



Introduction
Alzheimer’s disease (AD), a progressive neurodegenerative disease, is clinically characterized by decline in cognitive function [1]. Nowadays, the AD mechanism is still unknown. Extracellular senile plaques (SPs), intracellular neurofibrillary tangles (NFTs), and massive loss of neurons are primary hallmarks in AD patients’ brains [2–4]. SPs are formed by deposits of Amyloid beta (Aβ) peptide [5, 6]. Hyperphosphorylated tau aggregates and forms NFTs in the involved neurons [7]. Abnormal hyperphosphorylated tau is due to the overactivation of protein kinases or/and the inactivation of phosphates. SPs and NFTs all lead to structural damage of neurons and synapses, resulting in neuronal loose, ultimately decreased memory and learning of AD [7–9].
AD is mainly sporadic, and the pathogenesis is very complex [10]. Although scientists have made a great number of efforts, there has yet to be an ideal drug to cure or simply stop the development of AD. Gypenosides (GPs) have been used in Chinese folk medicine for many years. Accumulating data have shown that GPs possess neuroprotective effects, including anti-inflammation [11], antioxidative stress [12], and antineuronal apoptosis effects [13]. Moreover, GPs treatment has been a therapeutic strategy in stroke and traumatic brain injury in China [14–16]. We speculated that maybe GPs alleviate the symptoms of AD and slow down the progression. To explore this hypothesis, we intended to use Gypenoside IX (GP for short name in our experiments), one of the main extracts from Gynostemma to treat AD cell and animal models.
Staurosporine (STP) is commonly used as an inducer of apoptosis [17, 18]. Our previous study has shown that stereotactic injections in the hippocampus of rats with STP induced neuronal apoptosis and caused an increased tau phosphorylation [19], finally leading to cognitive impairments [20–22]. Therefore, in the present study, primary hippocampal neurons treated with STP were used as AD cell models, and rats injected with STP in bilateral hippocampi as AD animal models. Then, we investigated the effects of GP on STP treated cell and rat models. We found that GP rescues STP induced AD pathologies and cognitive impairments. Moreover, GP treatment recovered AKT/GSK-3β signaling pathway in STP cell and rats. Hence, our results reveal GP as a promising drug for reducing the symptoms of AD.
Results
GP attenuated cytotoxicity and apoptosis in primary hippocampal neurons treated with STP
To detect the toxic effects of STP on cells and select the appropriate concentration for cytotoxicity model, we treated primary hippocampal neurons with different concentrations of STP (Figure 1A). It showed that STP led to a decrease in cell viability. When the STP concentration was 0.4 μM, the cell viability approximately decreased by 50%. On the basis of previous report [23], we chose 0.4 μM STP as ideal concentration to make cell apoptosis in the following experiments. To test the effect of GP on neuronal toxicity, the primary hippocampal neurons were incubated with GP with or without STP for 24 h (Figure 1B). The CCK8 assay showed that 5 μM and 10 μM GP rescued cell viability decreased by STP, while GP had no obvious cytotoxic effect on hippocampal neurons. The result from TUNEL assay also supported that GP significantly attenuated STP-induced cell death (Figure 1C, 1D). We further checked the alterations of apoptotic proteins, and found that STP activated proapoptotic Bax and cleaved-caspase-3 [24, 25]. Meanwhile anti-apoptotic protein Bcl-2 was markedly decreased. Interestingly, GP10 recovered the expression levels of those above apoptotic proteins (Figure 1E–1H). Taking together, these findings showed that GP has neuroprotective effect on the neurons with reduced survival caused by STP, and reduces their apoptosis by restoring the expression of cleaved-caspase-3, Bax and Bcl-2.
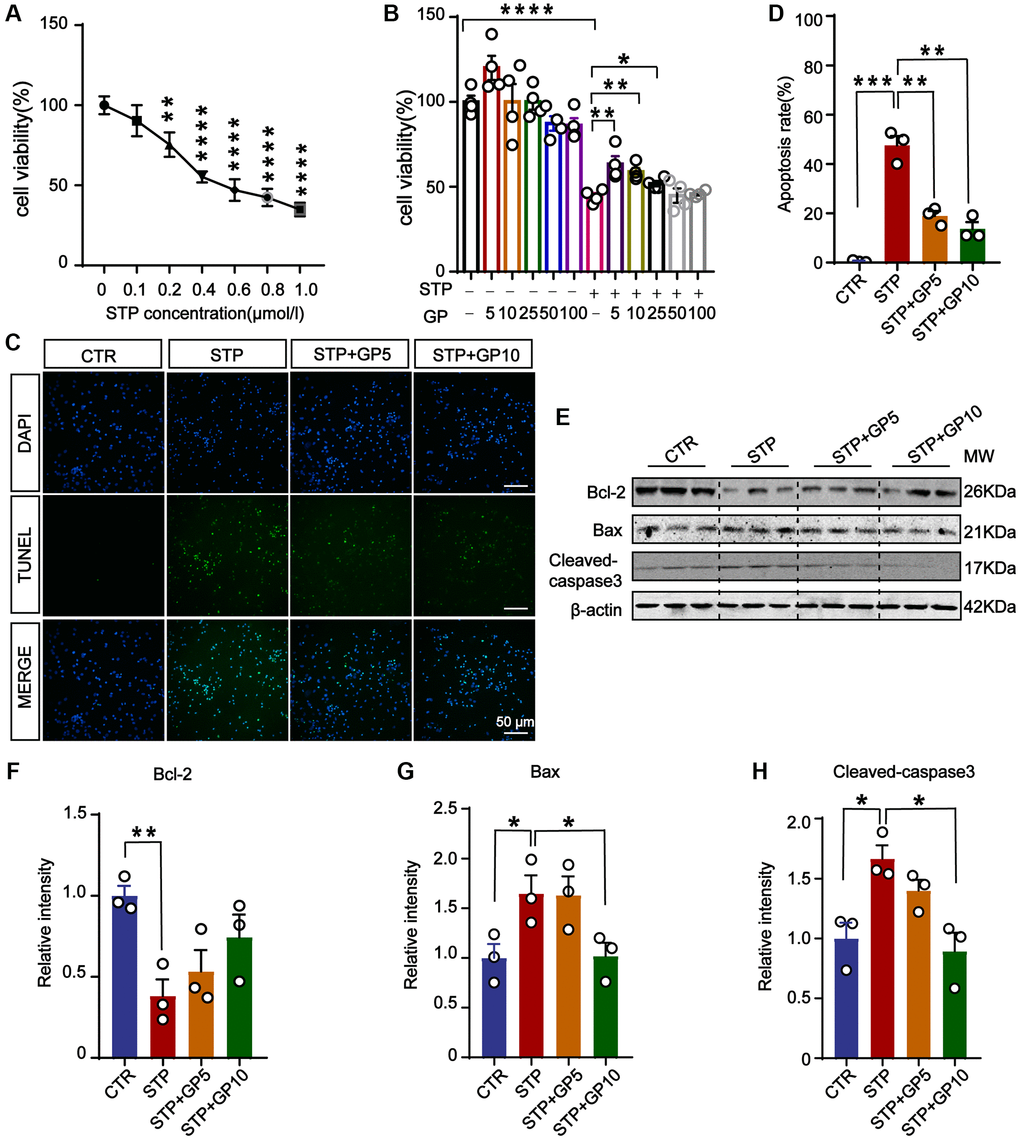
Figure 1. GP attenuated STP-induced cytotoxicity and inhibited apoptosis in primary hippocampal neurons. (A) STP inhibited cell viability in a dose-dependent manner. 0.4 μM of STP caused a decline of 50% in cell viability. n = 4. (B) GP rescued the cell viability with different concentrations (5–25 μM). n = 4. (C) TUNEL assay verified the inhibition of STP-induced (0.4 μM) apoptosis by GP (5 μM and 10 μM). Scale bar = 50 μm. (D) Quantification of the TUNEL/DAPI using ImageJ software. (E–H) Western blots and quantification of the relative protein expression levels (Bcl-2, Bax, and cleaved-caspase-3) after normalization to the β-actin signal. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
GP attenuated STP-induced tau phosphorylation and reduced Aβ production in primary hippocampus neurons
To investigate the effect of GP on tau phosphorylation, Western blot was carried out (Figure 2A). We found that tau phosphorylation levels at pS199, pS202/ pT205(AT8), pT231, pS262, pS396, and pS404 were significantly increased in STP-treated primary hippocampal neurons, and GP treatment effectively blocked STP-induced tau hyperphosphorylation at all above sites except pS404 (Figure 2B–2H). Together with previous studies showing GSK-3β association with tau phosphorylation at these sites [26, 27], we speculate that GP treatment probably alleviates STP-related tau phosphorylation by activation of GSK-3β.
