADPH, and ROS levels. Results showed that α-Hederin depleted cellular GSH and nicotinamide adenine dinucleotide phosphate (NADPH) concentration but stimulated the ROS levels (Figure 4C–4E). Besides, Immunofluorescence using C11-BODIPY also showed that α-Hederin induced lipid peroxidation in NSCLC cells (Figure 4F). Next, we evaluated ferroptosis markers SLC7A11 and GPX4. Western blot and qRT-PCR results revealed that α-Hederin reduced the protein and mRNA levels of SLC7A11 and GPX4 in a dose-dependent manner (Figure 4G–4I). Ferroptosis inhibitor Ferrostatin-1 (Fer-1) was used to verify the mechanism of ferroptosis in α-Hederin against NSCLC cisplatin resistance. As expected, Fer-1 reversed the anti-proliferation and pro-apoptotic effects of α-Hederin in NSCLC cells (Supplementary Figure 1A, 1B). Furthermore, Fer-1 inhibited lipid peroxidation stimulated by α-Hederin in NSCLC cells (Supplementary Figure 1C, 1D). Next, as shown in Supplementary Figure 1E, Fer-1 reversed the anti-proliferation effects of α-Hederin in parental NSCLC cells and inhibited lipid peroxidation stimulated by α-Hederin in parental NSCLC cells (Supplementary Figure 1F, 1G). Which indicated α-Hederin-induced ferroptosis affected cisplatin-resistant cells which is a common phenotype in both cisplatin-resistant cells and parental cells.
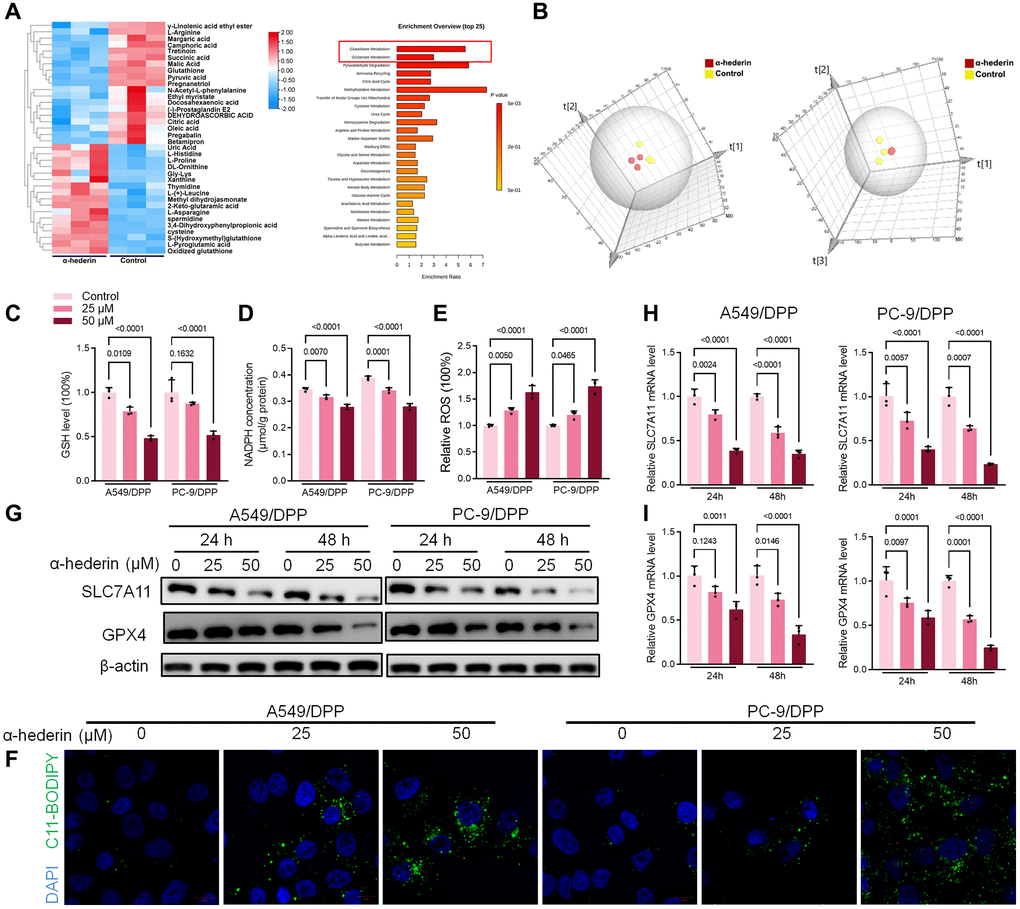
Figure 4. α-Hederin treatment activated ferroptosis in NSCLC. (A) OPLS-DA score plot showing differences between α-Hederin treatment and control groups in A549/DPP cells. (B) A heat map was used to identify metabolites in A549/DPP cells. (C) Cellular GSH level was detected in NSCLC cells after treatment with α-Hederin for 24 hours. (D) Cellular NADPH level was detected in NSCLC cells. (E) Cellular ROS level was detected in NSCLC cells. (F) Immunofluorescence assays were performed using C11-BODIPY (green) to assess oxidation and DAPI staining to visualize the Nuclei (blue) in NSCLC cells. (G) Western blot demonstrating SLC7A11 and GPX4 protein levels in NSCLC cells after treatment with α-Hederin for 24 hours. (H, I) qRT-PCR demonstrating SLC7A11 and GPX4 mRNA levels in NSCLC cells. (n = 3). Data are shown as mean ± SD, One-way ANOVA.
