



Introduction
Renal cell carcinoma (RCC) is a globally endemic cancer and the sixth most common malignancy in the United States, occurring in approximately 5% men and 3% women in the United States [1, 2]. The most common subtype of RCC is kidney renal clear cell carcinoma (KIRC), accounting for approximately 75% of all kidney cancers. When one kidney is damaged, both kidneys compensate for the damage; therefore, the loss of kidney function is often not detected at an early stage. Therefore, in terms of cancer development, about one-third of RCC patients present with metastases in addition to typical symptoms such as pain, lumps, and hematuria, this limits surgical treatment options [3, 4]. Despite early surgical treatment, up to 30% of patients develop recurrence and metastasis after surgery. Patients with advanced KIRC also have a poor prognosis due to their insensitivity to radiotherapy, chemotherapy, and drug resistance. This results in lower survival rates for patients with KIRC, with 5-year survival rates of only 10–20%. Therefore, owing to the dual difficulties in diagnosis and treatment, there is an urgent need to explore new therapeutic targets and prognostic markers for KIRC [5–7].
NF-κB was first described in 1986 when a “B cytokine” was discovered in the B cells that bind to a site encoding the enhancer region of the immunoglobulin kappa light chain gene and exerts biological effects. Nuclear factor kappa B (NF-κB) is a family of five transcription factors, including NF-κB1, NF-κB2, RelA, RelB, and c-Rel, which share a Rel homologous domain, which is responsible for its binding to DNA [8–10]. NF-κB is mainly present in the cytoplasm of most eukaryotic cells as dimers (p60/p50) and remains inactive in the cytoplasm due to binding to the inhibitory molecule, IκB (inhibitor of NF-κB). In the classical NF-κB-activated pathway, after the cell receives a foreign stimulus signal, IκB kinase in the cytoplasm is activated, IκBα is phosphorylated, and ultimately enabling IκBα to be specifically recognized by E3 ubiquitin ligase for ubiquitination and thereafter specifically recognized and degraded by the 26S proteasome. Then, the p60/p50 dimer is freed and translocated to the nucleus, where it acts as a nuclear transcription factor and participates in regulating the cell transcription process [11, 12]. Activation of NF-κB may be triggered by different signals, such as the binding of a variety of growth factors and cytokines to receptors on the cell membrane, including epidermal growth factor, insulin growth factor, and tumor necrosis factor family members. In addition, the activation of other signaling pathways, such as Ras/MAPK and PI3K/Akt, are also involved in NF-kB activation [13].
The stability of the NF-κB pathway is necessary for cell proliferation, differentiation, and body development. Moreover, the activated NF-κB signal, in addition to participating in cell proliferation and anti/proapoptotic signaling, also maintains the balance of immune regulation in epithelial tissue and inhibits the interference of inflammation with the internal environmental homeostasis of epithelial tissue; increasing evidence also indicates that it plays a key role in the occurrence and development of cancer [14–16]. For example, NF-κB is involved in the proliferation of the breast cancer cell line, T47D, by regulating cyclin D1 and inducing the upregulation of matrix metalloproteases (MMP) 2 and 9; metastasis of liver cancer cell lines, QGY-8024 and PLC97, was increased; and activation of the expression of VEGF, MMP9, and interleukin (IL)-8 in the human prostate cancer cell line, PC-3M, leading to the formation of new blood vessels and invasion of cancer cells. However, such reports in kidney cancer cells are rare [17–21].
In our study, 21 NF-κB-related genes were selected, and the Cancer Genome Atlas (TCGA) database and GSCALite website were used to analyze the copy number variation (CNV), single nucleotide variant (SNV), and messenger RNA (mRNA) expression of these genes in 32 human tumors, as well as their relationship with patient prognosis and anticancer drug sensitivity. Then, Gene Set Variation Analysis (GSVA) cluster analysis was used to divide KIRC patients into three groups to explore the correlation between the NF-κB pathway and occurrence, development, and clinical pathological features of KIRC, aiming to accurately interpret the mechanism of action of the NF-κB pathway in KIRC. Finally, 11 NF-κB-related genes were screened using least absolute shrinkage and selection operator (LASSO) regression to establish a KIRC prognostic model, and the accuracy of the model was further verified at the protein level. These results will guide the clinical diagnosis, treatment, and prognosis of patients with KIRC.
Results
NF-κB-related genes are differentially expressed in different cancers and associated with cellular pathways and drug sensitivity
To investigate variation and expression changes of NF-κB-related genes in a variety of human tumors, we measured the SNV of NF-κB-related genes in various tumors (Figure 1A and Supplementary Table 1), mRNA expression (Figure 1B and Supplementary Table 2) and frequency of CNV (Figure 1D) based on sample data from the TCGA database. In the mRNA expression profile, we can observe the expression level of NF-κB-related genes in various cancer types; particularly in patients with KIRC, TRADD, MYD88, TNFRSF1A, TNFRSF1B, MAP3K14, TNFAIP3, and NFKBIA were higher in KIRC tissues than in normal tissues, and the expression of IL1A, MAP3K1, CHUK, and IL1R1 were lower in the KIRC tissues than in normal tissues. The survival curve of patients with KIRC showed that the expression of most NF-κB-related genes was correlated with patient prognosis (Figure 1C). Such as CHUK, MAP3KI FADD TRAF6, TAB1 and RIPK1 raised related to the prognosis of patients with KIRC good genes, and IKBKB IL1R1, MAP3K7, IL1A, IKBKG raised KIRC patient’s prognosis related genes. CNV and SNV data obtained from the TCGA database and GSCALite website were analyzed, and the results showed that TRADD, TNFRSF1A, TNF, MAP3K14, IKBKG, and IKBKB showed CNV amplification in different types of tumors, whereas TRAF6, TNFRSF1B, TNFAIP3, TAB1, NFKB1, MYD88, MAP3K7, and CHUK showed CNV deletion. From the SNV results, it can be seen that NF-κB-related genes had different degrees of single-nucleotide variation in the 32 tumors. The methylation data of the NF-κB gene set obtained in pancancer through the GSCALite platform showed that the level of NF-κB gene methylation in a variety of cancers differed from that of normal samples, and the expression of MYD88, TNFRSF1A, TNFRSF1B, MAP3K14, and other genes had a strong correlation with the level of methylation, and all affected the survival of cancer patients, and the results were statistically different (Figure 2A). The relationship between NF-κB-related genes and classical cellular pathways, such as apoptosis, cell cycle, EMT, etc., showed that NF-κB plays a role in activation or inhibition, such as DNA damage response manifested as a high-intensity activation effect (Figure 2B. The expression of the NF-κB gene is also closely related to drug sensitivity, such as TNF expression is negatively correlated with the sensitivity of various targeted drugs, whereas TNFRSF1A is mainly positively correlated (Figure 2C). The u2-os cells are epithelial morphologic cell lines obtained from meso-differentiated sarcomas of the tibia of patients with osteosarcoma; due to the bone metastatic nature of KIRC, we selected the u2-os cell lines for verification, and based on the HPA website, expressions of IKBKB and IKBKG in u2-os cell lines were found. According to the immunofluorescence results, the target genes, IKBKB and IKBKG, were clearly expressed in the cytoplasm of the cells (Figure 2D).