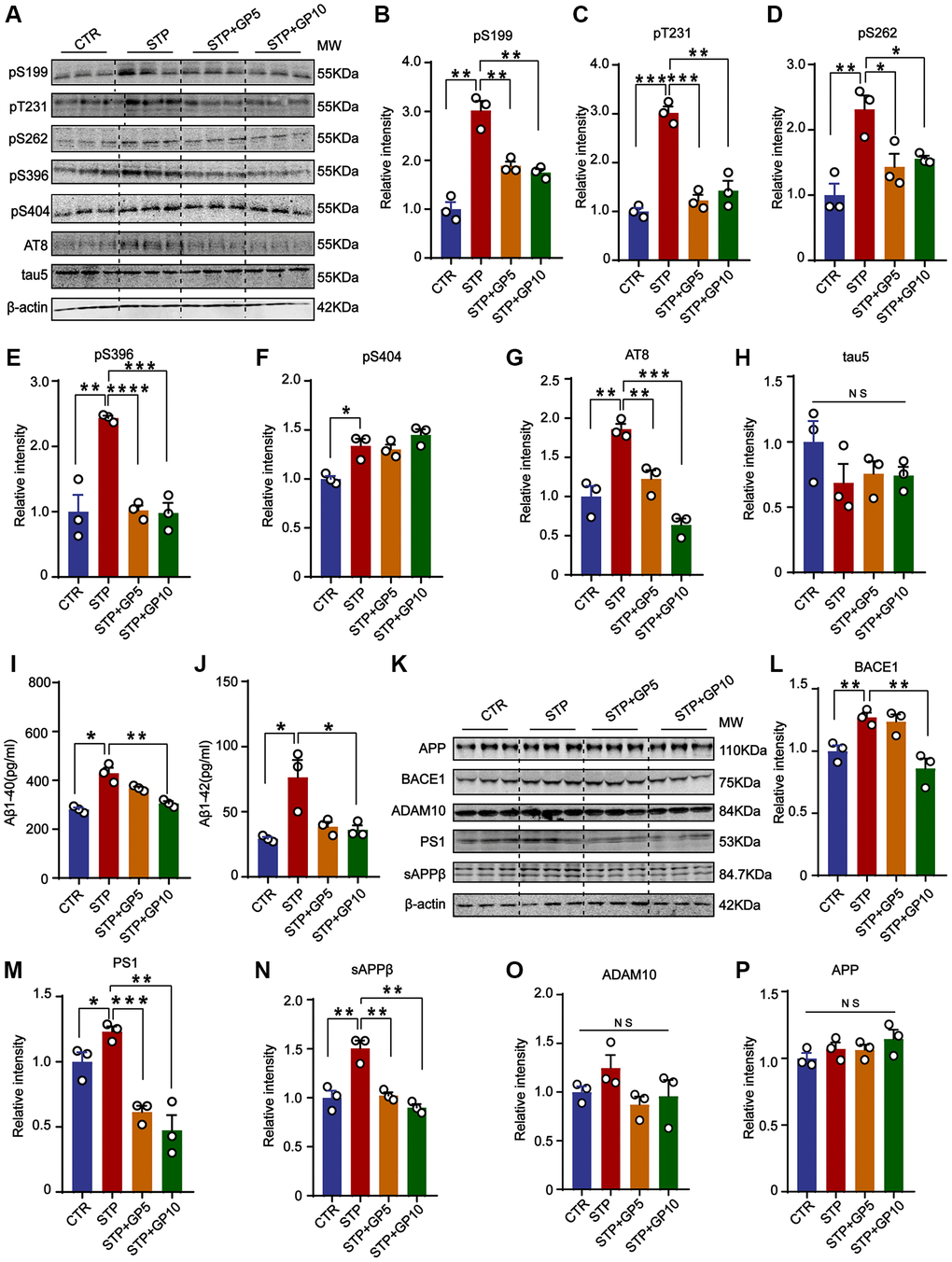
Figure 2. GP attenuated STP-induced tau phosphorylation and reduced Aβ generation in primary hippocampus neurons. (A) Western blot for tau phosphorylation levels at the sites of pS199, pT231, pS262, pS396, pS404, AT8 and total tau (Tau-5) in primary hippocampus neurons. (B–H) Quantification of the relative protein expression levels (pS199, pT231, pS262, pS396, pS404, AT8 and Tau-5) after normalization to the β-actin signal. (I, J) ELISA assay of the levels of Aβ40 and Aβ42 in rat primary hippocampal neurons. (K) Western blots for APP, BACE1, ADAM10, PS1 and sAPPβ in rat primary hippocampal neurons. (L–P) Quantification of the relative protein levels (BACE1, PS1, sAPPβ, ADAM10 and APP) after normalization to the β-actin signal. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Abbreviation: NS: no significance.
To further study the effect of GP on Aβ production, we detected the levels of Aβ40 and Aβ42. The ELISA showed that both Aβ40 and Aβ42 were obviously increased in STP group, and GP treatment restored the changes (Figure 2I, 2J). Aβ peptides are generated from APP cleavage by β-secretase (BACE1) and γ-secretase [28]. As the catalytic subunit of γ-secretase, Presenilin-1 (PS1) mediates cleavage of type I transmembrane proteins APP [29]. ADAM10 (alpha-secretase of APP) is mainly responsible for a non-toxic Aβ peptide generation [30]. Next, we measured the levels of BACE1, AMAD10, PS1, and sAPPβ (Figure 2K), and found that the levels of BACE1, PS1 and sAPPβ were significantly increased by STP, but not in GP treated primary hippocampus neurons. Only 5 μM GP did not inhibit the increasing of BACE1 level by STP (Figure 2L–2N). Meanwhile, the levels of APP and ADAM10 among these groups had no change (Figure 2O, 2P). The data showed that GP attenuated Aβ generation through partially regulating BACE1 and γ-secretase expression level in STP treated cell models.
GP restored the activity of AKT/GSK-3β signaling axis
Many previous researches revealed that AKT/GSK-3β signaling pathway had important functions related with apoptosis, neurogenesis, mitophagy, and synaptic plasticity in the central nervous system [31, 32]. Accumulating data reported that many natural products protect synaptic function, and inactive microglia through the AKT/GSK-3β axis in AD model [33, 34]. We here suppose that GP-mediated neuroprotection might be associated with AKT/GSK-3β pathway. In hippocampal neurons, we detected the active forms of AKT (phosphorylated AKT at Thr308 and Ser473) [35], and found that 5 μM and 10 μM, both concentration of GP rescued the STP-induced decline of p-AKT (473), but not p-AKT (308) (Figure 3A–3C). GSK-3β, a major kinase of tau [36], and also a regulator of Aβ generation [37], is inactive by phosphorylated at Ser9 [38]. Consistent with the change of p-AKT, STP promoted GSK-3β activity by decreasing the level of Ser9 phosphorylation, which was blocked by GP (Figure 3A, 3D). This result suggested that GP restored the Akt/GSK-3β axis and might be associated with an anti-apoptotic effect.
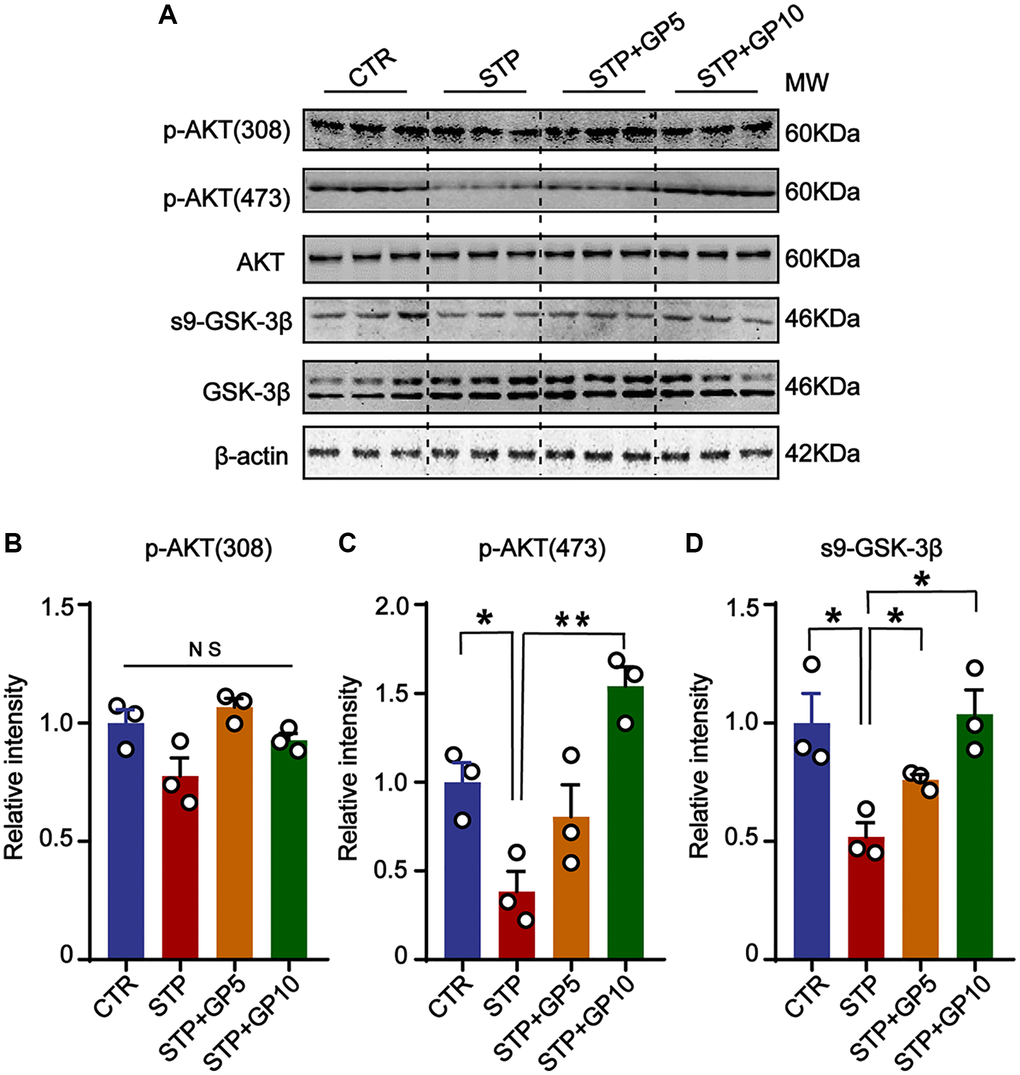
Figure 3. GP restored the activity of AKT/GSK-3β signaling axis. (A) Western blots for AKT/GSK-3β pathway-related proteins. (B–D) Quantification of the relative protein levels of p-AKT (308), p-AKT (473) and s9-GSK-3β after normalization to the β-actin signal. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3. *P < 0.05, **P < 0.01. Abbreviation: NS: no significance.
Effects of GP on neuronal dendrites and synaptic proteins in primary hippocampal neurons
The destruction of neuronal structure will affect the structure and function of synapses, and ultimately destroy neural networks. Next, to investigate whether GP could ameliorate synaptic damage induced by STP, we examined the morphology of cultured hippocampal neurons and the levels of synaptic proteins. Immunofluorescence staining and Sholl analysis showed that MAP-2 labeled neuronal dendrites and branches significantly decreased in STP groups, while GP treatment obviously alleviated the damage of neuronal dendrites in STP treated neurons (Figure 4A, 4B). Next, we detected the synaptophysin, PSD-95, GluR2 (Figure 4C). The levels of PSD-95 and GluR2 in STP group were markedly decreased when compared with control. But GP obviously elevated the levels of these proteins in STP+GP groups (Figure 4D, 4E), while the levels of synaptophysin proteins was unchanged (Figure 4F). This finding suggested that GP protected STP-induced synaptic dysfunction.