α-Hederin activated DDIT3/ATF3 pathway in NSCLC cells
Previous studies have reported that p53 represses SLC7A11 transcription and regulates cell ferroptosis [25]. PC-9 is a P53 mutant cell line [26]. Hence, we speculate that α-Hederin induced ferroptosis via a P53-independent pathway. RNA-seq was used to compare the mRNA differences between A549/DPP cells treated with or without 50 μM of α-Hederin for 24 hours. The results showed that DNA Damage Inducible Transcript 3 (DDIT3) and Activating Transcription Factor 3 (ATF3) were aberrantly expressed in cells treated with α-Hederin (Figure 5A, 5B). qRT-PCR and Western blot results were consistent with the RNA-seq results showing that α-Hederin activated the mRNA and protein levels of DDIT3 and ATF3 in a dose-dependent manner (Figure 5C, 5D). The TCGA database confirmed that the levels of DDIT3 and ATF3 in Lung squamous cell carcinoma (LUSC) and lung adenocarcinoma (LUAD) tumor tissues were significantly decreased compared with normal tissues. Besides, the ROC curves showed that DDIT3 and ATF3 had a promising ability to predict LUSC and LUAD in the TCGA cohort (Supplementary Figure 2A–2D). Furthermore, the Spearman rank correlation analysis showed a positive correlation between DDIT3 and ATF3 levels in LUSC and LUAD tumor tissues (Supplementary Figure 2E). Subsequently, A549/DPP and PC-9/DPP cell lines were transfected with DDIT3 small interfering RNA (si-DDIT3) for 24 hours. Figure 5E, 5F confirmed that si-DDIT3 significantly inhibited DITT3 and ATF3 levels in NSCLC cells. Previous research has reported that ATF3 regulates SLC7A11-mediated ferroptosis [27]. Hence, we assessed the role of DDIT3 in regulating ferroptosis. qRT-PCR and Western blot results showed that si-DDIT3 increased the mRNA and protein levels of ferroptosis markers SLC7A11 and GPX4 (Figure 5G, 5H). Downregulation of DDIT3 recovery cellular GSH and NADPH concentration and inhibited ROS level (Figure 5I–5K). Besides, Immunofluorescence using C11-BODIPY also showed a suppressive effect of si-DDIT3 on lipid peroxidation in NSCLC cells (Figure 5L). Furthermore, CCK8 and flow cytometry results suggested that si-DITT3 reversed the anti-proliferation and pro-apoptotic effect of α-Hederin in NSCLC cells (Figure 5M, 5N). Taken together, α-Hederin activated the DDIT3/ATF3 pathway and stimulated ferroptosis via inhibiting SLC7A11 and GPX4.
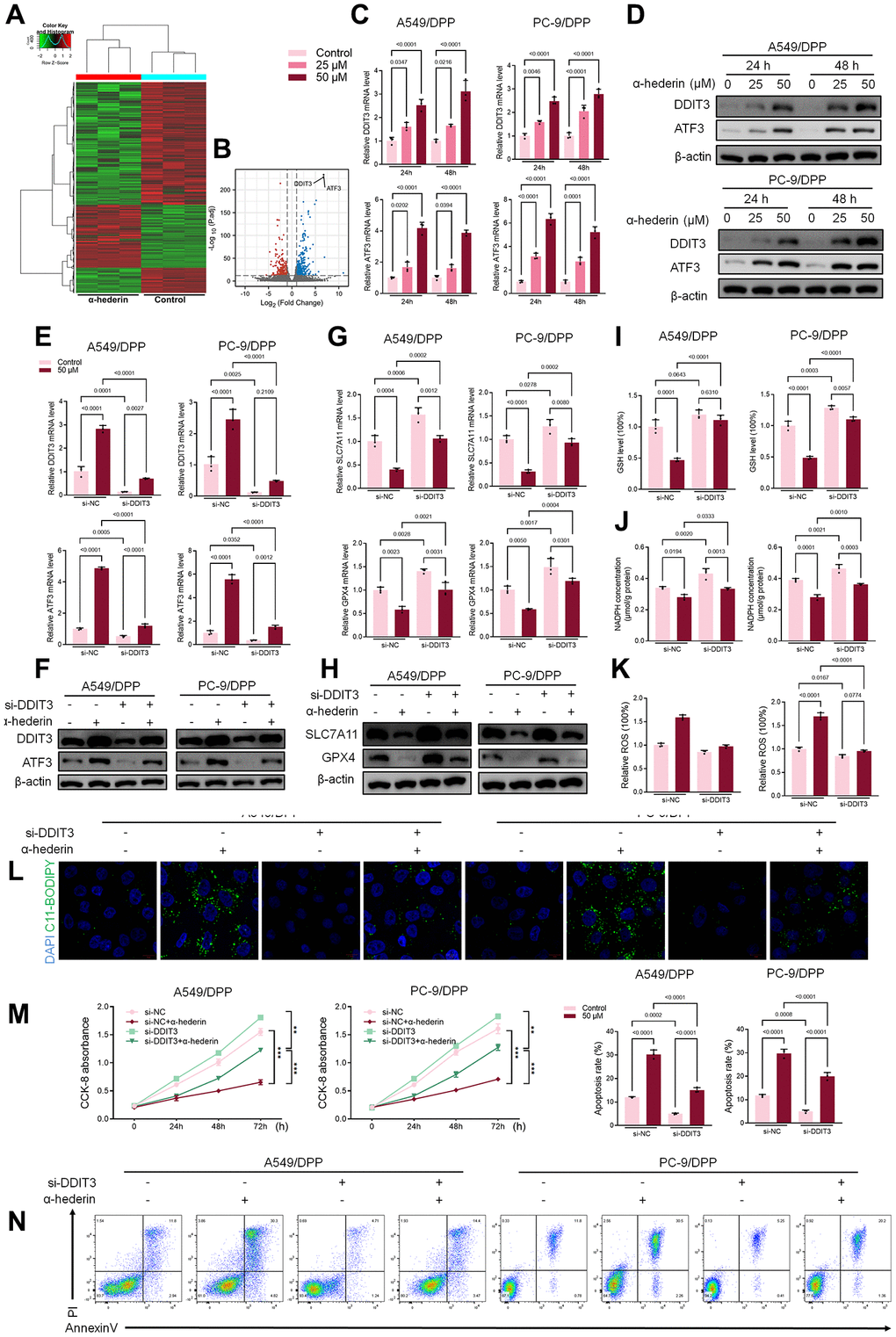
