



Introduction
Arising in the adenohypophysis, pituitary adenomas (PAs) are the second most common primary intracranial neoplasms and account for nearly 25% of central nervous system tumors in adults [1]. Although the majority of PAs are benign, 40% of PAs exhibit aggressive behaviors such as locoregional invasiveness, rapid growth, and a high tendency to recur, which represented a poor patient prognosis [2, 3]. At present, chemotherapy and surgical resection are major modalities for PA treatment [4]. However, aggressive PAs can develop resistance to these treatment modalities. It is reported that the rate of recurrence of PAs after resection is high with about 7~58% [5, 6]. Besides, the therapeutic effects of chemotherapy are not satisfactory, due to the strong cytotoxic effects of common anti-tumor drugs [7, 8]. Therefore, deep exploration of the pathogenesis of tumor invasion is desperately needed to identify new molecular targets and drugs for aggressive PAs.
Programmed cell death 10 (PDCD10), also termed “cerebral cavernous malformations 3” (CCM3) or “TF-1 cell apoptosis-related gene 15” (TFAR15), is widely expressed in various types of human cells [9]. Originally, PDCD10 was described as an apoptosis-related factor that is up-regulated upon apoptotic stimuli and inhibits natural cell death of HEK293 cells and fibroblast cells [10, 11]. Besides, PDCD10 was found to participate in the formation of a STRIPAK complex and regulates a wide variety of cellular processes [12]. For example, PDCD10 can recruit germinal centre kinase III (GCKIII) kinase to striatins (STRNs) to regulate cell proliferation and migration [13, 14]. As one of the CCM proteins family, PDCD10 also plays a critical role in angiogenesis in the central nervous system. Similar to CCM1 and CCM2, mutations in the PDCD10 gene were responsible for cerebral cavernous malformations which are characterized by aberrant angiogenesis in the brain [15, 16]. In recent years, the function of PDCD10 in tumors has been increasingly emphasized. Several studies reported that expression of PDCD10 was altered and associated with tumor progression in various types of cancers including ovarian cancer, breast cancer, hepatocellular carcinoma, and non-small cell lung cancer, while a study on glioblastoma indicated a dual role and disease-specificity of the function of PDCD10 [17–21]. Given these multiple functions in tumors, PCDC10 could be involved in PA pathology. However, the function of PDCD10 in PAs has not yet been described. A dataset from the GEO database shows that the expression level of PDCD10 was increased in PAs tissues compared with normal counterparts (Figure 1A), which suggests a pro-oncogenic effect of PDCD10 in PAs.
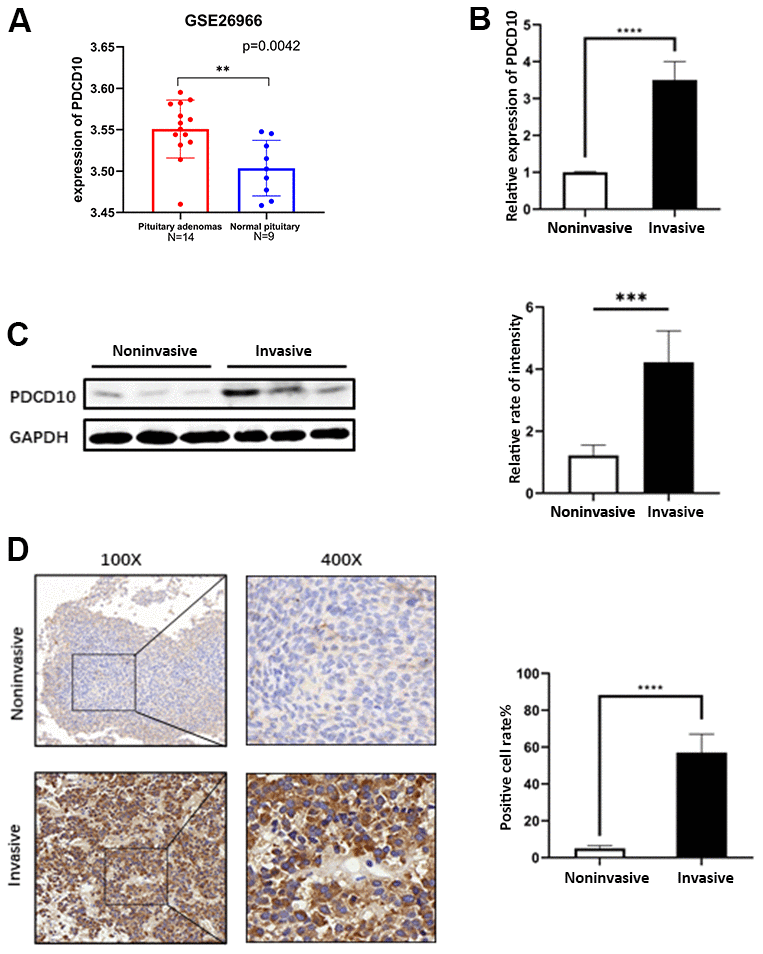
Figure 1. Expression of PDCD10 in human pituitary adenomas. (A) A dataset (GSE26966) was obtained from the GEO database to compare the mRNA expression level of PDCD10 between PA tumor tissues (N=14) and normal pituitary tissue (N=9). (B) Relative mRNA expression levels of PDCD10 by RT-qPCR in invasive (n=15) and non-invasive (n=15) pituitary adenomas. Non-invasive values were set to 1. (C) Representative Western blots showed the expression level of PCDC10 protein (25 kDa) in invasive and noninvasive pituitary adenomas. Band intensities were quantified and normalized to GAPDH. (D) Representative images of immunohistochemistry evaluated the expression of PDCD10 in invasive and non-invasive (n=15) pituitary adenomas. **P < 0.01; ***P < 0.001; ****P < 0.0001.