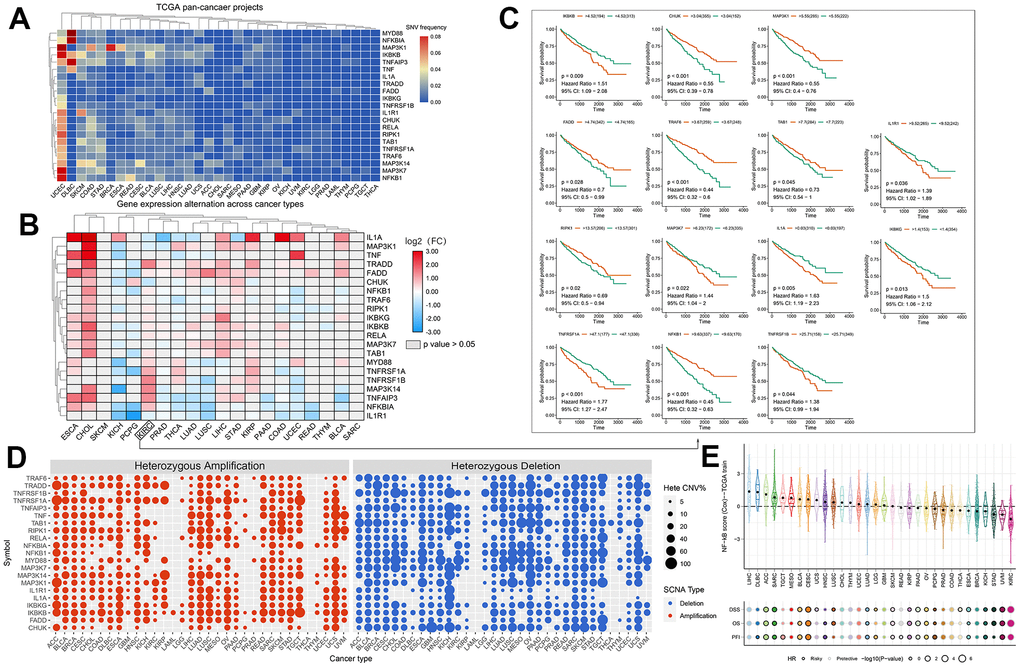
Figure 1. (A) SNV frequencies of 21 NF-κB pathway genes in 32 tumor types. Red and blue indicate high and low frequencies, respectively. (B) Expression levels of the NF-κB-related genes in 20 cancers. The color code bar shows the corresponding value of log2 (FC) on the right, with values ranging from 3.00 to -3.00 from red to blue. (C) Survival curve analysis of all statistically significant KIRC genes in TCGA samples. Red and green represent the high- and low-expression groups, respectively. (D) CNV frequencies of the 21 NF-κB pathway genes in 32 tumor types. Red and blue indicate amplification and loss of CNV, respectively. (E) Prognostic performance of the 11-gene NF-κB score in 32 types of cancers. The center color of the circle indicates the type of cancer, the color of the circle indicates “Risky/Protective”, and the size of the circle indicates statistical differences.
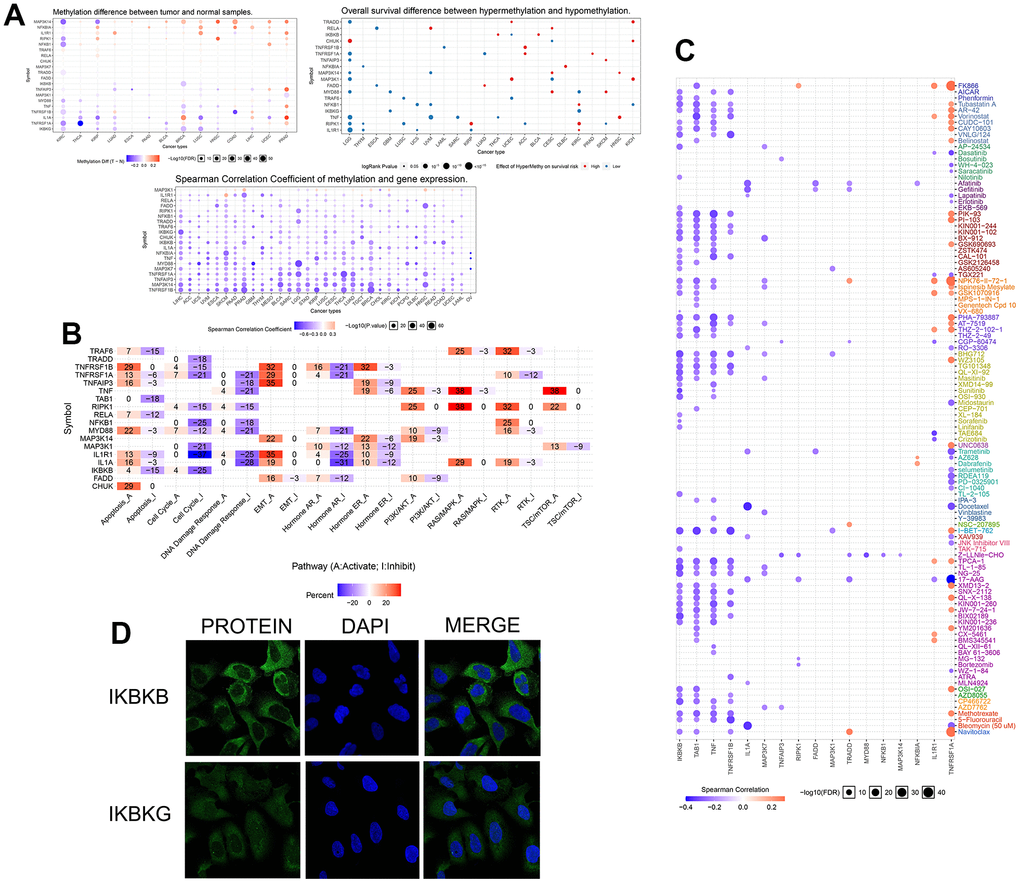
Figure 2. (A) The three figure parts show the different degrees of methylation of NF-κB-related genes in several human cancers, the relationship between methylation and mRNA expression level, and the correlation with patient prognosis. The colored circles indicate the value, and the size indicates the relationship with the p-value. (B) The relationship of NF-κB-related genes to 20 classical cellular pathways, “A” indicates activation and “I” indicates inhibition. (C) The relationship between NF-κB-related genes obtained in the GDSC database and drug sensitivity of individual targeted drugs, with blue indicating a positive correlation and orange indicating a negative correlation. (D) Immunofluorescence results from the HPA website showed the expression of IKBKB and IKBKG in the cytoplasm of u2os cells and the staining of the nucleus by DAPI.
Unsupervised hierarchical clustering and prognostic analysis
Based on the mRNA expression of the NF-κB-related genes, all TCGA samples of KIRC were divided into three clusters using the GSVA clustering analysis algorithm; the NF-κB score showed different expression levels of NF-κB related genes (Figure 3A), and the resulting C1, C2, and C3 clusters represented normal, active, and inactive scores, respectively. The violin plot (Figure 3B) showed the degree of difference in NF-κB expression between the three clusters, with the p-value <0.05, indicating that the difference between cluster groups, obtained by cluster analysis, was statistically significant. The survival curve highlighted the difference in prognosis among patients in the three clusters, indicating that the difference in NF-κB-related gene expression was related to prognosis; combined with the violin diagram results, it showed that patients in the C3 group, whose NF-κB pathway became inactive, had the worst prognosis (Figure 3C). The heat map showed that NF-κB-related gene expression in the three clusters is closely related to patient clinical data and survival outcomes (Figure 3D).
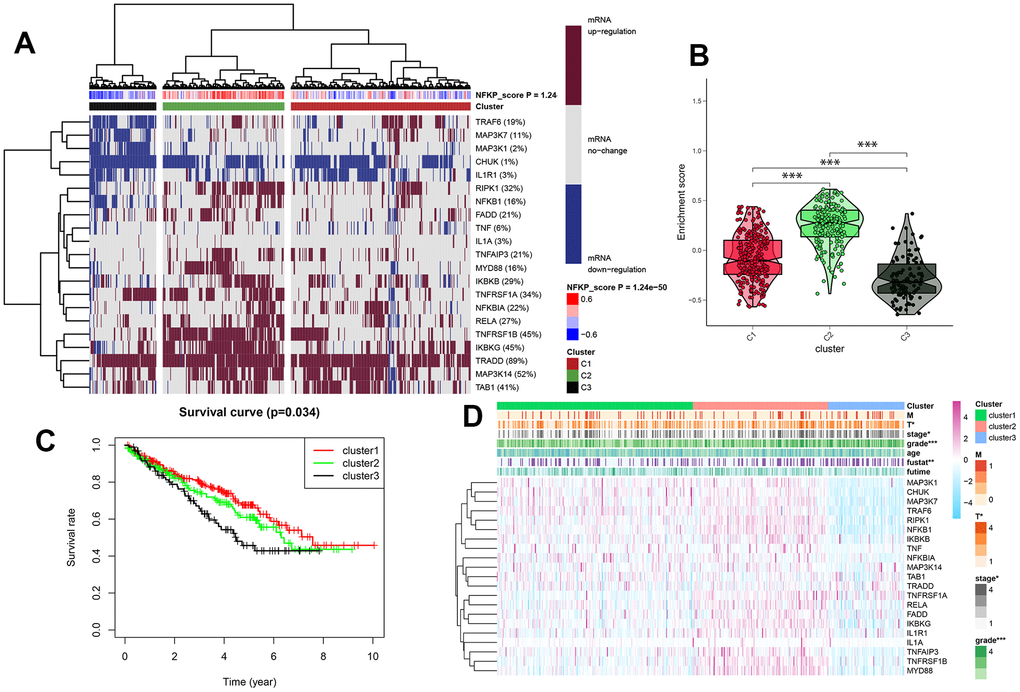
Figure 3. (A) All KIRC samples were divided into medium-, high-, and low-expression groups (clusters 1, 2, and 3), according to the level of NF-κB expression, and dark red and blue represent an increase and decrease in mRNA expression, respectively. The redder or bluer the color, the closer the NF-κB score is to 0.4 or -0.4, respectively. Cluster analysis is divided into three groups: red, green, and black for clusters 1, 2, and 3, respectively. (B) Enrichment and scores of the three clusters. (C) Survival curves for three cluster analyses. (D) Clinical pathological features of three clusters of KIRC patients. *: p<0.05, **: p<0.01, ***: p<0.001.