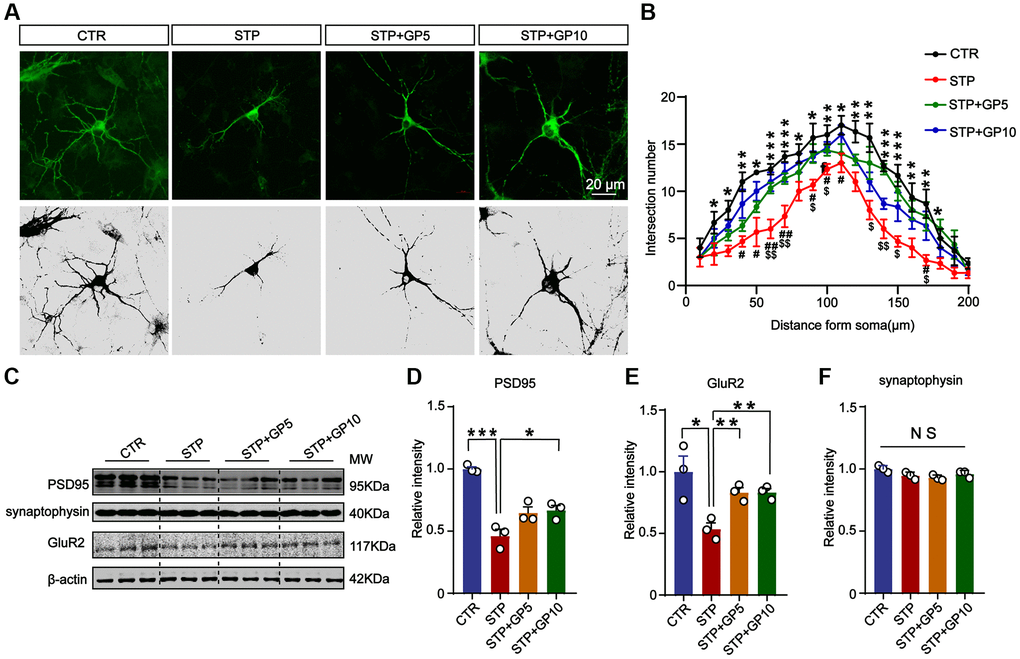
Figure 4. Effects of GP on neuronal dendrites and synaptic plasticity-related proteins in the hippocampus neurons. (A) Representative images of immunofluorescence staining for MAP-2 labeled neuronal dendrites; scale bar = 20 μm. (B) Sholl analysis of dendritic arborization of neurons in (A). (C) Representative Western blots of PSD-95, synaptophysin, and GluR2. (D–F) Quantitative analysis of PSD-95, GluR2, and synaptophysin after normalization to the β-actin signal. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3. *P < 0.05, ***P < 0.001 vs. control group, NS, no significance. #P < 0.05, ##P < 0.01 vs. GP10, $P < 0.05, $$P < 0.01 vs. GP5.
GP improves learning and memory impairment in STP-treated rats
It was demonstrated that GP could resist or reduce AD pathologies and synaptic dysfunction caused by STP in primary neurons. However, whether STP induces these harmful effects, or even leads to cognitive defects in vivo, and whether GP blocks them are unclear. To address these points, we designed the animal experiments, dividing the rats in four groups according to the schematic diagram in Figure 5A (see details in material and methods part).
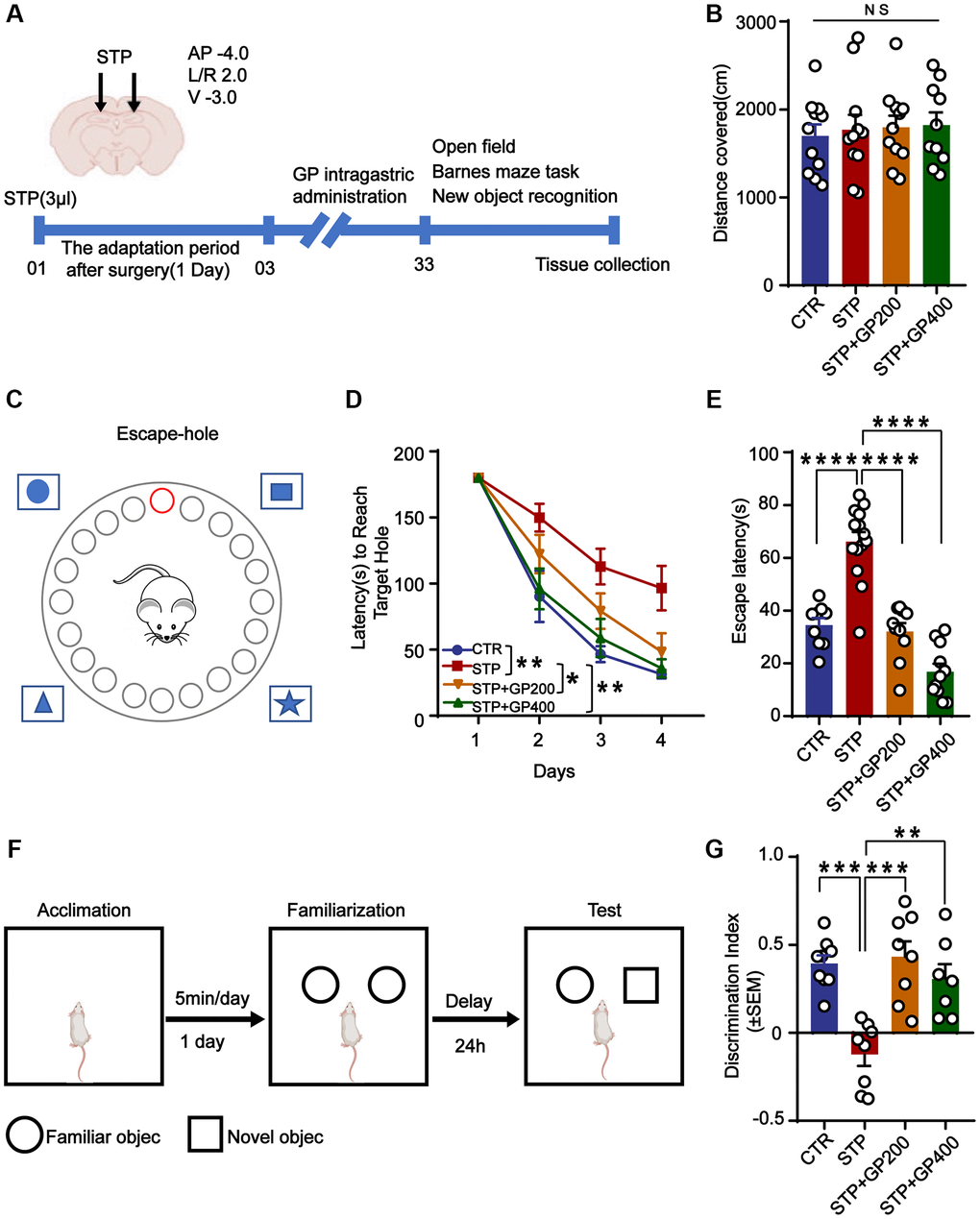
Figure 5. GP improves learning and memory impairment in STP-treated rats. (A) Study design timeline performed in the study. (B) The total distance was analyzed in the open-field test. (C–D) Escape latency in 4 days was analyzed in the Barnes maze test. (E) Escape latency on test day 5 was analyzed in the Barnes maze test. (F, G) The novel object recognition (NOR) test showed the measured recognition index of the new object within 24 hours. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA. *p < 0.05, **p < 0.01, ***P < 0.001 and ****P < 0.0001, Abbreviation: NS: no significance. n represents the number of rats in each group. For (B), Control n = 11, STP n = 11, STP+GP200 n = 11, STP+GP400 n = 10. For (D, E), Control n = 9, STP n = 14, STP+GP200 n = 10, STP+GP400 n = 11. For (B), Control n = 9, STP n = 8, STP+GP200 n = 8, STP+GP400 n = 7.
The open-field test showed no significance difference in total distance among these groups (Figure 5B), implying that neither STP nor GP affected locomotion activity. In the Barnes maze (the principle and method being shown in Figure 5C) experiment, as the training days increased, the escape latency of rats in all groups decreased, indicating that all the rats got certain memories as practicing for more days. However, the escape latency of rats in the STP group was markedly higher compared to that of control. Whereas the escape latency of rats treated with STP+GP was significantly reduced. (Figure 5D). Furthermore, STP group showed obviously increased exploration times before finding the hidden box, which was significantly decreased in the GP administration group on test day 5 (Figure 5E). Next, we carried out NOR and found that the curiosity toward exploring new things of STP rats was markedly reduced compared to control. However, GP blocked the decreased time spent for exploration of new object by STP (Figure 5F, 5G). Together, the data suggested that GP ameliorated cognitive impairment in STP rats.
Effect of GP on neuronal apoptosis in STP rats
To investigate the effect of GP on apoptosis in vivo, we performed the TUNEL staining to test the apoptotic rate in the hippocampi of four groups. As shown in Figure 6A, 6B, in the brain slices of the STP group, the neuronal apoptotic rate was higher when compared to control, especially in the CA3 of hippocampus. Interestingly, both GP200 and GP400 significantly reduced the apoptosis. Moreover, STP treatment upregulated Bax and cleaved caspase-3, and downregulated Bcl-2, which were blocked by GP (Figure 6C–6F). These data further supported that GP might prevent from apoptosis and protect the brain neurons in STP-induced AD rats.
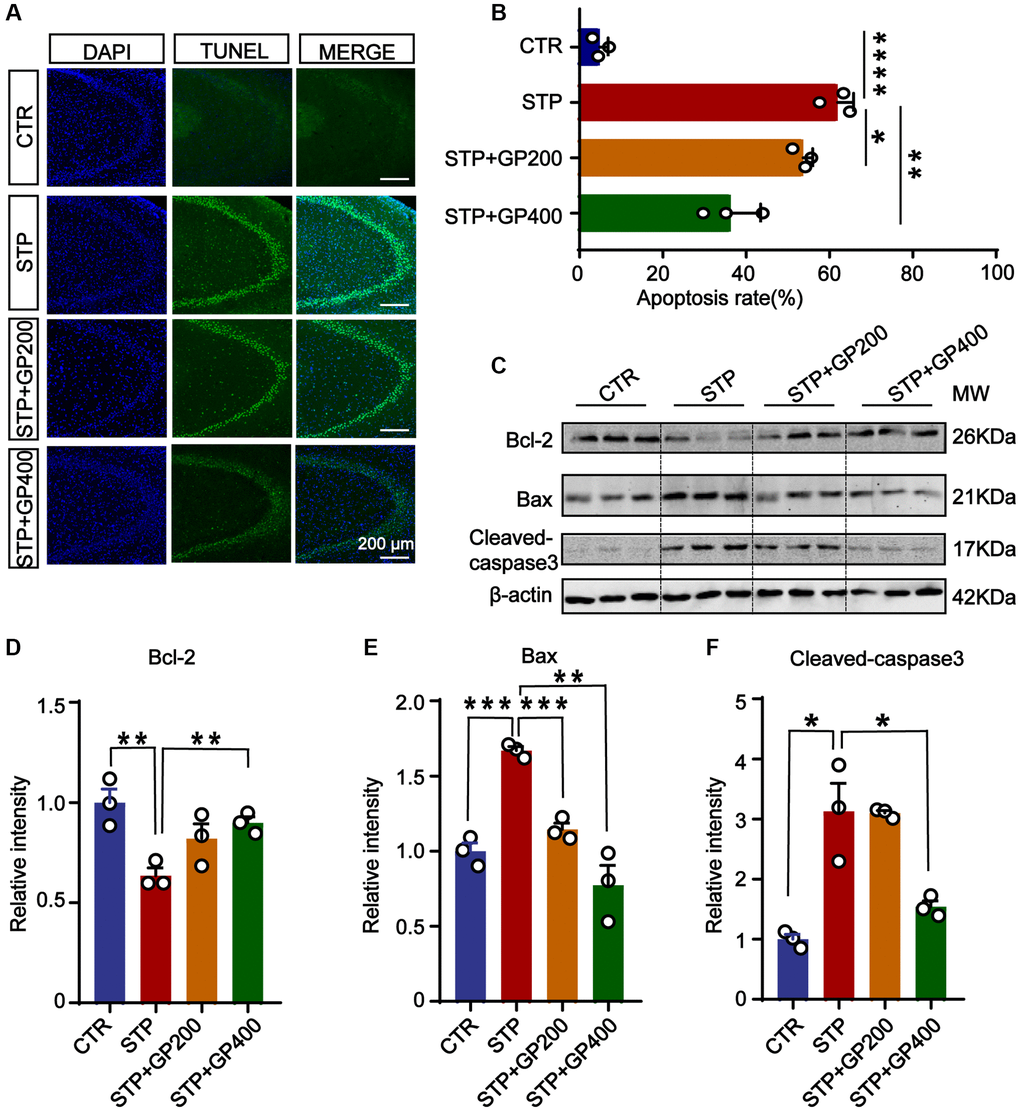
Figure 6. Effect of GP on neuronal apoptosis in the hippocampus of STP Rats. (A) Representative TUNEL-positive cells (green) in hippocampal CA3 region. Nuclei were stained with DAPI (in blue). Scale bar = 200 μm. (B) Quantification of the TUNEL/DAPI using ImageJ software. (C–F) Representative images and quantitative analysis of Western blotting for Bcl-2, Bax, and caspase-3 in the hippocampus after normalization to the β-actin signal. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3. *p < 0.05, **p < 0.01, ***P < 0.001 and ****P < 0.0001.