As a G-protein-coupled receptor, CXC motif chemokine receptor 2(CXCR2) is predominantly expressed on the surface of inflammatory cells including neutrophils, oligodendrocytes, mast cells, eosinophils, and monocytes [22]. However, CXCR2 has also been found on epithelial cells, endothelial cells, some neuroendocrine cells (e.g., pituitary), and various tumor cells [23, 24]. We previously showed that PDCD10 promotes tumor progression in vivo by enhancing CXCL2-CXCR2 signaling in glioblastoma [25]. In this study, we systematically explored the functional role of PDCD10 in PAs and demonstrated that PDCD10 could regulate activation of AKT/ERK signaling pathways by altering the protein expression level of CXCR2 to modulate cellular proliferation, migration, invasion and EMT in PAs.
Materials and Methods
Human pituitary adenoma samples
From July 2017 to October 2018, 15 invasive and 15 noninvasive PA samples were collected from patients who underwent surgery at the Department of Neurosurgery, Tongji Hospital affiliated with Tongji Medical College, Huazhong University of Science and Technology. Knosp grading scheme and intraoperative findings were employed to determine the invasiveness of the tumor [26]. The clinical characteristics of the patients were summarized in Table 1. The median age of patients at surgery was 43.5 years (range 23-68 years). Thirteen were male (43.3%), and 17 were female (56.7%) with a male to female ratio of 1:1.31. Tumor samples were used for immunohistochemical (IHC) staining, protein, and RNA extraction. The positive cell rate was quantified using ImageJ software (NIH).
Table 1. The clinical characteristics of the patients.
| Invasive PA | Noninvasive PA | |
| Age | ||
| Mean | 41.7 | 44.3 |
| Range | 23-68 | 28-65 |
| Gender | ||
| Male | 8 | 6 |
| Female | 7 | 9 |
| Knosp Grade | ||
| I | 0 | 7 |
| II | 0 | 8 |
| III | 5 | 0 |
| IV | 10 | 0 |
Cell culture and transfection
Att-20 cell line was established from mouse ACTH-secreting pituitary tumors by Buonassisi et al. [27], and TtT/GF is a murine folliculo-stellate-like cell line deriving from a thyrotrophic pituitary tumor [28]. The two mouse PA cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Carlsbad, USA) containing 10% fetal bovine serum (FBS, Gibco) and 1% Penicillin/Streptomycin and were placed in a thermostatic incubator at 37° C with 5% CO2. Recombinant mouse CXCL2 protein (R&D System) was added into the culture medium to study the signaling pathways in a subset of experiments as indicated.
Lentiviral shRNA vector for mouse PDCD10 (LV-PDCD10 71721) and control vector (Scramble shRNA, LVCON054) were purchased from GenePharma, Shanghai, China. Cells were seeded into 6-well plates to achieve 20-30% confluence before infection. And then 1 ml of serum-free medium containing lentivirus (1×109 TU/ml) was added to each well. After 72h, a culture medium containing 1 mg/ml puromycin was used for 3 weeks to select PA cells with stable knockdown of PDCD10. CDS-sequence of mouse PDCD10 was inserted into pcDNA-3.1 plasmids to gain PDCD10-overexpression vectors. Cells were transiently transfected with the pc3.1-PDCD10 plasmids and control plasmids (pc3.1) by using lipo3000 (Invitrogen) according to the manufacturer’s instructions. The knockdown or overexpression of PDCD10 was confirmed by RT2-PCR and western blotting.
Protein extraction and Western blotting
The protocols of cell protein extraction and western blotting were performed as previously [29]. Antibodies to PDCD10, E-cadherin, N-cadherin and Vimentin (VIM) were purchased from Proteintech Group. Antibody to GAPDH was purchased from Servicebio. Primary antibodies to CXCR2 and ki-67, and secondary antibodies for western blotting were purchased from Abcam. Antibodies to ERK1/2, p-ERK1/2 (phosphorylation site Thr202/Tyr204), STAT3, p-STAT3 (phosphorylation site Tyr705), AKT, and p-AKT (phosphorylation site Ser473) were obtained from Cell Signaling Technology, Inc.