The NF-κB pathway is closely related to the expression of histone-modified genes and classical oncogenes
Here, we first explored the relationship between the NF-κB pathway and expression of various classical protooncogenes, for example, the low-expression of BRAF, PTEN, KRAS, MTOR, and PIK3CA in the NF-κB downregulation group (C3), indicating that the interruption of the NF-κB pathway is related to tumor promotion. The high expression of CTNNB1, MYC, STAT3, TP53, etc. in the NF-κB upregulated group (C2) suggested that targeting these genes to treat cancer progression in the NF-κB upregulated group may be effective. In addition, acetylation and deacetylation of transcription factors have also been shown to be associated with a variety of kidney diseases, including diabetic nephropathy [22, 23], the analysis of histone acetylation-related genes showed that abnormal expression of SIRT and HDACs also had a strong correlation with abnormalities in the NF-κB pathway, the expression of HDACs in the NF-κB downregulated group (C3) was slightly lower than that in the other two groups (Figure 4C). Then, to elucidate the correlation between any two genes in the NF-κB pathway, we conducted a co-expression analysis of 21 NF-κB-related genes, and found that the NF-κB1 gene was highly correlated with most other genes in the pathway, such as RIPK1, TRAF6, and TNFAIP3, and most of the positive correlation. In addition, the correlation between CHUK and MAP3K1, TNFAIP3 and NFRSF1, IKBKG and MYD88 were also strong (Figure 4A).

Figure 4. (A) Co-expression analysis showed that 21 NF-κB-related genes were associated in tumor tissues, with R-value indicating correlation size, red indicating positive correlation, and blue indicating negative correlation, p<0.05 was statistically significant. The regression relationship of NFκB1 with the remaining genes is represented by a scatterplot. (B) Based on three clusters, the IC50 predictions of 12 common tumor-targeted drugs for drugs with KIRC cells were analyzed. (C) Association of acetylation-related genes (HDAC and SIRT) and classical tumor family genes with NF-κB scores.
Prediction of the efficacy of KIRC-targeted drugs
To further explore the potential value of the NF-κB pathway in the clinical treatment of patients with KIRC, we plotted box plots based on drug susceptibility predictions in the GDSC database to determine the effect of the NF-κB pathway on IC50 of 12 commonly renal carcinoma targeted drugs. In these drugs, sorafenib is the first targeted multi-kinase inhibitor and first-line chemical drug approved for the treatment of RCC, which blocks formation of tumor neovascularization and directly inhibits the proliferation of RCC cells by blocking RAF/MEK/ERK signaling pathway [24]; Sunitinib inhibits the development of tumor-associated vascular disorders and affects the infiltration of immune cells such as regulatory T cells and macrophages [25]; metformin inhibits RCC cell viability (including cell migration and invasion) and increases apoptosis by disrupting mitochondrial dynamics [26]; other drugs also show different mechanisms of action in cancer suppression. We found that sensitivity to sorafenib, gefitinib, and metformin was higher in the downregulated NF-κB group (C3) but not in the upregulated NF-κB group (C2). In contrast, the sensitivity of the upregulated NF-κB group (C2) to pazopanib, sunitinib, and bosutinib was higher than that of the other two groups (Figure 4B). These results may provide precise guidance for the development of KIRC-targeted drug therapies in the future.
Immune cell infiltration based on ssGSEA
In addition to targeted drugs, immunotherapy has gradually attracted widespread attention for cancer treatment. To examine the regulatory role of the NF-κB pathway in immunotherapy for KIRC, we investigated the relationship between immune cells and NF-κB. The bubble plot showed a correlation between classical immune infiltration-associated cells or functions and NF-κB (Figure 5A), it can be seen from the results in the figure that most of the immune cells were positively correlated with NF-κB score, and there was a statistical difference. Subsequently, the three immunomodulators most strongly correlated with NF-κB, namely Treg, CCR, and neutrophils, were selected for correlation analysis, and the results were all positively correlated, their correlation coefficients are 5.56, 0.60 and 0.51, respectively, all of them are statistically significant (Figure 5B–5D). Previous studies have also shown that the activity of Treg, CCR and neutrophils is closely related to the occurrence and development of KIRC, which is consistent with the conclusion of our study [27–29].
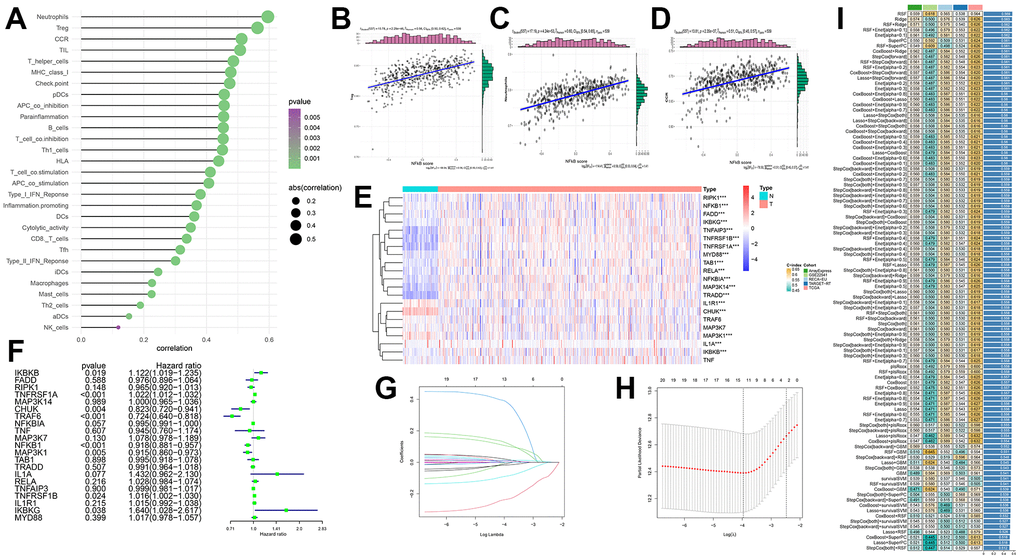
Figure 5. (A) Correlation of immune infiltration with NF-κB-related genes. The area of the circle represents ABS (correlation), and the color shows the p-value. (B–D) Scatterplots show the specific relationship between the three immune cells (Treg, neutrophils and CCR) and NF-κB score; they are all positively correlated. (E) Expression of NF-κB-related genes between the two samples. Red and blue represent up- and downregulation, respectively, N (green) is the normal sample, and T (red) represents the tumor sample (*: p<0.05, **: p<0.01, ***: p<0.001). (F) Forest plot showing hazard ratio analysis and p-values for 95% confidence intervals for 21 NF-κB-related genes. (G, H) LASSO coefficient spectra of NF-κB-related genes in patients with KIRC. LASSO Cox regression analysis was used to screen out the 11 genes. (I) Build 101 predictive models and calculate the C-index for each model on all validation datasets.
Establishing a prediction model using LASSO regression
By analyzing the NF-κB-related gene expression data in the normal control and KIRC groups in the TCGA database, we found that the 21 selected NF-κB pathway genes were significantly different between the two groups (Figure 5E). The figure shows that RIPK1, NFKB1, FADD IKBKG, TNFAIP3, TNFRSF1B, TNFRSF1A, MYD88, TAB1, RELA, NFKBIA and TRRAD genes expressed in tumor samples is higher. The expression of IL1R1, CHUK and MAP3K1 was higher in normal samples. The forest plot shows the results of the hazard ratio analysis for each gene, which were as follows: RIPK1, CHUK, TRAF6, NFKBIA, NFKB1, MAP3K1, and TRADD had a protective effect, whereas IKBKB, TNFPSF1A, MAP3K7, IL1A, RELA, TNFRSF1B, IL1R1, and IKBKG had a risk effect (Figure 5F). We then used LASSO regression analysis to select the appropriate genes to build a predictive model, (Figure 5G, 5H) and selected 11 genes: TRAF6, NFKBIA, NFKB1, MAP3K1, TRADD, IKBKB, TNFPSF1A, MAP3K7, RELA, TNFRSF1B, and IKBKG. In the TCGA dataset, we fit 101 prediction models through the LOOCV framework, and further calculate the C-index of each model on all validated datasets. If all the samples are paired and their outcomes are compared, the C-index refers to the proportion of combinations where the predicted outcome is consistent with the actual outcome. It can estimate the probability that the predicted results are consistent with the actual observed results, which is used to evaluate the predictive power of the model [30]. The model with the most accurate prediction was random survival forest (RSF), which had the highest average C-index (0.569), and this combined model led the C-index in all validated datasets (Figure 5I).