Figure 5. α-Hederin activated DDIT3/ATF3 pathway in NSCLC cells. (A) Heatmap of the inter-individual correlation of all mRNA transcripts (RNA-seq). (B) Volcano plot of gene expression. (C) qRT-PCR demonstrating DDIT3 and ATF3 mRNA levels in NSCLC cells after treatment with α-Hederin for 24 hours. (D) Western blot demonstrating DDIT3 and ATF3 protein levels in NSCLC cells after treatment with α-Hederin for 24 hours. (E, F) qRT-PCR and Western blot demonstrating DDIT3 and ATF3 levels in NSCLC cells after transfection with si-DDIT3 and treatment with α-Hederin for 24 hours. (G, H) qRT-PCR and Western blot demonstrating DDIT3, ATF3, SLC7A11 and GPX4 levels in NSCLC cells after transfection with si-DDIT3 and treatment with α-Hederin for 24 hours. (I–K) Cellular GSH, NADPH and ROS levels were detected in NSCLC cells after transfection with si-DDIT3 and treatment with α-Hederin for 24 hours. (L) Immunofluorescence assays were performed using C11-BODIPY (green) to determine oxidation and DAPI staining to detect the Nuclei (blue) in NSCLC cells. (M) CCK8 results showing the viability of NSCLC cells after transfection with si-DDIT3 and treatment with α-Hederin. (N) Flow cytometry detected NSCLC cells apoptosis after transfection with si-DDIT3 and treatment with α-Hederin for 24 hours. (n = 3). Data are shown as mean ± SD, One-way ANOVA, **P < 0.01, ***P < 0.001.
miR-96-5p directly binds to DDIT3 in NSCLC cells
Bioinformatics was used to predict the possible targets in determining the potential association between miR-96-5p and DDIT3. Results suggested that DDIT3 could be possible targets of miR-96-5p (Figure 6A). The dual-luciferase reporter assay results showed that overexpression of miR-96-5p reduced activity in NSCLC cells transfected with wild type (WT) DDIT3- vectors. However, no effect was seen in the cells transfected with the mutant type (MUT) DDIT3-vectors (Figure 6B). As shown in Figure 6A, the expression of miR-96-5p was blocked by α-Hederin treatment (Figure 6C). Besides, the TCGA database confirmed that the level of miR-96-5p in LUSC and LUAD tumor tissues was significantly increased compared with normal tissues (Supplementary Figure 2F, 2G). qRT-PCR and Western blot results showed that overexpression of miR-96-5p (miR-mimic) reduced the mRNA and protein levels of DDIT3. In contrast, inhibition of miR-96-5p (miR-inhibitor) increased the mRNA and protein levels of DDIT3 (Figure 6D, 6E).
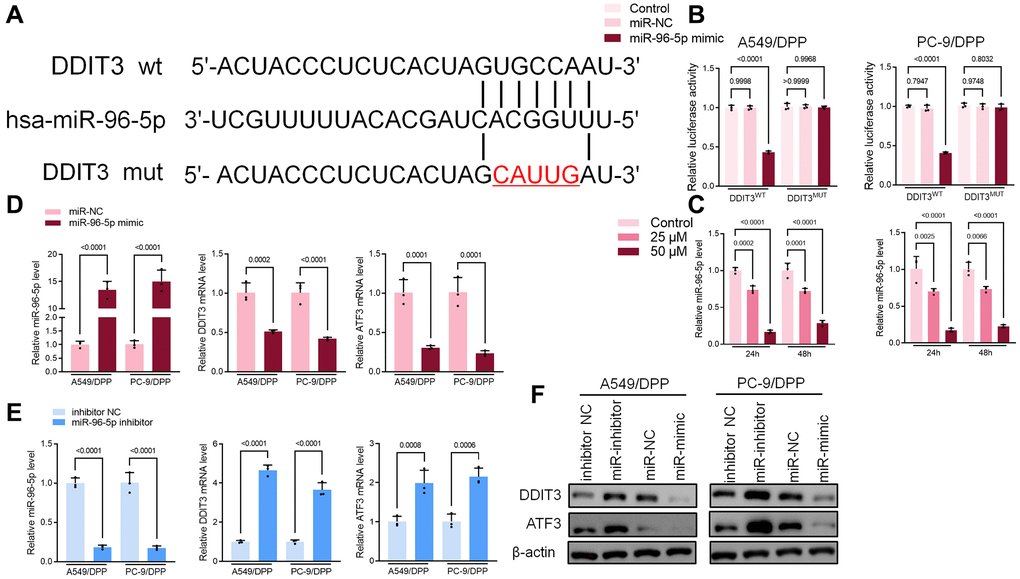
Figure 6. miR-96-5p directly binds to DDIT3 in NSCLC cells. (A) The predicted binding sites of miR-96-5p in DDIT3. The mutated (Mut) version of DDIT3 is also presented. (B) Relative luciferase activity in A549/DPP and PC-9/DPP cells was measured 48 h after transfection. (C) qRT-PCR demonstrating miR-96-5p levels in NSCLC cells after treatment with α-Hederin for 24 hours. (D, E) qRT-PCR demonstrating miR-96-5p, DDIT3, and ATF3 levels in NSCLC cells after transfection with miR-96-5p mimic or miR-96-5p inhibitor for 24 hours. (F) Western blot demonstrating DDIT3 and ATF3 levels in NSCLC cells after transfection with miR-96-5p mimic or miR-96-5p inhibitor for 24 hours. (n = 3). Data are shown as mean ± SD, One-way ANOVA.