Cell proliferation, migration, and invasion assay
4.0×103 cells were seeded into 96-well-plate in quintuplicate. After 72 hours of incubation, the absorbance was measured at 450 nm by adding a CCK8 reagent (Boster Biological Technology) to detect cell proliferation. Cell migration was evaluated by scratch assays as described previously [25]. After 16 hours of incubation, the migrated area was photographed and calculated by ImageJ software. For invasion assay, 8 mm pore transwell inserts (Corning Life Sciences) were coated with 75μl Matrigel matrix (Corning, 200 μg/mL) as upper chambers. 700μl DMEM medium with 20% FBS was added into the 24-well-plate. Meanwhile, 5.0×104 cells were suspended in a serum-free medium and were added to the inserts which were placed in a 24-well-plate. After 48h, the cells were fixed with 10% formalin and stained with 0.1% crystal violet solution. The numbers of invaded cells were counted at 200x magnification.
Xenograft experiments
Nude mice (blab/c-nu; SJA Laboratory Animal Co. Ltd, Hunan, China) were subcutaneously injected with 5 × 106 cells to establish the Att-20 and TtT/GF cell xenograft model. Tumor growth was monitored regularly and 20 days after injection, tumor sizes and weights were measured. For IHC staining, tumor samples were paraffin-embedded following 4% paraformaldehyde fixation. The IHC results were analyzed by ImageJ. For western blotting, the total protein of the tumor samples was extracted.
Statistical analysis
Statistical software GraphPad Prism Ver.8.0 was employed for performing Statistical analysis and graphing. The student’s t-test and ANOVA were appropriately used for statistical analyses. Statistical significance was established as p<0.05.
Results
Expression of PDCD10 in human pituitary adenomas
A dataset (GSE26966) from the GEO database shows that the mRNA expression level of PDCD10 was increased in PAs tissues compared with normal counterparts (Figure 1A). Furthermore, the expression of PDCD10 was examined between non-invasive and invasive PA tumor samples which were collected from the Neurosurgery department at Tongji Hospital. In contrast to non-invasive PAs, the mRNA and protein expression levels of PDCD10 were significantly upregulated in invasive PAs on the mRNA (Figure 1B), protein level (Figure 1C), and following IHC staining with anti-PDCD10 (Figure 1D).
PDCD10 silencing suppresses the proliferation, migration, invasion and EMT of PA cells