Based on the median risk score calculated from patients with KIRC, patients can be divided into low- and high-risk groups. The survival curves of both groups initially showed the predictive performance of the predictive model (Figure 6A and Supplementary Table 3). ROC curves for four different survival time nodes (3, 5, 7, and 10 years) (Figure 6B–6E) indicated that AUC values for all observed survival time nodes were greater than 0.7 (0.7 and above are considered to have predictive value). To further study the association between NF-κB-related genes and KIRC, we used the Kaplan–Meier “survival” software package to calculate the optimal cut-off value of the risk score, reclassified the cancer patient samples into low- and high-risk groups, and displayed the clinical data and genetic characteristics of the 11 genes from the two groups in the form of heat maps. The results showed that the risk score was strongly correlated with clinical features. The M and T stages, grade, and survival of patients in the high-risk group were worse than those of patients in the low-risk group. IKBKB, RELA, and TNFRSF1A showed a significant upregulation trend in the high-risk group, whereas TRAF6 and NFBIA showed a significant upregulation trend in the low-risk group (Figure 6F). The univariate Cox regression analysis showed that age, grade, stage, tumor size (T), tumor metastasis (M), and survival model risk scores were associated with Overall Survival (OS) in patients with KIRC; the multivariate Cox regression analysis showed that age, grade, stage, and survival model risk scores were independent risk factors affecting the prognosis of patients with KIRC (Figure 6G, 6H and Supplementary Tables 4, 5).
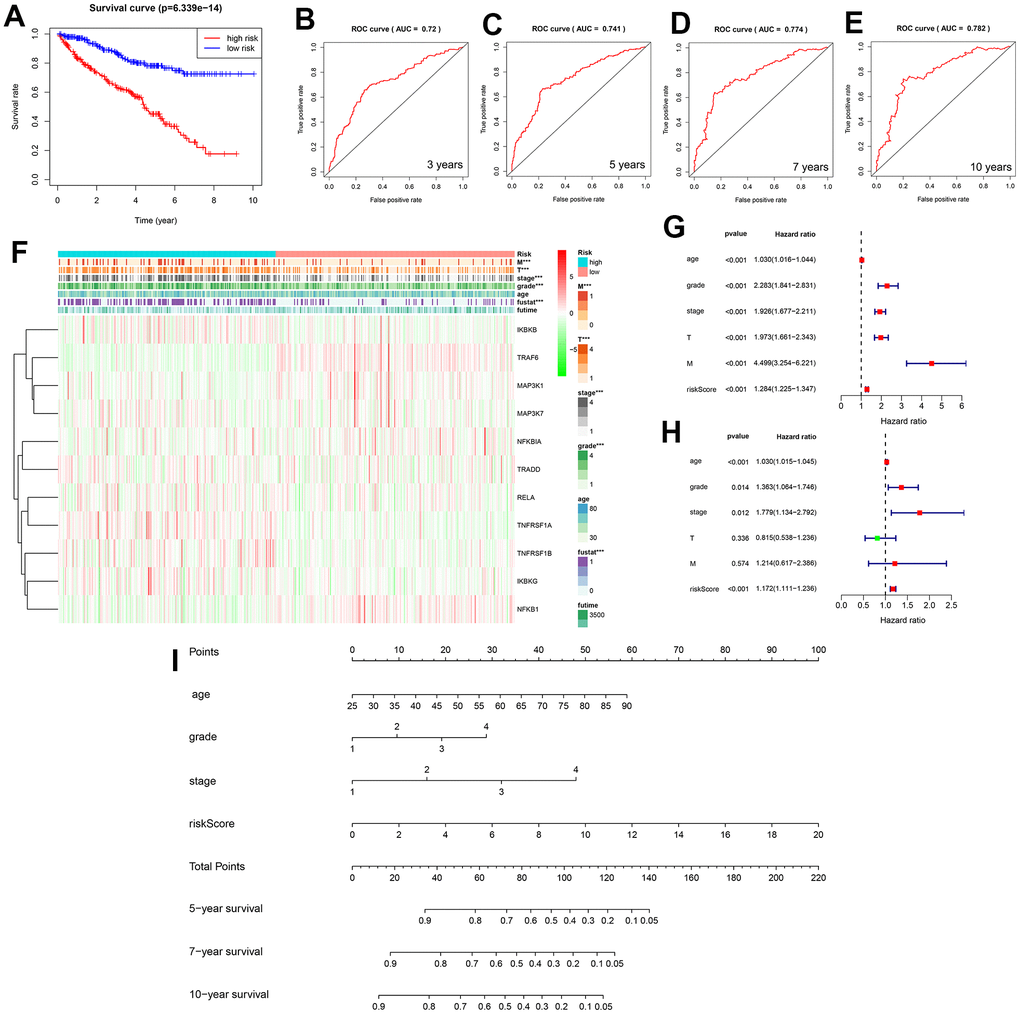
Figure 6. (A) Two survival curves based on the model. Blue and red represent the low- and high-risk groups, respectively. (B–E) Three-, 5-, 7-, and 10-year receiver operating characteristic curves, area under the curve values of 0.72, 0.741, 0.774, and 0.782. (F) Heat map showing the correlation between the 11 selected genes and clinicopathological features of the two groups of samples. The two-colored bars indicate gene expression; red and green represent upregulation and downregulation, respectively. (G) Forest plot for the univariate Cox regression analysis. (H) Forest plot for the multivariate Cox regression analysis. (I) Nomogram of the prediction model was used to calculate the total score to obtain the 5-, 7-, and 10-year survival rates of patients with KIRC.
Predictive model analysis and validation
We used a nomogram to predict the prognosis of patients with KIRC, and 10 parallel lines were generated; each successive line represented the score, age, grade, stage, and risk score, and the total score calculated by adding the scores of age, grade, stage, and risk score, respectively, after which we could easily estimate the survival rate of patients with KIRC at 5, 7, and 10 years (Figure 6I). The NF-κB scoring system also showed significant organ specificity in the prognosis of different cancers, tumors from the liver, adrenal glands, and testes generally had higher NF-κB scores, while tumors from the uvea, stomach, and breast generally had lower NF-κB scores. Based on the analysis of DSS, OS and PFI, it is suggested that NF-κB-related pathways play different roles in the prognosis of different cancers. The expression of NF-κB in LIHC, MESO, CHOL, and THYM is a protective trend, while the expression of NF-κB pathway in ACC, SARC, SKCM, and UVM is a risk trend (Figure 1E).
Several additional analyses were conducted to verify the accuracy and validity of the model; we selected three genes for protein level verification, namely CHUK, IKGGB, and IKBKG. Among them, IKGGB and IKBKG are protective genes used to build the model, while CHUK is a risk gene associated with the prognosis of KIRC patients but not used to build the model. Firstly, the differential expression of CHUK, IKGGB, and IKBKG between normal and cancerous tissues was analyzed at the protein level using the UALCAN database, and the results were consistent with the mRNA expression results (Figure 7A–7C). Subsequently, using the HPA database, we downloaded immunohistochemical images of IKBKG and IKBKB from normal and cancerous tissues and found that their expression of IKBKG and IKBKB in renal cancer tissues was higher than that in normal kidney tissues (Figure 7D). Finally, we demonstrated the correlation between high- and low-risk scores and the level of immune cell invasion using various immune infiltration algorithms, such as the low-expression of the endothelial cells and CD4+ T cells in the high-risk group in the EPIC algorithm (Figure 7E). These results can be used to identify and characterize the immune microenvironment in patients with different KIRCs and to further evaluate the efficacy of precision therapy, including immunotherapy.