GP blocks tau hyperphosphorylation and Aβ overproduction in STP rats
Tau hyperphosphorylation and Aβ toxicity contribute to cognitive impairment in AD. In the primary hippocampal neurons experiment, we found that GP reduced tau phosphorylation levels and inhibited STP-induced Aβ overproduction (Figure 2). Thereby, we further detected tau phosphorylation in vivo. The results indicated that compared to that of control rats, tau phosphorylation level in the hippocampus of the STP rats was increased at Ser199 (pS199), Thr231 (pT231), Ser262 (pS262), and AT8 (Ser202, Thr205), but not at Ser396 (pS396) and Ser404 (pS404) site (with only an increasing trend). Supplementation with GP in STP rats effectively restored tau phosphorylation (Figure 7A–7H).
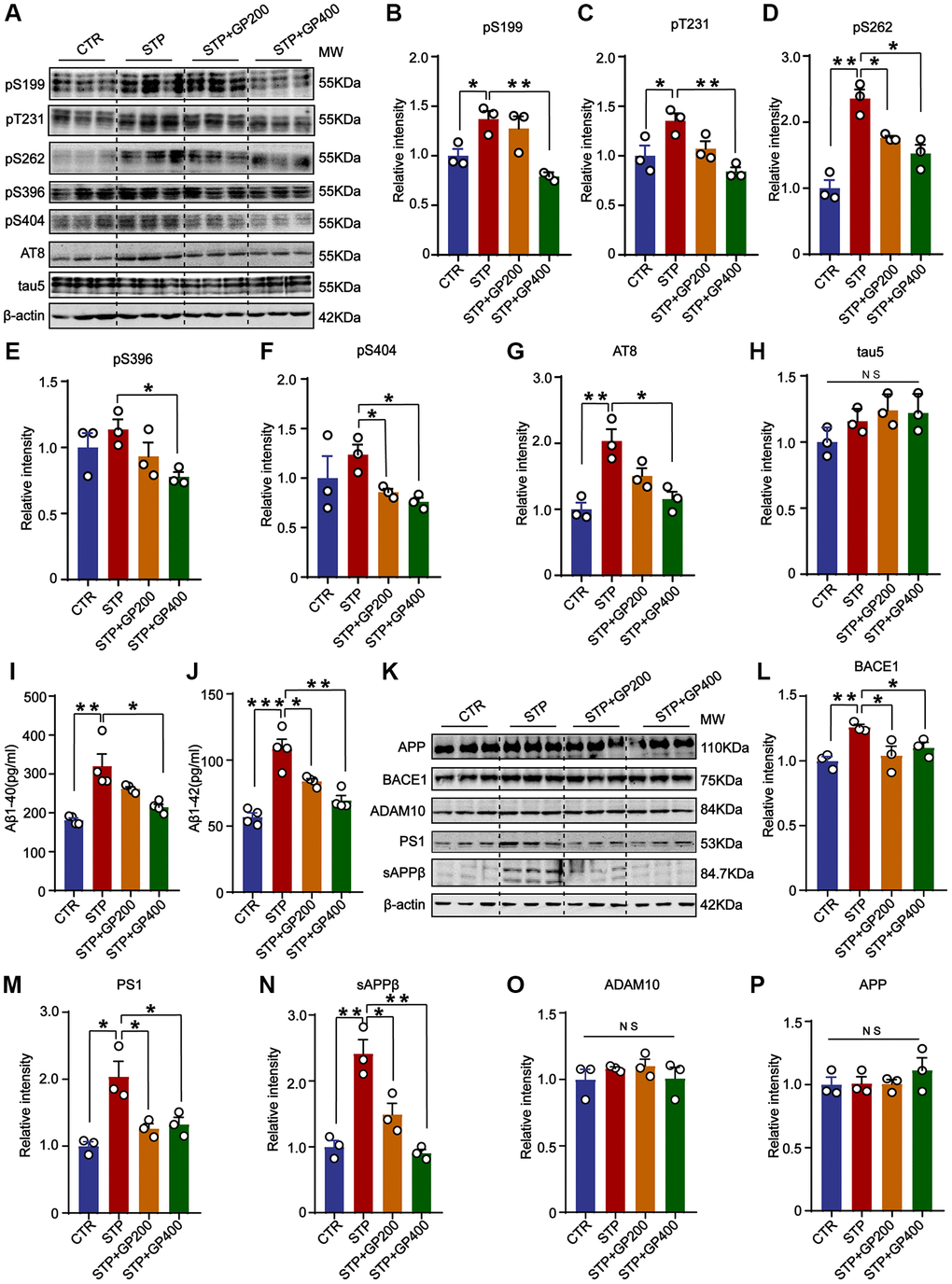
Figure 7. GP blocks tau hyperphosphorylation and Aβ overproduction in STP rats. (A) Western blot for tau phosphorylation levels at the sites of pS199, pT231, pS262, pS396, pS404, AT8 and total tau (tau-5) in rat hippocampus. (B–H) Quantification of the relative protein expression levels (pS199, pT231, pS262, pS396, pS404, AT8 and tau-5) after normalization to the β-actin signal. (I, J) ELISA assay of the levels of Aβ40 and Aβ42 in the rat hippocampus. (K) Western blots for APP, BACE1, ADAM10, PS1 and sAPPβ in rat hippocampus. (L–P) Quantification of the relative protein levels (BACE1, PS1, sAPPβ, ADAM10 and APP) after normalization to the β-actin signal. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3. *p < 0.05, **p < 0.01 and ***P < 0.001, Abbreviation: NS: no significance.
Next, we studied the effect of GP on APP metabolism in STP rat. Firstly, we carried out the ELISA and found that GP200 and GP400 decreased the Aβ40 and Aβ42 overproduction induced by STP (Figure 7I, 7J). Then, we investigated the possible reason why GP reduces Aβ production, we detected the impact of GP on APP processing and found that there was no significant increase in APP level in the hippocampus of STP rats compared to control (Figure 7K, 7P). The APP cleavage enzymes and cleavage fragments were detected in rat brain (Figure 7K). Compared to control, the protein levels of BACE1, PS1, and sAPPβ in the hippocampus of STP rats were significantly higher, which was blocked by GP treatment. However, ADAM10 was not differentially expressed in the hippocampus of the four groups of rats (Figure 7L–7O). These data further supported that GP significantly inhibited STP-induced tau hyperphosphorylation and Aβ overproduction.
GP reactivates AKT/GSK-3β pathway in STP rats
To further examine if GP affects AKT/GSK-3β activity in vivo, we detected p-AKT (308), p-AKT (473) and s9-GSK-3β expression in four groups. The levels of p-AKT (T308/S473) and s9-GSK-3β in STP rats were markedly decreased compared to that of control, while the expressions of p-AKT (T308/S473) and s9-GSK-3β were increased after treatment with GP (Figure 8A–8D). These finding suggested that the AKT/GSK-3β axis might be associated with the STP-related AD pathologies and cognitive impairments.