To explore the functional role of PDCD10 in PAs, we first examined the impact of PDCD10 silencing on cell proliferation, migration, and invasion in PA cell lines. As shown in Figure 2A, 2B, western blotting confirmed the knockdown efficiency of PDCD10-shRNA lentivirus. Besides, PDCD10 silencing significantly increased the epithelial marker expression (E-cadherin) but reduced the mesenchymal marker expression (N-cadherin and VIM), demonstrating a significant inhibition of EMT process in PA cells (Figure 2A, 2B). The results of the CCK-8 assay showed significant suppression of cell proliferation after PDCD10 silencing in Att-20 and TtT/GF cells (Figure 2C, 2D).
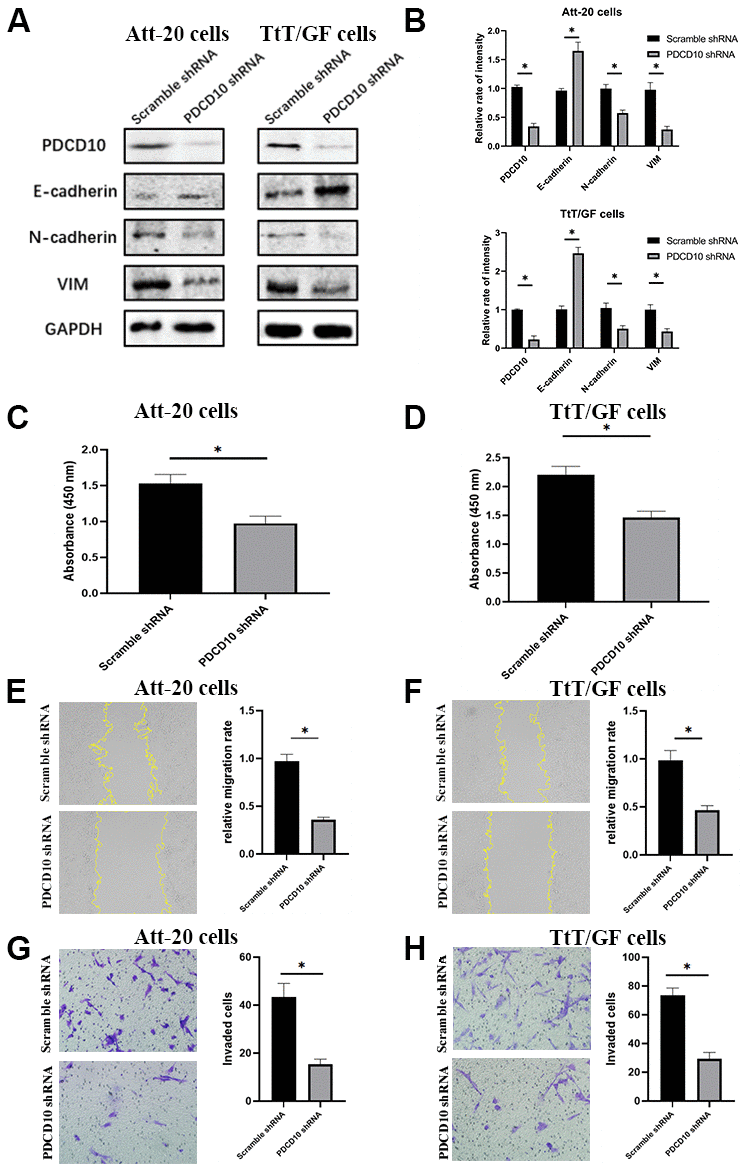
Figure 2. PDCD10 silencing suppresses the proliferation, migration, invasion and EMT of PA cells. (A) Western blotting was performed to detect the impact of PDCD10 silencing on the expression of EMT markers in Att-20 cells and TtT/GF cells. (B) Band intensities were quantified and normalized to GAPDH. (C, D) CCK-8 assay was used to assess cell proliferation capacity after PDCD10 silencing in Att-20 and TtT/GF cells. (E, F) Scratch assay was used to examine the relative migration rates of Att-20 and TtT/GF cells (magnification:100x). (G, H) Transwell invasion assay was used to analyze the invasion potential of Att-20 and TtT/GF cells (magnification: 200x). * P < 0.05.
According to the scratch assay, the relative migration rate of Att-20 and TtT/GF cells in the PDCD10-shRNA group was 35.2±4.3% and 45.7±6.4%, respectively (Figure 2E, 2F). In addition, transwell invasion assays revealed that PDCD10 silencing reduced numbers of invaded cells in Att-20 (65.2±11.6%) and TtT/GF (60.3±8.5%) cells (Figure 2G, 2H).
Overexpression of PDCD10 promotes the proliferation, migration, invasion and EMT of PA cells
We further sought to verify the effect of PDCD10 in a gain-of-function setting, PDCD10-overexpressing PA cell lines were generated by transfection of pcDNA 3.1 plasmids containing the full-length coding sequence of murine PDCD10. The transfection efficiency was confirmed by western blotting (Figure 3A, 3B). Overexpression of PDCD10 reduced E-cadherin expression but increased the expression of N-cadherin and VIM, indicating a remarkable promotion of EMT process in PA cells (Figure 3A, 3B). Besides, up-regulation of PDCD10 remarkably promoted the capacities of proliferation (Figure 3C, 3D). The relative migration rate of Att-20 and TtT/GF cells in PDCD10 overexpression group was 168.7±7.4% and 163.8±6.3%, respectively (Figure 3E, 3F). Overexpression of PDCD10 increased numbers of invaded cells in Att-20 (56.2±13.2%) and TtT/GF (79.5±15.8%) cells (Figure 3G, 3H). Altogether, these results suggest that PDCD10 exerts a pro-oncogenic effect in PA cells.
