



Introduction
Lung cancer (LC), one of the most common malignancies worldwide, was the leading cause of cancer death with 18.0% of total cancer deaths for both sexes combined in 2020 [1]. For men, LC has ranked first in both incidence and mortality, accounting for 14.3% of total new cases and 21.5% of total cancer deaths, respectively [1]. Especially in some countries with a higher human development index (HDI), the incidence and mortality of LC are about 4-fold and 3-fold higher than those in lower HDI countries [1]. The burden of LC incidence and mortality is rapidly growing globally. However, the overall survival (OS) of LC patients remains poor and the overall 5-year survival rate is only about 19% for all stages combined, much lower than 98% of prostate cancer and 90% of female breast cancer [2].
Non-small cell lung cancer (NSCLC) is the most common histological subtype and accounts for about 85% of all LC patients [3], of which lung adenocarcinoma (LUAD) is the major subtype and accounts for greater than 40% of LC cases and its relative frequency is increasing [3, 4]. Currently, the LUAD TNM (tumor-node-metastasis) staging system remains the prevailing method and the most important factor to predict the OS of LUAD patients [5]. However, due to the high heterogeneity of LUAD, the prognoses of LUAD patients within the same TNM group presented heterogeneous outcomes [6], which suggests that some new prognostic methods should be developed and used to refine risk stratification, such as molecular prognostic marker [6]. In recent years, with the progress of molecular biology, some molecular prognostic markers including protein, mRNA, miRNA, lncRNA, and oncogene were identified [6–10]. In addition, liquid biopsies based on circulating tumor cells (CTCs) and circulating free DNA (cfDNA) and tumor microenvironment (TME) based on tumor-infiltrating immune cells (TICs) are of much interest to many scientists in predicting the survival, and some valuable results have been obtained [6, 11, 12].
Epigenetic regulation is an essential mechanism for normal development and maintenance of gene expression patterns in mammals [13]. Growing evidence has shown that epigenetic modification plays a critical role in the regulation of all DNA-based processes, and epigenetic alterations can lead to induction and maintenance of various cancers and are increasingly recognized as a cancer hallmark [14]. Chromatin regulators (CRs) including DNA methylators, histone modifiers and chromatin remodelers, as indispensable regulatory factors, play critical roles in driving epigenetic modification [15]. Some studies have shown that the dysregulation or dysfunction of CRs was correlated with various cancers and some CRs have been used as key drug targets against cancers [16–19]. For example, DNA methylator DNMT1 has been shown to play an oncogenic role in promoting cell proliferation, migration and invasion by suppressing cell differentiation in pancreatic cancer [20]. Chromatin remodeler CTCF has been shown to be upregulated in colorectal cancer tissue, and the overexpression of CTCF promoted colorectal cancer cell proliferation [21]. A few recent studies reported that CRs were correlated with the prognoses of cancer patients [22, 23]. For instance, the CBX7 expression resulted in a poor prognosis in ovarian clear cell adenocarcinoma [22]. Similarly, the CDK1 overexpression was associated with a poor prognosis in colorectal cancer [23]. In addition, several studies reported that some CRs were associated with LC such as CDK1 and MGMT [24, 25]. Despite some CRs advances, little is known about the roles of CRs in LUAD biology and a better understanding of CRs associated with the prognosis urgently requires identification.
In this study, we first constructed and evaluated two molecular prognostic subtypes (MPSs) based on the prognostic CRs. Subsequently, we established a six-gene prognostic signature based on two MPSs. Furthermore, we investigated the clinical and molecular features between differing MPSs and between differing risk subgroups, especially exploring the relationships of MPS and risk signature with TICs. This study will be helpful for better understanding the prognostic and immunological role of CRs in LUAD.
Results
This study was divided into 5 research modules sequentially and the flow chart was presented in Figure 1: (1) Data collection. LUAD-related gene expression datasets and clinical data were retrieved from the public databases The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO). The TCGA-LUAD dataset was used to construct prognostic subtypes and prognostic gene signature. The GEO-LUAD datasets were used to evaluate the validity of prognostic subtype and the robustness of prognostic gene signature. (2) Prognostic subtype construction. Two MPSs were constructed based on 27 differentially expressed chromatin regulators (DECRs) with significant prognostic value using the TCGA-LUAD dataset and the validity of MPSs was evaluated using 4 independent GEO-LUAD datasets. (3) Key prognostic gene identification. Six differentially expressed genes (DEGs) between two MPSs were identified to be key prognostic DEGs based on the univariate Cox regression analysis (UCRA), multivariate Cox regression analysis (MCRA) and least absolute shrinkage and selection operator (LASSO) analysis. (4) Prognostic signature establishment. A six-gene prognostic signature was established using the TCGA-LUAD dataset and its robustness was assessed using 8 independent GEO-LUAD datasets. Subsequently, the predictive ability of six-gene prognostic signature was evaluated by comparing with 19 reported prognostic signatures. (5) Clinical and immunological features analyses. The associations of MPS, key prognostic genes and risk score with clinical and immunological features were investigated. Potential responses of LUAD patients to immunotherapy and chemotherapy were predicted in different MPSs and in different risk subgroups, respectively.
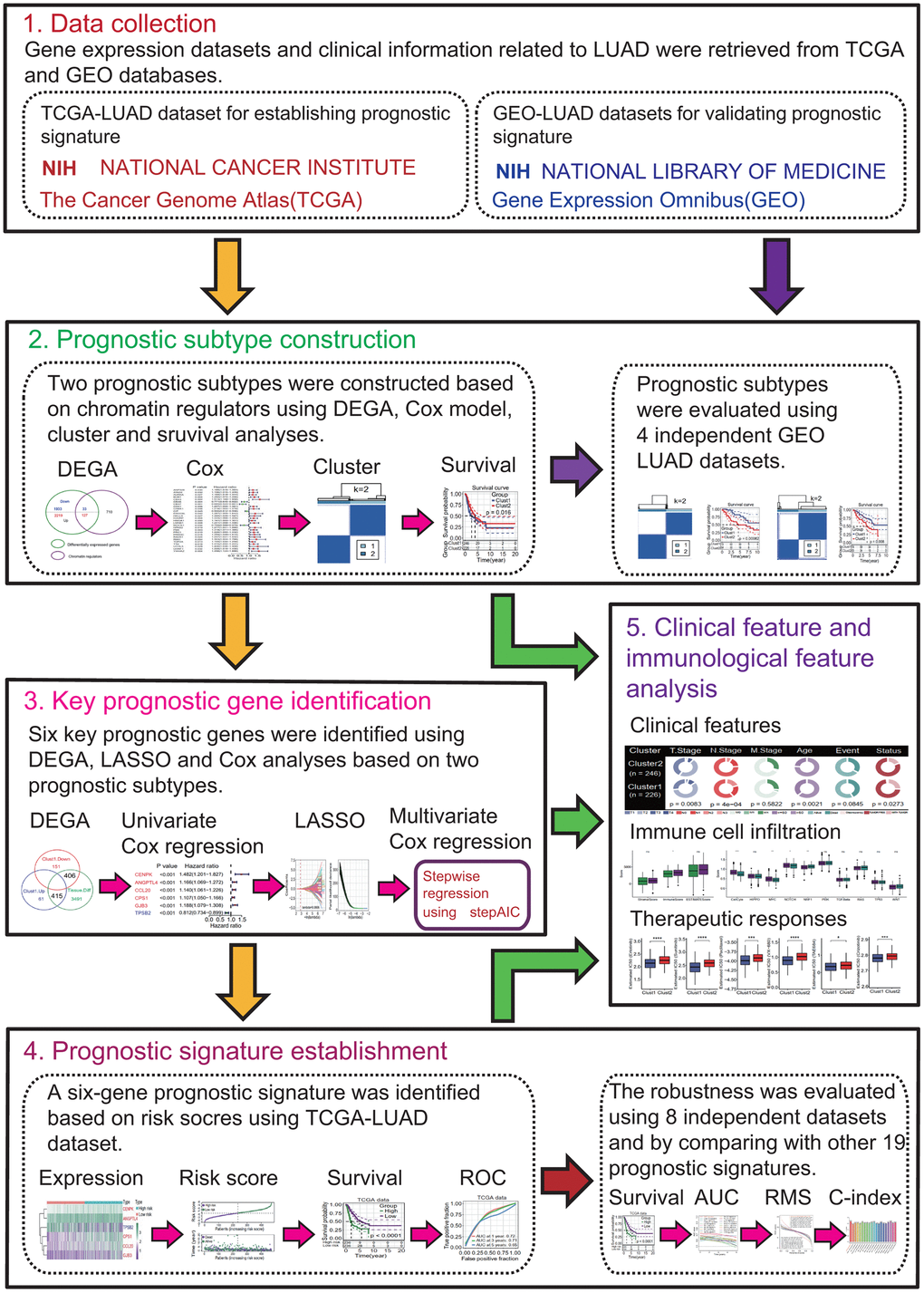
Figure 1. Flow chart of the present study. This study was divided into 5 research modules sequentially. (1) Data collection. Lung adenocarcinoma (LUAD) gene expression datasets were retrieved from the public gene expression omnibus (GEO) and the cancer genome atlas (TCGA) databases, respectively. (2) Prognostic subtype construction. Prognostic subtypes were constructed based on prognostic chromatin regulators. (3) Key prognostic gene identification. Key prognostic genes were identified by Cox regression and the least absolute shrinkage and selection operator (LASSO) analyses. (4) Prognostic signature establishment. Prognostic signature was established and evaluated. (5) Clinical feature and immunological feature analyses. The relationships of prognostic subtypes, prognostic genes and signature with clinical characteristics and immunological features were analyzed.
Twenty-seven DECRs were correlated with the prognosis in LUAD
To identify key prognostic CRs, a DEGA and a UCRA were performed. The DEGA result showed that totals of 2346 upregulated and 1966 downregulated genes were identified in LUAD tissue compared to normal lung tissue (Figure 2A and Supplementary Table 1). Among 127 upregulated and 33 downregulated genes were DECRs (Figure 2B and Supplementary Table 2). These DECRs, especially 127 upregulated DECRs, mainly involved in chromatin organization (GO:0051276, p = 8.37e-49), cell cycle (hsa04110, p = 2.87e-07), chromatin modifying enzymes (HAS-3247509, p = 2.67e-15), etc. (Figure 2B). The disease ontology (DO) result showed that 127 upregulated DECRs were mainly associated with cancer (DOID:162, p = 0.0334) (Figure 2B). The UCRA result showed that 27 of 160 DECRs had significant prognostic values (p < 0.05, Figure 2C). The 27 prognostic DECRs include 16 histone modifiers, 4 chromatin remodelers, 2 DNA methylators and 5 unknown types, respectively (Table 1). Except for CBX6, CBX7, CIT and MOCS1, the other 23 DECRs were risk factors (Figure 2C) and their expressions showed strong positive correlations with each other (most p < 0.001, Figure 2D). Except for CBX7, CBX6 and MOCS1, the other 24 DECRs were significantly upregulated in LUAD tissues (adjusted p < 0.05, Figure 2E).
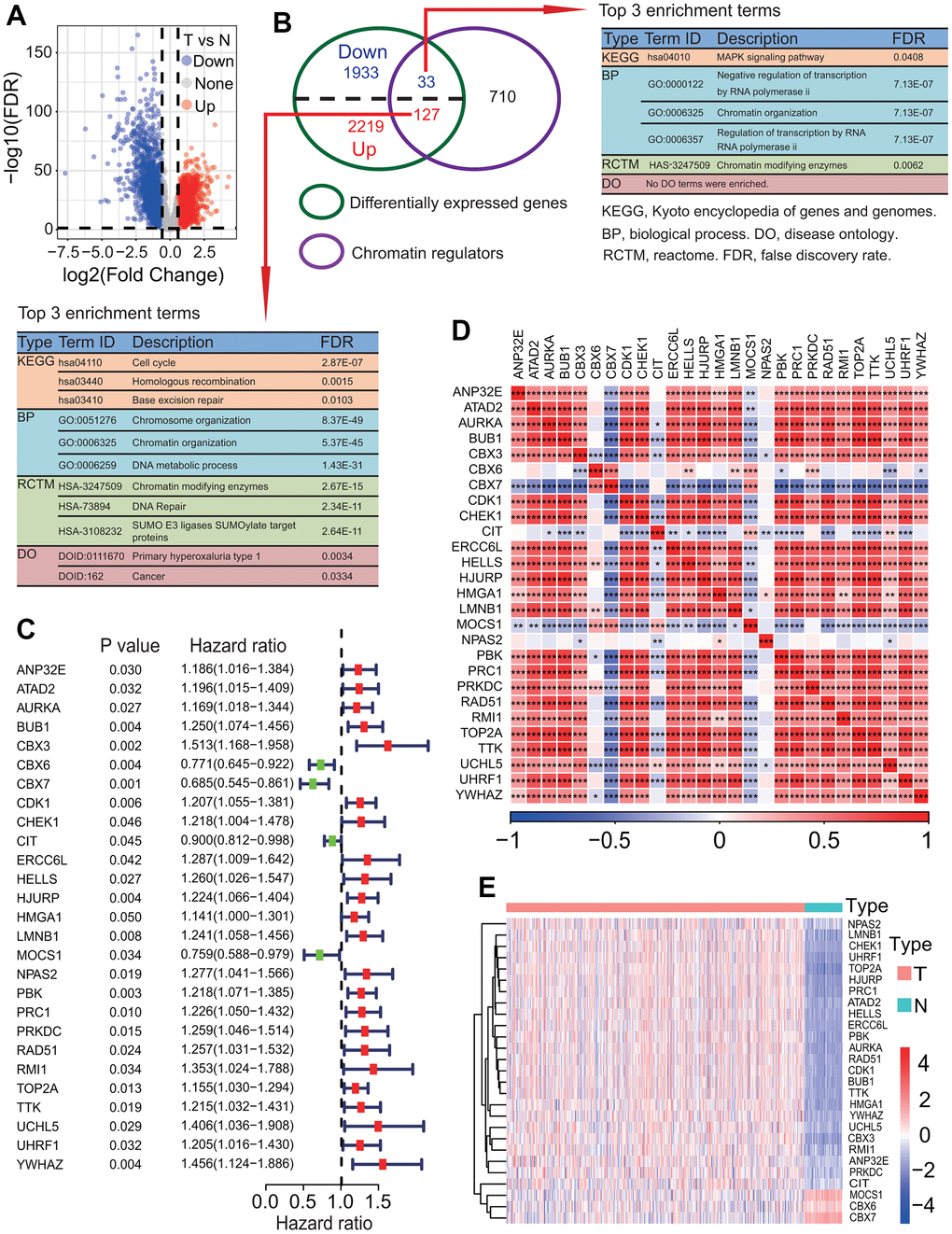
Figure 2. Identification of key differentially expressed chromatin regulators associated with survival. (A) Distribution of differentially expressed genes (DEGs) between lung adenocarcinoma (LUAD) and normal lung tissues. Totals of 2346 upregulated and 1966 downregulated DEGs were identified in LUAD tissue. (B) Identification of differentially expressed chromatin regulators (DECRs). Totals of 127 upregulated and 33 downregulated DECRs were identified in LUAD tissue. (C) Identification of key DECRs associated with overall survival (OS) rate of patients with LUAD. Univariate Cox regression analysis showed that 27 DECRs were associated with the OS of patients with LUAD. (D) Correlations between key prognostic DECRs in expression. Except three DECRs including MOCS1, CBX7 and CIT, the expressions of the other 24 DECRs showed strong positive correlations on the whole. (E) Heat map of key prognostic DECRs in expression. Except three DECRs including MOCS1, CBX7 and CBX6, the expression of the other 24 DECRs were upregulated in LUAD tissue.
Table 1. Basic functional information of 27 prognostic chromatin regulators.
| Symbol | Type | Histone type | Methytor type | Function |
| AURKA | Histone Modifier | Writer | / | Histone modification write (Histone phosphorylation) |
| BUB1 | Histone Modifier | Writer | / | Histone modification write (Histone phosphorylation) |
| PBK | Histone Modifier | Writer | / | Histone modification write (Histone phosphorylation) |
| PRKDC | Histone Modifier | Writer | / | Histone modification write (Histone phosphorylation) |
| TTK | Histone Modifier | Writer | / | Histone modification write cofactor (Histone phosphorylation) |
| CDK1 | Histone Modifier | Writer | / | Histone modification write (Histone phosphorylation) |
| CHEK1 | Histone Modifier | Writer | / | Histone modification write (Histone phosphorylation) |
| CIT | Histone Modifier | Writer | / | Histone modification write cofactor (Histone phosphorylation) |
| ANP32E | Histone Modifier | Reader | / | Histone chaperone, Histone modification read |
| CBX3 | Histone Modifier | Reader | / | Histone modification read |
| CBX6 | Histone Modifier | Reader | / | Histone modification read |
| CBX7 | Histone Modifier | Reader | / | Histone modification read |
| YWHAZ | Histone Modifier | Reader | / | Histone modification read |
| RAD51 | Histone Modifier | Eraser | / | Histone modification erase (Histone ubiquitination) |
| UCHL5 | Histone Modifier | Eraser | / | Histone modification erase cofactor (Histone ubiquitination) |
| HJURP | Histone Modifier | / | / | Histone chaperone |
| ATAD2 | Chromatin Remodeler | / | / | Chromatin remodelling |
| HELLS | Chromatin Remodeler | / | / | Chromatin remodelling |
| NPAS2 | Chromatin Remodeler | / | / | Chromatin remodelling |
| TOP2A | Chromatin Remodeler | / | / | Chromatin remodelling |
| RMI1 | DNA Methylator | / | / | DNA modification |
| UHRF1 | DNA Methylator Histone Modifier | Reader, Writer | Reader | Histone modification read, Histone modification write cofactor, DNA modification |
| LMNB1 | / | / | / | Global heterochromatic changes |
| ERCC6L | / | / | / | / |
| HMGA1 | / | / | / | / |
| MOCS1 | / | / | / | / |
| PRC1 | / | / | / | / |
To further investigate the expression levels of 27 prognostic DECRs between LUAD and normal lung tissues, the gene expressions of these 27 DECRs were compared between LUAD and normal lung tissues from 10 LUAD patients using a paired t-test method. The results showed that the expression levels of these 27 DECRs were highly consistent with the TCGA-LUAD results (Figure 3A and Supplementary Table 3).