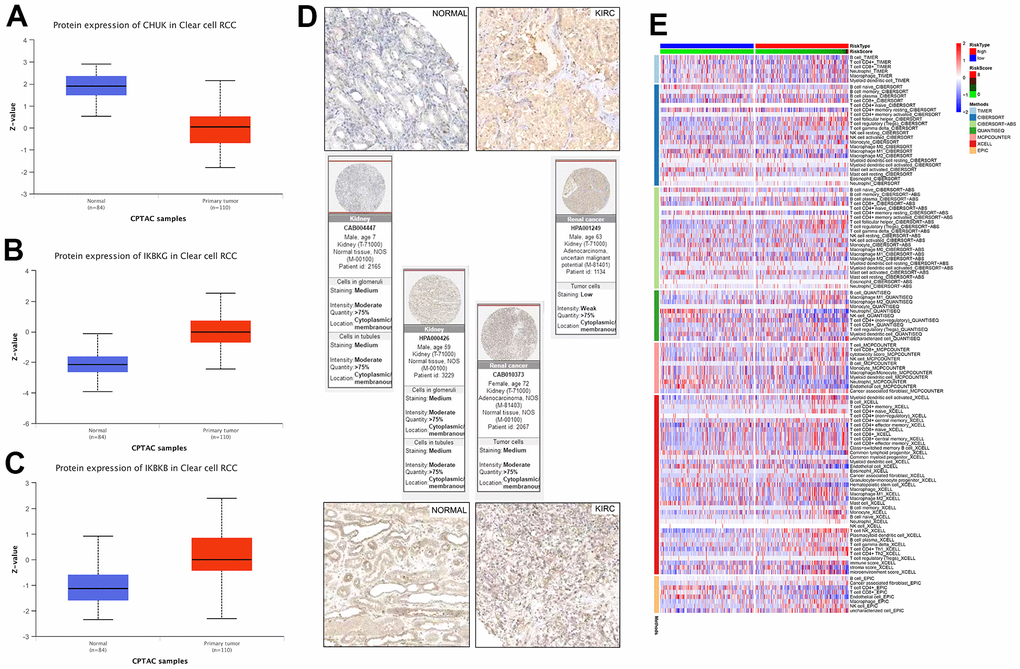
Figure 7. (A–C) Link between the NF-κB-related genes and KIRC was confirmed at the protein level. (D) Immunohistochemical comparison of IKBKG and IKBKB. (E) Based on different algorithms, showing the heat map of the response of immune cells in the high- and low-risk groups. Red and blue represent high and low infiltration levels, respectively, and different algorithms are represented by different colored area bars.
Discussion
NF-κB family transcription factors play an important role as stressors in the cellular environment, involved in several physiological processes, such as immunity, inflammation, cell proliferation, and death. NF-κB proteins are located in the cytoplasm and can be activated by various stimulus signals. There are two pathways for NF-κB activation, the typical and atypical pathways, and the activated NF-κB translocate to the nucleus, where gene expression is regulated [31]. In terms of cell proliferation and death, NF-κB participates in the expression of cell cycle regulators, including cyclin A, cyclin D1, and CDK6, and the activation of NF-κB can protect cells from TNF-α-induced apoptosis [32]. During cancer development, uncontrolled proliferation and insensitivity to cell death are often accompanied by the activation of NF-κB signaling [33], this is reflected in a variety of cancers, including breast cancer [34], prostate cancer [35], esophageal squamous cell carcinoma [36], lymphoma [37], lung adenocarcinoma [38], etc. Abnormal NF-κB signaling also frequently leads to abnormal changes in tumor resistance, local inflammatory response, and immune microenvironment [38–41].
As the most common subtype of renal cell carcinoma, increased activity of NF-κB in KIRC has been shown to be associated with upregulation of angiogenic markers, and knockout of NF-κB leads to downregulation of IL-6 [42]. Unfortunately, similar studies of NF-kappa B's mechanism in KIRC are currently known to be superficial. In this study, we first analyzed the mutations and expression of NF-κB-related genes in various human cancers by studying the correlation between gene expression and mutations using clinical report information in the TCGA database. To determine whether NF-κB may be a potential target for KIRC therapy, we compared the effects of NF-κB-related genes on prognosis and drug sensitivity in patients with KIRC; interestingly, we found that the genes involved in the NF-κB pathway are both protective and hazardous.
Then, we divided the KIRC samples into three clusters according to the RNA expression levels of NF-κB-related genes: high, normal, and low expression of NF-κB-related genes, respectively. Based on these three clusters, we constructed a survival curve for NF-κB-related genes. We found that patients with downregulated NF-κB pathways had the worst survival rates. In order to confirm the role of commonly used tumor-targeted therapy drugs in the treatment of KIRC, we conducted GDSC analysis, and the results showed that the IC50 of most targeted drugs for KIRC treatment is related to the level of gene expression in the NF-κB pathway, which indicates that the selection of different drugs according to the characteristics of patients can achieve better efficacy or appropriately reduce drug concentrations to reduce the side-effects of drugs. In addition, acetylation and deacetylation are common epigenetic modifications that play a vital role in the formation and development of tumors. By analyzing the expression of classical oncogenes, such as EGFR and mTOR, and acetylation-related genes in the three clusters, we found that the expression of these genes is mainly related to NF-κB pathways, such as SIRT1, HDAC9, CTNNBI, KRAS, and PIK3CA. Therefore, it may be more efficient to select different targets in different NF-κB characteristic groups.
Tumors infiltrating immune cells (TIICs) are effective targets for drug improvement in tumor therapy [43]. Studies have shown that patients with KIRC have typical characteristics of immunogenic tumors, and TIICs in KIRC, including CD4 T cells, CD8 T cells, natural killer molecules, and dendritic cells, are inhibited to varying degrees, leading to antitumor immune disorders and successful evasion of immune recognition [44]. Additionally, the microenvironment contains a large number of tumor-infiltrating T lymphocytes and cytotoxic T cells that recognize and selectively destroy tumor cells [45]. It has been proved that the number of Treg cell subsets in peripheral blood can help predict the prognosis of immunotherapy, and can reflect the anti-tumor immune status in KIRC patients [46]. In addition, infiltrating neutrophils promote KIRC migration and invasion through VEGFa/HIF2α and estrogen receptor β signaling [47]. Chemokines and their receptor proteins have also been shown to regulate a variety of biological processes [48]. Exploring the relationship between NF-κB and immunity will help us gain a deeper understanding of immunotherapy. Our results show that NF-κB-related genes are closely related to immune cell infiltration, and three of these immunomodulatory factors, Treg, CCR, and neutrophils, are strongly positively correlated with the NF-κB pathway. These analyses will allow patients to receive personalized treatments and provide new ideas for the development of novel targeted therapies.
Finally, 11 genes in the NF-κB pathway were screened out using LASSO regression, namely TRAF6, NFKBIA, NFKB1, MAP3K1, TRADD, IKBKB, TNFPSF1A, MAP3K7, RELA, TNFRSF1B, and IKBKG, among them, IKBKB, TNFPSF1A, MAP3K7, IL1A, RELA, TNFRSF1B, IL1R1, and IKBKG are risk genes for KIRC patients, and RIPK1, CHUK, TRAF6, NFKBIA, NFKB1, MAP3K1, and TRADD are protective genes for KIRC patients. We also constructed a survival prediction model to predict the survival rate of KIRC patients at 5, 7, and 10 years. The area under the ROC curve obtained using the model showed a high predictive value. The multivariate Cox regression analysis revealed that the survival model risk score was an independent risk factor affecting the prognosis of patients with KIRC. The link between NF-κB-related genes and KIRC was confirmed at the protein level. We believe that this model has great value in future clinical research on the treatment and prognosis of KIRC. Specifically, using our survival model and existing clinical data, we can clearly predict the prognosis of KIRC patients. In addition, based on differences in the expression of NF-κB-related genes, each KIRC patient can receive personalized targeted drug therapy, which will greatly reduce the drug resistance of patients and improve the effectiveness of postoperative chemotherapy.
There are few studies on the role of the genes screened in KIRC, all of which are consistent with our conclusions. For example, one study showed that the expression level of IKBKB in KIRC is decreased, and the upregulation of IKBKB protein levels is associated with an increase in the tumor nuclei grade and a significantly shortened survival period, suggesting that the gene plays a carcinogenic role in KIRC [49]. RIPK1 is highly expressed in KIRC and is upregulated by TNF-α, which further induces necrotic apoptosis of cancer cells through the RIPK1/RIPK3/MLKL/Drp1 axis [50]. Another study showed that the transcriptional activity of TRAF6 is regulated by miR146b-5p, and that inhibition of miR146b-5p can increase inflammatory cytokine secretion and TRAF6 expression in renal tumor mouse models, further inhibiting orthotropic tumor cell growth [51].