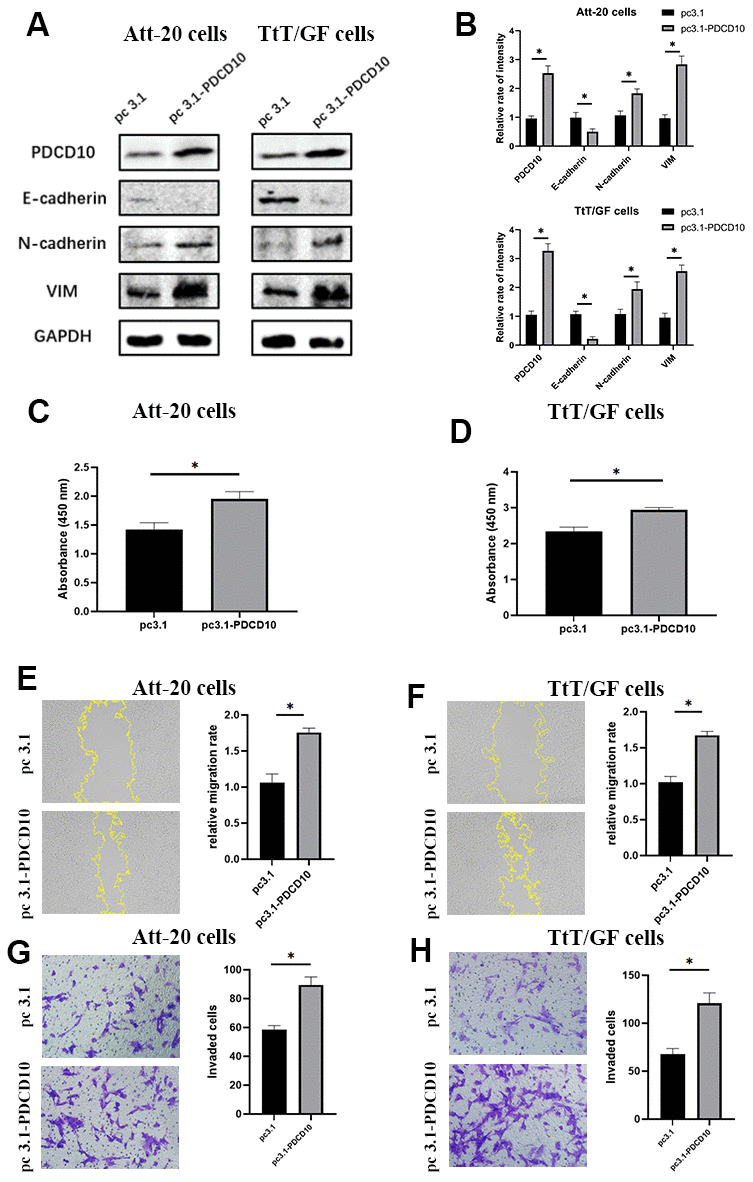
Figure 3. Overexpression of PDCD10 promotes the proliferation, migration, invasion and EMT of PA cells. (A, B) Western blotting was performed to detect the impact of PDCD10 overexpression on the expression levels of EMT markers in Att-20 cells and TtT/GF cells. Band intensities were quantified and normalized to GAPDH. (C, D) CCK-8 assay was used to assess cell proliferation potential after PDCD10 overexpression in Att-20 and TtT/GF cells. (E, F) Scratch assay was employed to examine the relative migration rates of Att-20 and TtT/GF cells after PDCD10 overexpression (magnification:100x). (G, H) Transwell invasion assay was used to detect the invasion potential of Att-20 and TtT/GF cells after PDCD10 overexpression (magnification: 200x). * P < 0.05.
PDCD10 alters the protein expression level of CXCR2 and regulates the activation of downstream AKT/ERK signal pathways
According to our previous study, CXCL2-CXCR2 signaling mediated by PDCD10 participates in the crosstalk between glioblastoma cells and microglia/macrophages and promotes tumor growth [25]. Therefore, we analyzed expression levels of CXCR2 in PDCD10-silencing and PDCD10-overexpressing PA cells. Figure 4 revealed that PDCD10 silencing reduced the protein expression levels of CXCR2, while CXCR2 expression was elevated in the PDCD10-overexpression group of Att-20 and TtT/GF cells. Meanwhile, the activation state of downstream signaling pathways including AKT, ERK, and STAT3 was examined to further explore the underlying mechanism of PDCD10. As shown in Figure 4, PDCD10 silencing significantly inhibited phosphorylation of AKT and ERK1/2 but not STAT3 in Att-20 and TtT/GF cells. In addition, compared with the control group, simultaneous activation of AKT and ERK1/2 was observed in PDCD10-overexpression Att-20 and TtT/GF cells.