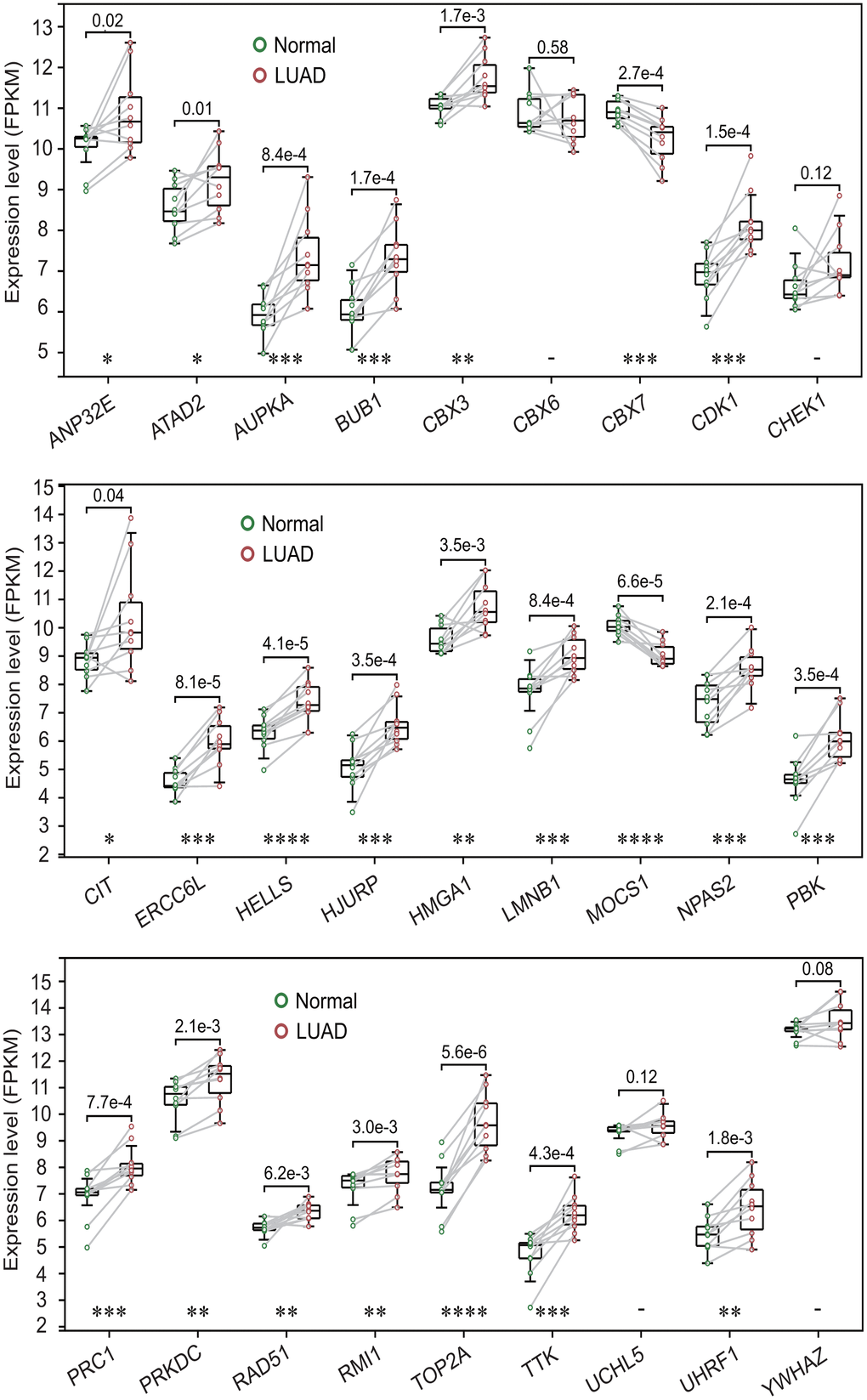
Figure 3. Expression levels of 27 prognostic chromatin regulators between lung adenocarcinoma (LUAD) and normal lung tissues. The expression levels of 27 prognostic chromatin regulators were compared between LUAD and normal lung tissues by a paired t-test method using 10 pairs of samples from 10 LUAD patients. The result was highly consistent with the TCGA-LUAD result.
Two MPSs constructed by 27 prognostic DECRs were valid
To investigate the relationships of 27 prognostic DECRs with LUAD prognostic subtype, a consensus clustering for the TCGA-LUAD samples was performed based on the gene expression profiles of 27 prognostic DECRs. According to clustering stabilities increasing from k = 2 to 9 (Figure 4A), k = 2 was selected for sample cluster and LUAD samples were clustered into two clusters called Cluster1 (C1 subtype) and Cluster2 (C2 subtype) (Figure 4B). The LUAD patients in the C2 subtype had a higher OS rate than those in the C1 subtype (p = 0.016, Figure 4C).
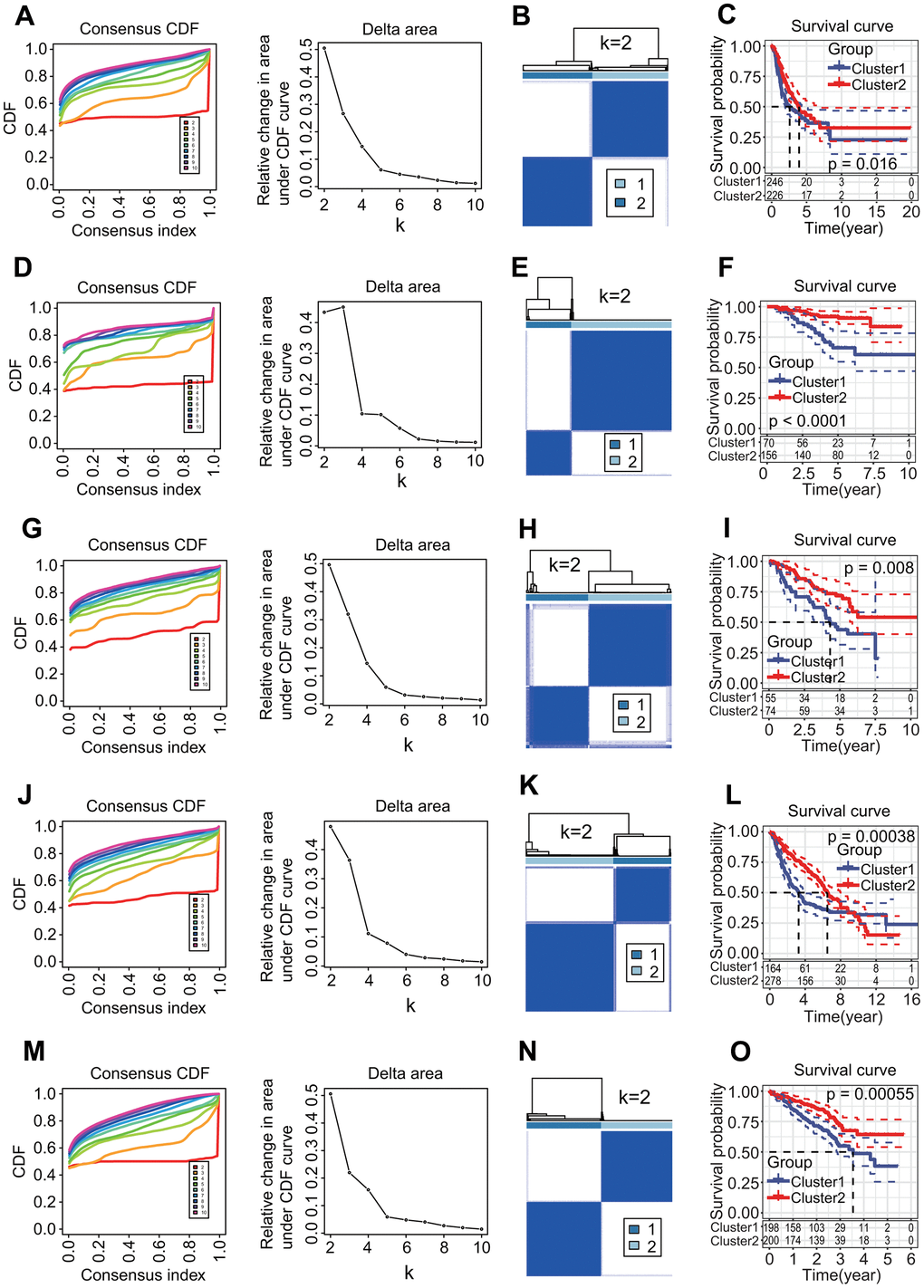
Figure 4. Molecular prognostic subtypes construction based on 27 key prognostic differentially expressed chromatin regulators (DECRs) and the associations with overall survival (OS) of patients in lung adenocarcinoma (LUAD). (A–C) TCGA-LUAD. (D–F) GSE31210. (G–I) GSE50081. (J–L) GSE68465. (M–O) GSE72094. (A, D, G, J, M) Cumulative distribution function (CDF) curve and CDF delta area curve. Clustering stability increasing curves were plotted from k = 2 to 9 for five gene expression datasets including TCGA-LUAD, GSE31210, GSE50081, GSE68465 and GSE72094. (B, E, H, K, N) Consensus clustering heat map. The k = 2 was selected to cluster samples for each of five gene expression datasets, and LUAD patients were divided into two clusters. (C, F, I, L, O) Survival curve. Two clusters were significantly associated with the OS of LUAD patients in five independent datasets (p = 0.016, < 0.0001, = 0.008, = 0.00038, = 0.00055, respectively).
To evaluate the validity of two MPSs, four independent GEO-LUAD gene expression datasets including GSE31210, GSE50081, GSE68465 and GSE72094 were clustered using the same clustering method, respectively. The k = 2 was the optimal parameter for each of four datasets (Figure 4D, 4G, 4J, 4M) and LUAD samples were clearly divided into two clusters (Figure 4E, 4H, 4K, 4N). There was a significant difference in the OS rate between two clusters (p < 0.0001, = 0.008, = 0.00038, = 0.00055, respectively, Figure 4F, 4I, 4L, 4O).
The expression analysis showed that 26 of 27 prognostic DECRs had significant differences between two clusters except for NPAS2 (Figure 5A), and 4 DECRs including CBX6, CBX7, MOCS1 and CIT were highly expressed in the C2 subtype. Among the 26 DECRs, except for CIT, the expression levels of 25 DECRs between the C1 subtype and LUAD group showed the same trends, and those between the C2 subtype and normal lung tissue, respectively (Figures 2E, 5A).
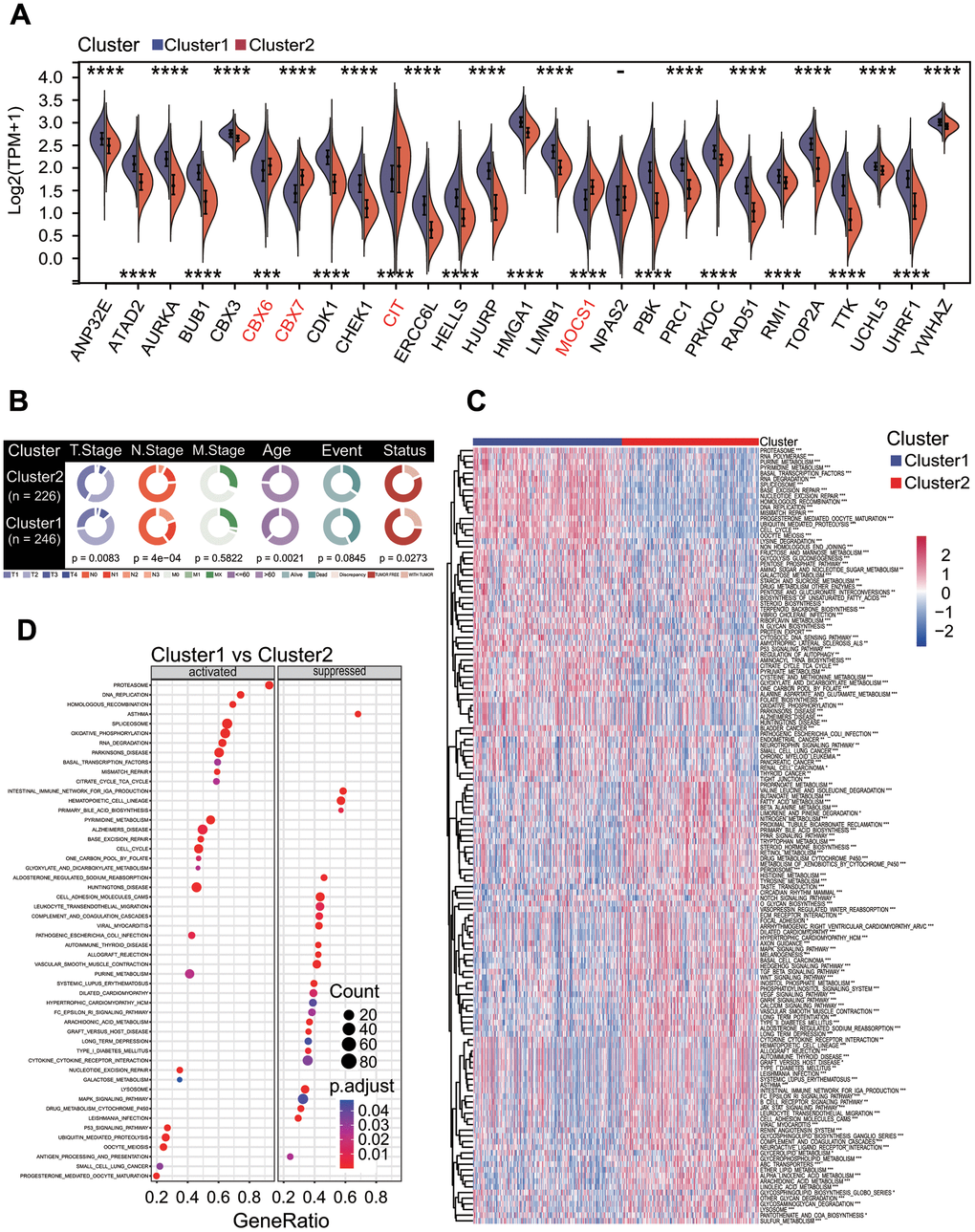
Figure 5. Correlations of molecular prognostic subtype with clinical features and KEGG pathway enrichment. (A) Expression of 27 prognostic chromatin regulators between two clusters. (B) Comparison of clinical features between two clusters. Chi-square test showed the significant differences in T staging (p = 0.0083), N staging (p = 4e-04), age (p = 0.0021) and cancer status (p = 0.0273). (C) Gene set variation analysis. Totals of 184 KEGG pathways were significantly enriched between two clusters. (D) Gene set enrichment analysis. The top 26 significantly enriched KEGG pathways were shown between two clusters.
MPSs were correlated with clinical and biological features
To explore the correlations of MPSs with clinical features, clinical features between C1 and C2 subtypes were compared. The results showed that there were significant differences in the T staging (chi-square p = 0.0083), N staging (chi-square p = 4e-04), age (chi-square p = 0.0021) and cancer status (chi-square p = 0.0273) between these two MPSs (Figure 5B). The LUAD patients in the C2 subtype had a lower T staging and N staging than those in the C1 subtype.
To investigate the correlations of MPSs with biological features, functional enrichment analyses were performed to discover potential functional differences between these two MPSs by using GSVA and GSEA methods. The GSVA result showed that a total of 184 KEGG pathways was significantly enriched (p < 0.05, Figure 5C). Among these KEGG pathways, some key oncogenic signaling pathways (OSPs) in the C2 subtype were lower active than those in the C1 subtype, such as cell cycle and P53 signaling pathway (both p < 0.001, Figure 5C). The GSEA result was consistent with that obtained by the GSVA method (Figure 5D) and some OSPs including cell cycle (p = 4.60e-09) and P53 signaling pathway (p = 2.12e-04) were significantly downregulated in the C2 subtype (Figure 5D).
To further investigate the characteristics of genomic variations between two MPSs, a somatic mutation analysis was performed. The result showed that the aneuploidy score, non-silent mutation rate, fraction altered, number of segments and homologous recombination defects in the C2 subtype were significantly lower than those in the C1 subtype (Wilcoxon test all p < 0.0001, Figure 6A). The mutation rate and main mutation type of top 10 altered genes were significantly different between two MPSs (Figure 6B).
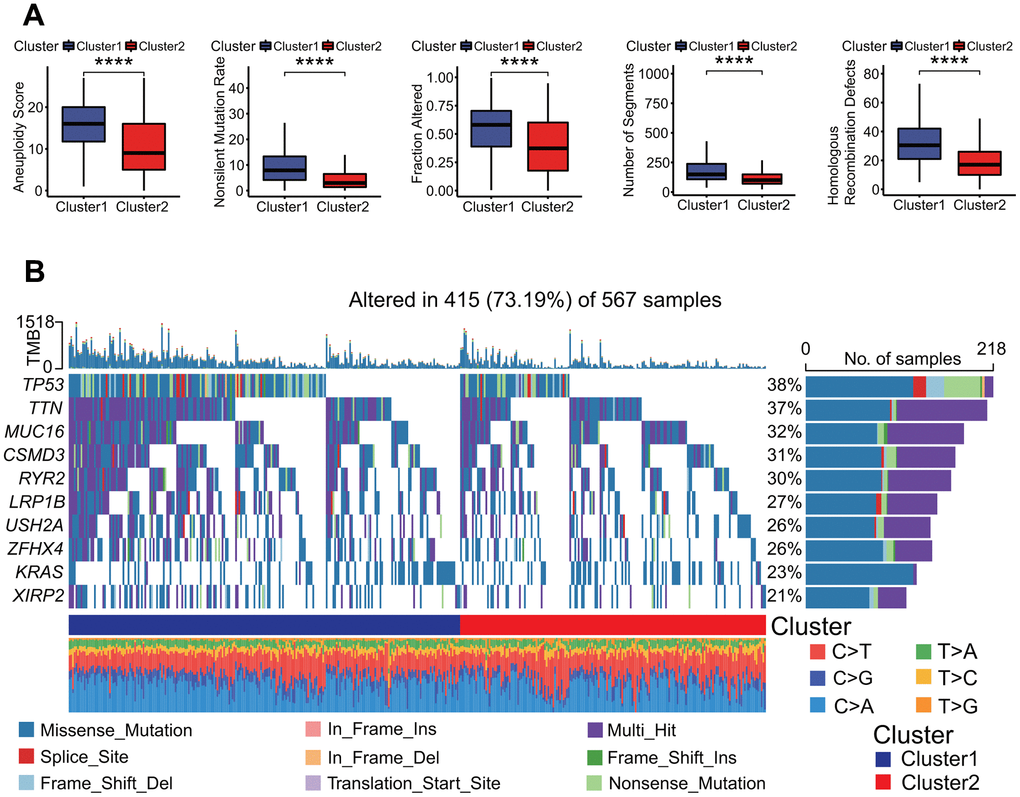
Figure 6. Genomes alterations between two clusters. (A) Molecular features of genome alterations. There was higher aneuploidy score, nonsilent mutation rate, fraction altered of genome, number of segments and homologous recombination defects in the cluster 1. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. (B) Gene mutation profiles between two clusters. The mutation frequencies of top 10 genes including TP53, TTN and MUC16 and so on had significant differences in two clusters, and there was higher mutation frequency in the cluster 1.