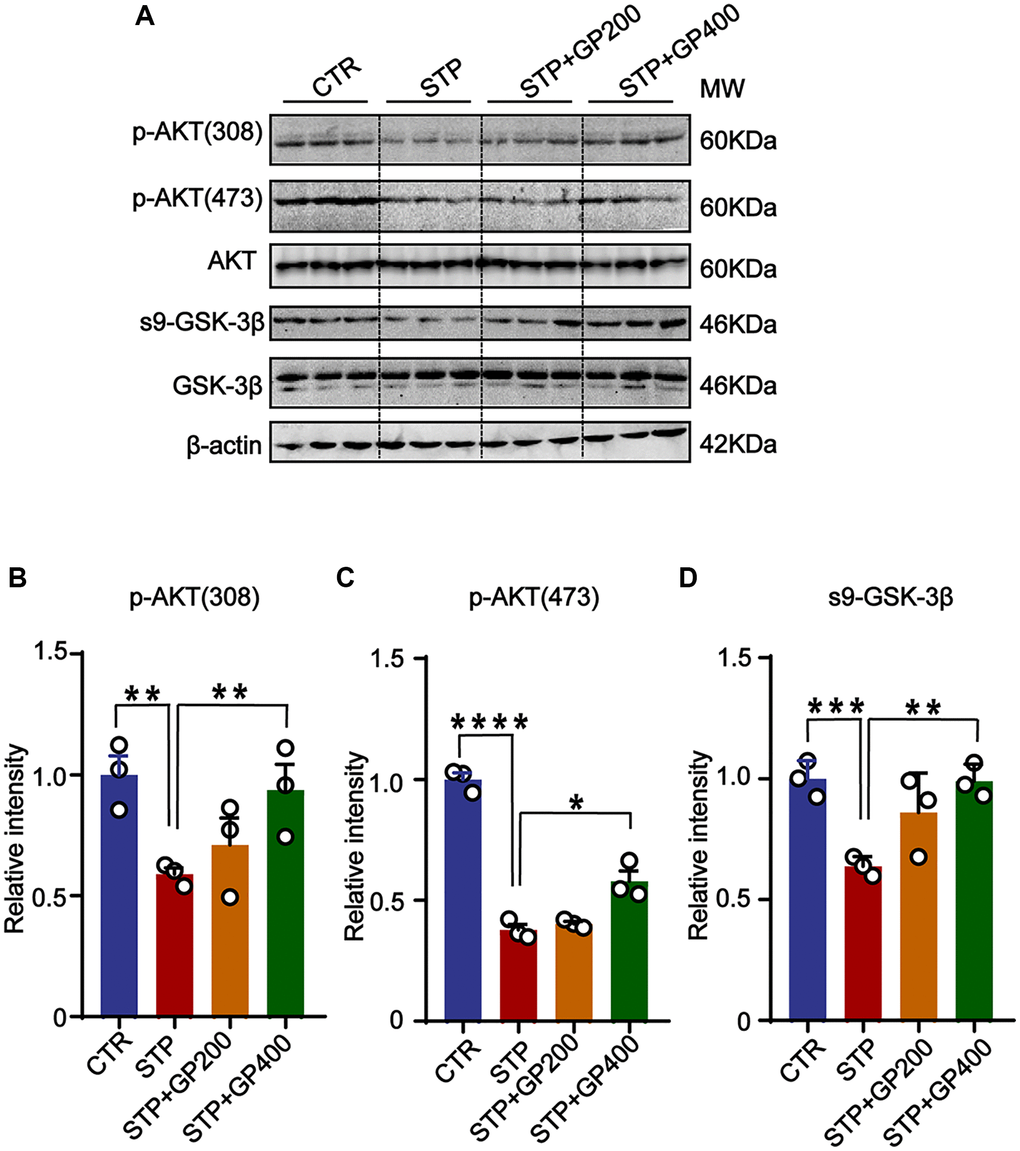
Figure 8. GP reactivates AKT/GSK-3β pathway in STP rats. (A) Western blots for AKT/GSK-3β pathway-related proteins. (B–D) Quantification of the relative protein levels (p-AKT (308), p-AKT (473), s9-GSK-3β) after normalization to the β-actin signal. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
GP recovers synaptic deficit in STP rats
Since synaptic deficit is a pathological phenotype in AD [39], we evaluated the neuron and dendritic spine (where synapses being formed) lose in four groups of rats. Firstly, in CA3 of hippocampus in STP group, neuronal loss increased revealed by Nissl staining (Figure 9A, 9B). Meanwhile, STP treatment led to dendritic spine loss in the same region, and GP200 or GP400 obviously attenuated the spine loss (Figure 9C, 9D). Moreover, the levels of synaptic proteins including PSD95, synaptophysin and GluR2 were reduced in STP rats after treatment with GP200 or GP400 (Figure 9E–9H). These findings indicated that GP rescued synaptic damage in STP rats through recovering dendritic morphology and postsynaptic related protein.
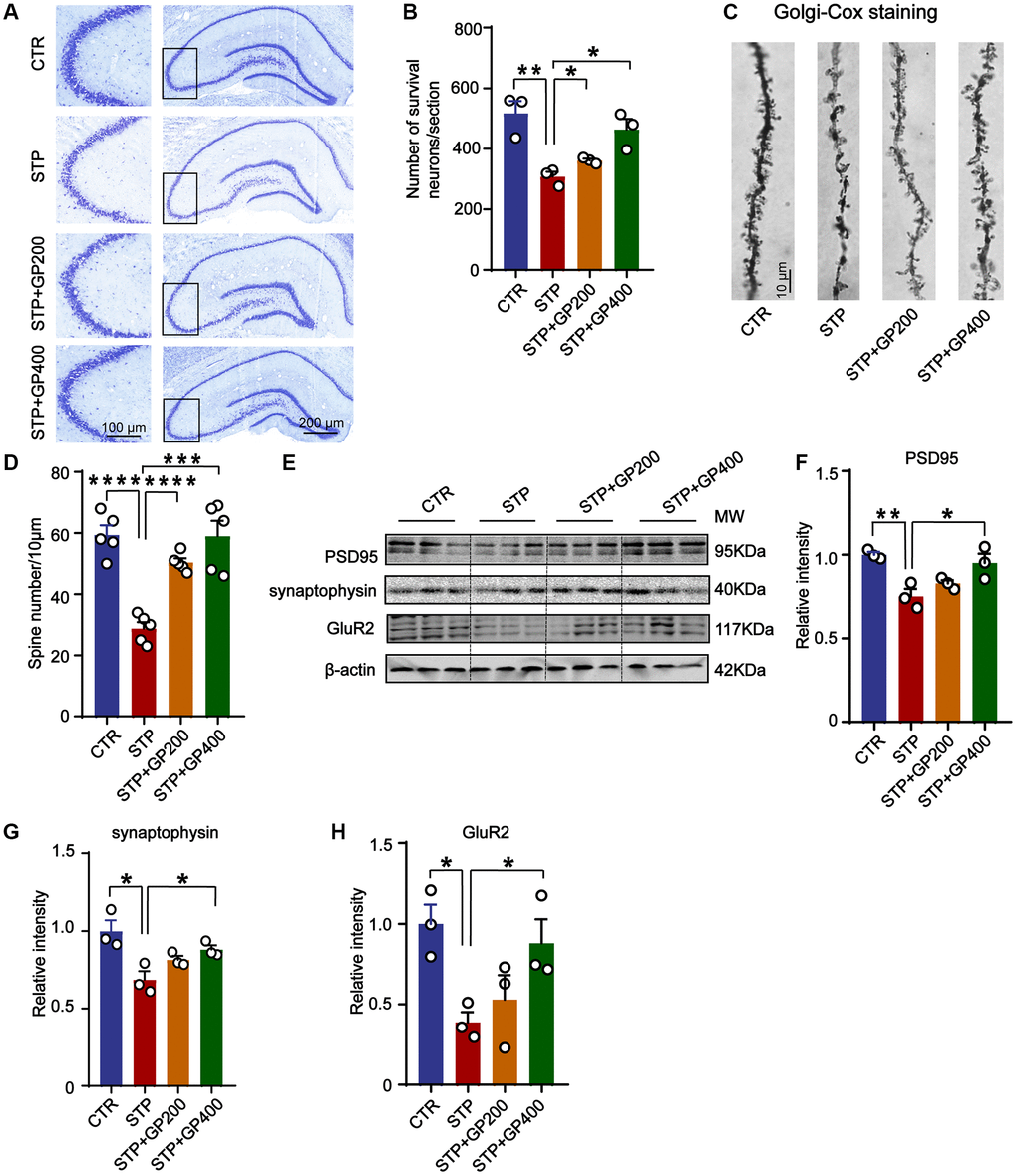
Figure 9. GP reverses dendritic spine loss and recovers postsynaptic proteins. (A, B) Representative Nissl staining images and the quantification of neuron number in the CA3 region of rat hippocampus, scale bar = 200 and 100 μm for low and high magnifications respectively. (C, D) Representative Golgi staining images and statistics analysis of spine number in the dentate gyrus region of rat hippocampus (scale bar, 10 μm). n = 5. (E–H) Western blots and quantitatively analysis for synaptic proteins (PSD95, GluR2 and synaptophysin) levels in rat hippocampus. Data represent mean ± SEM, p-value significance is calculated from one-way ANOVA, n = 3, *p < 0.05, **p < 0.01, ***P < 0.001 and ****P < 0.0001.
Discussion
Alzheimer’s disease is a most common neurodegenerative disease [1, 6, 40]. It is classified as familial or sporadic. Dominant familial or autosomal dominant inheritance accounts for 1–5% of the clinic cases. It is classified as early-onset (EOAD; age <65 years) and manifests as mutations in the PSEN1, PSEN2 or APP genes [41]. The characteristic pathological changes in Alzheimer’s disease include neuronal loss, extracellular deposition of Aβ plaques, and neuronal fiber tangles formed by intracellular tau protein hyperphosphorylation [42, 43]. The pathogenesis of AD is very complex, which leads to the slow development of drugs for its treatment. Several drugs targeting Aβ and phosphorylation of tau were discontinued due to poor efficacy [44, 45]. Using herbal medicine acts as an important therapeutic strategy in China. It may be hopeful to find useful agents from natural herbs to treat or stop the progression of AD. Gynostemma mainly distributed in China in Hunan, Hubei, Yunnan, Guangxi and other provinces, known as the “southern ginseng”, growing in the south of the Gynostemma medicinal content is relatively high, the folk said it is a magical “longevity herb”, which has been used widely for nervous system diseases [46]. The effect of Gypenosides extracted from Gynostemma in AD was explored by several researches. Some studies have reported that Gypenoside attenuates Aβ toxicity such as Aβ-induced inflammation, parallel autophagic and apoptotic cell death [47, 48]. By network pharmacology analysis, one work revealed that three compounds (GPM2, 7 and 9) in 13 active compounds of Gynostemma constructed network with eighteen intersecting targets of AD, in which HIF-1 signaling pathway and cytokine-cytokine receptor interaction were involved in the therapeutic effect [49]. These studies have identified the promising value of Gynostemma in the treatment of AD, but have not made it clear whether it can alleviate the major pathological changes and cognitive impairments in AD. Hence, in the current study we try to evaluate the effect of GP (Gypenoside IX, one of the main extracts from Gynostemma) on AD models.
Previous studies have found that STP can cause neuronal apoptosis, tau hyperphosphorylation, most important pathological characteristics in AD [19, 50, 51]. Therefore, in our experiments, we treated primary hippocampal neurons with STP and injected STP into rat hippocampus respectively, to mimic multiple AD pathological features simultaneously.