However, our study had some limitations. Although we predicted some of the results through bioinformatics analysis, this conclusion lacked support from the experimental data. In other words, although we conducted a preliminary verification of protein levels using the database, we hope that future experiments will further validate the results of our analysis because the role of NF-κB in the pathogenesis of KIRC has not been fully established. In addition, kidney cancer is a cancer prone to metastasis, but the samples we studied lacked lung, liver, brain and other metastatic sites, making this study incomplete. This part will also be an important link in our future research plan.
In summary, our study found that most NF-κB-related genes differ significantly in KIRC expression compared with normal kidney tissue and can be used as risk or protective factors affecting KIRC treatment and prognosis and are closely related to drug sensitivity, immune cell infiltration, classical oncogenes, and histone modification. Classifying patients with KIRC according to NF-κB score is of great significance for evaluating the prognosis of patients and finding new targets, and the prognostic model we constructed may provide more comprehensive suggestions for the development of personalized treatment for patients with KIRC.
Materials and Methods
Data acquisition and pancancer analysis
The “Biocarta” dataset was found using the Gene Set Enrichment Analysis (GSEA) website (http://www.gsea-msigdb.org/gsea/index.jsp) [52], and 21 genes closely related to the NF-κB pathway were selected for further analysis. The TCGA database is a genome-wide gene expression collection established through large-scale gene sequencing and multidimensional analysis. We used the TCGA database (https://portal.gdc.cancer.gov) to download the changes in SNV and mRNA levels of NF-κB-related genes in 32 cancers [53]. Data were analyzed using Perl, and visual analysis was performed using the “TBtools” software package. Data on CNV, methylation, and classical cellular pathways of genes associated with the 32 cancers were obtained from the GSCALite website (http://bioinfo.life.hust.edu.cn/web/GSCALite/) [54]. Genomics of Drug Sensitivity in Cancer (GDSC) is a publicly accessible database (https://www.cancerrxgene.org/) that provides information regarding drugs, genes, and tumors [55]; based on the GDSC database, we also analyzed the relationship between relevant genes and drug susceptibility of various chemotherapy drugs.
Cluster analysis based on NF-κB score
GSVA is an analytical method that transforms the expression matrix of genes between different samples into gene sets by unsupervised classification of samples [56]. We used the GSVA algorithm to calculate the NF-κB enrichment score of KIRC patients, and the following three cluster samples were obtained by cluster analysis according to the NF-κB enrichment score of each sample: cluster 1, cluster 2, and cluster 3, which represented the normal, low expression, and high expression of NF-κB pathway-related genes in KIRC patients, respectively. The accuracy of the three clusters was verified with a violin plot and survival curve, and a heat map showing the relationship between gene expression levels and clinical pathological features was generated. A box plot was created using the “pRRophetic” software package in R software to depict the 50% inhibiting concentration (IC50) prediction of the three cluster samples for targeted drugs to treat KIRC [57]. Statistical significance was set at p<0.05.
Immune cell infiltration
Single-sample GSEA (ssGSEA) can be applied to gene signals expressed by immune cells in a single sample; we used ssGSEA combined with the expression of relevant genes in the TCGA database to quantify immune cells [61]. Based on the ssGSEA results, we showed correlations between NF-κB scores and 28 types of immune cells, where the area of the spheroids indicated the degree of correlation. Color represents the p-value. The R software packages “ggplot2”, “dplyr”, “data.table”, “tidyr”, and “ggstatsplot” were used for analysis and plotting [62–65]. We then selected three classic immunomodulators: regulatory T cell (Treg), chemokine receptor (CCR), and neutrophil and generated scatter plots using the “ggdisterstats” package to show their correlation with NF-κB scores [66].
Construction of predictive models
We used the “pheatmap” software package to create heat maps to describe differences in NF-κB-related gene expression levels in KIRC and normal tissues [67]. The univariate Cox regression was used to analyze the relationship between NF-κB pathway-related genes and risk indicators (stage, grade, etc.) in KIRC patients. The “glmnet” package was used to construct the LASSO regression model, risk score = ∑Ni = 1 (Expi * Coei); where N, Coei, and Expi were the number of genes, regression correlation coefficient, and gene expression level obtained using LASSO regression analysis, respectively (Supplementary Materials: Table Coef). The cut-off value of the KIRC risk score was calculated using the “survival” package, according to which the samples were divided into high- and low-risk groups, and the survival curve was plotted. Concretely speaking, we calculated the risk score of each sample based on the “survival” package, thus obtaining the median score of all samples, and further divided all samples into high- and low-risk groups according to the median value and the score of each sample. Finally, the receiver operating characteristic (ROC) curve was plotted using the “survival ROC” package to obtain area under the curve (AUC) values for the 3-, 5-, 7-, and 10-year survival rates. AUC is the area value under the ROC curve used to measure the performance of the classifier. The closer the AUC value is to 1, the better the classifier performance, and the closer the AUC value is to 0, the worse the classifier performance. Heat map showed the clinicopathological features of the high- and low-risk groups. Statistical significance level was set at p < 0.05. We also investigated the role of NF-κB-based risk scores in survival of patients with different cancers. We used prognostic models to assess the association of NF-κB scores with disease-specific survival (DSS) in patients with the TCGA pancancer series.
Validation of the prediction model and nomogram
We also integrated 10 machine learning algorithms and 101 algorithm combinations to develop prognostic models with high accuracy, algorithms include Random survival Forest (RSF), Elastic Network (Enet), Lasso, Ridge, stepwise Cox, and Cox boost, Cox Partial least squares regression (plsRcox), supervised Principal Component (SuperPC), Generalized Enhanced regression modeling (GBM), and survival support vector Machine (Survival-SVM) [68]. Univariate and multivariate Cox regression analyses were used to show the correlation between age, stage, grade, T stage, M stage, and risk scores in the model, with a p-value < 0.05 considered statistically significant. RStudio was used for data analysis. To confirm our conclusions, we performed controlled immunohistochemistry experiments on two key molecules involved in the model, IKBKG and IKBKB, using clinical KIRC specimens. The UALCAN database uses data obtained from TCGA to assess the expression of protein-coding genes and their impact on the survival of patients with 33 types of cancer. The HPA database is a tool based on proteomics, transcriptomics, and systems biology data, which is used to map tissues, cells, organs, etc. [69–71]. We obtained information on protein levels from the HPA (https://www.proteinatlas.org/) and UALCAN databases (http://ualcan.path.uab.edu/). IKBKB and IKBKG protein immunofluorescence assays were performed using the u2-os cell line. The nomogram was plotted using the “rms” package in R [72].
Author Contributions
Sun is the first author, responsible for collecting information, drafting and revising the manuscript. Wu and Chen design research direction, provide writing guidance, participate in the revision and supplement of manuscripts. All authors agree to the manuscript version for this release and have agreed to work on their respective aspects.
Acknowledgments
We thank The Cancer Genome Atlas (TCGA) project for providing us with the publicly available data. We would like to thank Editage (https://www.editage.cn) for English language editing.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Ethical Statement
Our study is based on open-source data provided by TCGA and other public databases. So, there are no ethical issues.
Funding
This project is supported by Dalian Young Stars of Science and Technology Fund (No. 2022RQ010).