MPSs were correlated with immunological feature and therapeutic response
To elucidate the relationships of MPSs with immunological features, a comprehensive immunological analysis was implemented. A CIBERSORT-based analysis showed that the relative abundances of 14 of 22 immune cell types had significant differences between two MPSs (Wilcoxon test p < 0.05 or < 0.01 or < 0.001, Figure 7A). Among these 14 immune cells types, the abundances of 6 immune cell types including B cells memory (p < 0.001), T cells CD4 memory resting (p < 0.001), monocytes (p < 0.001), dendritic cells resting (p < 0.001), dendritic cells activated (p < 0.01) and mast cells resting (p < 0.001) were significantly higher in the C2 subtype than those in the C1 subtype (Figure 7A). Inversely, the abundances of 8 immune cell types including T cells CD8 (p < 0.05), T cells CD4 memory activated (p < 0.001), T cells follicular helper (p < 0.05), T cells gamma delta (p < 0.05), NK cells resting (p < 0.001), macrophages M0 (p < 0.001), macrophages M1 (p < 0.001) and mast cells activated (p < 0.001) were significantly lower in the C2 subtype (Figure 7A). The stromal score (p < 0.001), immune score (p < 0.01) and ESTIMAT score (p < 0.001) in the C2 subtype were significantly higher than those in the C1 subtype (Figure 7B). Two OSPs including cell cycle (p < 0.001) and MYC (p < 0.001) in the C2 subtype were less active than those in the C1 subtype (Figure 7C), and 4 OSPs containing NRF1 (p < 0.001), TGF-beta (p < 0.001), RAS (p < 0.001) and WNT (p < 0.001) in the C2 subtype were more active than those in the C1 subtype (Figure 7C). The TIDE result showed that the TIDE score (p < 0.001), IFNG score (p < 0.01), exclusion score (p < 0.001) and MDSC score (p < 0.001) were significantly lower in the C2 subtype, and the dysfunction score (p < 0.001) and TAM.M2 score (p < 0.001) were significantly higher in the C2 subtype (Figure 7D). The estimated IC50 values for 6 conventional chemotherapy agents including erlotinib (p < 0.0001), sunitinib (p < 0.0001), paclitaxel (p < 0.001), VX-680 (p < 0.0001), TAE684 (p < 0.05) and crizotinib (p < 0.001) in the C2 subtype were significantly higher than those in the C1 subtype (Figure 7E).
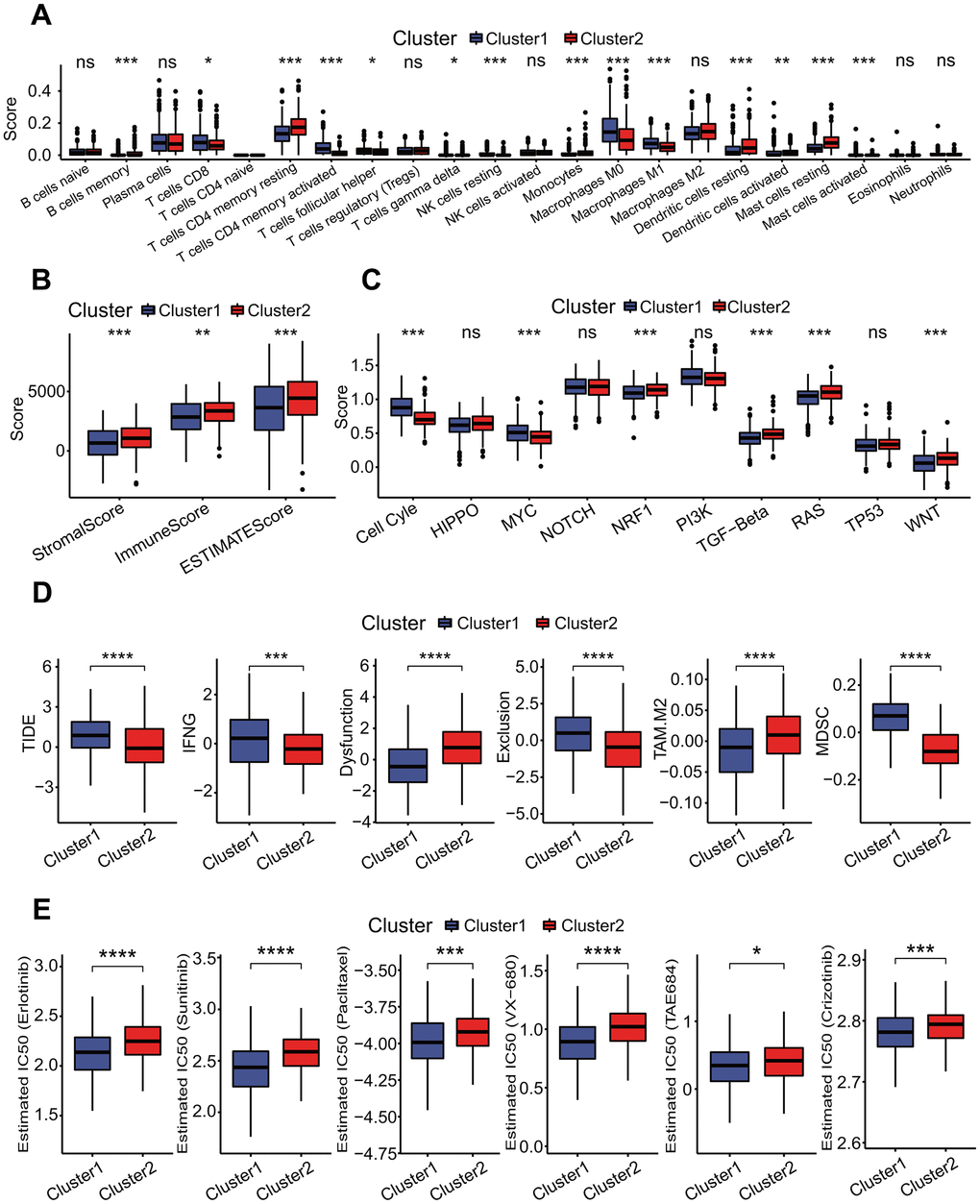
Figure 7. Correlations of molecular prognostic subtypes with immunological features. (A) Infiltration comparisons of 22 immune cell types. The scores of some immune cell types had significant differences between two clusters, such as T cells CD4 memory resting, mast cells resting and dendritic cells resting with higher scores in the cluster 2. *p < 0.05, **p < 0.01 and ***p < 0.001. (B) Infiltration comparison of stromal and immune cells. The cluster 2 had higher stromal score, immune score and ESTIMATE score. *p < 0.05, **p < 0.01 and ***p < 0.001. (C) Comparisons of activities of 10 oncogenic signaling pathways. The scores of cell cycle, MYC, NRF1, TGF-beta, RAS and WNT pathways had significant differences between two clusters, and these oncogenic signaling pathways had lower activities in the cluster 2. *p < 0.05, **p < 0.01, ***p < 0.001. (D) Comparisons of tumor immune dysfunction and exclusion (TIDE) score, interferon-gamma (IFNG), T cell dysfunction (Dysfunction), T cell exclusion (Exclusion), tumor-associated macrophages M2 (TAM.M2) and myeloid-derived suppressor cells (MDSC). These terms had significant differences between two clusters, and TIDE, IFNG, Exclusion and MDSC in the cluster 1 were significantly higher than those in the cluster 2. Dysfunction and TAM. M2 in the cluster 1 were significantly lower than those in the cluster 2. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. (E) Comparisons of estimated IC50 values for 6 conventional chemotherapy agents. The estimated IC50 values for erlotinib, sunitinib, paclitaxel, VX-680, TAE684 and crizotinib in the cluster 1 were significantly lower than those in the cluster 2. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Six key DEGs between two MPSs were correlated with the prognosis in LUAD
To identify key DEGs associated with the survival of LUAD patients between two MPSs, a comprehensive analysis including DEGA, UCRA, LASSO and MCRA was performed. The DEGA result showed that totals of 476 upregulated and 557 downregulated genes were identified in the C1 subtype compared with the C2 subtype (Figure 8A and Supplementary Table 4). Compared with the DEGs obtained from LUAD and normal lung tissues, a total of 821 dysregulated DEGs was common (Figure 8B and Supplementary Table 5). The UCRA showed that 258 of 821 DEGs had significant prognostic values (Supplementary Table 6). The LASSO analysis showed that 10 (CENPH, RHOV, ANLN, MDFI, TPSB2, CPS1, ANGPTL4, CCL20, CENPK, GJB3) of 258 DEGs with UCRA prognostic value had the most important feature when lambda was equal to 0.0606 (Figure 8C). The MCRA by AIC criterion showed that 6 (TPSB2, CPS1, ANGPTL4, CCL20, CENPK, GJB3) of 10 DEGs with the most important feature had the greatest fitting degree. Except for TPSB2, the expressions of the other five prognostic DEGs (CPS1, ANGPTL4, CCL20, CENPK, GJB3) were significantly negatively correlated with the survival of LUAD patients (all p < 0.001, Figure 8D) and their expressions were significantly upregulated in LUAD tissues (all p < 0.001, Figure 8E) and in the C1 subtype (all p < 0.001, Figure 8F). The expression analysis based on the GEPIA (gene expression profiling interactive analysis), real transcriptome data and GSE19804 datasets showed that the expression levels of 6 prognostic DEGs were consistent with those from the TCGA dataset between LUAD and normal lung tissues (Figure 8G–8I and Supplementary Table 3). Except for CPS1, each of the other five prognostic DEGs had a low mutation rate (Figure 9A) and a lower co-occurrence and mutually exclusive (Figure 9B).
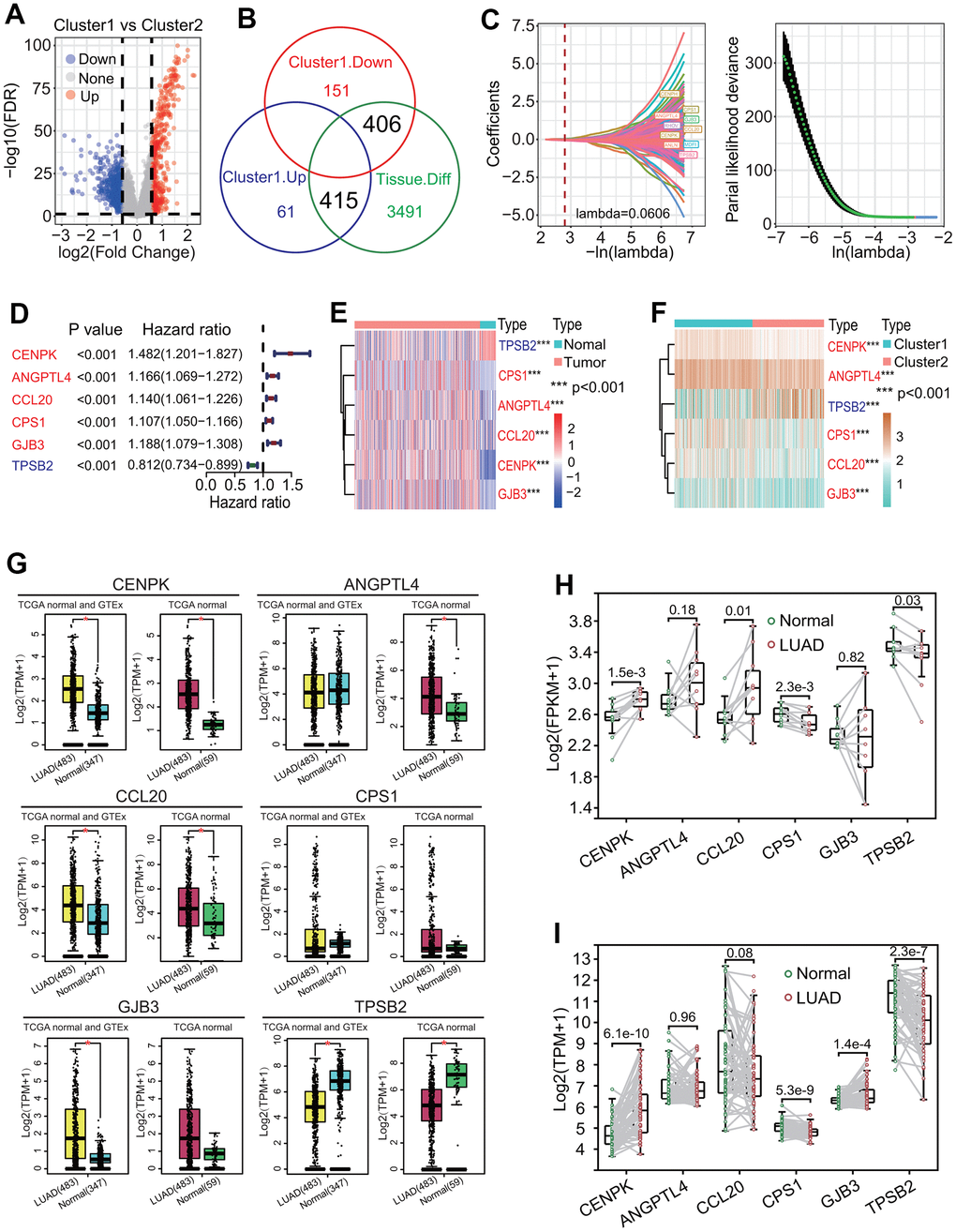
Figure 8. Identification of key differentially expressed genes (DEGs) between two clusters. (A) Distribution of DEGs between two clusters. Totals of 466 upregulated and 557 downregulated DEGs were identified in the cluster 1. (B) Identification of key DEGs between two clusters. An overlap analysis showed that totals of 821 dysregulated DEGs were important between two clusters. (C) LASSO Cox analysis. A 10-round cross validation was performed to prevent overfitting and 10 DEGs (CENPH, RHOV, ANLN, MDFI, TPSB2, CPS1, ANGPTL4, CCL20, CENPK, GJB3) had the most important feature when lambda was equal to 0.0606. (D) Univariate Cox regression analysis. Six DEGs including TPSB2, CPS1, ANGPTL4, CCL20, CENPK and GJB3 had significant prognostic values (all p < 0.001). (E) Heat map of expressions of six DEGs between lung adenocarcinoma (LUAD) and normal lung tissues. Five DEGs including CPS1, ANGPTL4, CCL20, CENPK and GJB3 were upregulated expressed and TPSB2 was downregulated expressed in LUAD tissue. (F) Heat map of expressions of six DEGs between two clusters. Five DEGs including CPS1, ANGPTL4, CCL20, CENPK and GJB3 were upregulated expressed and TPSB2 was downregulated expressed in the cluster 1. (G–I) The expression levels of 6 prognostic genes between LUAD and normal lung tissues based on GEPIA (gene expression profiling interactive analysis), real RNA-seq and GSE19804 datasets.