α-Hederin promoted EGR1 nuclear translocation and directly repressed miR-96-5p in NSCLC cells
TransmiR v2.0 database (http://www.cuilab.cn/transmir/) was used to predict miR-96-5p associated transcription factor Early Growth Response 1 (EGR1). The EGR1 binding motif was predicted from JASPAR matrix models (Figure 7A). Next, we mutated the potential binding promoter site of miR-96-5p (−1363~−1350) (Figure 7B). An EGR1 expression vector containing a 3 kb fragment upstream of the miR-96-5p stem-loop was co-transfected into NSCLC cells. EGR1 significantly repressed the luciferase activity of miR-96-5p. However, miR-96-5p mutations showed no effect on EGR1 (Figure 7C). Molecular docking revealed that α-Hederin interacted with the active site of EGR1 (Figure 7D). Western blot results showed that α-Hederin significantly promoted EGR1 nuclear translocation in A549/DPP and PC-9/DPP cell lines (Figure 7E). Besides, the TCGA database confirmed that the level of EGR1 in LUSC and LUAD tumor tissues was significantly decreased compared with normal tissues (Supplementary Figure 2H, 2I). To elucidate the mechanism of EGR1 in NSCLC cells, we transfected EGR1 small interfering RNA (si-EGR1) into NSCLC cells. Figure 8A demonstrates that downregulation of EGR1 significantly suppressed the level of DDIT3 and ATF3 but enhanced SLC7A11 and GPX4. Further, we co-transfected the NSCLC cells with si-EGR1 and miR-96-5p inhibitors. The CCK-8 analysis results revealed that the downregulation of EGR1 increased cell viability. However, the miR-96-5p inhibitor abrogated this enhancement (Figure 8B). Furthermore, A549/DPP and PC-9/DPP cell lines were treated with 50 μM α-Hederin, co-transfected with si-EGR1 and miR-96-5p inhibitor, and cultured for 24 hours. As shown in Figure 8C–8H, downregulation of EGR1 reverses the effects of α-Hederin on cell proliferation, apoptosis, migration, invasion, and sphere-formation in A549/DPP and PC-9/DPP cell lines. At the same time, the inhibition of miR-96-5p abolished the effect of si-EGR1. Western blot assay results were consistent with these findings (Figure 8H). Besides, ROS results confirmed that overexpression of miR-96-5p or downregulation of EGR1 affects α-Hederin induced lipid peroxidation (Figure 8I). Therefore, these results demonstrate that α-Hederin promoted EGR1 nuclear translocation and directly repressed miR-96-5p in A549/DPP and PC-9/DPP cell lines.
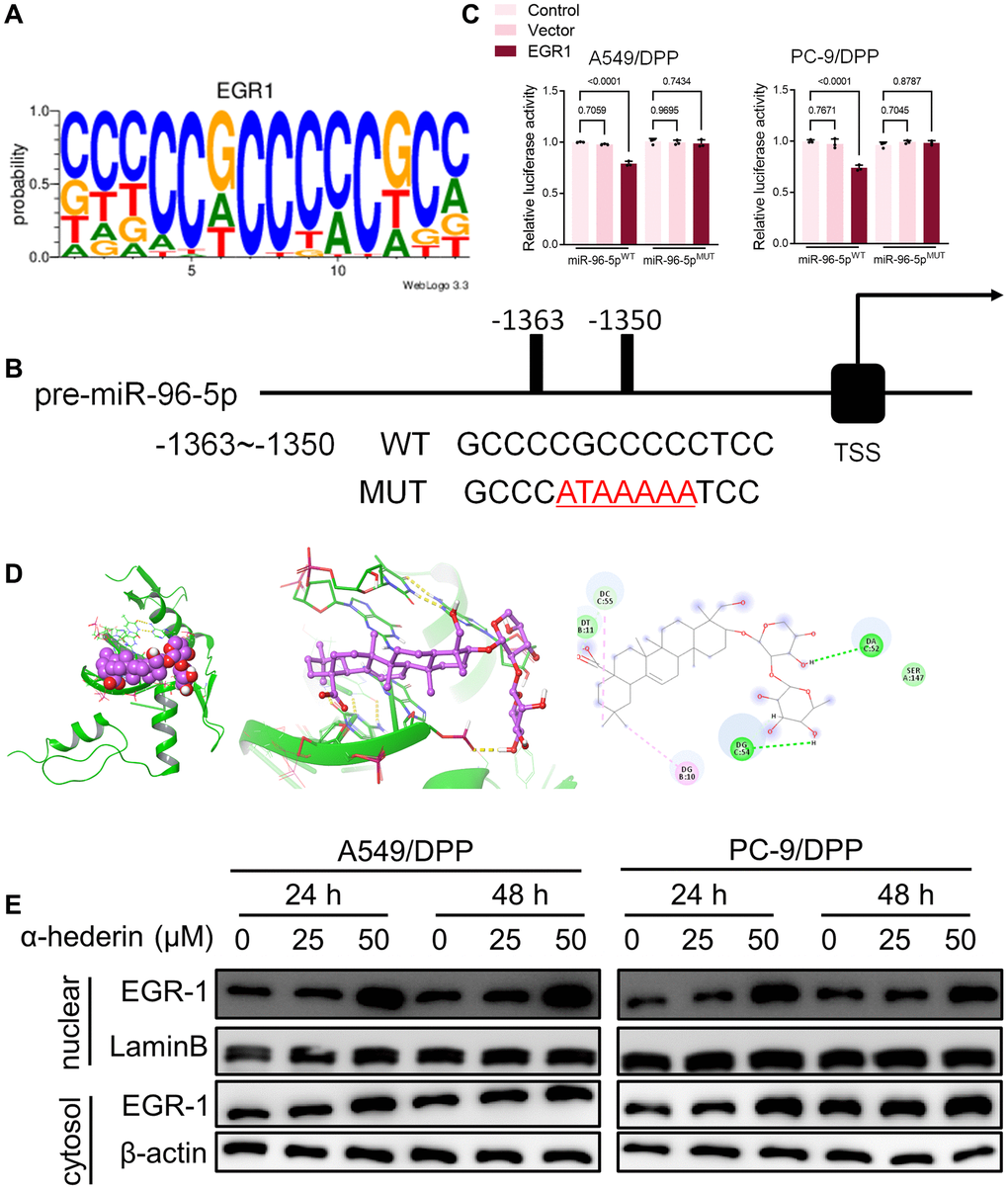
Figure 7. EGR1 directly binds to the miR-96-5p promoter region. (A) Sequence logo representing the consensus EGR1 binding motif from JASPAR database (http://jaspar.genereg.net/). (B) miR-96-5p promoter region was analyzed from the TSS. The mutation was seen from -1363~-1350. (C) The luciferase activity was detected in NSCLC cells after transfection with the EGR1-expressing vector. The luciferase reporter plasmid contained a 2 kb fragment upstream of the miR-96-5p stem-loop. (D) Theoretical binding mode of α-Hederin at the binding site of EGR1. (E) Western blot demonstrating EGR1 protein levels in nuclear and cytosol in NSCLC cells after treatment with α-Hederin for 24 hours. (n = 3). Data are shown as mean ± SD, One-way ANOVA.