References
- 1. Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009; 373:1119–32. https://doi.org/10.1016/S0140-6736(09)60229-4 [PubMed]
- 2. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007; 57:43–66. https://doi.org/10.3322/canjclin.57.1.43 [PubMed]
- 3. Lam JS, Leppert JT, Belldegrun AS, Figlin RA. Novel approaches in the therapy of metastatic renal cell carcinoma. World J Urol. 2005; 23:202–12. https://doi.org/10.1007/s00345-004-0466-0 [PubMed]
- 4. Rajandram R, Bennett NC, Morais C, Johnson DW, Gobe GC. Renal cell carcinoma: resistance to therapy, role of apoptosis, and the prognostic and therapeutic target potential of TRAF proteins. Med Hypotheses. 2012; 78:330–6. https://doi.org/10.1016/j.mehy.2011.11.014 [PubMed]
- 5. Makhov P, Joshi S, Ghatalia P, Kutikov A, Uzzo RG, Kolenko VM. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol Cancer Ther. 2018; 17:1355–64. https://doi.org/10.1158/1535-7163.MCT-17-1299 [PubMed]
- 6. Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev. 2008; 34:193–205. https://doi.org/10.1016/j.ctrv.2007.12.001 [PubMed]
- 7. Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho TH, Cheng EH. Chromosome 3p Loss-Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J Clin Oncol. 2018; 36:JCO2018792549. https://doi.org/10.1200/JCO.2018.79.2549 [PubMed]
- 8. Baldwin AS
Jr . The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996; 14:649–83. https://doi.org/10.1146/annurev.immunol.14.1.649 [PubMed] - 9. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008; 132:344–62. https://doi.org/10.1016/j.cell.2008.01.020 [PubMed]
- 10. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986; 46:705–16. https://doi.org/10.1016/0092-8674(86)90346-6 [PubMed]
- 11. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013; 12:86. https://doi.org/10.1186/1476-4598-12-86 [PubMed]
- 12. Motolani A, Martin M, Sun M, Lu T. Phosphorylation of the Regulators, a Complex Facet of NF-κB Signaling in Cancer. Biomolecules. 2020; 11:15. https://doi.org/10.3390/biom11010015 [PubMed]
- 13. Patel M, Horgan PG, McMillan DC, Edwards J. NF-κB pathways in the development and progression of colorectal cancer. Transl Res. 2018; 197:43–56. https://doi.org/10.1016/j.trsl.2018.02.002 [PubMed]
- 14. Viatour P, Bentires-Alj M, Chariot A, Deregowski V, de Leval L, Merville MP, Bours V. NF- kappa B2/p100 induces Bcl-2 expression. Leukemia. 2003; 17:1349–56. https://doi.org/10.1038/sj.leu.2402982 [PubMed]
- 15. Freudlsperger C, Bian Y, Contag Wise S, Burnett J, Coupar J, Yang X, Chen Z, Van Waes C. TGF-β and NF-κB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013; 32:1549–59. https://doi.org/10.1038/onc.2012.171 [PubMed]
- 16. Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer Immunol Res. 2014; 2:823–30. https://doi.org/10.1158/2326-6066.CIR-14-0112 [PubMed]
- 17. Chen E, Li CC. Association of Cdk2/cyclin E and NF-kappa B complexes at G1/S phase. Biochem Biophys Res Commun. 1998; 249:728–34. https://doi.org/10.1006/bbrc.1998.9224 [PubMed]
- 18. Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol. 1999; 19:2690–8. https://doi.org/10.1128/MCB.19.4.2690 [PubMed]
- 19. Park MH, Hong JT. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells. 2016; 5:15. https://doi.org/10.3390/cells5020015 [PubMed]
- 20. Li J, Lau GK, Chen L, Dong SS, Lan HY, Huang XR, Li Y, Luk JM, Yuan YF, Guan XY. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-kB induced matrix metalloproteinases 2 and 9 expression. PLoS One. 2011; 6:e21816. https://doi.org/10.1371/journal.pone.0021816 [PubMed]
- 21. Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene. 2001; 20:4188–97. https://doi.org/10.1038/sj.onc.1204535 [PubMed]
- 22. Ji J, Tao P, Wang Q, Li L, Xu Y. SIRT1: Mechanism and Protective Effect in Diabetic Nephropathy. Endocr Metab Immune Disord Drug Targets. 2021; 21:835–42. https://doi.org/10.2174/1871530320666201029143606 [PubMed]
- 23. Tao P, Ji J, Wang Q, Cui M, Cao M, Xu Y. The role and mechanism of gut microbiota-derived short-chain fatty in the prevention and treatment of diabetic kidney disease. Front Immunol. 2022; 13:1080456. https://doi.org/10.3389/fimmu.2022.1080456 [PubMed]
- 24. He Y, Luo Y, Huang L, Zhang D, Wang X, Ji J, Liang S. New frontiers against sorafenib resistance in renal cell carcinoma: From molecular mechanisms to predictive biomarkers. Pharmacol Res. 2021; 170:105732. https://doi.org/10.1016/j.phrs.2021.105732 [PubMed]
- 25. Peng Y, Dong S, Song Y, Hou D, Wang L, Li B, Wang H. Key sunitinib-related biomarkers for renal cell carcinoma. Cancer Med. 2021; 10:6917–30. https://doi.org/10.1002/cam4.4206 [PubMed]
- 26. Hong SH, Lee KS, Hwang HJ, Park SY, Han WK, Yoon YE. Synergic Effect of Metformin and Everolimus on Mitochondrial Dynamics of Renal Cell Carcinoma. Genes (Basel). 2022; 13:1211. https://doi.org/10.3390/genes13071211 [PubMed]
- 27. Xu W, Wu Y, Liu W, Anwaier A, Tian X, Su J, Huang H, Wei G, Qu Y, Zhang H, Ye D. Tumor-associated macrophage-derived chemokine CCL5 facilitates the progression and immunosuppressive tumor microenvironment of clear cell renal cell carcinoma. Int J Biol Sci. 2022; 18:4884–900. https://doi.org/10.7150/ijbs.74647 [PubMed]
- 28. Nishida J, Momoi Y, Miyakuni K, Tamura Y, Takahashi K, Koinuma D, Miyazono K, Ehata S. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat Cell Biol. 2020; 22:465–75. https://doi.org/10.1038/s41556-020-0491-2 [PubMed]
- 29. Zhang Z, Tang Y, Li L, Yang W, Xu Y, Zhou J, Ma K, Zhang K, Zhuang H, Gong Y, Gong K. Downregulation of FXYD2 Is Associated with Poor Prognosis and Increased Regulatory T Cell Infiltration in Clear Cell Renal Cell Carcinoma. J Immunol Res. 2022; 2022:4946197. https://doi.org/10.1155/2022/4946197 [PubMed]
- 30. Su W, He B, Zhang YD, Yin G. C-index regression for recurrent event data. Contemp Clin Trials. 2022; 118:106787. https://doi.org/10.1016/j.cct.2022.106787 [PubMed]
- 31. Zinatizadeh MR, Schock B, Chalbatani GM, Zarandi PK, Jalali SA, Miri SR. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2020; 8:287–97. https://doi.org/10.1016/j.gendis.2020.06.005 [PubMed]
- 32. Zinatizadeh MR, Momeni SA, Zarandi PK, Chalbatani GM, Dana H, Mirzaei HR, Akbari ME, Miri SR. The Role and Function of Ras-association domain family in Cancer: A Review. Genes Dis. 2019; 6:378–84. https://doi.org/10.1016/j.gendis.2019.07.008 [PubMed]
- 33. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005; 446:475–82. https://doi.org/10.1007/s00428-005-1264-9 [PubMed]
- 34. Zhang Y, Yang F. FAM84B promotes breast cancer tumorigenesis through activation of the NF-κB and death receptor signaling pathways. Pathol Res Pract. 2023; 249:154785. https://doi.org/10.1016/j.prp.2023.154785 [PubMed]
- 35. Zhao W, Jiang Y, Jia X, Wang X, Guo Y. Berbamine Inhibits the Biological Activities of Prostate Cancer Cells by Modulating the ROS/NF-κB Axis. Anticancer Agents Med Chem. 2023; 23:1626–33. https://doi.org/10.2174/1871520623666230503094540 [PubMed]
- 36. Han G, Zhou S, Shen J, Yang Y, Bian X, Li Y, Ling R, Liang R, Tao M. The role of TMEM26 in disrupting tight junctions and activating NF-κB signaling to promote epithelial-mesenchymal transition in esophageal squamous cell carcinoma. Clinics (Sao Paulo). 2023; 78:100276. https://doi.org/10.1016/j.clinsp.2023.100276 [PubMed]
- 37. Vashisht M, Ge H, John J, McKelvey HA, Chen J, Chen Z, Wang JH. TRAF2/3 deficient B cells resist DNA damage-induced apoptosis via NF-κB2/XIAP/cIAP2 axis and IAP antagonist sensitizes mutant lymphomas to chemotherapeutic drugs. Cell Death Dis. 2023; 14:599. https://doi.org/10.1038/s41419-023-06122-2 [PubMed]