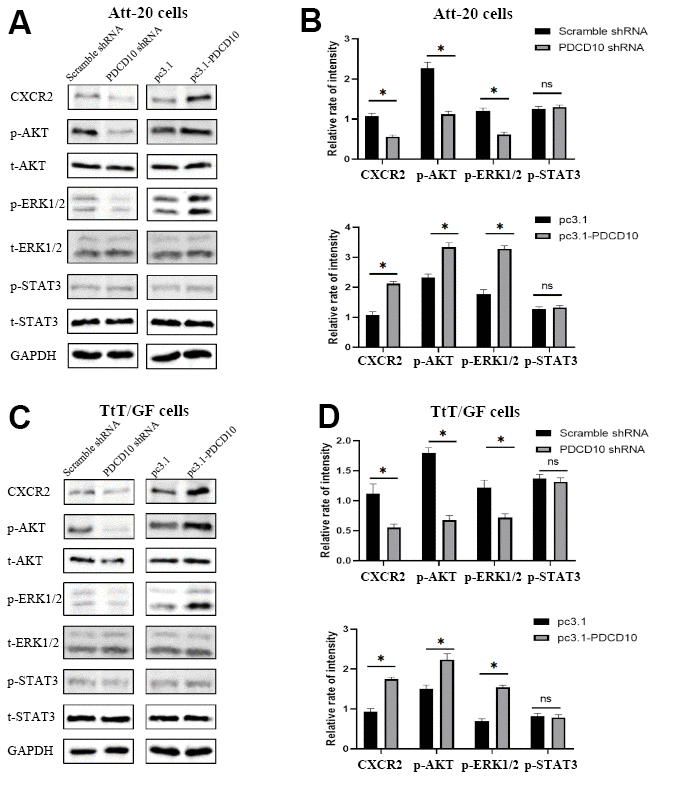
Figure 4. PDCD10 alters the protein expression level of CXCR2 and regulates the activation of downstream AKT/ERK signal pathways. (A, B) Western blotting was used to detect the expression level of CXCR2 and phosphorylation level of AKT, ERK1/2 and STAT3 in Att-20 cells after PDCD10 silencing or overexpression. Band intensities were quantified and normalized to GAPDH. * P < 0.05. (C, D) In TtT/GF cells, western blotting was performed to examine the expression level of CXCR2 and phosphorylation level of AKT, ERK1/2 and STAT3 after PDCD10 silencing or overexpression. Band intensities were quantified and normalized to GAPDH. * P < 0.05.
Activation of CXCR2 rescues the inactivation of AKT/ERK signaling and the tumor-suppressive effects induced by PDCD10 silencing
Based on the above results, we presumed that CXCR2 may participate in the regulation of PDCD10 in the aggressive behavior of PA cells. To verify the hypothesis, PDCD10-silencing cells were treated with recombinant mouse CXCL2 protein to induce the activation of CXCR2, followed by analyses of cellular proliferation, migration, and invasion.
After treatment with CXCL2, the phosphorylation levels of AKT and ERK1/2 were significantly increased (Figure 5A, 5B). Moreover, the inhibition of cellular proliferation induced by PDCD10 silencing was reverted via activation of CXCR2 by CXCL2 administration in Att-20 and TtT/GF cells (Figure 5C, 5D). CXCL2 treatment in PDCD10-silencing group also increased migration area (87.8±8.2% and 147.4±13.6%, respectively) and numbers of invaded cells (79.2±15.8% and 110±13.7%, respectively) in PA cell lines (Figure 5E–5H). These results suggest that PCDC10 and CXCL2 are part of the same signal cascade.
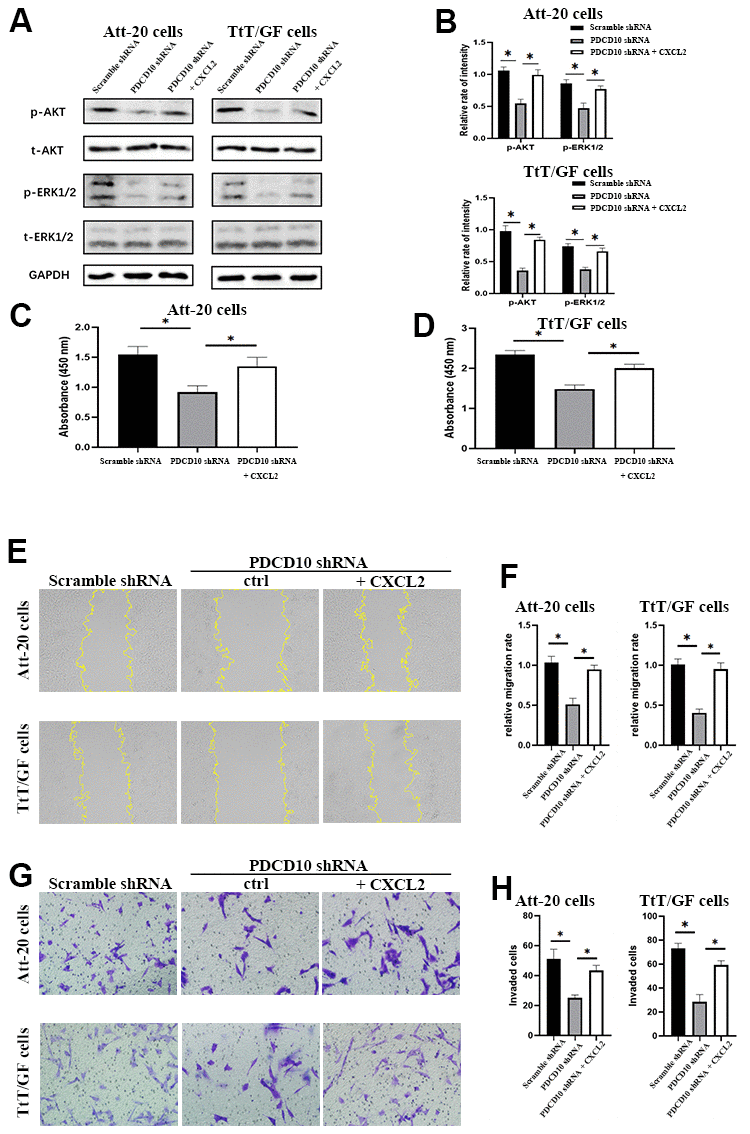
Figure 5. Activation of CXCR2 rescues the inactivation of AKT/ERK signaling and the tumor-suppressive effects induced by PDCD10 silencing. (A, B) Western blotting was performed to examine the phosphorylation levels of AKT and ERK1/2 after recombinant CXCL2 administration (200ng/ml) in Att-20 and TtT/GF cells with PDCD10 silencing. (C, D) CCK-8 assay was used to evaluate cell proliferation potential after CXCL2 administration in Att-20 and TtT/GF cells with PDCD10 silencing. (E, F) Relative migration rate of ATT-20 and TtT/GF cells with PDCD10 silencing was analyzed by scratch assay after CXCL2 administration (magnification: 100x). (G, H) Transwell invasion assay was employed to assess the invasion capacity of Att-20 and TtT/GF cells with PDCD10 silencing after CXCL2 administration (magnification: 200x). * P < 0.05.