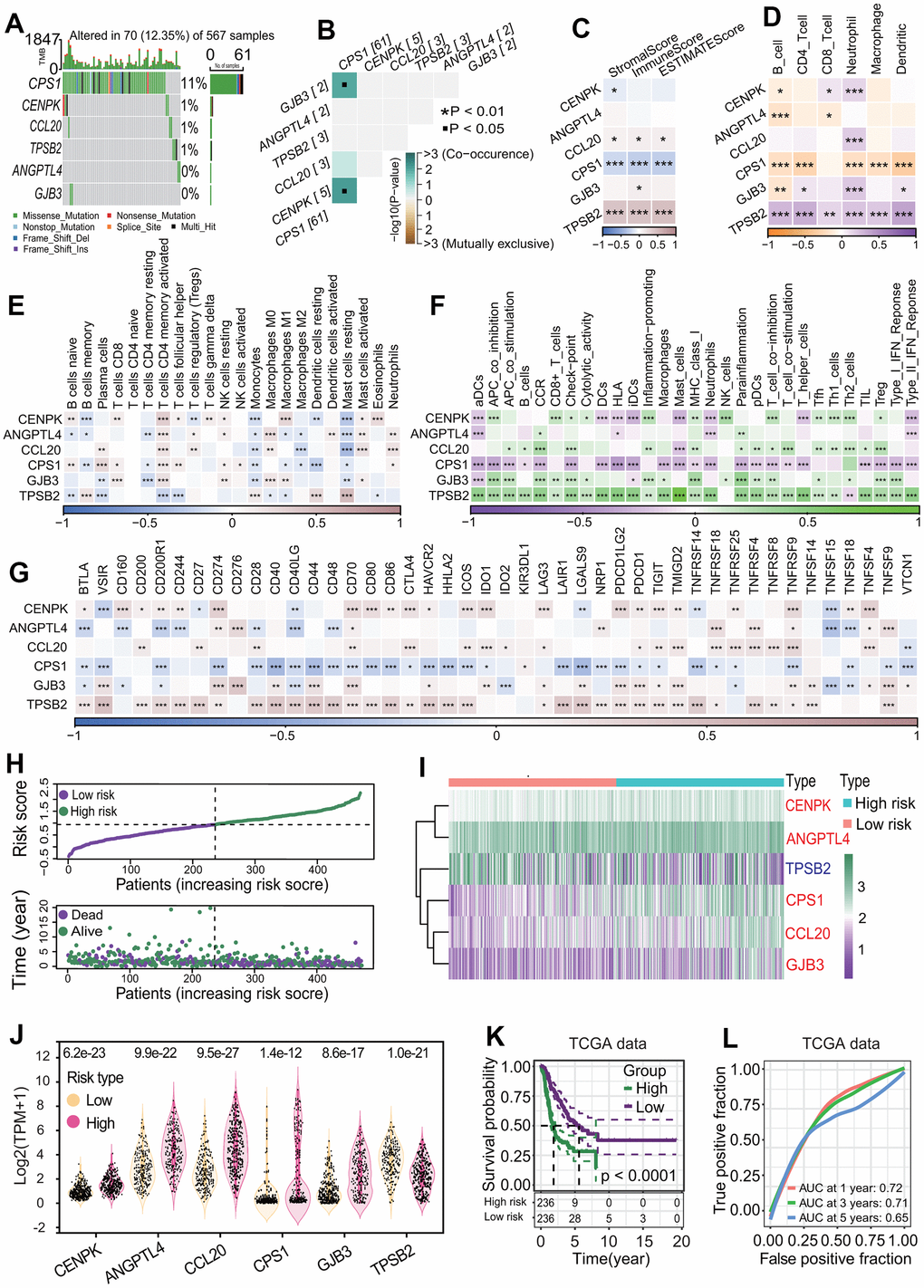
Figure 9. Correlation of six differentially expressed genes (DEGs) with immunity and establishment of six-gene prognostic signature. (A) Gene mutation profiles of six DEGs. Only 12.35% of LUAD samples had one or more mutations in six DEGs (CPS1, ANGPTL4, CCL20, CENPK, GJB3, TPSB2). (B) Co-occurrence and mutually exclusive of gene mutation. Six DEGs had lower co-occurrence and are mutually exclusive. (C) Correlation of six DEGs with stromal and immune cell infiltration. Two genes including CPS1 and TPSB2 were strongly negatively and positively correlated with three infiltration scores of two cells including stromal and immune cells, respectively. *p < 0.05, **p < 0.01 and ***p < 0.001. (D) Correlations of six DEGs with infiltrations of six immune cells. The expressions of six DEGs had significant correlations with one or more types of immune cells. In particular, CPS1 and TPSB2 were strongly negatively and positively correlated with immune infiltrations of six types of immune cells, respectively. *p < 0.05, **p < 0.01 and ***p < 0.001. (E) Correlations of six DEGs with 22 immune cell types. In general, the expressions of 6 DEGs (CPS1, ANGPTL4, CCL20, CENPK, GJB3, TPSB2) were strongly correlated with 22 immune cell types. *p < 0.05, **p < 0.01 and ***p < 0.001. (F) Correlations of six DEGs with 29 immune cell types. The expressions of six genes had very strong correlations with multiple immune cell types. In particular, CPS1 and TPSB2 were strongly negatively and positively correlated with immune infiltrations of 29 types of immune cells, respectively. *p < 0.05, **p < 0.01 and ***p < 0.001. (G) Correlations of six DEGs with 44 immune checkpoint genes. The expressions of six DEGs had very strong correlations with the expressions of multiple checkpoint genes. Especially, CPS1 and TPSB2 were strongly negatively and positively correlated with most checkpoint genes, respectively. *p < 0.05, **p < 0.01 and ***p < 0.001. (H) Risk score distribution and survival overview of LUAD patients. According to the median risk score, LUAD patients were divided into high- and low-risk subgroups. (I, J) Expression levels of six prognostic genes between two risk subgroups. Five genes including CENPK, ANGPTL4, CPS1, CCL20 and GJB3 were highly expressed and TPSB2 gene were lowly expressed in the high-risk subgroup. (K) Survival curve. LUAD patients in the low-risk subgroup had a higher OS rate than that in the high-risk subgroup (p < 0.0001). (L) Receiver operating characteristic (ROC) curve. The areas under the curve (AUCs) associated with 1-year, 3-year and 5-year survival were 0.72, 0.71 and 0.65, respectively.
Six-gene prognostic signature is robust in predicting the prognosis in LUAD
To further explore the correlation of six key prognostic DEGs with the survival, a six-gene prognostic model was constructed using the TCGA-LUAD dataset. In the light of the MCRA method by AIC criterion, the regression coefficient of each of six DEGs was calculated and the risk score was formulated:
The risk score of each patient was calculated by the risk score formula. According to the median risk score, LUAD patients were divided into high-risk and low-risk subgroups (Figure 9H). Except for TPSB2, the other five prognostic DEGs including CENPK, ANGPTL4, CPS1, CCL20 and GJB3 were significantly upregulated in the high-risk subgroup (all p < 0.05, Figure 9I, 9J). The LUAD patients in the high-risk subgroup had a poorer OS rate than those in the low-risk subgroup (p < 0.0001, Figure 9K), and the AUCs of 1-year, 3-year and 5-year correlated with the survival were separately 0.72, 0.71 and 0.65 in the ROC curve (Figure 9L). Survival analysis based on eight independent GEO-LUAD datasets showed that the OS rate of LUAD patients in the low-risk subgroup was significantly higher than that in the high-risk subgroup for each GEO-LUAD dataset (GSE3141, p = 0.0045, Figure 10A; GSE72094, p < 0.0001, Figure 10B; GSE26939, p = 0.0038, Figure 10C; GSE30219, p = 0.021, Figure 10D; GSE31210, p = 0.016, Figure 10E; GSE37745, p = 0.0099, Figure 10F; GSE50081, p < 0.0001, Figure 10G; GSE29016, p = 0.00013, Figure 10H). The AUCs of 5-year associated with the survival were 0.67 (GSE3141, Figure 10A), 0.87 (GSE72094, Figure 10B), 0.65 (GSE26939, Figure 10C), 0.7 (GSE30219, Figure 10D), 0.67 (GSE31210, Figure 10E), 0.64 (GSE37745, Figure 10F), 0.7 (GSE50081, Figure 10G) and 0.78 (GSE29016, Figure 10H) for eight independent datasets, respectively.
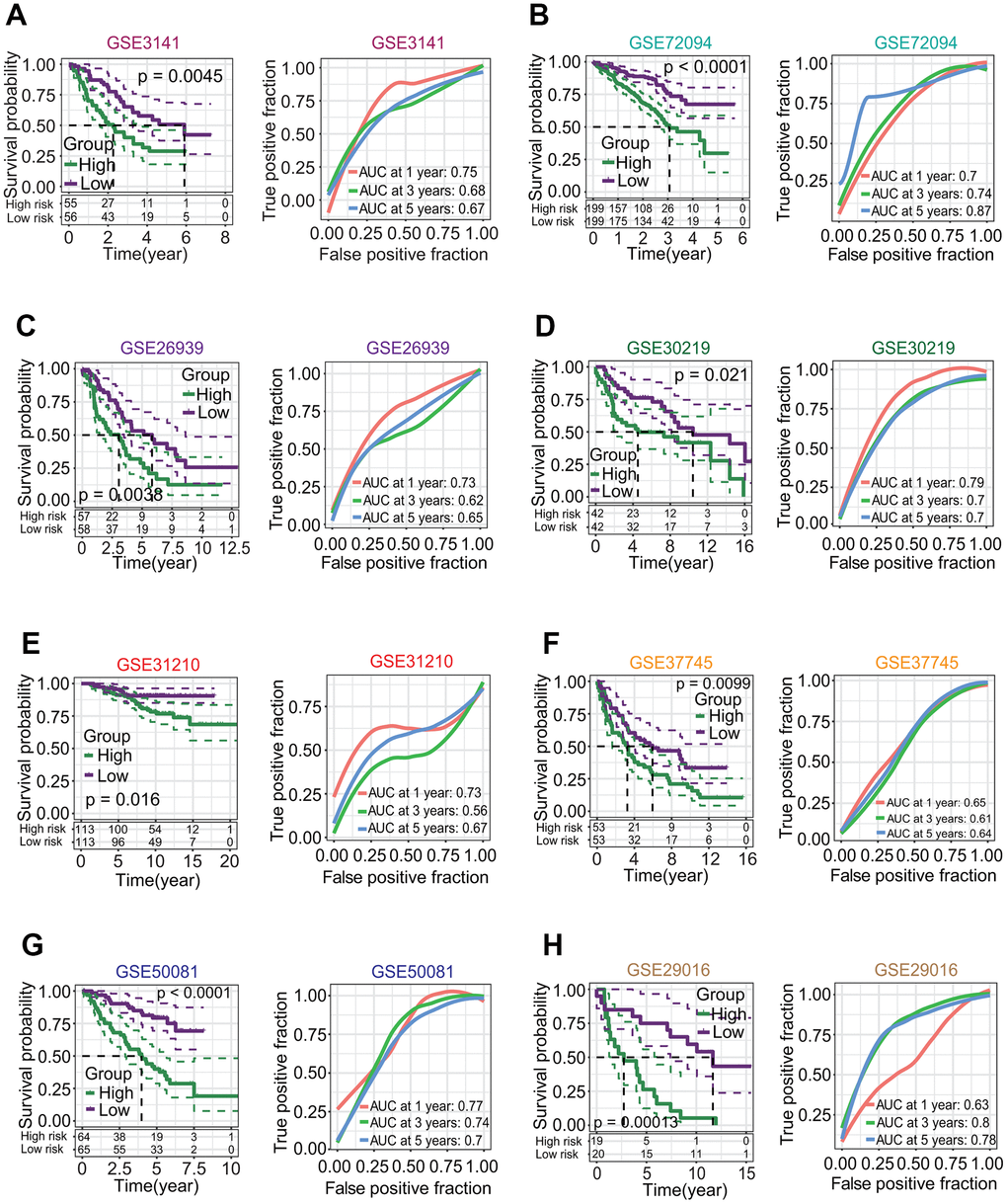
Figure 10. Correlations of six-gene signature with overall survival (OS) of patients in lung adenocarcinoma (LUAD). (A–H) Survival analysis based on eight independent LUAD datasets from the gene expression omnibus (GEO) database. Eight GEO-LUAD datasets were GSE3141, GSE72094, GSE26939, GSE30219, GSE31210, GSE37745, GSE50081 and GSE29016. For each dataset, LUAD patients in the low-risk subgroup had a higher OS rate than those in high-risk subgroup (p = 0.0045, < 0.0001, = 0.0038, = 0.021, = 0.016, = 0.0099, < 0.0001, = 0.00013, respectively), and the AUCs associated with 5-year survival were 0.67, 0.87, 0.65, 0.7, 0.67, 0.64, 0.7 and 0.78, respectively.
To further evaluate the robustness of six-gene prognostic signature in predicting the prognosis, we compared the prognostic significance and predictive performance of six-gene prognostic signature with 19 reported prognostic signatures [26–45] (Supplementary Figures 1–3). The results showed that the six-gene prognostic signature in this study had the highest AUC values associated with 1-year and 3-year survival, and the next highest AUC value associated with 5-year survival (Figure 11A). Six-gene prognostic signature had the highest concordance index (C-index) 0.673 (Figure 11B) and hazard ratio (HR) 2.718 of restricted mean survival (RMS) time (Figure 11C).
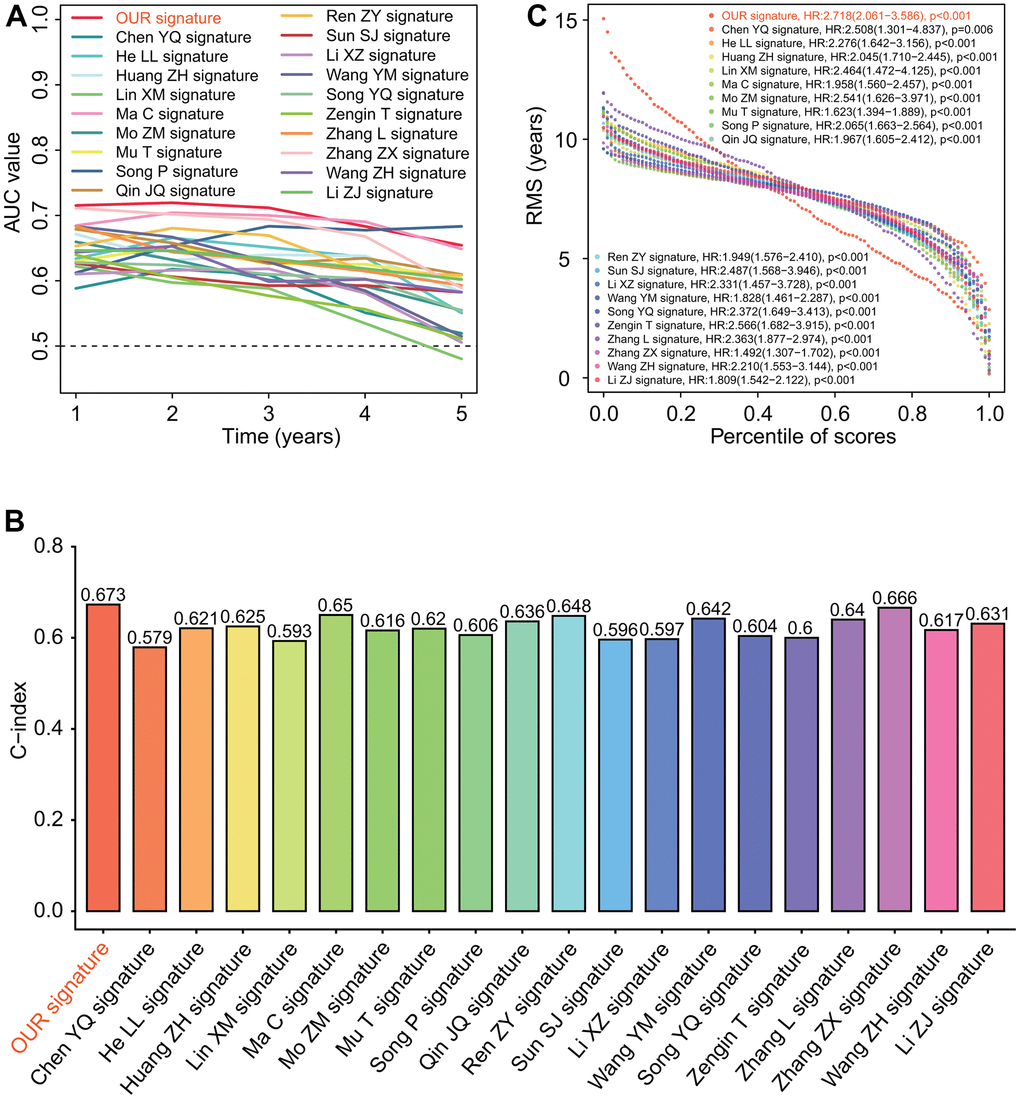
Figure 11. Predictive performances of 20 prognostic signatures. (A) AUC (area under the curve of receiver operating characteristic) change curve. Six-gene prognostic signature had the highest AUC values associated with 1-year and 3-year survivals and the next highest AUC value associated with 5-year survival. (B) Concordance index (C-index). Six-gene prognostic signature had the highest C-index 0.673 among 20 prognostic signatures. (C) Restricted mean survival (RMS) curve. Six-gene prognostic signature we developed had the highest hazard rate among 20 prognostic signatures.
Risk score was correlated with clinical and biological features
To investigate the relationships of risk score with clinical and biological features, these features between the high- and low-risk subgroups were compared. The results showed that there were significant differences in the MPS (chi-square p = 0), T staging (chi-square p = 1e-04), N staging (chi-square p = 1e-04), survival status (chi-square p = 0) and cancer status (chi-square p = 0.0023) between the two risk subgroups (Figure 12A). The risk scores for LUAD patients in the C2 subtype were significantly lower than those in the C1 subtype (p < 0.0001, Figure 12B). The risk scores for LUAD patients with stage N0 were significantly lower than those for patients with stage N1 and patients with stage N2 (p < 0.001 and < 0.0001, respectively, Figure 12C). Similarly, the risk scores for LUAD patients with stage T1 were significantly lower than those for patients with stage T2 and patients with stage T3 (both p < 0.0001, Figure 12F). The risk scores for LUAD patients in the living subgroup were significantly lower than those in the dead subgroup (p < 0.0001, Figure 12G). The risk scores for patients with tumor free were significantly lower than those for patients with tumor and patients with discrepancy (p < 0.0001 and < 0.05, respectively, Figure 12H).