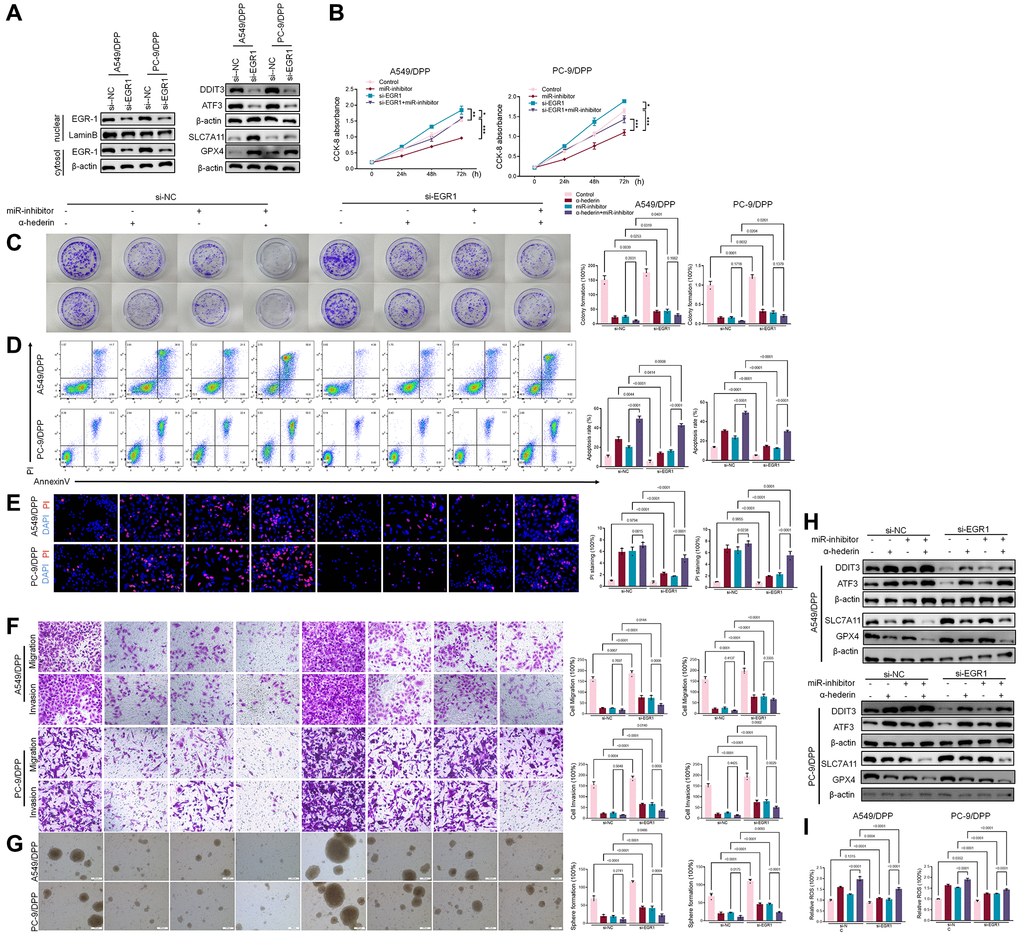
Figure 8. α-Hederin promoted EGR1 nuclear translocation and directly repressed miR-96-5p in NSCLC. (A) Western blot demonstrating EGR1 protein levels in nuclear and cytosol. DDIT3, ATF3, GPX4, and SLC7A11 protein levels are shown following transfection with si-EGR1 for 24 hours. (B) CCK8 results showing the viability of NSCLC cells after transfection with si-EGR1 or miR-96-5p mimic for 24 hours. (C) Colony formation assay showing proliferation in NSCLC cells after treatment with α-Hederin (50 μM) for 24 hours and transfection with si-EGR1 or miR-96-5p mimic for 24 hours. (D) Cell apoptosis was detected by flow cytometry. (E) Propidium iodide (PI) staining (red) indicates apoptotic/necrotic cells. (F) Cell migration and invasion were detected by Transwell assay. (G) Spheroid formation ability assessed in NSCLC cells. (H) Western blot demonstrating EGR1, DDIT3, ATF3, GPX4, and SLC7A11 protein levels in nuclear and cytosol of NSCLC cells. (I) Cellular ROS levels were detected in NSCLC cells after treatment with α-Hederin (50 μM) for 24 hours and transfection with si-EGR1 or miR-96-5p mimic for 24 hours. (n = 3). Data are shown as mean ± SD, One-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Currently, platinum-based chemotherapy remains the mainstay treatment in patients with advanced non-small-cell lung cancer without driver mutations. However, chemotherapy resistance in NSCLC greatly limits therapeutic effectiveness [7]. Ferroptosis is a novel form of cell death that was first defined in 2012. It is characterized by iron-dependent lipid peroxidation. Activation of lipid peroxidation depends on the inactivation of the GPX4 enzyme and the inhibition of GSH. GSH is a key molecule that inhibits cisplatin binding to DNA. Ferroptosis could be involved in regulating cisplatin resistance in tumor cells. Studies have demonstrated that artemisinin and its derivatives can effectively reverse cisplatin resistance in head and neck cancer, associated with ferroptosis induction [11]. Jin et al. showed that Solasonine, a natural compound, promoted ferroptosis of HCC cells in vitro and in vivo via GPX4 [28]. Besides, ferroptosis agonist erastin enhanced the sensitivity of NSCLC cells to CDDP via NRF2 [29]. This study found that α-Hederin inhibited GSH and activated lipid peroxidation in drug-resistant cell lines. Therefore, we hypothesized that induction of ferroptosis by α-Hederin plays a role in inhibiting NSCLC drug resistance.