In the cell experiment, we found that STP significantly increased the apoptosis rate of primary hippocampal neurons through TUNEL method, and GP decreased the apoptosis rate. By Western blotting, it showed that GP reduced the increased Bax and cleaved caspase-3 caused by STP, but increased the decreased Bcl-2, further supporting that GP blocks STP-induced apoptosis. At the same time, STP treatment resulted in hyperphosphorylation of multiple sites of tau protein, increasing intracellular Aβ production and enzymes associated with APP cleavage, while GP treatment could obviously reverse these pathological changes. Consistent with previous studies reported that both tau hyperphosphorylation and apoptosis are associated with synaptic damage [52, 53], we also found that STP-induced tau hyperphosphorylation and apoptosis were accompanied with dendritic spine loss and synaptic proteins deficiency. Moreover, all STP-induced harmful effects were significantly mitigated by GP treatment in cultured primary neurons and rats. Behavioral tests also verified that GP improved the learning and memory in STP-caused AD rats. However, we found that GP+200 and GP+400 in vivo experiments could not completely reverse the neuronal apoptosis caused by STP, while the GP+400 animals took markedly shorter time to find the platform than the control group. We speculate two things might be relevant. The first is the complexity of drug interactions on higher concentration which should be clarified in the future study. The second possibility is that the loss of nerve cells due to apoptosis is not enough to cause cognitive dysfunction. Akt/GSK-3β pathway is closely to take part in many physiological processes. Dysregulation of this pathway has been implicated in various diseases, including cancer, neurodegenerative disorders, and metabolic disorders [54]. Akt phosphorylates GSK-3β at Serine 9 and in turn inactivates GSK-3β [54, 55]. Activation of GSK-3β is engaged in the regulation of amyloidogenic processing of APP, which generates Aβ [37, 56, 57]. As a therapeutic target, blocking GSK-3β attenuates the generation of Aβ peptides and its toxicity in neurons [58, 59]. As a tau kinase, the increase of GSK-3β in STP mice and rat promotes tau phosphorylation [19], and the blockage of GSK-3β attenuates tau phosphorylation [60, 61]. We here investigate the Akt/GSK-3β signaling axis, and found that both pS473-Akt and pS9-GSK-3β were significantly decreased in the brain of STP rats. GP treatment restored the levels of pS473-Akt and pS9-GSK-3β in STP-treated neurons and rats, supporting that GP attenuates STP-induced tau hyperphosphorylation and Aβ production might be associated with restoration of Akt/GSK-3β axis.
Conclusion
In summary, our data demonstrate that STP dysregulation of Akt/GSK-3β pathway is associated with apoptosis, tau hyperphosphorylation, Aβ overproduction, synaptic dysfunction and cognitive decline. Restoration of Akt/GSK-3β pathway by GP might correct AD-like pathological changes and ameliorate cognitive deficits. Our study provides a promising application of GP for AD therapeutic strategy.
Materials and Methods
Reagents
GP (Gynostemma Extract: Gypenoside IX, #CAS 80321-63-7, purity ≥99.92%) was provided by MedChemExpress (USA). STP is a cell inhibitor. It has been reported that STP can induce apoptosis at concentrations ranging from 0.2 to 1 μM [62, 63], which was provided by Abcam (ab146588, Cambridge, MA, USA). Table 1 lists the primary antibodies used in this study. IRDye 800CW-Conjugated secondary antibody (#ab216773, dilution 1:8000), used in Western blotting, was provided by Abcam.
Table 1. Primary antibodies.
| Antibodies | Specific | Type | Dilution | Source | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bcl-2 | Anti-Bcl-2 | pAb | 1:1000 for WB | ABclonal (A21592) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cleaved-caspase-3 | Anti-cleaved-caspase-3 | pAb | 1:1000 for WB | Cell Signaling (#9661) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bax | Anti-Bax | pAb | 1:1000 for WB | Abcam (A12009) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| p-AKT (473) | Anti-Phosphorylated AKT at Ser473 | pAb | 1:1000 for WB | ABclonal (AP1208) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| p-AKT (308) | Anti-Phosphorylated tau at Tyr 308 | mAb | 1:1000 for WB | Santa cruz (sc-271964) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AKT | Anti-Total AKT | mAb | 1:1000 for WB | Cell Signaling (#2920) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| s9-GSK-3β | Anti-PhosphorylatedGSK-3βat Ser9 | pAb | 1:1000 for WB | ABclonal (AP0039) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GSK-3β | Anti-Glycogen synthase kinase-3β | pAb | 1:1000 for WB | ABclonal (A11731) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pS199 | Anti-Phosphorylated tau at Ser199 | pAb | 1:1000 for WB | ThermoFisher (44-734G) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pT231 | Anti-Phosphorylated tau at Thr231 | pAb | 1:1000 for WB | ZENBIO (R381181) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pS262 | Anti-Phosphorylated tau at Ser262 | pAb | 1:1000 for WB | Signalway Antibody (#21100) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pS396 | Anti-Phosphorylated tau at Ser396 | pAb | 1:1000 for WB | Signalway Antibody (#21093) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pS404 | Anti-Phosphorylated tau at Ser404 | pAb | 1:1000 for WB | Signalway Antibody (#21001) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AT8 | Anti-Phosphorylated tau at Ser202/Thr205 | mAb | 1:1000 for WB | Thermo (MN1020) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tau5 | Anti-Total tau | mAb | 1:1000 for WB | Abcam (ab80579) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| β-actin | Anti-β-actin | pAb | 1:1000 for WB | ABclonal (AC026) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| APP | Anti-C-terminal of APP | pAb | 1:1000 for WB | Cell Signaling (E8B3O) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BACE1 | Anti-BACE1 | pAb | 1:1000 for WB | Zen-BioScience (R381865) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MAP-2 | Anti-MAP-2 | pAb | 1:200 for IF | Sigma-Aldrich (#M9942) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ADAM10 | Anti-ADAM10 | pAb | 1:1000 for WB | Zen-BioScience (R381790) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PS1 | Anti-PS1 | pAb | 1:1000 for WB | Zen-BioScience (R25440) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| sAPPβ | Anti-sAPPβ | pAb | 1:1000 for WB | Abcam (ab126732) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSD95 | Anti-PSD9 | mAb | 1:1000 for WB | Merck (ZMS1068) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| synaptophysin | Synaptophysin (aa 250-350) | pAb | 1:1000 for WB | Proteintech (17785-1-AP) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GluR2 | Anti-Phosphorylated GluR2 at Ser 880 | pAb | 1:1000 for WB | Absin (abs123742) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: mAb: monoclonal antibody; pAb: polyclonal antibody; WB: Western blotting; IF: Immunofluoresence. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Primary hippocampal neurons
Rat primary hippocampal neurons were obtained as described in our previous paper [64, 65].
For drug treatments, STP (dissolved in DMSO) was added to the culture medium and incubated with neurons for 24 h, and then the neurons were treated with GP (diluted according to the instruction) in different concentrations for 24 h.
CCK8 assay
Cell Counting Kit 8 assay (CCK-8 assay) was employed to check the cell viability. About 5 × 103 cells in a volume of 100 μL per well of 96-well plate were cultured in medium with 10% FBS, for four replicates. The cells were treated with different concentrations of STP without or with GP for different time. 10 μL CCK-8 reagent (ab228554, Abcam, Cambridge, MA, USA) was added into 90 μL DMEM to make the working solution. 100 μL working solution was added into per well and incubated for 2 h, then optical density detection.
Animals and grouping
The study used male SD rats that were 2 months old and weighed around 250 ± 20 g. These rats were obtained from the Laboratory Animal Centre of Tongji Medical College, Huazhong University of Science and Technology. The rats were housed under standard laboratory conditions, including appropriate temperature and a 12/12-hour light/dark cycle. They had free access to water and food throughout the study. After a week of adapting to the environment, the rats were randomly divided into four groups including control group (CTR), STP injection group (STP group), GP treatment groups (STP+GP200, STP+GP400) (STP injection plus GP treatment at 200 or 400 mg/kg [66]). On the 1st day for surgery, rats were anesthetized with sodium pentobarbital at a dose of 0.15 ml per 100 g body weight. The sodium pentobarbital used was obtained from Sigma (Darmstadt, Germany). After anesthesia, the rats were placed on a stereotactic apparatus manufactured by Stoelting Corporation (Wood Dale, IL, USA). The STP group, and the STP+GP groups of the rats were injected with 3 μL STP (500 μM, dissolved in DMSO) in each side, 6 μL total in the bilateral hippocampi with a micro-syringe at a rate of 0.5 μL/min, at coordinates from bregma and dura of AP-4.0, L/R-2.0 and V-3.0 (in mm) [19]. The needle was left for 10 min after injection and then withdrawn slowly. Control rats were injected with the same volume DMSO. On the 3rd day, GP treatment groups were given 200 mg/kg and 400 mg/kg GP (dissolved in 0.9% saline) by gavage once a day for 30 days. The same volume of 0.9% sterile saline was used in the other groups. On the 33rd day, the rats were assessed by behavioral assessment of their learning and memory abilities. The brain samples were then collected for morphological testing, ELISA or Western blot analysis.
Animal behavior tests
Open-field
Spontaneous exploratory motor activity and anxiety behaviors of rats were checked using open field (OF), as previously mentioned [67]. An open-ended black wooden box (with an internal field size of 100 × 100 cm × 70) is used. Each rat is tested once by placing it face to the wall and allowed to explore the environment for five minutes freely. At the end of each rat test, a wet cloth was used to wash and wipe the site and dry it with a dry cloth. We tested rat motor function by total movement distance.
Barnes maze task
The Barnes maze is a commonly used test to evaluate animal spatial learning and memory [68]. In this test, animals are trained to find a hidden escape chamber located beneath one of the holes on the perimeter of a disk. The disk is brightly illuminated to create an aversive stimulus. During the experiment, the animals are trained twice a day for four consecutive days. At the beginning of each trial, the animals are placed in the center of the maze, covered by a cylindrical starting chamber. After a 10-second delay, the starter chamber is lifted and the animal begins to search for the escape chamber. The experiment ends when the animal enters the escape chamber or after the predetermined 3-minute time limit. Before and after each experiment, all surfaces of the maze are cleaned to remove any potential olfactory cues left by the previous animals.
New object recognition
The novel object recognition test is used to assess learning and memory. The rats were put into a container with dimensions of 100 cm × 100 cm × 70 cm and allowed to freely explore the arena for five minutes to become acquainted with the environment. The next day, the rats are placed back into the arena from the same starting point. They are given five minutes to explore and familiarize themselves with two objects, object A and object B. After a 24-hour interval, object B (the familiar object) is replaced with a new object, object C. The rats are then given five minutes to explore both objects, object A and object C. The behavior of the rats during the exploration is recorded. The recognition index is calculated based on the time spent exploring the objects. The formula for calculating the recognition index is as follows: TA/(TA + TB): TB/(TA + TB): TC/(TA + TC).
TUNEL assay
TUNEL staining was carried out to assess apoptosis using the Annexin V FITC/PI kit (Abbkine, China). Primary hippocampal neurons cultured on coverslips or brain slices were fixed in 4% paraformaldehyde for 30 minutes. The fixed cells or brain slices were then incubated in a solution of 0.5% Triton X-100 diluted in PBS for 10 minutes. The cells or brain slices in each group were incubated with the TUNEL solution at 4°C for 1 hour. After TUNEL staining, the cells or brain slices were incubated with DAPI for nuclear staining. The stained samples were visualized using a fluorescence microscope (Olympus, Tokyo, Japan). The resulting data were analyzed by counting the number of TUNEL-positive cells, which are indicative of apoptotic cells, as well as the total number of cells. The apoptosis rate was then calculated by dividing the number of TUNEL-positive cells by the total number of cells and multiplying the result by 100%.