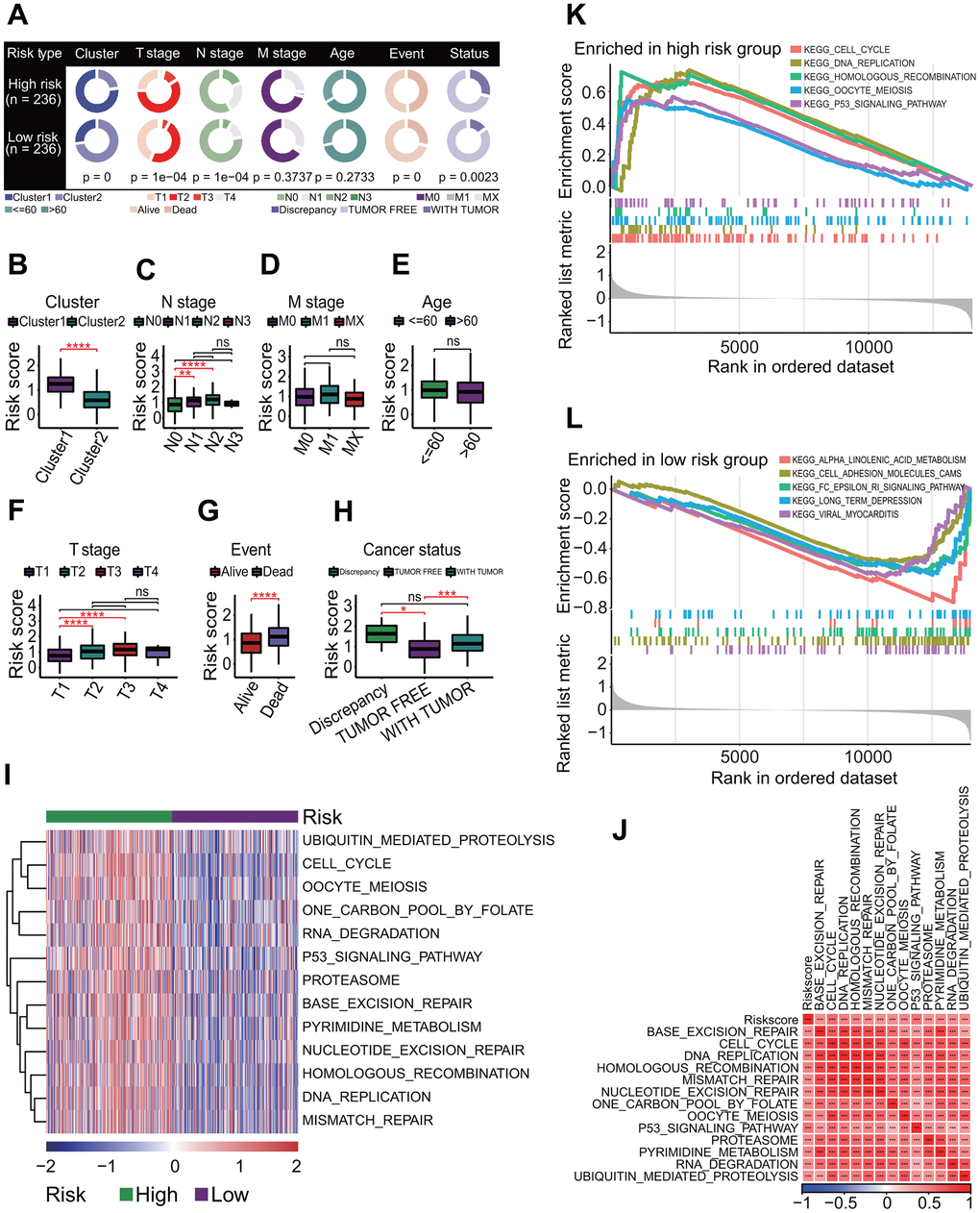
Figure 12. Correlations of risk score with clinical characteristics and biological pathways. (A) Correlations of risk score with clinical characteristics. Risk score was significantly correlated with prognostic cluster (p = 0), T staging (p = 1e-04), N staging (p = 1e-04), survival status (p = 0) and cancer status (p = 0.0023). (B) Comparisons of risk scores for patients between two clusters. Risk score in the cluster 1 was significantly higher than that in the cluster 2. ****p < 0.0001. (C) Comparisons of risk scores for patients between four N stages. Risk scores of patients with stage N0 were significantly lower than those with stage N1 and those with stage N2. **p < 0.01 and ***p < 0.001. (D) Comparisons of risk scores for patients between three M stages. The risk scores had no significant differences between three M stages. (E) Comparison of risk scores for patients between <= 60 and > 60 age subgroups. The risk scores had no significant differences between two age subgroups. (F) Comparisons of risk scores for patients between four T stages. Risk scores of patients with stage T1 were significantly lower than those with stage T2 and those with stage T3. ****p < 0.0001. (G) Comparison of risk scores for patients between two survival statuses. The risk scores for the living LUAD patients were significantly lower than those for dead patients. ****p < 0.0001. (H) Comparisons of risk scores for patients between three cancer statuses. The risk scores for patients with tumor free were significantly lower than those with discrepancy tumor. *p < 0.05 and ***p < 0.001. (I) Gene set variation analysis. At the correlation of risk score with KEGG pathway > 0.4 or < -0.4 and p < 0.001, thirteen KEGG pathways were positively correlated with risk score. (J) Correlations of risk score with KEGG pathways. There was a strong positive correlation between risk score and 13 KEGG pathways. ***p < 0.001. (K) Top 5 KEGG pathways enriched in the high-risk group. Five KEGG pathways were separately cell cycle, DNA replication, homologous recombination, oocyte meiosis and P53 signaling pathway. (L) Top 5 KEGG pathways enriched in the low-risk group. Five KEGG pathways were separately ALPHA linolenic acid metabolism, cell adhesion molecules (CAMs), Fc epsilon RI signaling pathway, long term depression and viral myocarditis.
The GSVA result showed that 121 KEGG pathways associated with risk score were significantly enriched (p < 0.01, Supplementary Table 7). Thirteen KEGG pathways with a |cor| > 0.4 were found to be more active in the high-risk subgroups, including two key OSPs cell cycle (p = 0 and cor = 0.5975) and P53 signaling pathway (p = 0 and cor = 0.5202, Figure 12I). There were extremely strong positive correlations between the risk score and the activities of 13 KEGG pathways (all p < 0.001, Figure 12J). The GSEA result showed that 17 KEGG pathways were significantly enriched (adjusted p < 0.05, Supplementary Table 8). Among these pathways, 9 and 8 KEGG pathways in the high-risk subgroup were significantly upregulated and downregulated, respectively (Supplementary Table 8). Except for upregulated cytokine-cytokine receptor interaction pathway, the other 16 pathways were observed in the pathway list enriched by the GSVA method (Supplementary Tables 7, 8). Top 5 upregulated pathways were cell cycle (adjusted p = 1.82e-08), DNA replication (adjusted p = 0.001377), homologous recombination (adjusted p = 0.023061), oocyte meiosis (adjusted p = 0.004126) and P53 signaling pathway (adjusted p = 0.023061) (Figure 12K), and the top 5 downregulated pathways were alpha-linolenic acid metabolism (adjusted p = 0.030427), cell adhesion molecules (CAMs, adjusted p = 0.030427), Fc epsilon RI signaling pathway (adjusted p = 0.030427), long-term depression (adjusted p = 0.030855) and viral myocarditis (adjusted p = 0.022938) (Figure 12L).
Risk score is an independent prognostic factor in LUAD
To evaluate the independent predictive capability of six-gene signature in predicting the survival of LUAD patients, an independent prognostic analysis was performed. A UCRA-based analysis showed that the risk score (p = 0.000, HR = 2.631, 95% CI = 1.927-3.541), cluster (subtype) (p = 0.016, HR = 0.698, 95% CI = 0.521-0.936), T staging (p = 0.002, HR = 1.904, 95% CI = 1.262-2.872) and N staging (p = 0.001, HR = 1.638, 95% CI = 1.217-2.205) had significant prognostic values (Figure 13A). Furthermore, a MCRA-based analysis showed risk score (p = 0.000, HR = 2.57, 95% CI = 1.825-3.621), T staging (p = 0.041, HR = 1.548, 95% CI = 1.018-2.352) and N staging (p = 0.048, HR = 1.366, 95% CI = 1.033-1.862) had significant prognostic values (Figure 13B).
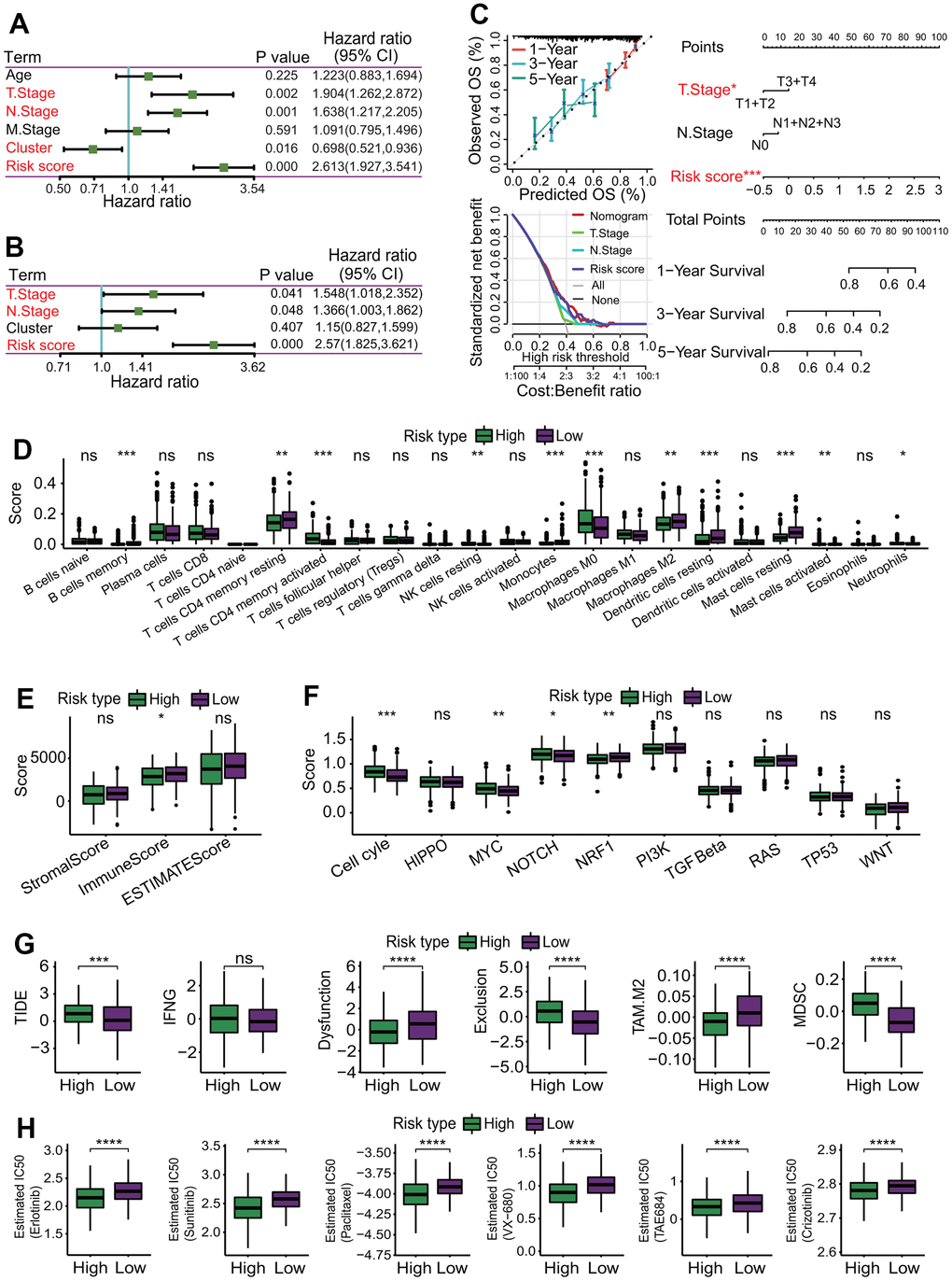
Figure 13. Independent prognostic analysis and correlations of risk score with immunological features. (A) Independent prognostic analysis for risk score using univariate Cox regression analysis. Risk score was significantly correlated with the overall survival of LUAD patients, and could serve as an independent prognosticator in predicting the survival of LUAD patients. (B) Independent prognostic analysis for risk score using multivariate Cox regression analysis. Risk score was significantly correlated with the overall survival of LUAD patients, and could serve as an independent prognosticator in predicting the survival of LUAD patients. (C) Construction of a nomogram predicting the survival. A nomogram for predicting 1-year, 3-year and 5-year overall survival was constructed. (D) Comparisons of 22 immune cell types between high- and low-risk subgroups. Some immune cell types had significant differences between two risk subgroups. *p < 0.05, **p < 0.01 and ***p < 0.001. (E) Comparison of immune infiltration score. LUAD patients in the low-risk subgroup had higher immune scores than those in the high-risk subgroup. *p < 0.05. (F) Comparisons of activities of 10 oncogenic signaling pathways between two risk subgroups. The activities of cell cycle, MYC, NOTCH and NRF1 pathways had significant differences, and three oncogenic signaling pathways (cell cycle, MYC, NOTCH) had higher activities in the high-risk subgroup. *p < 0.05, **p < 0.01 and ***p < 0.001. (G) Comparisons of tumor immune dysfunction and exclusion (TIDE) score, interferon-gamma (IFNG), T cell dysfunction (Dysfunction), T cell exclusion (Exclusion), tumor-associated macrophages M2 (TAM.M2) and myeloid-derived suppressor cells (MDSC). Except IFNG, those terms had significant differences between two risk subgroups, and TIDE, Exclusion and MDSC in the high-risk subgroup were significantly higher than those in the low-risk subgroup. Dysfunction and TAM. M2 in the high-risk subgroup were significantly lower than those in the low-risk subgroup. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. (H) Comparisons of estimated IC50 values for 6 conventional chemotherapy agents. The estimated IC50 values for 6 chemotherapy agents including erlotinib, sunitinib, paclitaxel, VX-680, TAE684 and crizotinib in the high-risk subgroup were significantly lower than those in the low-risk subgroup. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
To better assess the survival of LUAD patients by incorporating various prognostic factors, a nomogram including the risk score, T staging and N staging was constructed to predict the 1-year, 3-year and 5-year OS rate of LUAD patients (Figure 13C). The calibration curve showed that the actual OS rate was basically in line with the predicted value (Figure 13C). The decision curve showed that the nomogram model had a better predictive ability in predicting the survival of LUAD patients (Figure 13C).
Quantitative real-time PCR validated the expression of prognostic genes
To validate the expression of prognostic genes between LUAD and normal lung tissues, three prognostic genes including CIT, CPS1 and TPSB2 were selected to perform the expression analysis using quantitative real-time PCR (qPCR) method. The result showed that the expression of three genes had a consistent trend with RNA-seq result (Figures 3, 8H, 14A).
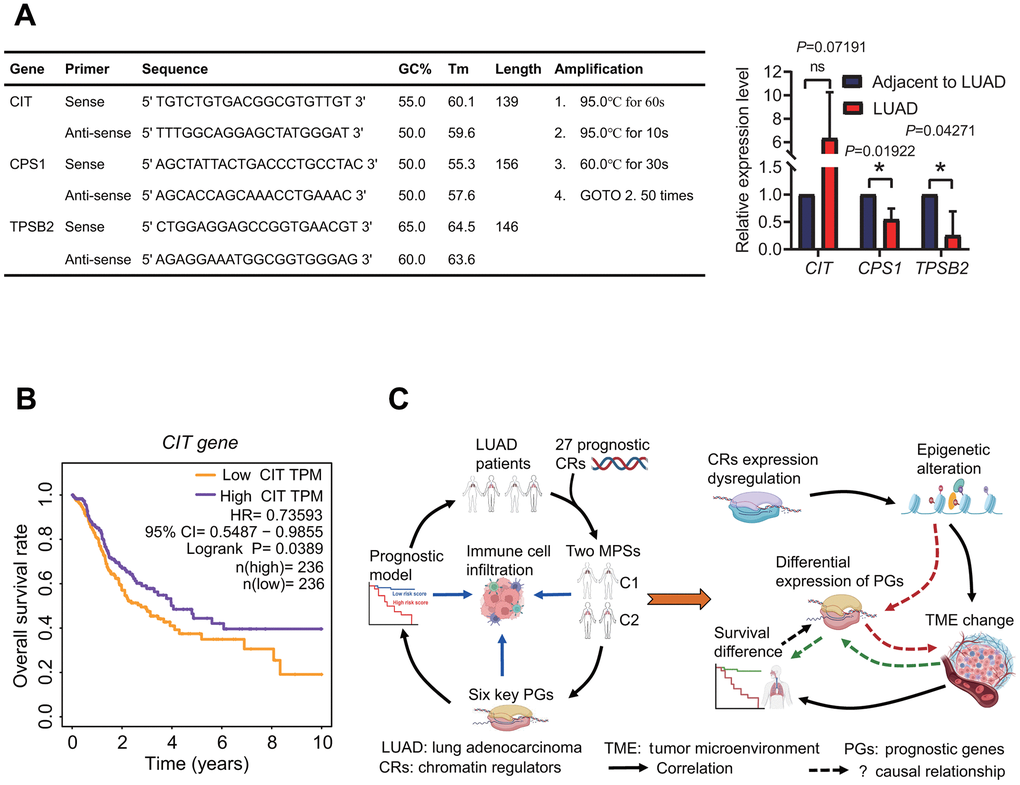
Figure 14. Expression of prognostic genes and underlying prognostic mechanism. (A) Expression of prognostic genes. CPS1 and TPSB2 genes were lowly expressed in LUAD tissues by a quantitative real-time PCR method. (B) Survival curve. The high expression of CIT gene resulted in a better overall survival rate in patients with lung adenocarcinoma. (C) Prognostic results and potential prognostic mechanism based on prognostic chromatin regulators. According to the prognostic results and existing literature, the potential prognostic mechanism was delineated.
Discussion
Developing a robust prognostic signature is very important to effectively predict the survival of LUAD patients. In recent years, many types of molecular prognostic signatures have been developed, including protein, mRNA, miRNA and lncRNA [6–10]. Some signatures have also showed better predictive capability [38, 39, 42, 45]. However, the high heterogeneity of LUAD suggests that some new prognostic subtypes and signatures must be developed to refine the risk stratification. CRs, as the key participators and even drivers in tumorigenesis, have been shown to have the potential as a robust prognostic signature in some cancers, such as bladder cancer and hepatocellular carcinoma [46, 47]. However, little is known about the roles of CRs in LUAD biology and few studies have investigated the prognostic role in LUAD by a comprehensive analysis. In this study, we first constructed and evaluated two MPSs based on CRs, and established and assessed a six-gene prognostic signature based on two MPSs. Subsequently, the correlations of MPS, key prognostic DEGs and risk score with TICs were systematically investigated. Last, the correlations of MPS and risk score with immunotherapeutic and chemotherapeutic responses were predicted.
Through a comprehensive analysis, 27 DECRs were identified to have association with the survival of LUAD patients and used to construct MPSs. Among these 27 prognostic DECRs, 23 risk and 4 protective CRs were identified. At present, many of these 27 prognostic CRs we identified have been shown to have correlations with prognosis in some cancers [48–52]. For example, CBX6 and CBX7 were identified as prognostic biomarkers in bladder cancer [48]. PBK was found to be associated with glioblastoma and oral squamous cell carcinoma treated with radiotherapy [49, 50]. TTK was identified as a prognostic biomarker in NSCLC [51]. BUB1 was found as a prognostic factor for hepatocellular carcinoma [52]. In addition, some new prognostic CRs have also been identified in LUAD, such as NPAS2 and HMGA2 [53, 54]. In this study, an interesting finding is that CIT was highly expressed as a protective gene in LUAD tissue, and the high expression of CIT was significantly associated with a high OS rate (Figure 14B). CIT was further found to be significantly overexpressed in the C2 subtype with a high OS rate (Figures 4, 5A). Despite this, we have a little confusion about the correlation between the CIT expression and the OS in LUAD patients. CIT (Citron Rho-Interacting Serine/Threonine Kinase) is a gene encoding a serine/threonine-protein kinase that functions in cell division. Several recent studies have reported the potential role of CIT gene in the initiation and progression of some tumors [55–57]. The upregulation of CIT could promote the growth of cancer cells [56, 57], and was associated with a poor outcome in some cancers such as bladder cancer and pancreatic ductal adenocarcinomas [58, 59]. On the contrary, some studies showed that the upregulation of CIT resulted in a better time to progression in some other cancers such as ovarian carcinomas [60]. From the above, we speculated that whether CIT is a risk gene or a protective gene might be related to cancer type. In this study, although we identified that CIT was a protective gene in LUAD, the prognostic mechanism still needs to be further revealed.