DDIT3, a key transcription factor in the endoplasmic reticulum stress pathway, regulates apoptosis and inflammation [30]. Previous research showed that DDIT3 induced apoptosis by promoting the transcription of apoptosis-related molecules through the regulation of ATF family members such as ATF3 [31]. ATF3-mediated ferroptosis has been shown to play a key role in a variety of diseases such as gastric cancer [32], hepatocellular carcinoma [33], acute kidney injury [34], among others. Results of this study showed that α-Hederin activated the DDIT3/ATF3 pathway, thus promoting ferroptosis. In contrast, silencing DDIT3 suppressed ATF3 and abrogated ferroptosis in NSCLC cells. P53 mutations are present in 50% of all patients with non-small cell lung cancer and are the most common mutations associated with primary resistance to targeted therapy [35]. Hence, α-Hederin could activate ferroptosis through a P53-dependent pathway.
microRNAs (miRNAs) are small non-coding RNAs with approximately 18–22 nucleotides [36]. miRNAs have a key role in the post-transcriptional regulation of gene expression. By binding to specific sites in the 3′-UTRs of targeted mRNAs, miRNAs mediate their degradation and translational regulation. Deng et al. reported that miR-324 directly inhibited GPX4 and enhanced ferroptosis in cisplatin-resistant NSCLC cells. Besides, it has been demonstrated that miR-4443 repressed FSP1-mediated ferroptosis and promoted cisplatin resistance in NSCLC [37]. EGR1 is a member of the early growth response gene family whose expression is regulated by many extracellular signaling molecules and whose biological roles are closely related to cell proliferation, apoptosis, migration, invasion, and differentiation [38]. EGR1 expression was elevated in gastric cancer, prostate cancer, and glioblastoma compared to normal tissues. Increased EGRI expression significantly increased the proliferation, activity, and metastasis of cancer cells. High expression of EGR1 was also associated with poor prognosis in cancer patients [39–41].
EGR1 has a highly conserved DNA-binding domain, composed of three GysHis2 zinc finger junctions that specifically identify and bind the promoter regions of downstream genes, thereby acting as a transcriptional regulator [42]. EGR1 has been shown to target multiple miRNAs and influence the effects of these miRNAs on downstream target genes [43]. miR-152, an autophagy-regulated miRNA, was shown to be down-regulated in ovarian cancer resistant cells A2780/CP70. EGR1 has been shown to regulate the transcription of miR-152 by binding to the miR-152 promoter region. Up-regulation of both EGR1 and miR-152 inhibit the downstream target genes of miR-152, and ATG14, thus suppressing cisplatin resistance in ovarian cancer cells by preventing protective autophagy in ovarian cancer cells [44]. Our data revealed that α-Hederin promotes EGR1 translocation to the nucleus and directly binds to the miR-96-5p promoter region, thus suppressing miR-96-5p and sensitizing the NSCLC cells to cisplatin.
Conclusion
This study demonstrated that α-Hederin was able to reverse cisplatin resistance in NSCLC in vitro and in vivo. This effect was mediated by activation of nuclear translocation of EGRI and direct repression of miR-96-5p which promoted DDIT3/ATF3-mediated ferroptosis.
Author Contributions
Guarantor of integrity of the entire study: Shugao Han. Study concepts: Qian Yang and Jing Zhuang. Study design: Shugao Han. Definition of intellectual content: Shugao Han and Qian Yang. Literature research: Qian Yang and Qin Zhou. Experimental studies: Qian Yang, Jing Zhuang, Lixin Ru and Furong Niu. Data acquisition: Qian Yang and Jingjing Wang. Data analysis: Qian Yang and Wei Mao. Statistical analysis: Qian Yang and Wei Mao. Manuscript preparation: Qian Yang and Jing Zhuang. Manuscript editing: Qin Zhou. Manuscript review: Shugao Han.
Acknowledgments
We thank HOME for Researchers (http://www.home-for-researchers.com/static/index.html#/retouch_draw) for editing this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
All animal experiments and this study were approved by the Experimental Animal Management and Ethics Committee of Huzhou Central Hospital with protocol number 202311005.
Funding
This work was supported by the Public Welfare Technology Application Research Program of Huzhou (No. 2018GZ36) and the Zhejiang Province Public Welfare Technology Application Research Project (No. LGF22H090037 and No. LGF22H290002).