- 38. Lin X, Yang S, Zhou C, Ao C, Sun D. The NEDD8-activating enzyme E1 UBA3 orchestrates the immunosuppressive microenvironment in lung adenocarcinoma via the NF-кB pathway. Med Oncol. 2023; 40:286. https://doi.org/10.1007/s12032-023-02162-y [PubMed]
- 39. Shari K, El Gedaily RA, Allam RM, Meselhy KM, Khaleel AE, Abdel-Sattar E. Jatrophone: a cytotoxic macrocylic diterpene targeting PI3K/AKT/NF-κB pathway, inducing apoptosis and autophagy in resistant breast cancer cells. BMC Complement Med Ther. 2023; 23:293. https://doi.org/10.1186/s12906-023-04113-6 [PubMed]
- 40. Tas SW, Bryant VL, Cook MC. Editorial: Non-canonical NF-κB signaling in immune-mediated inflammatory diseases and malignancies. Front Immunol. 2023; 14:1252939. https://doi.org/10.3389/fimmu.2023.1252939 [PubMed]
- 41. Cao M, Ren Y, Li Y, Deng J, Su X, Tang Y, Yuan F, Deng H, Yang G, He Z, Liu B, Yao Z, Deng M. Lnc-ZEB2-19 Inhibits the Progression and Lenvatinib Resistance of Hepatocellular Carcinoma by Attenuating the NF-κB Signaling Pathway through the TRA2A/RSPH14 Axis. Int J Biol Sci. 2023; 19:3678–93. https://doi.org/10.7150/ijbs.85270 [PubMed]
- 42. S Teixeira LF, Bellini MH. NF-ĸB1 knockout reduces IL6 expression under hypoxia in renal cell carcinoma. Cell Mol Biol (Noisy-le-grand). 2023; 69:8–14. https://doi.org/10.14715/cmb/2023.69.6.2 [PubMed]
- 43. Zhang S, Zhang E, Long J, Hu Z, Peng J, Liu L, Tang F, Li L, Ouyang Y, Zeng Z. Immune infiltration in renal cell carcinoma. Cancer Sci. 2019; 110:1564–72. https://doi.org/10.1111/cas.13996 [PubMed]
- 44. Nukui A, Masuda A, Abe H, Arai K, Yoshida KI, Kamai T. Increased serum level of soluble interleukin-2 receptor is associated with a worse response of metastatic clear cell renal cell carcinoma to interferon alpha and sequential VEGF-targeting therapy. BMC Cancer. 2017; 17:372. https://doi.org/10.1186/s12885-017-3369-3 [PubMed]
- 45. Tan P, Chen H, Huang Z, Huang M, Du Y, Li T, Chen Z, Liu Y, Fu W. MMP25-AS1/hsa-miR-10a-5p/SERPINE1 axis as a novel prognostic biomarker associated with immune cell infiltration in KIRC. Mol Ther Oncolytics. 2021; 22:307–25. https://doi.org/10.1016/j.omto.2021.07.008 [PubMed]
- 46. Zhang CH, Huang Y. Effect of Peripheral Blood CD4 + CD25 + Regulatory T Cell on Postoperative Immunotherapy for Patients with Renal Carcinoma. Asian Pac J Cancer Prev. 2016; 17:2027–30. https://doi.org/10.7314/apjcp.2016.17.4.2027 [PubMed]
- 47. Song W, Yeh CR, He D, Wang Y, Xie H, Pang ST, Chang LS, Li L, Yeh S. Infiltrating neutrophils promote renal cell carcinoma progression via VEGFa/HIF2α and estrogen receptor β signals. Oncotarget. 2015; 6:19290–304. https://doi.org/10.18632/oncotarget.4478 [PubMed]
- 48. Li L, Du Z, Rong B, Zhao D, Wang A, Xu Y, Zhang H, Bai X, Zhong J. Foam cells promote atherosclerosis progression by releasing CXCL12. Biosci Rep. 2020; 40:BSR20193267. https://doi.org/10.1042/BSR20193267 [PubMed]
- 49. Krazinski BE, Kowalczyk AE, Sliwinska-Jewsiewicka A, Grzegrzolka J, Godlewski J, Kwiatkowski P, Dziegiel P, Kmiec Z, Kiewisz J. IKBKB expression in clear cell renal cell carcinoma is associated with tumor grade and patient outcomes. Oncol Rep. 2019; 41:1189–97. https://doi.org/10.3892/or.2018.6872 [PubMed]
- 50. Al-Lamki RS, Lu W, Manalo P, Wang J, Warren AY, Tolkovsky AM, Pober JS, Bradley JR. Tubular epithelial cells in renal clear cell carcinoma express high RIPK1/3 and show increased susceptibility to TNF receptor 1-induced necroptosis. Cell Death Dis. 2016; 7:e2287. https://doi.org/10.1038/cddis.2016.184 [PubMed]
- 51. Meng G, Li G, Yang X, Xiao N. Inhibition of miR146b-5p suppresses CT-guided renal cell carcinoma by targeting TRAF6. J Cell Biochem. 2019; 120:2382–90. https://doi.org/10.1002/jcb.27566 [PubMed]
- 52. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–50. https://doi.org/10.1073/pnas.0506580102 [PubMed]
- 53. Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015; 19:A68–77. https://doi.org/10.5114/wo.2014.47136 [PubMed]
- 54. Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018; 34:3771–2. https://doi.org/10.1093/bioinformatics/bty411 [PubMed]
- 55. Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, Ramaswamy S, Futreal PA, Haber DA, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013; 41:D955–61. https://doi.org/10.1093/nar/gks1111 [PubMed]
- 56. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. https://doi.org/10.1186/1471-2105-14-7 [PubMed]
- 57. Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 2014; 9:e107468. https://doi.org/10.1371/journal.pone.0107468 [PubMed]
- 58. Carafa V, Altucci L, Nebbioso A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front Pharmacol. 2019; 10:38. https://doi.org/10.3389/fphar.2019.00038 [PubMed]
- 59. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009; 10:32–42. https://doi.org/10.1038/nrg2485 [PubMed]
- 60. Wang L, Wang D, Yang L, Zeng X, Zhang Q, Liu G, Pan Y. Cuproptosis related genes associated with Jab1 shapes tumor microenvironment and pharmacological profile in nasopharyngeal carcinoma. Front Immunol. 2022; 13:989286. https://doi.org/10.3389/fimmu.2022.989286 [PubMed]
- 61. Zhang L, Zhao Y, Dai Y, Cheng JN, Gong Z, Feng Y, Sun C, Jia Q, Zhu B. Immune Landscape of Colorectal Cancer Tumor Microenvironment from Different Primary Tumor Location. Front Immunol. 2018; 9:1578. https://doi.org/10.3389/fimmu.2018.01578 [PubMed]
- 62. Ito K, Murphy D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacometrics Syst Pharmacol. 2013; 2:e79. https://doi.org/10.1038/psp.2013.56 [PubMed]
- 63. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, Fu X, Liu S, Bo X, Yu G. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2021; 2:100141. https://doi.org/10.1016/j.xinn.2021.100141 [PubMed]
- 64. Román Palacios C, Wright A, Uyeda J. treedata.table: a wrapper for data.table that enables fast manipulation of large phylogenetic trees matched to data. PeerJ. 2021; 9:e12450. https://doi.org/10.7717/peerj.12450 [PubMed]
- 65. Mangiola S, Doyle MA, Papenfuss AT. Interfacing Seurat with the R tidy universe. Bioinformatics. 2021; 37:4100–7. https://doi.org/10.1093/bioinformatics/btab404 [PubMed]
- 66. Che X, Li J, Xu Y, Wang Q, Wu G. Analysis of genomes and transcriptomes of clear cell renal cell carcinomas identifies mutations and gene expression changes in the TGF-beta pathway. Front Genet. 2022; 13:953322. https://doi.org/10.3389/fgene.2022.953322 [PubMed]
- 67. Hu K. Become Competent in Generating RNA-Seq Heat Maps in One Day for Novices Without Prior R Experience. Methods Mol Biol. 2021; 2239:269–303. https://doi.org/10.1007/978-1-0716-1084-8_17 [PubMed]
- 68. Liu Z, Liu L, Weng S, Guo C, Dang Q, Xu H, Wang L, Lu T, Zhang Y, Sun Z, Han X. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat Commun. 2022; 13:816. https://doi.org/10.1038/s41467-022-28421-6 [PubMed]
- 69. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, et al. A pathology atlas of the human cancer transcriptome. Science. 2017; 357:eaan2507. https://doi.org/10.1126/science.aan2507 [PubMed]
- 70. Chandrashekar DS, Karthikeyan SK, Korla PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne U, Creighton CJ, Varambally S. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia. 2022; 25:18–27. https://doi.org/10.1016/j.neo.2022.01.001 [PubMed]
- 71. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BV, Varambally S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017; 19:649–58. https://doi.org/10.1016/j.neo.2017.05.002 [PubMed]
- 72. Liu TT, Li R, Huo C, Li JP, Yao J, Ji XL, Qu YQ. Identification of CDK2-Related Immune Forecast Model and ceRNA in Lung Adenocarcinoma, a Pan-Cancer Analysis. Front Cell Dev Biol. 2021; 9:682002. https://doi.org/10.3389/fcell.2021.682002 [PubMed]