In the six-gene prognostic signature, CENPK (Centromere Protein K) is a subunit of CENP-H-I complex that maintains the proper kinetochore function and mitotic progression [61]. In the last five years, the role of CENPK has been gradually discovered in cancers and become a research hotspot. A few recent studies have reported that CENPK played roles in cervical cancer, thyroid cancer, ovarian cancer, etc. [62–64]. The overexpression of CENPK promoted cell proliferation and migration that was correlated with a poor prognosis [63, 64], and the expression downregulation of CENPK suppressed cell growth and inhibited cancer progression [65, 66]. So far, only a few studies were found to report that CENPK was upregulated in LUAD, and the overexpression of CENPK was associated with advanced LUAD and poor prognosis of LUAD patients [67]. Our result was consistent with those of the above findings, which further confirmed the prognostic role of CENPK in LUAD. ANGPTL4 (Angiopoietin like 4) encodes a glycosylated protein that plays roles in regulating lipid metabolism, glucose homeostasis and insulin sensitivity. In some cancers, the ANGPTL4 expression promoted venous invasion and cancer progression in breast cancer, thyroid cancer, colorectal cancer and gastric cancer [68–71], and was associated with a poor prognosis in breast cancer [68]. In addition, some studies have also showed that ANGPTL4 had tumor-suppressive role, even dual roles in some cancers such as urothelial carcinoma [72]. So far, little is known about the roles of ANGPTL4 in LUAD. Our result showed that ANGPTL4 was significantly upregulated in LUAD and was associated with a poor prognosis. CPS1 (Carbamoyl-Phosphate Synthase 1) encodes a crucial mitochondrial enzyme that catalyzes the synthesis of carbamoyl phosphate in the urea cycle. Several recent studies showed the associations of CPS1 with some cancers such as gastric cancer and ovarian cancer [73, 74]. Two recent published studies showed that the expression level of CPS1 was upregulated in LUAD tissue, and its high expression resulted in a poor survival rate [75, 76]. Some additional studies showed that lncRNA CPS1 intronic transcript 1 (CPS1-IT1) as a positive regulatory factor suppressed cell invasion and metastasis in colorectal cancer, ovarian cancer and LC [77–79]. The above studies indicated that CPS1 as a risk factor might play an important role and serve as a potential prognostic marker in LUAD. Our result was consistent with the above results, which supports the opinion of CPS1 as a potential prognosticator. CCL20 (C-C Motif Chemokine Ligand 20), belonging to the subfamily of small cytokine CC genes, plays important roles in the chemotaxis of some immune cells including dendritic cells, effector/memory T cells and B cells by the ligand-receptor pair CCL20-CCR6. Some recent published studies have demonstrated that CCL20 promoted cancer progression by some pathways such as by activating p38 pathway in laryngeal cancer [80], through PI3K pathway in LC and via NF-kappa B pathway in thyroid cancer [81, 82]. GJB3 (Gap Junction Protein Beta 3) belonging to the connexin gene family, encodes the Connexin 31. The mutations of GJB3 are mainly associated with non-syndromic deafness or erythrokeratodermia variabilis. To date, only a recent study was found to report that GJB3 as a predictor of a 9-mRNA prognostic signature was associated with the survival of LUAD patients and was identified as a protective factor [83]. What’s different was that our result showed that GJB3 was a risk gene in the 6-mRNA prognostic signature we identified. The reasons for the difference remain unclear, which forces us to explore more deeply the association of GJB3 with LUAD in the future. TPSB2 (Tryptase Beta 2) is the only protective factor in the prognostic signature established in this study. TPSB2 encoding the beta-tryptase, is mainly expressed in mast cells and is a common marker for mast cell. Several previous studies showed that TPSB2 was abnormally expressed or somatic genomic alterations occurred in some cancers including breast cancer and gastric cancer [84, 85]. A growing evidence now shows that TPSB2 expression is a potential prognostic marker in cancers including gastric cancer [85].
Notably, the gene expressions in the six-gene signature were significantly correlated with immune cell infiltration. Especially, the CPS1 expression was strongly negatively correlated with immune cell infiltration, while the TPSB2 expression was strongly positively correlated with immune cell infiltration. Furthermore, the risk score based on the six-gene signature was significantly correlated with immunological status of LUAD patients. It is well known that tumor immune microenvironment (TIME) plays important roles in cancer biology, and patients with differing TIME present differing immunotherapeutic responses and clinical outcomes [86–88]. Tumor immunophenotype has been gradually recognized as an independent prognostic factor to estimate the prognosis [89]. As a whole, tumor patients with higher immune infiltration levels had a higher OS rate [90, 91]. In this study, LUAD patients in the low-risk subgroup had lower risk scores and higher infiltration levels of some immune cell types including B cells memory, mast cells resting, dendritic cells resting, which was consistent with the results from previous published researches [90–92]. From the above researches, we speculate that the abnormal expression of six prognostic genes may have an influence on immune cell infiltration or immune infiltration affects their expression. However, we still do not know the potential causal relationship between them by reviewing a large amount of literature. Despite the important role of TIME in tumor biology, the potential correlations of immune cells infiltration with the prognosis remain ambiguous. Even the associations between the infiltration levels of some immune cells with the prognosis are paradoxical in different studies or subpopulations. For example, the correlations of increased mast cells with either good or poor prognosis depended on some factors such as tumor type and stage [93]. So, it is important to reveal the underlying prognostic mechanism of prognostic signature, which not only contributes to explaining the biological mechanism of cancer development and progression, but also offers a great help for evaluating the survival of tumor patients more accurately. In view of this, we summarized the results to delineate the underlying relationship between CRs, six-gene prognostic model, immunological features and the prognosis (Figure 14C). Now, it is clear that the epigenetics regulation and tumor microenvironment are two important factors affecting tumor formation and development, and there is a complex interactive relationship among cancer, epigenetics and immunity. CRs as indispensable upstream regulatory factors of epigenetics, drive the epigenetics alterations by acting as “writers”, “readers”, “erasers” of histone and DNA, or “remodelers” of chromatin. CRs have been shown to play key roles in driving cancer by numerous studies [16–19]. In this study, we hypothesize that the expression dysregulation of 27 CRs causes the epigenetic alteration in LUAD patients, which results in the TME changes. Different TMEs led to the differences in the survival and the expression of six prognostic genes in LUAD patients. Of course, the underlying prognostic mechanism must be verified on the basis of the correlations of the expressions between these genes, and of their expressions with immune cell infiltration and the OS rate of LUAD patients in the future.
In addition, a lot of signaling pathways were identified to have higher activities in LUAD patients with a poor prognosis by the GSAV and GSEA methods, including cell cycle and p53 signaling pathways. At present, a large number of studies have demonstrated that the dysregulated activity of many signaling pathways was closely associated with the occurrence and progression of tumor, and the landscape of pathway alterations in 33 cancer types have been charted in detail, including LUAD [94]. As far as we know, the cell cycle plays critical role in the regulation of mitotic cell cycle progression [95], and its alterations were the most common in many tumors, except for rare alterations in uveal melanoma, thymoma, testicular cancer and acute myeloid leukemia [94]. Some genes in the cell cycle have been identified to be associated with LC, and serve as potential prognostic markers including BUB1B, ZWINT, CDC20, etc. [96–98]. The high expression of these genes was often associated with a poor prognosis in some cancers including LUAD [96, 99–102]. Like the cell cycle, the p53 signaling pathway is one of the most common oncogenic pathways across various cancers [94], and one of its key functions is to regulate the cell cycle [103]. In fact, the p53 signaling pathway is connected to its downstream pathway cell cycle (https://www.kegg.jp/kegg/pathway.html). The high activities of the cell cycle and p53 pathways were associated with a worse prognosis in LUAD patients, indicating that these two signaling pathways contribute to LUAD progression. In addition, some pathways involved in the replication and repair were identified to be negatively correlated with the survival of LUAD patients, showing that the upregulation of these pathways are identical to the upregulation of the cell cycle and p53 signaling pathways. Currently, some therapeutic approaches targeting signaling pathways have been investigating or have been approved for therapy, such as cell cycle, PI3K, WNT, RAS and p53 signaling pathways [94]. And some therapeutic targets have been investigating and new drugs against these targets are being designed, such as cyclin-dependent kinases (CDKs) [104]. Understanding the alterations is critical for the development of new therapeutic approaches in the oncogenic signaling pathways, which will contribute to improve the care of LUAD patients. Meanwhile, the identification of oncogenic signaling pathways related to the survival will provide a valuable resource for LUAD precision medicine.
Although a six-gene prognostic signature was established based on MPSs constructed by CRs and the validity of MPSs and the robustness of the prognostic model have been well evaluated using some other independent datasets, some limitations should be mentioned. First, the current study was a retrospective study and required further validation in prospective clinical studies. Second, the underlying mechanism by which CRs predict the survival of LUAD patients through six prognostic genes needs further investigation.
Conclusions
In conclusion, the current study developed two MPSs based on CRs and established a new six-gene prognostic signature based on MPSs in LUAD. The MPS and risk score were significantly correlated with the TIME. These findings provide novel insights into the biological mechanism of CRs in LUAD biology, which may offer a help for building a more accurate prognosis assessment system and personalized immunotherapy system.
Materials and Methods
Collection of samples and clinical data
The gene expression datasets and clinical data for LUAD were retrieved from the public databases The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). The TCGA-LUAD dataset was used to construct the MPS and prognostic signature. The GEO-LUAD datasets including GSE31210, GSE30219, GSE37745, GSE50081, GSE42127, GSE3141, GSE26939, GSE29016, GSE68465, GSE72094 and GSE19804 were used to test the validity of MPS and the robustness of prognostic model. The main information for these datasets was listed in Table 2, including sample number, data platform, gender and survival status. All datasets have been approved by the Institutional Review Board of relevant participating institutions and no additional approval was required in this study.
Table 2. Main clinical information of datasets in this study.
| Datasets | Platform | Number | Gender | Age | Stage | Survival status | Application | |||||||
| Female | Male | <=60 | >60 | I and II | III and IV | Alive | Dead | |||||||
| TCGA | HT-seq | 472 | 255 | 217 | 146 | 326 | 378 | 86(8?) | 289 | 183 | Model construction | |||
| GSE31210 | GPL570 | 226 | 121 | 105 | 108 | 118 | 226 | 0 | 191 | 35 | Model evaluation | |||
| GSE30219 | GPL570 | 85 | 19 | 66 | 43 | 42 | / | / | 40 | 45 | Model evaluation | |||
| GSE37745 | GPL570 | 106 | 60 | 46 | 46 | 60 | 89 | 17 | 29 | 77 | Model evaluation | |||
| GSE50081 | GPL570 | 129 | 62 | 67 | 19 | 110 | 62 | 67 | 76 | 53 | Model evaluation | |||
| GSE42127 | GPL6884 | 133 | 65 | 68 | 38 | 95 | 111 | 22 | 90 | 43 | Model evaluation | |||
| GSE3141 | GPL570 | 111 | / | / | / | / | / | / | 52 | 59 | Model evaluation | |||
| GSE26939 | GPL9503 | 116 | 51 | 65(16?) | 40 | 76 | 71 | 45(30?) | 49 | 66(1?) | Model evaluation | |||
| GSE29016 | GPL6947 | 39 | 21 | 18 | 12 | 27 | 36 | 3(1?) | 10 | 29 | Model evaluation | |||
| GSE68465 | GPL96 | 443 | 220 | 223 | 147 | 296 | / | / | 207 | 236 | Model evaluation | |||
| GSE72094 | GPL15048 | 442 | 240 | 202 | 70 | 372(21?) | 334 | 108(28?) | 298 | 144(22?) | Model evaluation | |||
| GSE19804 | GPL570 | 60 pairs | 60 | 0 | 29 | 31 | 47 | 13 | / | / | Expression analysis | |||
| GEPIA database | HT-seq | 820 | / | / | / | / | / | / | / | / | Expression analysis | |||
| Real sequencing data | Hiseq2000 | 10 pairs | 5 | 5 | 10 | 0 | 10 | 0 | / | / | Expression analysis | |||
Production of transcriptome sequencing data
The transcriptomes of LUAD and normal lung tissues from 10 LUAD patients (5 male and 5 female) of 35-50 years old were sequenced using the Illumina sequencing platform with the paired-end method by Beijing Novogene Technology Co., Ltd. (China). All tissues were collected by The First People’s Hospital of Yunnan Province and informed consent was obtained from all LUAD patients. This study was approved by the Institutional Review Board of The First People’s Hospital of Yunnan Province (No. 2017YY227). The transcriptome sequencing data were used to evaluate the expression levels of prognostic genes between LUAD and normal lung tissues.
Acquisition of gene set
Totals of 870 CRs and 10 oncogenic signaling pathways (OSPs) were downloaded from the previous studies by Lu et al. [15] and by Sanchez-Vega et al. [94], respectively.
Identification of differentially expressed gene and functional analysis
Differentially expressed genes (DEGs) were identified using differentially expressed gene analysis (DEGA) based on the limma package (version 3.52.2) in the Bioconductor project (version 3.15, http://www.bioconductor.org/) [105]. An empirical Bayes (eBayes) method was used to assess the gene expression change by a moderated t-test. The Benjamini-Hochberg correction was used to adjust p-value for multiple testing.
Functional analysis based on KEGG pathway set was implemented using the gene set variation analysis (GSVA) method in the GSVA package (version 1.44.2) [106] and the gene set enrichment analysis (GSEA) method in the clusterProfiler package (version 3.14.3) [107], respectively. GSVA is a non-parametric and unsupervised gene set enrichment (GSE) method. GSVA estimates pathway activity variation over a sample population by characterizing pathways from a gene expression profiling. The non-parametric kernel estimation of the cumulative density function for each gene expression profile was performed using the Gaussian kernel. The enrichment score for each sample was calculated using GSVA method. The maximum and minimum number of genes was set at 100 and 10 in an output gene set, respectively. GSEA was performed using the default parameters. The maximum and minimum number of genes was set at 500 and 10, respectively. The permutation number was set at 1000 and multiple testing was corrected using the Benjamini-Hochberg method. The Wilcoxon’s test method was used to compare the difference of enrichment pathways between differing subgroups.
Construction of prognostic model
A univariate Cox regression analysis (UCRA) was applied to identify genes associated with the prognosis using the R survival package (version 3.2.13). The least absolute shrinkage and selection operator (LASSO) Cox regression analysis was used to regularize the genes with significant UCRA prognostic value. The basic idea behind the LASSO algorithm is that an added L1-norm regularization term is used to penalize the weight of variables in a linear model. The LASSO regularization term is defined as:
where
where Coe(Genei) is the MCRA Cox coefficient of the i gene and Exp(Genei) is the mRNA level of the i gene. On the basis of the median risk score, LUAD patients were divided into high-risk and low-risk subgroups. The Kaplan-Meier (KM) estimator and log-rank test were used to assess the OS rate of LUAD patients between two risk subgroups based on the R survival package (version 3.2.13). The receiver operating characteristic (ROC) analysis was used to evaluate the predictive capability for prognostic model using the R survivalROC package (version 1.0.3), and the area under curve (AUC) indicated the predictive accuracy of prognostic model in the ROC curve.
Identification of MPSs based on prognostic CRs
MPSs were identified based on the differentially expressed CRs (DECRs) with the UCRA prognostic value using an unsupervised consensus clustering algorithm. The consensus clustering method is one of the most commonly used methods for classifying cancer subtypes by adopting resampling strategy of different omics data sets. In this study, the partitioning around medoid (PAM) algorithm with the Spearman correlation distance metric was applied to the consensus clustering based on the gene expression profiles of 27 prognostic DECRs from 472 LUAD patients in the TCGA database, and performed 500 bootstraps each 80% resampling of LUAD patients. The unsupervised consensus clustering was performed using the R ConsensusClusterPlus package (version 1.60.0) [108]. The maximum number of clusters (k) is set to 10, and the optimal k value was assessed on the basis of the cumulative distribution function and consensus matrix.
TICs analysis
First, the immune infiltrations of 22 types of TICs were estimated between differing MPSs and between differing risk subgroups using the CIBERSORT (cell-type identification by estimating relative subsets of RNA transcript) method [109]. On the basis of gene expression profiles, the proportions of 22 types of TICs were quantified using the LM22 signature and 100 permutations by the CIBERSORT algorithm. The relative abundances of 22 types of TICs between differing subgroups were compared using the Wilcoxon rank sum test.
Second, the infiltration of immune and stromal cells between differing MPSs and between differing risk subgroups was assessed using the ESTIMATE (estimation of stromal and immune cells in malignant tumors) algorithm in the R estimate package (version 1.0.13) [110]. The fraction of immune and stromal cells was inferred using the gene expression signatures in LUAD samples and compared using the Wilcoxon rank sum test.
Last, tumor immune estimation resource (TIMER, http://cistrome.org/TIMER/) was used to assess the immune infiltrations of 6 types of TICs including dendritic cells, macrophages, neutrophils, CD8 T cells, CD4 T cells and B cells [111], and the correlations of TICs with risk genes were measured using the Pearson correlation coefficient.
Prediction of therapeutic response
The potential response of immune checkpoint blockade (ICB) therapy between differing subgroups was predicted using the Tumor Immune Dysfunction and Exclusion (TIDE) algorithm (http://tide.dfci.harvard.edu/) [112]. The TIDE score, interferon-gamma (IFNG) score, T cell dysfunction (Dysfunction) score, T cell exclusion (Exclusion) score, tumor-associated macrophages M2 (TAM.M2) score and myeloid-derived suppressor cells (MDSC) score were used to evaluate potential therapeutic responses. To further investigate potential responses of ICB therapy, the correlations of 44 immune checkpoint genes (ICGs) with risk genes were measured using the Pearson correlation coefficient [113].