PDCD10 silencing impairs the tumorigenesis and reduces CXCR2 expression of PA cells in vivo
Finally, in vivo experiments were implemented to verify the effect of PDCD10 on tumorigenesis in PAs by establishing Att-20 and TtT/GF cell xenograft nude mice model with stable PDCD10-silencing. Consistent with the results from the in vitro experiments, PDCD10 silencing significantly reduced the tumor sizes and weights of xenograft tumors (Att-20 cell line: 0.94±0.07g vs. 0.38±0.06g, and TtT/GF cell line:1.04±0.11g vs. 0.34g±0.12g) (Figures 6A, 5B). Besides, the Ki-67 staining, a proliferation indicator, was also decreased in PDCD10-silencing tumor samples according to IHC results (Figure 6C–6F). IHC staining and western blotting also further confirmed that PDCD10 silencing decreased CXCR2 expression level in vivo (Figure 6C–6H). These data confirm that PDCD10 promotes tumor growth by regulating the expression of CXCR2 in PAs.
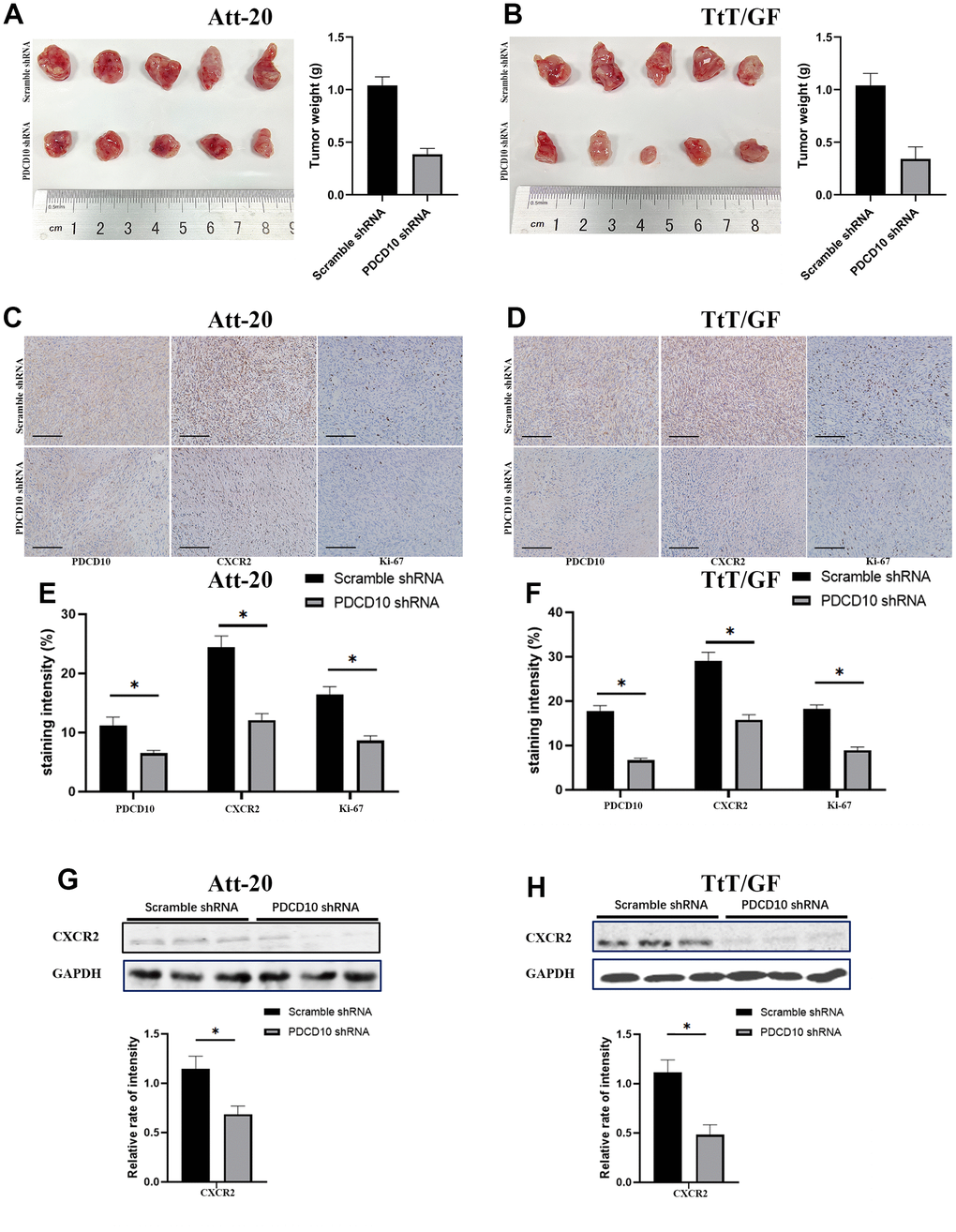
Figure 6. PDCD10 silencing impairs the tumorigenesis and reduces CXCR2 expression of PA cells in vivo. (A, B) Images for Att-20 and TtT/GF xenografts from nude mice (Left). Statistical analysis of xenograft tumor weights (Right). (C, D) Representative IHC images of Att-20 and TtT/GF xenograft tumor tissues for PDCD10, CXCR2 and Ki-67 staining (200x). Scale bars: 100μm. (E, F) Intensity of PDCD10, CXCR2 and Ki-67 staining were analyzed by IHC-Profiler. (G, H) Western blotting was performed to examine the expression of CXCR2 in Att-20 and TtT/GF xenograft tumor samples. Band intensities were quantified and normalized to GAPDH. * P < 0.05.
Discussion
As the second most common type of primary CNS tumors, high recurrence rates, and increased mortality as a consequence of aggressive PAs remain a great clinical challenge. The strong abilities of proliferation and infiltration/invasion of tumor cells result in large tumor size and severe destruction of adjacent normal structures, which is the major cause for incomplete removal of PAs and relapse [30]. However, the molecular and cellular mechanisms underlying the aggressive behaviors of PA cells need to be elucidated.