References
- 1. Lam KC, Mok TS. Targeted therapy: an evolving world of lung cancer. Respirology. 2011; 16:13–21. https://doi.org/10.1111/j.1440-1843.2010.01821.x [PubMed]
- 2. Zhou C. Lung cancer molecular epidemiology in China: recent trends. Transl Lung Cancer Res. 2014; 3:270–9. https://doi.org/10.3978/j.issn.2218-6751.2014.09.01 [PubMed]
- 3. McGuire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv Nutr. 2016; 7:418–9. https://doi.org/10.3945/an.116.012211 [PubMed]
- 4. Almodovar T, Teixeira E, Barroso A, Soares M, Queiroga HJ, Cavaco-Silva J, Barata F. Elderly patients with advanced NSCLC: The value of geriatric evaluation and the feasibility of CGA alternatives in predicting chemotherapy toxicity. Pulmonology. 2019; 25:40–50. https://doi.org/10.1016/j.pulmoe.2018.07.004 [PubMed]
- 5. Custodio A, Méndez M, Provencio M. Targeted therapies for advanced non-small-cell lung cancer: current status and future implications. Cancer Treat Rev. 2012; 38:36–53. https://doi.org/10.1016/j.ctrv.2011.04.001 [PubMed]
- 6. Tartari F, Santoni M, Burattini L, Mazzanti P, Onofri A, Berardi R. Economic sustainability of anti-PD-1 agents nivolumab and pembrolizumab in cancer patients: Recent insights and future challenges. Cancer Treat Rev. 2016; 48:20–4. https://doi.org/10.1016/j.ctrv.2016.06.002 [PubMed]
- 7. Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O'Flaherty JD, Fennell DA, Richard D, O'Leary JJ, O'Byrne KJ. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS One. 2013; 8:e54193. https://doi.org/10.1371/journal.pone.0054193 [PubMed]
- 8. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016; 23:369–79. https://doi.org/10.1038/cdd.2015.158 [PubMed]
- 9. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016; 73:2195–209. https://doi.org/10.1007/s00018-016-2194-1 [PubMed]
- 10. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, Zhang Q, Lin D, Ge S, Bai M, Wang X, Zhang L, Li H, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020; 19:43. https://doi.org/10.1186/s12943-020-01168-8 [PubMed]
- 11. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017; 11:254–62. https://doi.org/10.1016/j.redox.2016.12.010 [PubMed]
- 12. Zhang J, Liang X, Li J, Yin H, Liu F, Hu C, Li L. Apigenin Attenuates Acetaminophen-Induced Hepatotoxicity by Activating AMP-Activated Protein Kinase/Carnitine Palmitoyltransferase I Pathway. Front Pharmacol. 2020; 11:549057. https://doi.org/10.3389/fphar.2020.549057 [PubMed]
- 13. Hu C, Chen Y, Cao Y, Jia Y, Zhang J. Metabolomics analysis reveals the protective effect of quercetin-3-O-galactoside (Hyperoside) on liver injury in mice induced by acetaminophen. J Food Biochem. 2020. [Epub ahead of print]. https://doi.org/10.1111/jfbc.13420 [PubMed]
- 14. Zhao L, Zhang J, Pan L, Chen L, Wang Y, Liu X, You L, Jia Y, Hu C. Protective effect of 7,3',4'-flavon-3-ol (fisetin) on acetaminophen-induced hepatotoxicity in vitro and in vivo. Phytomedicine. 2019; 58:152865. https://doi.org/10.1016/j.phymed.2019.152865 [PubMed]
- 15. Keyhanmanesh R, Saadat S, Mohammadi M, Shahbazfar AA, Fallahi M. The Protective Effect of α-Hederin, the Active Constituent of Nigella sativa, on Lung Inflammation and Blood Cytokines in Ovalbumin Sensitized Guinea Pigs. Phytother Res. 2015; 29:1761–7. https://doi.org/10.1002/ptr.5429 [PubMed]
- 16. Wolf A, Gosens R, Meurs H, Häberlein H. Pre-treatment with α-hederin increases β-adrenoceptor mediated relaxation of airway smooth muscle. Phytomedicine. 2011; 18:214–8. https://doi.org/10.1016/j.phymed.2010.05.010 [PubMed]
- 17. Mendel M, Chłopecka M, Dziekan N, Karlik W, Wiechetek M. Participation of extracellular calcium in α-hederin-induced contractions of rat isolated stomach strips. J Ethnopharmacol. 2013; 146:423–6. https://doi.org/10.1016/j.jep.2012.12.023 [PubMed]
- 18. Lorent J, Lins L, Domenech Ò, Quetin-Leclercq J, Brasseur R, Mingeot-Leclercq MP. Domain formation and permeabilization induced by the saponin α-hederin and its aglycone hederagenin in a cholesterol-containing bilayer. Langmuir. 2014; 30:4556–69. https://doi.org/10.1021/la4049902 [PubMed]
- 19. Rooney S, Ryan MF. Modes of action of alpha-hederin and thymoquinone, active constituents of Nigella sativa, against HEp-2 cancer cells. Anticancer Res. 2005; 25:4255–9. [PubMed]
- 20. Zhan Y, Wang K, Li Q, Zou Y, Chen B, Gong Q, Ho HI, Yin T, Zhang F, Lu Y, Wu W, Zhang Y, Tan Y, et al. The Novel Autophagy Inhibitor Alpha-Hederin Promoted Paclitaxel Cytotoxicity by Increasing Reactive Oxygen Species Accumulation in Non-Small Cell Lung Cancer Cells. Int J Mol Sci. 2018; 19:3221. https://doi.org/10.3390/ijms19103221 [PubMed]
- 21. Danloy S, Quetin-Leclercq J, Coucke P, De Pauw-Gillet MC, Elias R, Balansard G, Angenot L, Bassleer R. Effects of alpha-hederin, a saponin extracted from Hedera helix, on cells cultured in vitro. Planta Med. 1994; 60:45–9. https://doi.org/10.1055/s-2006-959406 [PubMed]