Immunofluorescence and dendrite morphological analysis
The cultured cells were prepared in the same way in TUNEL assay. The fixed cells were incubated in PBS containing 0.1% Triton X-100 and 5% BSA for 30 min at room temperature. The cells were then incubated with the primary antibody at 4°C overnight, followed by rinse for three times (5 min/time) in 0.01 M PBS. Next, the cells were incubated with the secondary antibodies from Jackson ImmunoResearch, a company based in West Grove, PA, USA, which were diluted at a 1:200 in PBS for 1.5 hours at room temperature, then the cells were rinsed three times in PBS. Finally, the coverslips were mounted onto the slides with DAPI to stain the nuclei of the cells. Laser confocal microscope (Zeiss 710, Germany) was used for image acquisition.
Neuronal dendrite morphology was analyzed with ImageJ software. The saved grayscale image was opened in ImageJ, and the ruler was set. The “Neuron J” patch was used to trace the dendrites and outline the neuronal structure, and then, the picture was saved. Lastly, the saved picture was opened again, draw the distance from the cell body to the farthest dendrite, and click “Sholl Analysis” to get the relevant measurements of the neuron dendrites. The data were saved for subsequent analysis.
Western blotting
Protein samples of primary hippocampal neurons and tissues were prepared according to the method in our previous paper [65]. 10 μg protein from hippocampal tissue and primary hippocampal neurons were separated using SDS-PAGE and transferred onto a nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). Next, the membranes were subjected to overnight incubation at 4°C with primary antibodies. After rinsing with TBST for three times, the membranes were incubated with secondary antibodies. The results were shown using the detection reagent and analyzed using ImageJ software (Rawak Software, Stuttgart, Germany).
ELISA for Aβ1–42 and Aβ40 levels
Aβ40 and Aβ42 levels are commonly performed using a sandwich ELISA kit. Primary hippocampal neurons or hippocampal tissues were homogenized using PBS with the addition of a protease inhibitor PMSF at a concentration of 100 mg/ml. After homogenization by repeated freezing and thawing to break down the cells and tissues, the samples were centrifuged at 2.5 × 103 g rpm for 20 min. The supernatant was then incorporated into a 96-well plate. The ELISA assay followed the protocol provided by the manufacturer, Elab-science Biotechnology, located in Wuhan, China.
Nissl staining
After perfusion and fixation, the brains were sectioned into slices with a thickness of 30 μm. These sections were then mounted on slides that had been coated with gelatin. To visualize the neurons, Nissl staining solution was used. The staining procedure followed the instructions provided by the manufacturer, BeoTeam Biotechnology Co., Ltd., located in Shanghai, China.
Golgi staining
Golgi staining is a technique employed to visualize dendritic spines. The FD Rapid Golgi Staining Kit PK401, developed by FD Neuro Technologies, Inc. in Columbia, MO, USA, is one of the commonly used kits for this purpose. Brain sections were stained following the manufacturer’s instructions provided. The hippocampus slices were imaged by 10×, 20× and 40× objectives using Olympus VS120.
Statistical analyses
Statistical analyses in the study were carried out by GraphPad Prism 9 statistical software. The data were presented as mean ± SEM (standard error of the mean). One-way analysis of variance (ANOVA) was used to assess the differences among groups, P-value was equal to or less than 0.05, the observed differences were considered statistically significant.
Data availability statement
All data used in this study are available from the corresponding authors on reasonable request.
Author Contributions
X.W. and H.-L.L. designed and organized all experiments, and wrote the manuscript; L.L. performed all experiments; Y.L. and D.K. performed in vitro cell culture and biochemical analysis; R.L. and J.-Z.W. analyzed the data.
Acknowledgments
The authors are grateful to Mr. Dan Ke and Ms. Qun Wang for helpful technical suggestions during the conduct of this study.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
All animal handling and uses abided by the protocol approved by the Institutional Animal Care and Use Committee of Tongji Medical School of Huazhong University of Science and Technology (IACUC Number: 3308).
Funding
This work was supported by grants from the NSFC (82071440, 31929002, and 92049107), Science, Technology and Innovation Commission of Shenzhen Municipality (JCYJ20210324141405014), Guangdong Basic and Applied Basic Research Foundation (2020B1515120017).
References
- 1. Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol. 2018; 25:59–70. https://doi.org/10.1111/ene.13439 [PubMed]
- 2. Lee VM. Regulation of tau phosphorylation in Alzheimer's disease. Ann N Y Acad Sci. 1996; 777:107–13. https://doi.org/10.1111/j.1749-6632.1996.tb34408.x [PubMed]
- 3. He H, Dong W, Huang F. Anti-amyloidogenic and anti-apoptotic role of melatonin in Alzheimer disease. Curr Neuropharmacol. 2010; 8:211–7. https://doi.org/10.2174/157015910792246137 [PubMed]
- 4. Andrade-Moraes CH, Oliveira-Pinto AV, Castro-Fonseca E, da Silva CG, Guimarães DM, Szczupak D, Parente-Bruno DR, Carvalho LR, Polichiso L, Gomes BV, Oliveira LM, Rodriguez RD, Leite RE, et al. Cell number changes in Alzheimer's disease relate to dementia, not to plaques and tangles. Brain. 2013; 136:3738–52. https://doi.org/10.1093/brain/awt273 [PubMed]
- 5. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011; 34:185–204. https://doi.org/10.1146/annurev-neuro-061010-113613 [PubMed]
- 6. Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer's disease. Lancet. 2016; 388:505–17. https://doi.org/10.1016/S0140-6736(15)01124-1 [PubMed]
- 7. Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer's disease. Trends Mol Med. 2015; 21:394–402. https://doi.org/10.1016/j.molmed.2015.03.003 [PubMed]
- 8. Lv ZY, Tan CC, Yu JT, Tan L. Spreading of Pathology in Alzheimer's Disease. Neurotox Res. 2017; 32:707–22. https://doi.org/10.1007/s12640-017-9765-2 [PubMed]
- 9. Kimura N. Diabetes Mellitus Induces Alzheimer's Disease Pathology: Histopathological Evidence from Animal Models. Int J Mol Sci. 2016; 17:503. https://doi.org/10.3390/ijms17040503 [PubMed]
- 10. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, Nixon RA, Jones DT. Alzheimer disease. Nat Rev Dis Primers. 2021; 7:33. https://doi.org/10.1038/s41572-021-00269-y [PubMed]
- 11. Attawish A, Chivapat S, Phadungpat S, Bansiddhi J, Techadamrongsin Y, Mitrijit O, Chaorai B, Chavalittumrong P. Chronic toxicity of Gynostemma pentaphyllum. Fitoterapia. 2004; 75:539–51. https://doi.org/10.1016/j.fitote.2004.04.010 [PubMed]
- 12. Li K, Du Y, Fan Q, Tang CY, He JF. Gypenosides might have neuroprotective and immunomodulatory effects on optic neuritis. Med Hypotheses. 2014; 82:636–8. https://doi.org/10.1016/j.mehy.2014.02.030 [PubMed]
- 13. Zhang HK, Ye Y, Li KJ, Zhao ZN, He JF. Gypenosides Prevent H2O2-Induced Retinal Ganglion Cell Apoptosis by Concurrently Suppressing the Neuronal Oxidative Stress and Inflammatory Response. J Mol Neurosci. 2020; 70:618–30. https://doi.org/10.1007/s12031-019-01468-9 [PubMed]
- 14. Jia D, Rao C, Xue S, Lei J. Purification, characterization and neuroprotective effects of a polysaccharide from Gynostemma pentaphyllum. Carbohydr Polym. 2015; 122:93–100. https://doi.org/10.1016/j.carbpol.2014.12.032 [PubMed]
- 15. Wang J, Zhao M, Cheng X, Han Y, Zhao T, Fan M, Zhu L, Yang JL. Dammarane-Type Saponins from Gynostemma pentaphyllum Prevent Hypoxia-Induced Neural Injury through Activation of ERK, Akt, and CREB Pathways. J Agric Food Chem. 2020; 68:193–205. https://doi.org/10.1021/acs.jafc.9b06659 [PubMed]
- 16. Wang XJ, Sun T, Kong L, Shang ZH, Yang KQ, Zhang QY, Jing FM, Dong L, Xu XF, Liu JX, Xin H, Chen ZY. Gypenosides pre-treatment protects the brain against cerebral ischemia and increases neural stem cells/progenitors in the subventricular zone. Int J Dev Neurosci. 2014; 33:49–56. https://doi.org/10.1016/j.ijdevneu.2013.12.001 [PubMed]
- 17. Krohn AJ, Preis E, Prehn JH. Staurosporine-induced apoptosis of cultured rat hippocampal neurons involves caspase-1-like proteases as upstream initiators and increased production of superoxide as a main downstream effector. J Neurosci. 1998; 18:8186–97. https://doi.org/10.1523/JNEUROSCI.18-20-08186.1998 [PubMed]
- 18. Ashutosh, Chao C, Borgmann K, Brew K, Ghorpade A. Tissue inhibitor of metalloproteinases-1 protects human neurons from staurosporine and HIV-1-induced apoptosis: mechanisms and relevance to HIV-1-associated dementia. Cell Death Dis. 2012; 3:e332. https://doi.org/10.1038/cddis.2012.54 [PubMed]
- 19. Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ, Yang Y, Zhang JY, Wang Q, Xu H, Liao FF, Wang JZ. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci U S A. 2007; 104:3591–6. https://doi.org/10.1073/pnas.0609303104 [PubMed]
- 20. Biundo F, Del Prete D, Zhang H, Arancio O, D'Adamio L. A role for tau in learning, memory and synaptic plasticity. Sci Rep. 2018; 8:3184. https://doi.org/10.1038/s41598-018-21596-3 [PubMed]
- 21. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986; 83:4913–7. https://doi.org/10.1073/pnas.83.13.4913 [PubMed]