Potential clinical chemotherapeutic responses of LUAD patients to conventional chemotherapy agents were predicted using the R pRRophetic package (version 0.5) [114]. The half-maximal inhibitory concentration (IC50) was used to evaluate the drug sensitivity.
Activity assessment of 10 OSPs
The activities of 10 OSPs including 335 genes between differing MPSs and between differing risk subgroups were assessed using the single sample gene set enrichment analysis (ssGSEA) in the R GSVA package (version 1.44.2) [106]. The 10 OSPs were separate cell cycle (15 genes), HIPPO (38 genes), MYC (13 genes), NOTCH (71 genes), NRF1 (3 genes), PI3K (29 genes), TGF-beta (7 genes), RAS (85 genes), TP53 (6 genes) and WNT (68 genes). A list of 335 genes can be downloaded by referring to a published article [94].
ssGSEA
The ssGSEA was used to assess the immune cell infiltration in LUAD tissues. A marker gene set for 29 types of immune cells was obtained from a published article [115]. The relative abundances of 29 types of immune cells were quantified using the ssGSEA method in the R GSVA package (version 1.44.2) [106].
Statistical analysis
All the statistical analyses of the data in this study were performed based on the open-source R software (version 4.1.3). The DEGA was performed to identify DEGs using a moderated t-test method, and a gene with an adjusted p < 0.05 and a |log2(FC)| > log2(1.5) was considered to significantly change in expression. The GSVA and GSEA were performed to investigate the function of DEGs using the Wilcoxon’s test method, and an adjusted p < 0.05 was considered to have significant functional change. An unsupervised consensus clustering algorithm was applied to identify MPSs. The UCRA was performed to identify genes associated with the prognosis, and a gene with a p < 0.05 was considered to have significant association with the survival. The association of prognostic signature with the OS was evaluated using the KM method, and a p < 0.05 was considered to have a significant association. Immune cell infiltrations between differing MPSs and between differing risk subgroups were compared using the Wilcoxon’s test method, and a p < 0.05 was considered to have a significant difference. The correlations of MPS and risk score with clinical features were compared using a chi-square test method, and a p < 0.05 was considered to have a significant correlation. The expression levels of prognostic genes were validated by a paired t test method between LUAD and normal lung tissues using transcriptome sequencing data, and a p < 0.05 was considered to have a significant change. The accuracy of transcriptome sequencing data was evaluated using the qPCR method, and the relative expression levels of genes were compared using an unpaired t test method and a p < 0.05 was considered to have a significant change in expression.
Supplementary Materials
Author Contributions
QC and JH conceived and designed the study. QC and HZ analyzed the data. JH provided experimental samples. QC wrote the paper. All authors have read and approved the final manuscript.
Acknowledgments
The authors acknowledge the GEO and TCGA databases for providing their platforms and contributors for providing their LUAD-related datasets.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Statement and Consent
The studies involving human participants were reviewed and approved by the Institutional Review Board of First People’s Hospital of Yunnan Province (No. 2017YY227) and informed consents were obtained from all LUAD patients. All datasets have been approved by the Institutional Review Board of relevant participating institutions.
Funding
This work was partially supported by the Yunnan Province Applied Basic Research Project (2016FB146) and the Fund from Health Commission of Yunnan Province (2017NS261). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71:209–49. https://doi.org/10.3322/caac.21660 [PubMed]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020; 70:7–30. https://doi.org/10.3322/caac.21590 [PubMed]
- 3. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018; 553:446–54. https://doi.org/10.1038/nature25183 [PubMed]
- 4. Hutchinson BD, Shroff GS, Truong MT, Ko JP. Spectrum of Lung Adenocarcinoma. Semin Ultrasound CT MR. 2019; 40:255–64. https://doi.org/10.1053/j.sult.2018.11.009 [PubMed]
- 5. Lim W, Ridge CA, Nicholson AG, Mirsadraee S. The 8th lung cancer TNM classification and clinical staging system: review of the changes and clinical implications. Quant Imaging Med Surg. 2018; 8:709–18. https://doi.org/10.21037/qims.2018.08.02 [PubMed]
- 6. Woodard GA, Jones KD, Jablons DM. Lung Cancer Staging and Prognosis. Cancer Treat Res. 2016; 170:47–75. https://doi.org/10.1007/978-3-319-40389-2_3 [PubMed]
- 7. Friboulet L, Olaussen KA, Pignon JP, Shepherd FA, Tsao MS, Graziano S, Kratzke R, Douillard JY, Seymour L, Pirker R, Filipits M, André F, Solary E, et al. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N Engl J Med. 2013; 368:1101–10. https://doi.org/10.1056/NEJMoa1214271 [PubMed]
- 8. Hu J, Wang T, Chen Q. Competitive endogenous RNA network identifies four long non-coding RNA signature as a candidate prognostic biomarker for lung adenocarcinoma. Transl Cancer Res. 2019; 8:1046–64. https://doi.org/10.21037/tcr.2019.06.09 [PubMed]
- 9. Chen Q, Wang X, Hu J. Systematically integrative analysis identifies diagnostic and prognostic candidates and small-molecule drugs for lung adenocarcinoma. Transl Cancer Res. 2021; 10:3619–46. https://doi.org/10.21037/tcr-21-526 [PubMed]
- 10. Kosaka T, Yatabe Y, Onozato R, Kuwano H, Mitsudomi T. Prognostic implication of EGFR, KRAS, and TP53 gene mutations in a large cohort of Japanese patients with surgically treated lung adenocarcinoma. J Thorac Oncol. 2009; 4:22–9. https://doi.org/10.1097/JTO.0b013e3181914111 [PubMed]
- 11. Dowler Nygaard A, Spindler KL, Pallisgaard N, Andersen RF, Jakobsen A. Levels of cell-free DNA and plasma KRAS during treatment of advanced NSCLC. Oncol Rep. 2014; 31:969–74. https://doi.org/10.3892/or.2013.2906 [PubMed]
- 12. Stankovic B, Bjørhovde HAK, Skarshaug R, Aamodt H, Frafjord A, Müller E, Hammarström C, Beraki K, Bækkevold ES, Woldbæk PR, Helland Å, Brustugun OT, Øynebråten I, Corthay A. Immune Cell Composition in Human Non-small Cell Lung Cancer. Front Immunol. 2019; 9:3101. https://doi.org/10.3389/fimmu.2018.03101 [PubMed]
- 13. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010; 31:27–36. https://doi.org/10.1093/carcin/bgp220 [PubMed]
- 14. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012; 150:12–27. https://doi.org/10.1016/j.cell.2012.06.013 [PubMed]
- 15. Lu J, Xu J, Li J, Pan T, Bai J, Wang L, Jin X, Lin X, Zhang Y, Li Y, Sahni N, Li X. FACER: comprehensive molecular and functional characterization of epigenetic chromatin regulators. Nucleic Acids Res. 2018; 46:10019–33. https://doi.org/10.1093/nar/gky679 [PubMed]
- 16. Mardinian K, Adashek JJ, Botta GP, Kato S, Kurzrock R. SMARCA4: Implications of an Altered Chromatin-Remodeling Gene for Cancer Development and Therapy. Mol Cancer Ther. 2021; 20:2341–51. https://doi.org/10.1158/1535-7163.MCT-21-0433 [PubMed]
- 17. Zhang Z, Wang Q, Chen F, Liu J. Elevated expression of HMGA1 correlates with the malignant status and prognosis of non-small cell lung cancer. Tumour Biol. 2015; 36:1213–9. https://doi.org/10.1007/s13277-014-2749-4 [PubMed]
- 18. Debaugny RE, Skok JA. CTCF and CTCFL in cancer. Curr Opin Genet Dev. 2020; 61:44–52. https://doi.org/10.1016/j.gde.2020.02.021 [PubMed]
- 19. Wong KK. DNMT1: A key drug target in triple-negative breast cancer. Semin Cancer Biol. 2021; 72:198–213. https://doi.org/10.1016/j.semcancer.2020.05.010 [PubMed]
- 20. Wong KK. DNMT1 as a therapeutic target in pancreatic cancer: mechanisms and clinical implications. Cell Oncol (Dordr). 2020; 43:779–92. https://doi.org/10.1007/s13402-020-00526-4 [PubMed]
- 21. Lai Q, Li Q, He C, Fang Y, Lin S, Cai J, Ding J, Zhong Q, Zhang Y, Wu C, Wang X, He J, Liu Y, et al. CTCF promotes colorectal cancer cell proliferation and chemotherapy resistance to 5-FU via the P53-Hedgehog axis. Aging (Albany NY). 2020; 12:16270–93. https://doi.org/10.18632/aging.103648 [PubMed]
- 22. Shinjo K, Yamashita Y, Yamamoto E, Akatsuka S, Uno N, Kamiya A, Niimi K, Sakaguchi Y, Nagasaka T, Takahashi T, Shibata K, Kajiyama H, Kikkawa F, Toyokuni S. Expression of chromobox homolog 7 (CBX7) is associated with poor prognosis in ovarian clear cell adenocarcinoma via TRAIL-induced apoptotic pathway regulation. Int J Cancer. 2014; 135:308–18. https://doi.org/10.1002/ijc.28692 [PubMed]
- 23. Li J, Wang Y, Wang X, Yang Q. CDK1 and CDC20 overexpression in patients with colorectal cancer are associated with poor prognosis: evidence from integrated bioinformatics analysis. World J Surg Oncol. 2020; 18:50. https://doi.org/10.1186/s12957-020-01817-8 [PubMed]
- 24. Deng H, Hang Q, Shen D, Ying H, Zhang Y, Qian X, Chen M. High Expression Levels of CDK1 and CDC20 in Patients With Lung Squamous Cell Carcinoma are Associated With Worse Prognosis. Front Mol Biosci. 2021; 8:653805. https://doi.org/10.3389/fmolb.2021.653805 [PubMed]
- 25. Chen C, Hua H, Han C, Cheng Y, Cheng Y, Wang Z, Bao J. Prognosis value of MGMT promoter methylation for patients with lung cancer: a meta-analysis. Int J Clin Exp Pathol. 2015; 8:11560–4. [PubMed]
- 26. Chen Y, Tang L, Huang W, Zhang Y, Abisola FH, Li L. Identification and validation of a novel cuproptosis-related signature as a prognostic model for lung adenocarcinoma. Front Endocrinol (Lausanne). 2022; 13:963220. https://doi.org/10.3389/fendo.2022.963220 [PubMed]
- 27. He L, Chen J, Xu F, Li J, Li J. Prognostic Implication of a Metabolism-Associated Gene Signature in Lung Adenocarcinoma. Mol Ther Oncolytics. 2020; 19:265–77. https://doi.org/10.1016/j.omto.2020.09.011 [PubMed]
- 28. Huang Z, Shi M, Zhou H, Wang J, Zhang HJ, Shi J-. Prognostic signature of lung adenocarcinoma based on stem cell-related genes. Sci Rep. 2021; 11:1687. https://doi.org/10.1038/s41598-020-80453-4 [PubMed]
- 29. Lin X, Zhou T, Hu S, Yang L, Yang Z, Pang H, Zhou X, Zhong R, Fang X, Yu Z, Hu K. Prognostic significance of pyroptosis-related factors in lung adenocarcinoma. J Thorac Dis. 2022; 14:654–67. https://doi.org/10.21037/jtd-22-86 [PubMed]
- 30. Ma C, Li F, Wang Z, Luo H. A Novel Immune-Related Gene Signature Predicts Prognosis of Lung Adenocarcinoma. Biomed Res Int. 2022; 2022:4995874. https://doi.org/10.1155/2022/4995874 [PubMed]
- 31. Mo Z, Yu L, Cao Z, Hu H, Luo S, Zhang S. Identification of a Hypoxia-Associated Signature for Lung Adenocarcinoma. Front Genet. 2020; 11:647. https://doi.org/10.3389/fgene.2020.00647 [PubMed]
- 32. Mu T, Li H, Li X. Prognostic Implication of Energy Metabolism-Related Gene Signatures in Lung Adenocarcinoma. Front Oncol. 2022; 12:867470. https://doi.org/10.3389/fonc.2022.867470 [PubMed]
- 33. Song P, Li W, Guo L, Ying J, Gao S, He J. Identification and Validation of a Novel Signature Based on NK Cell Marker Genes to Predict Prognosis and Immunotherapy Response in Lung Adenocarcinoma by Integrated Analysis of Single-Cell and Bulk RNA-Sequencing. Front Immunol. 2022; 13:850745. https://doi.org/10.3389/fimmu.2022.850745 [PubMed]
- 34. Qin J, Xu Z, Deng K, Qin F, Wei J, Yuan L, Sun Y, Zheng T, Li S. Development of a gene signature associated with iron metabolism in lung adenocarcinoma. Bioengineered. 2021; 12:4556–68. https://doi.org/10.1080/21655979.2021.1954840 [PubMed]
- 35. Ren Z, Hu M, Wang Z, Ge J, Zhou X, Zhang G, Zheng H. Ferroptosis-Related Genes in Lung Adenocarcinoma: Prognostic Signature and Immune, Drug Resistance, Mutation Analysis. Front Genet. 2021; 12:672904. https://doi.org/10.3389/fgene.2021.672904 [PubMed]
- 36. Sun S, Guo W, Wang Z, Wang X, Zhang G, Zhang H, Li R, Gao Y, Qiu B, Tan F, Gao Y, Xue Q, Gao S, He J. Development and validation of an immune-related prognostic signature in lung adenocarcinoma. Cancer Med. 2020; 9:5960–75. https://doi.org/10.1002/cam4.3240 [PubMed]
- 37. Li X, Dai Z, Wu X, Zhang N, Zhang H, Wang Z, Zhang X, Liang X, Luo P, Zhang J, Liu Z, Zhou Y, Cheng Q, Chang R. The Comprehensive Analysis Identified an Autophagy Signature for the Prognosis and the Immunotherapy Efficiency Prediction in Lung Adenocarcinoma. Front Immunol. 2022; 13:749241. https://doi.org/10.3389/fimmu.2022.749241 [PubMed]
- 38. Wang Y, Zhang G, Wang R. Six CT83-related Genes-based Prognostic Signature for Lung Adenocarcinoma. Comb Chem High Throughput Screen. 2022; 25:1565–75. https://doi.org/10.2174/1871520621666210713112630 [PubMed]
- 39. Song Y, Zhang J, Fang L, Liu W. Prognostic necroptosis-related gene signature aids immunotherapy in lung adenocarcinoma. Front Genet. 2022; 13:1027741. https://doi.org/10.3389/fgene.2022.1027741 [PubMed]
- 40. Zengin T, Önal-Süzek T. Analysis of genomic and transcriptomic variations as prognostic signature for lung adenocarcinoma. BMC Bioinformatics. 2020 (Suppl 14); 21:368. https://doi.org/10.1186/s12859-020-03691-3 [PubMed]
- 41. Zhang L, Zhang Z, Yu Z. Identification of a novel glycolysis-related gene signature for predicting metastasis and survival in patients with lung adenocarcinoma. J Transl Med. 2019; 17:423. https://doi.org/10.1186/s12967-019-02173-2 [PubMed]
- 42. Zhang Z, Zhu H, Wang X, Lin S, Ruan C, Wang Q. A novel basement membrane-related gene signature for prognosis of lung adenocarcinomas. Comput Biol Med. 2023; 154:106597. https://doi.org/10.1016/j.compbiomed.2023.106597 [PubMed]
- 43. Wang Z, Embaye KS, Yang Q, Qin L, Zhang C, Liu L, Zhan X, Zhang F, Wang X, Qin S. Establishment and validation of a prognostic signature for lung adenocarcinoma based on metabolism-related genes. Cancer Cell Int. 2021; 21:219. https://doi.org/10.1186/s12935-021-01915-x [PubMed]
- 44. Li Z, Zeng T, Zhou C, Chen Y, Yin W. A prognostic signature model for unveiling tumor progression in lung adenocarcinoma. Front Oncol. 2022; 12:1019442. https://doi.org/10.3389/fonc.2022.1019442 [PubMed]
- 45. Zhang M, Ma J, Guo Q, Ding S, Wang Y, Pu H. CD8+ T Cell-Associated Gene Signature Correlates With Prognosis Risk and Immunotherapy Response in Patients With Lung Adenocarcinoma. Front Immunol. 2022; 13:806877. https://doi.org/10.3389/fimmu.2022.806877 [PubMed]
- 46. Zhu K, Liu X, Deng W, Wang G, Fu B. Identification of a chromatin regulator signature and potential candidate drugs for bladder cancer. Hereditas. 2022; 159:13. https://doi.org/10.1186/s41065-021-00212-x [PubMed]
- 47. Chen J, Chen X, Li T, Wang L, Lin G. Identification of chromatin organization-related gene signature for hepatocellular carcinoma prognosis and predicting immunotherapy response. Int Immunopharmacol. 2022; 109:108866. https://doi.org/10.1016/j.intimp.2022.108866 [PubMed]
- 48. Li X, Li L, Xiong X, Kuang Q, Peng M, Zhu K, Luo P. Identification of the Prognostic Biomarkers CBX6 and CBX7 in Bladder Cancer. Diagnostics (Basel). 2023; 13:1393. https://doi.org/10.3390/diagnostics13081393 [PubMed]
- 49. Yu WN, Lin HF, Lee YI, Shia WC, Sung WW, Yeh CM, Lin YM. PBK Expression Is Associated With Prognosis of Patients With Oral Squamous Cell Carcinoma Treated With Radiotherapy: A Retrospective Study. Anticancer Res. 2021; 41:2177–82. https://doi.org/10.21873/anticanres.14991 [PubMed]