- 22. Sun D, Shen W, Zhang F, Fan H, Xu C, Li L, Tan J, Miao Y, Zhang H, Yang Y, Cheng H. α-Hederin inhibits interleukin 6-induced epithelial-to-mesenchymal transition associated with disruption of JAK2/STAT3 signaling in colon cancer cells. Biomed Pharmacother. 2018; 101:107–14. https://doi.org/10.1016/j.biopha.2018.02.062 [PubMed]
- 23. Cheng L, Xia TS, Wang YF, Zhou W, Liang XQ, Xue JQ, Shi L, Wang Y, Ding Q, Wang M. The anticancer effect and mechanism of α-hederin on breast cancer cells. Int J Oncol. 2014; 45:757–63. https://doi.org/10.3892/ijo.2014.2449 [PubMed]
- 24. Li J, Wu DD, Zhang JX, Wang J, Ma JJ, Hu X, Dong WG. Mitochondrial pathway mediated by reactive oxygen species involvement in α-hederin-induced apoptosis in hepatocellular carcinoma cells. World J Gastroenterol. 2018; 24:1901–10. https://doi.org/10.3748/wjg.v24.i17.1901 [PubMed]
- 25. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015; 520:57–62. https://doi.org/10.1038/nature14344 [PubMed]
- 26. Kashii T, Mizushima Y, Monno S, Nakagawa K, Kobayashi M. Gene analysis of K-, H-ras, p53, and retinoblastoma susceptibility genes in human lung cancer cell lines by the polymerase chain reaction/single-strand conformation polymorphism method. J Cancer Res Clin Oncol. 1994; 120:143–8. https://doi.org/10.1007/BF01202192 [PubMed]
- 27. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, Ding HF, Zhang J, Wang H, Chen X, Yan C. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. 2020; 27:662–75. https://doi.org/10.1038/s41418-019-0380-z [PubMed]
- 28. Jin M, Shi C, Li T, Wu Y, Hu C, Huang G. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4-induced destruction of the glutathione redox system. Biomed Pharmacother. 2020; 129:110282. https://doi.org/10.1016/j.biopha.2020.110282 [PubMed]
- 29. Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett. 2020; 19:323–33. https://doi.org/10.3892/ol.2019.11066 [PubMed]
- 30. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010; 140:900–17. https://doi.org/10.1016/j.cell.2010.02.034 [PubMed]
- 31. Zieba BJ, Artamonov MV, Jin L, Momotani K, Ho R, Franke AS, Neppl RL, Stevenson AS, Khromov AS, Chrzanowska-Wodnicka M, Somlyo AV. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J Biol Chem. 2011; 286:16681–92. https://doi.org/10.1074/jbc.M110.205062 [PubMed]
- 32. Fu D, Wang C, Yu L, Yu R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol Biol Lett. 2021; 26:26. https://doi.org/10.1186/s11658-021-00271-y [PubMed]
- 33. Fei Z, Lijuan Y, Jing Z, Xi Y, Yuefen P, Shuwen H. Molecular characteristics associated with ferroptosis in hepatocellular carcinoma progression. Hum Cell. 2021; 34:177–86. https://doi.org/10.1007/s13577-020-00431-w [PubMed]
- 34. Wang Y, Quan F, Cao Q, Lin Y, Yue C, Bi R, Cui X, Yang H, Yang Y, Birnbaumer L, Li X, Gao X. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res. 2020; 28:231–43. https://doi.org/10.1016/j.jare.2020.07.007 [PubMed]
- 35. Ahrendt SA, Hu Y, Buta M, McDermott MP, Benoit N, Yang SC, Wu L, Sidransky D. p53 mutations and survival in stage I non-small-cell lung cancer: results of a prospective study. J Natl Cancer Inst. 2003; 95:961–70. https://doi.org/10.1093/jnci/95.13.961 [PubMed]
- 36. Takahashi RU, Prieto-Vila M, Hironaka A, Ochiya T. The role of extracellular vesicle microRNAs in cancer biology. Clin Chem Lab Med. 2017; 55:648–56. https://doi.org/10.1515/cclm-2016-0708 [PubMed]
- 37. Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021; 276:119399. https://doi.org/10.1016/j.lfs.2021.119399 [PubMed]
- 38. Min IM, Pietramaggiori G, Kim FS, Passegué E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell. 2008; 2:380–91. https://doi.org/10.1016/j.stem.2008.01.015 [PubMed]
- 39. Chen DG, Zhu B, Lv SQ, Zhu H, Tang J, Huang C, Li Q, Zhou P, Wang DL, Li GH. Inhibition of EGR1 inhibits glioma proliferation by targeting CCND1 promoter. J Exp Clin Cancer Res. 2017; 36:186. https://doi.org/10.1186/s13046-017-0656-4 [PubMed]
- 40. Li L, Ameri AH, Wang S, Jansson KH, Casey OM, Yang Q, Beshiri ML, Fang L, Lake RG, Agarwal S, Alilin AN, Xu W, Yin J, Kelly K. EGR1 regulates angiogenic and osteoclastogenic factors in prostate cancer and promotes metastasis. Oncogene. 2019; 38:6241–55. https://doi.org/10.1038/s41388-019-0873-8 [PubMed]
- 41. Liu HT, Liu S, Liu L, Ma RR, Gao P. EGR1-Mediated Transcription of lncRNA-HNF1A-AS1 Promotes Cell-Cycle Progression in Gastric Cancer. Cancer Res. 2018; 78:5877–90. https://doi.org/10.1158/0008-5472.CAN-18-1011 [PubMed]
- 42. Wang B, Guo H, Yu H, Chen Y, Xu H, Zhao G. The Role of the Transcription Factor EGR1 in Cancer. Front Oncol. 2021; 11:642547. https://doi.org/10.3389/fonc.2021.642547 [PubMed]
- 43. Wang W, Zhou D, Shi X, Tang C, Xie X, Tu J, Ge Q, Lu Z. Global Egr1-miRNAs binding analysis in PMA-induced K562 cells using ChIP-Seq. J Biomed Biotechnol. 2010; 2010:867517. https://doi.org/10.1155/2010/867517 [PubMed]
- 44. He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu LZ, Jiang BH. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy. 2015; 11:373–84. https://doi.org/10.1080/15548627.2015.1009781 [PubMed]