- 22. Puzzo D, Piacentini R, Fá M, Gulisano W, Li Puma DD, Staniszewski A, Zhang H, Tropea MR, Cocco S, Palmeri A, Fraser P, D'Adamio L, Grassi C, Arancio O. LTP and memory impairment caused by extracellular Aβ and Tau oligomers is APP-dependent. Elife. 2017; 6:e26991. https://doi.org/10.7554/eLife.26991 [PubMed]
- 23. Qin XY, Lv JH, Cui J, Fang X, Zhang Y. Curcumin protects against staurosporine toxicity in rat neurons. Neurosci Bull. 2012; 28:606–10. https://doi.org/10.1007/s12264-012-1275-x [PubMed]
- 24. Kumari S, Dhapola R, Reddy DH. Apoptosis in Alzheimer's disease: insight into the signaling pathways and therapeutic avenues. Apoptosis. 2023; 28:943–57. https://doi.org/10.1007/s10495-023-01848-y [PubMed]
- 25. Sethi G, Rath P, Chauhan A, Ranjan A, Choudhary R, Ramniwas S, Sak K, Aggarwal D, Rani I, Tuli HS. Apoptotic Mechanisms of Quercetin in Liver Cancer: Recent Trends and Advancements. Pharmaceutics. 2023; 15:712. https://doi.org/10.3390/pharmaceutics15020712 [PubMed]
- 26. Qian W, Shi J, Yin X, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimers Dis. 2010; 19:1221–9. https://doi.org/10.3233/JAD-2010-1317 [PubMed]
- 27. Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013 (Suppl 1); 33:S123–39. https://doi.org/10.3233/JAD-2012-129031 [PubMed]
- 28. Viola KL, Klein WL. Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015; 129:183–206. https://doi.org/10.1007/s00401-015-1386-3 [PubMed]
- 29. Cheng YL, Choi Y, Sobey CG, Arumugam TV, Jo DG. Emerging roles of the γ-secretase-notch axis in inflammation. Pharmacol Ther. 2015; 147:80–90. https://doi.org/10.1016/j.pharmthera.2014.11.005 [PubMed]
- 30. Endres K, Deller T. Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms. Front Mol Neurosci. 2017; 10:56. https://doi.org/10.3389/fnmol.2017.00056 [PubMed]
- 31. Matsuda S, Ikeda Y, Murakami M, Nakagawa Y, Tsuji A, Kitagishi Y. Roles of PI3K/AKT/GSK3 Pathway Involved in Psychiatric Illnesses. Diseases. 2019; 7:22. https://doi.org/10.3390/diseases7010022 [PubMed]
- 32. Kumar M, Bansal N. Implications of Phosphoinositide 3-Kinase-Akt (PI3K-Akt) Pathway in the Pathogenesis of Alzheimer's Disease. Mol Neurobiol. 2022; 59:354–85. https://doi.org/10.1007/s12035-021-02611-7 [PubMed]
- 33. Long HZ, Cheng Y, Zhou ZW, Luo HY, Wen DD, Gao LC. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer's Disease and Parkinson's Disease. Front Pharmacol. 2021; 12:648636. https://doi.org/10.3389/fphar.2021.648636 [PubMed]
- 34. Ding MR, Qu YJ, Hu B, An HM. Signal pathways in the treatment of Alzheimer's disease with traditional Chinese medicine. Biomed Pharmacother. 2022; 152:113208. https://doi.org/10.1016/j.biopha.2022.113208 [PubMed]
- 35. Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers. 2008; 23:1–9. https://doi.org/10.1177/172460080802300101 [PubMed]
- 36. Hernandez F, Lucas JJ, Avila J. GSK3 and tau: two convergence points in Alzheimer's disease. J Alzheimers Dis. 2013 (Suppl 1); 33:S141–4. https://doi.org/10.3233/JAD-2012-129025 [PubMed]
- 37. Sofola O, Kerr F, Rogers I, Killick R, Augustin H, Gandy C, Allen MJ, Hardy J, Lovestone S, Partridge L. Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer's disease. PLoS Genet. 2010; 6:e1001087. https://doi.org/10.1371/journal.pgen.1001087 [PubMed]
- 38. Liao S, Wu J, Liu R, Wang S, Luo J, Yang Y, Qin Y, Li T, Zheng X, Song J, Zhao X, Xiao C, Zhang Y, et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: Role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2 activation. Redox Biol. 2020; 36:101644. https://doi.org/10.1016/j.redox.2020.101644 [PubMed]
- 39. Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic Impairment in Alzheimer's Disease: A Dysregulated Symphony. Trends Neurosci. 2017; 40:347–57. https://doi.org/10.1016/j.tins.2017.04.002 [PubMed]
- 40. Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer's disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomedicine. 2019; 14:5541–54. https://doi.org/10.2147/IJN.S200490 [PubMed]
- 41. Kowalska A, Pruchnik-Wolińska D, Florczak J, Modestowicz R, Szczech J, Kozubski W, Rossa G, Wender M. Genetic study of familial cases of Alzheimer's disease. Acta Biochim Pol. 2004; 51:245–52. [PubMed]
- 42. Revi M. Alzheimer's Disease Therapeutic Approaches. Adv Exp Med Biol. 2020; 1195:105–16. https://doi.org/10.1007/978-3-030-32633-3_15 [PubMed]
- 43. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001; 81:741–66. https://doi.org/10.1152/physrev.2001.81.2.741 [PubMed]
- 44. Tatulian SA. Challenges and hopes for Alzheimer's disease. Drug Discov Today. 2022; 27:1027–43. https://doi.org/10.1016/j.drudis.2022.01.016 [PubMed]
- 45. Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018; 14:399–415. https://doi.org/10.1038/s41582-018-0013-z [PubMed]
- 46. Nguyen NH, Ha TKQ, Yang JL, Pham HTT, Oh WK. Triterpenoids from the genus Gynostemma: Chemistry and pharmacological activities. J Ethnopharmacol. 2021; 268:113574. https://doi.org/10.1016/j.jep.2020.113574 [PubMed]
- 47. Cai H, Liang Q, Ge G. Gypenoside Attenuates β Amyloid-Induced Inflammation in N9 Microglial Cells via SOCS1 Signaling. Neural Plast. 2016; 2016:6362707. https://doi.org/10.1155/2016/6362707 [PubMed]
- 48. Meng X, Wang M, Sun G, Ye J, Zhou Y, Dong X, Wang T, Lu S, Sun X. Attenuation of Aβ25-35-induced parallel autophagic and apoptotic cell death by gypenoside XVII through the estrogen receptor-dependent activation of Nrf2/ARE pathways. Toxicol Appl Pharmacol. 2014; 279:63–75. https://doi.org/10.1016/j.taap.2014.03.026 [PubMed]
- 49. Wang J, Shi J, Jia N, Sun Q. Network pharmacology analysis reveals neuroprotection of Gynostemma pentaphyllum (Thunb.) Makino in Alzheimer' disease. BMC Complement Med Ther. 2022; 22:57. https://doi.org/10.1186/s12906-022-03534-z [PubMed]
- 50. Prehn JH, Jordán J, Ghadge GD, Preis E, Galindo MF, Roos RP, Krieglstein J, Miller RJ. Ca2+ and reactive oxygen species in staurosporine-induced neuronal apoptosis. J Neurochem. 1997; 68:1679–85. https://doi.org/10.1046/j.1471-4159.1997.68041679.x [PubMed]
- 51. Hansson CA, Popescu BO, Laudon H, Cedazo-Minguez A, Popescu LM, Winblad B, Ankarcrona M. Caspase cleaved presenilin-1 is part of active gamma-secretase complexes. J Neurochem. 2006; 97:356–64. https://doi.org/10.1111/j.1471-4159.2006.03735.x [PubMed]
- 52. Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014; 71:505–8. https://doi.org/10.1001/jamaneurol.2013.5847 [PubMed]
- 53. Wang H, Shang Y, Wang E, Xu X, Zhang Q, Qian C, Yang Z, Wu S, Zhang T. MST1 mediates neuronal loss and cognitive deficits: A novel therapeutic target for Alzheimer's disease. Prog Neurobiol. 2022; 214:102280. https://doi.org/10.1016/j.pneurobio.2022.102280 [PubMed]
- 54. Kitagishi Y, Nakanishi A, Ogura Y, Matsuda S. Dietary regulation of PI3K/AKT/GSK-3β pathway in Alzheimer's disease. Alzheimers Res Ther. 2014; 6:35. https://doi.org/10.1186/alzrt265 [PubMed]
- 55. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017; 169:381–405. https://doi.org/10.1016/j.cell.2017.04.001 [PubMed]
- 56. Llorens-Martín M, Jurado J, Hernández F, Avila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014; 7:46. https://doi.org/10.3389/fnmol.2014.00046 [PubMed]
- 57. Cai Z, Zhao Y, Zhao B. Roles of glycogen synthase kinase 3 in Alzheimer's disease. Curr Alzheimer Res. 2012; 9:864–79. https://doi.org/10.2174/156720512802455386 [PubMed]
- 58. Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013; 123:224–35. https://doi.org/10.1172/JCI64516 [PubMed]
- 59. Koh SH, Noh MY, Kim SH. Amyloid-beta-induced neurotoxicity is reduced by inhibition of glycogen synthase kinase-3. Brain Res. 2008; 1188:254–62. https://doi.org/10.1016/j.brainres.2007.10.064 [PubMed]
- 60. Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005; 102:6990–5. https://doi.org/10.1073/pnas.0500466102 [PubMed]
- 61. Pérez M, Hernández F, Lim F, Díaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis. 2003; 5:301–8. https://doi.org/10.3233/jad-2003-5405 [PubMed]
- 62. Couldwell WT, Hinton DR, He S, Chen TC, Sebat I, Weiss MH, Law RE. Protein kinase C inhibitors induce apoptosis in human malignant glioma cell lines. FEBS Lett. 1994; 345:43–6. https://doi.org/10.1016/0014-5793(94)00415-3 [PubMed]
- 63. Yang JR, Ren TT, Lan R, Qin XY. Tea polyphenols attenuate staurosporine-induced cytotoxicity and apoptosis by modulating BDNF-TrkB/Akt and Erk1/2 signaling axis in hippocampal neurons. IBRO Rep. 2020; 8:115–21. https://doi.org/10.1016/j.ibror.2020.04.002 [PubMed]
- 64. Seibenhener ML, Wooten MW. Isolation and culture of hippocampal neurons from prenatal mice. J Vis Exp. 2012; 3634. https://doi.org/10.3791/3634 [PubMed]
- 65. Lei L, Feng J, Wu G, Wei Z, Wang JZ, Zhang B, Liu R, Liu F, Wang X, Li HL. HIF-1α Causes LCMT1/PP2A Deficiency and Mediates Tau Hyperphosphorylation and Cognitive Dysfunction during Chronic Hypoxia. Int J Mol Sci. 2022; 23:16140. https://doi.org/10.3390/ijms232416140 [PubMed]
- 66. Zhang G, Zhao Z, Gao L, Deng J, Wang B, Xu D, Liu B, Qu Y, Yu J, Li J, Gao G. Gypenoside attenuates white matter lesions induced by chronic cerebral hypoperfusion in rats. Pharmacol Biochem Behav. 2011; 99:42–51. https://doi.org/10.1016/j.pbb.2011.03.019 [PubMed]
- 67. Seibenhener ML, Wooten MC. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp. 2015; e52434. https://doi.org/10.3791/52434 [PubMed]
- 68. Wu C, Yang L, Tucker D, Dong Y, Zhu L, Duan R, Liu TC, Zhang Q. Beneficial Effects of Exercise Pretreatment in a Sporadic Alzheimer's Rat Model. Med Sci Sports Exerc. 2018; 50:945–56. https://doi.org/10.1249/MSS.0000000000001519 [PubMed]