- 50. Dong C, Fan W, Fang S. PBK as a Potential Biomarker Associated with Prognosis of Glioblastoma. J Mol Neurosci. 2020; 70:56–64. https://doi.org/10.1007/s12031-019-01400-1 [PubMed]
- 51. Chen J, Wu R, Xuan Y, Jiang M, Zeng Y. Bioinformatics analysis and experimental validation of TTK as a biomarker for prognosis in non-small cell lung cancer. Biosci Rep. 2020; 40:BSR20202711. https://doi.org/10.1042/BSR20202711 [PubMed]
- 52. Qi W, Bai Y, Wang Y, Liu L, Zhang Y, Yu Y, Chen H. BUB1 predicts poor prognosis and immune status in liver hepatocellular carcinoma. APMIS. 2022; 130:371–82. https://doi.org/10.1111/apm.13219 [PubMed]
- 53. Yang B, Rong X, Jiang C, Long M, Liu A, Chen Q. Comprehensive analyses reveal the prognosis and biological function roles of chromatin regulators in lung adenocarcinoma. Aging (Albany NY). 2023; 15:3598–620. https://doi.org/10.18632/aging.204693 [PubMed]
- 54. Shi Q, Han S, Liu X, Wang S, Ma H. Integrated single-cell and transcriptome sequencing analyses determines a chromatin regulator-based signature for evaluating prognosis in lung adenocarcinoma. Front Oncol. 2022; 12:1031728. https://doi.org/10.3389/fonc.2022.1031728 [PubMed]
- 55. Hanicinec V, Brynychova V, Rosendorf J, Palek R, Liska V, Oliverius M, Kala Z, Mohelnikova-Duchonova B, Krus I, Soucek P. Gene expression of cytokinesis regulators PRC1, KIF14 and CIT has no prognostic role in colorectal and pancreatic cancer. Oncol Lett. 2021; 22:598. https://doi.org/10.3892/ol.2021.12859 [PubMed]
- 56. Wu Z, Zhu X, Xu W, Zhang Y, Chen L, Qiu F, Zhang B, Wu L, Peng Z, Tang H. Up-regulation of CIT promotes the growth of colon cancer cells. Oncotarget. 2017; 8:71954–64. https://doi.org/10.18632/oncotarget.18615 [PubMed]
- 57. Liu Z, Yan H, Yang Y, Wei L, Xia S, Xiu Y. Down-regulation of CIT can inhibit the growth of human bladder cancer cells. Biomed Pharmacother. 2020; 124:109830. https://doi.org/10.1016/j.biopha.2020.109830 [PubMed]
- 58. Shou J, Yu C, Zhang D, Zhang Q. Overexpression of Citron Rho-Interacting Serine/Threonine Kinase Associated with Poor Outcome in Bladder Cancer. J Cancer. 2020; 11:4173–80. https://doi.org/10.7150/jca.43435 [PubMed]
- 59. Cong L, Bai Z, Du Y, Cheng Y. Citron Rho-Interacting Serine/Threonine Kinase Promotes HIF1a-CypA Signaling and Growth of Human Pancreatic Adenocarcinoma. Biomed Res Int. 2020; 2020:9210891. https://doi.org/10.1155/2020/9210891 [PubMed]
- 60. Ehrlichova M, Mohelnikova-Duchonova B, Hrdy J, Brynychova V, Mrhalova M, Kodet R, Rob L, Pluta M, Gut I, Soucek P, Vaclavikova R. The association of taxane resistance genes with the clinical course of ovarian carcinoma. Genomics. 2013; 102:96–101. https://doi.org/10.1016/j.ygeno.2013.03.005 [PubMed]
- 61. Okada M, Cheeseman IM, Hori T, Okawa K, McLeod IX, Yates JR 3rd, Desai A, Fukagawa T. The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat Cell Biol. 2006; 8:446–57. https://doi.org/10.1038/ncb1396 [PubMed]
- 62. Lin X, Wang F, Chen J, Liu J, Lin YB, Li L, Chen CB, Xu Q. N6-methyladenosine modification of CENPK mRNA by ZC3H13 promotes cervical cancer stemness and chemoresistance. Mil Med Res. 2022; 9:19. https://doi.org/10.1186/s40779-022-00378-z [PubMed]
- 63. Li Q, Liang J, Zhang S, An N, Xu L, Ye C. Overexpression of centromere protein K (CENPK) gene in Differentiated Thyroid Carcinoma promote cell Proliferation and Migration. Bioengineered. 2021; 12:1299–310. https://doi.org/10.1080/21655979.2021.1911533 [PubMed]
- 64. Lee YC, Huang CC, Lin DY, Chang WC, Lee KH. Overexpression of centromere protein K (CENPK) in ovarian cancer is correlated with poor patient survival and associated with predictive and prognostic relevance. PeerJ. 2015; 3:e1386. https://doi.org/10.7717/peerj.1386 [PubMed]
- 65. Wang J, Li H, Xia C, Yang X, Dai B, Tao K, Dou K. Downregulation of CENPK suppresses hepatocellular carcinoma malignant progression through regulating YAP1. Onco Targets Ther. 2019; 12:869–82. https://doi.org/10.2147/OTT.S190061 [PubMed]
- 66. Wu S, Cao L, Ke L, Yan Y, Luo H, Hu X, Niu J, Li H, Xu H, Chen W, Pan Y, He Y. Knockdown of CENPK inhibits cell growth and facilitates apoptosis via PTEN-PI3K-AKT signalling pathway in gastric cancer. J Cell Mol Med. 2021; 25:8890–903. https://doi.org/10.1111/jcmm.16850 [PubMed]
- 67. Ma J, Chen X, Lin M, Wang Z, Wu Y, Li J. Bioinformatics analysis combined with experiments predicts CENPK as a potential prognostic factor for lung adenocarcinoma. Cancer Cell Int. 2021; 21:65. https://doi.org/10.1186/s12935-021-01760-y [PubMed]
- 68. Zhao J, Liu J, Wu N, Zhang H, Zhang S, Li L, Wang M. ANGPTL4 overexpression is associated with progression and poor prognosis in breast cancer. Oncol Lett. 2020; 20:2499–505. https://doi.org/10.3892/ol.2020.11768 [PubMed]
- 69. Yang L, Wang Y, Sun R, Zhang Y, Fu Y, Zheng Z, Ji Z, Zhao D. ANGPTL4 Promotes the Proliferation of Papillary Thyroid Cancer via AKT Pathway. Onco Targets Ther. 2020; 13:2299–309. https://doi.org/10.2147/OTT.S237751 [PubMed]
- 70. Nakayama T, Hirakawa H, Shibata K, Nazneen A, Abe K, Nagayasu T, Taguchi T. Expression of angiopoietin-like 4 (ANGPTL4) in human colorectal cancer: ANGPTL4 promotes venous invasion and distant metastasis. Oncol Rep. 2011; 25:929–35. https://doi.org/10.3892/or.2011.1176 [PubMed]
- 71. Nakayama T, Hirakawa H, Shibata K, Abe K, Nagayasu T, Taguchi T. Expression of angiopoietin-like 4 in human gastric cancer: ANGPTL4 promotes venous invasion. Oncol Rep. 2010; 24:599–606. https://doi.org/10.3892/or_00000897 [PubMed]
- 72. Stone L. Bladder cancer: Context is key: dual roles of ANGPTL4. Nat Rev Urol. 2017; 14:702. https://doi.org/10.1038/nrurol.2017.191 [PubMed]
- 73. Seborova K, Kloudova-Spalenkova A, Koucka K, Holy P, Ehrlichova M, Wang C, Ojima I, Voleska I, Daniel P, Balusikova K, Jelinek M, Kovar J, Rob L, et al. The Role of TRIP6, ABCC3 and CPS1 Expression in Resistance of Ovarian Cancer to Taxanes. Int J Mol Sci. 2021; 23:73. https://doi.org/10.3390/ijms23010073 [PubMed]
- 74. Fang X, Wu X, Xiang E, Luo F, Li Q, Ma Q, Yuan F, Chen P. Expression profiling of CPS1 in Correa’s cascade and its association with gastric cancer prognosis. Oncol Lett. 2021; 21:441. https://doi.org/10.3892/ol.2021.12702 [PubMed]
- 75. Wu G, Zhao Z, Yan Y, Zhou Y, Wei J, Chen X, Lin W, Ou C, Li J, Wang X, Xiong K, Zhou J, Xu Z. CPS1 expression and its prognostic significance in lung adenocarcinoma. Ann Transl Med. 2020; 8:341. https://doi.org/10.21037/atm.2020.02.146 [PubMed]
- 76. Gong X, Li N, Sun C, Li Z, Xie H. A Four-Gene Prognostic Signature Based on the TEAD4 Differential Expression Predicts Overall Survival and Immune Microenvironment Estimation in Lung Adenocarcinoma. Front Pharmacol. 2022; 13:874780. https://doi.org/10.3389/fphar.2022.874780 [PubMed]
- 77. Xiaoguang Z, Meirong L, Jingjing Z, Ruishen Z, Qing Z, Xiaofeng T. Long Noncoding RNA CPS1-IT1 Suppresses Cell Proliferation and Metastasis in Human Lung Cancer. Oncol Res. 2017; 25:373–80. https://doi.org/10.3727/096504016X14741486659473 [PubMed]
- 78. Wang YS, Ma LN, Sun JX, Liu N, Wang H. Long non-coding RNA CPS1-IT1 is a positive prognostic factor and inhibits epithelial ovarian cancer tumorigenesis. Eur Rev Med Pharmacol Sci. 2017; 21:3169–75. [PubMed]
- 79. Zhang W, Yuan W, Song J, Wang S, Gu X. LncRNA CPS1-IT1 suppresses EMT and metastasis of colorectal cancer by inhibiting hypoxia-induced autophagy through inactivation of HIF-1α. Biochimie. 2018; 144:21–7. https://doi.org/10.1016/j.biochi.2017.10.002 [PubMed]
- 80. Lu E, Su J, Zhou Y, Zhang C, Wang Y. CCL20/CCR6 promotes cell proliferation and metastasis in laryngeal cancer by activating p38 pathway. Biomed Pharmacother. 2017; 85:486–92. https://doi.org/10.1016/j.biopha.2016.11.055 [PubMed]
- 81. Wang B, Shi L, Sun X, Wang L, Wang X, Chen C. Production of CCL20 from lung cancer cells induces the cell migration and proliferation through PI3K pathway. J Cell Mol Med. 2016; 20:920–9. https://doi.org/10.1111/jcmm.12781 [PubMed]
- 82. Zeng W, Chang H, Ma M, Li Y. CCL20/CCR6 promotes the invasion and migration of thyroid cancer cells via NF-kappa B signaling-induced MMP-3 production. Exp Mol Pathol. 2014; 97:184–90. https://doi.org/10.1016/j.yexmp.2014.06.012 [PubMed]
- 83. Wu Y, Fu L, Wang B, Li Z, Wei D, Wang H, Zhang C, Ma Z, Zhu T, Yu G. Construction of a prognostic risk assessment model for lung adenocarcinoma based on Integrin β family-related genes. J Clin Lab Anal. 2022; 36:e24419. https://doi.org/10.1002/jcla.24419 [PubMed]
- 84. Hao Y, Yang W, Zheng W, Chen X, Wang H, Zhao L, Xu J, Guo X. Tumor elastography and its association with cell-free tumor DNA in the plasma of breast tumor patients: a pilot study. Quant Imaging Med Surg. 2021; 11:3518–34. https://doi.org/10.21037/qims-20-443 [PubMed]
- 85. Lin C, Liu H, Zhang H, Cao Y, Li R, Wu S, Li H, He H, Xu J, Sun Y. Tryptase expression as a prognostic marker in patients with resected gastric cancer. Br J Surg. 2017; 104:1037–44. https://doi.org/10.1002/bjs.10546 [PubMed]
- 86. Chen Q, Ma J, Wang X, Zhu X. Identification of prognostic candidate signatures by systematically revealing transcriptome characteristics in lung adenocarcinoma with differing tumor microenvironment immune phenotypes. Aging (Albany NY). 2022; 14:4786–818. https://doi.org/10.18632/aging.204112 [PubMed]
- 87. Saleh K, Cheminant M, Chiron D, Burroni B, Ribrag V, Sarkozy C. Tumor Microenvironment and Immunotherapy-Based Approaches in Mantle Cell Lymphoma. Cancers (Basel). 2022; 14:3229. https://doi.org/10.3390/cancers14133229 [PubMed]
- 88. Mackenzie NJ, Nicholls C, Templeton AR, Perera MP, Jeffery PL, Zimmermann K, Kulasinghe A, Kenna TJ, Vela I, Williams ED, Thomas PB. Modelling the tumor immune microenvironment for precision immunotherapy. Clin Transl Immunology. 2022; 11:e1400. https://doi.org/10.1002/cti2.1400 [PubMed]
- 89. Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, Zlobec I, Berger A, Bifulco C, Botti G, Tatangelo F, Britten CM, Kreiter S, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med. 2012; 10:205. https://doi.org/10.1186/1479-5876-10-205 [PubMed]
- 90. Ren C, Li J, Zhou Y, Zhang S, Wang Q. Typical tumor immune microenvironment status determine prognosis in lung adenocarcinoma. Transl Oncol. 2022; 18:101367. https://doi.org/10.1016/j.tranon.2022.101367 [PubMed]
- 91. Zhou H, Zhang H, Shi M, Wang J, Huang Z, Shi J. A robust signature associated with patient prognosis and tumor immune microenvironment based on immune-related genes in lung squamous cell carcinoma. Int Immunopharmacol. 2020; 88:106856. https://doi.org/10.1016/j.intimp.2020.106856 [PubMed]
- 92. Ma C, Luo H, Cao J, Zheng X, Zhang J, Zhang Y, Fu Z. Identification of a Novel Tumor Microenvironment-Associated Eight-Gene Signature for Prognosis Prediction in Lung Adenocarcinoma. Front Mol Biosci. 2020; 7:571641. https://doi.org/10.3389/fmolb.2020.571641 [PubMed]
- 93. Aponte-López A, Muñoz-Cruz S. Mast Cells in the Tumor Microenvironment. Adv Exp Med Biol. 2020; 1273:159–73. https://doi.org/10.1007/978-3-030-49270-0_9 [PubMed]
- 94. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, et al, and Cancer Genome Atlas Research Network. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018; 173:321–37.e10. https://doi.org/10.1016/j.cell.2018.03.035 [PubMed]
- 95. Matthews HK, Bertoli C, de Bruin RA. Cell cycle control in cancer. Nat Rev Mol Cell Biol. 2022; 23:74–88. https://doi.org/10.1038/s41580-021-00404-3 [PubMed]
- 96. Peng F, Li Q, Niu SQ, Shen GP, Luo Y, Chen M, Bao Y. ZWINT is the next potential target for lung cancer therapy. J Cancer Res Clin Oncol. 2019; 145:661–73. https://doi.org/10.1007/s00432-018-2823-1 [PubMed]
- 97. Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, Sanders D, Thomas DG, Doherty G, Hammer G. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res. 2009; 15:668–76. https://doi.org/10.1158/1078-0432.CCR-08-1067 [PubMed]
- 98. Huang R, Gao L. Identification of potential diagnostic and prognostic biomarkers in non-small cell lung cancer based on microarray data. Oncol Lett. 2018; 15:6436–42. https://doi.org/10.3892/ol.2018.8153 [PubMed]
- 99. Shao MT, Hu YZ, Ding H, Wu Q, Pan JH, Zhao XX, Pan YL. The overexpression of ZWINT in integrated bioinformatics analysis forecasts poor prognosis in breast cancer. Transl Cancer Res. 2020; 9:187–93. https://doi.org/10.21037/tcr.2019.12.66 [PubMed]
- 100. Ying H, Xu Z, Chen M, Zhou S, Liang X, Cai X. Overexpression of Zwint predicts poor prognosis and promotes the proliferation of hepatocellular carcinoma by regulating cell-cycle-related proteins. Onco Targets Ther. 2018; 11:689–702. https://doi.org/10.2147/OTT.S152138 [PubMed]
- 101. Zhang MY, Liu XX, Li H, Li R, Liu X, Qu YQ. Elevated mRNA Levels of AURKA, CDC20 and TPX2 are associated with poor prognosis of smoking related lung adenocarcinoma using bioinformatics analysis. Int J Med Sci. 2018; 15:1676–85. https://doi.org/10.7150/ijms.28728 [PubMed]
- 102. Long Z, Wu T, Tian Q, Carlson LA, Wang W, Wu G. Expression and prognosis analyses of BUB1, BUB1B and BUB3 in human sarcoma. Aging (Albany NY). 2021; 13:12395–409. https://doi.org/10.18632/aging.202944 [PubMed]
- 103. Zhao Y, Cai J, Shi K, Li H, Du J, Hu D, Liu Z, Wang W. Germacrone induces lung cancer cell apoptosis and cell cycle arrest via the Akt/MDM2/p53 signaling pathway. Mol Med Rep. 2021; 23:452. https://doi.org/10.3892/mmr.2021.12091 [PubMed]
- 104. Selvaraj C. Therapeutic targets in cancer treatment: Cell cycle proteins. Adv Protein Chem Struct Biol. 2023; 135:313–42. https://doi.org/10.1016/bs.apcsb.2023.02.003 [PubMed]
- 105. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. https://doi.org/10.1093/nar/gkv007 [PubMed]
- 106. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. https://doi.org/10.1186/1471-2105-14-7 [PubMed]
- 107. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–7. https://doi.org/10.1089/omi.2011.0118 [PubMed]
- 108. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010; 26:1572–3. https://doi.org/10.1093/bioinformatics/btq170 [PubMed]
- 109. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019; 37:773–82. https://doi.org/10.1038/s41587-019-0114-2 [PubMed]
- 110. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013; 4:2612. https://doi.org/10.1038/ncomms3612 [PubMed]
- 111. Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, Signoretti S, Liu JS, Liu XS. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016; 17:174. https://doi.org/10.1186/s13059-016-1028-7 [PubMed]
- 112. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, Liu J, Freeman GJ, Brown MA, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018; 24:1550–8. https://doi.org/10.1038/s41591-018-0136-1 [PubMed]
- 113. Danilova L, Ho WJ, Zhu Q, Vithayathil T, De Jesus-Acosta A, Azad NS, Laheru DA, Fertig EJ, Anders R, Jaffee EM, Yarchoan M. Programmed Cell Death Ligand-1 (PD-L1) and CD8 Expression Profiling Identify an Immunologic Subtype of Pancreatic Ductal Adenocarcinomas with Favorable Survival. Cancer Immunol Res. 2019; 7:886–95. https://doi.org/10.1158/2326-6066.CIR-18-0822 [PubMed]
- 114. Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 2014; 9:e107468. https://doi.org/10.1371/journal.pone.0107468 [PubMed]
- 115. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013; 39:782–95. https://doi.org/10.1016/j.immuni.2013.10.003 [PubMed]