PDCD10 is an evolutionarily conserved protein and is widely distributed in a variety of tissues. According to a dataset from the GEO database, the expression levels of PDCD10 are increased in PA tissues compared to normal pituitaries (Figure 1A), suggesting that PDCD10 may exert a pro-tumorigenic effect in PAs.
Using loss-of-function as well as gain-of-function approaches, we demonstrate the pro-oncogenic effect of PDCD10 in Att-20 and TtT/GF cells: when expressed at high levels, PDCD10 enhanced the abilities of cellular proliferation, migration, invasion and epithelial to mesenchymal transition (EMT) in PAs. Some studies have indicated that PDCD10 plays an essential role in cell survival and migration. For example, Fidalgo et al. found PDCD10 can protect cells exposed to reactive oxygen species from death by mediating phosphorylation of the Ezrin/Radixin/Moesin proteins [31]. Proteomic analysis identified some proteins which regulate the cell cycle as interactors of PDCD10 [16, 32]. Under physiological conditions, PDCD10 is indispensable for neuronal migration during development [33]. In recent years, accumulating evidence has shown that dysregulation of PDCD10 was closely related to tumorigenesis and progression in diverse tumor types. Initially, PDCD10 was found to be associated with protein phosphatase-2A, a regulator of mitogenesis, and to enhance proliferation in malignant T cells [34]. Sun et al. reported that PDCD10 promotes the progression of hepatocellular carcinoma by interacting with PP2Ac to increase activation of YAP [21]. In ovarian cancer cells, PDCD10 upregulates Wnt/β-catenin signaling thereby augmenting migration of tumor cells [17]. Genome-wide analysis also showed that increased copy number and high expression level of PDCD10 are associated with tumor grade, nodal involvement and advanced FIGO stage in ovarian cancer [35]. In breast cancer, overexpression of PDCD10 induced the inactivation of the ROCK/Rho signal pathway to suppress cell adhesion and promote cell migration and invasion [20]. PDCD10 also participates in regulation of stemness of breast cancer cell [36]. Besides, PDCD10 was identified as the target of CircSMARCA5-miR-432 axis, and was found to promote proliferation, metastasis and glycolysis of prostate cancer cells [37]. The pro-tumorigenic effect of PDCD10 was also recently reported in lung cancer and bladder cancer [19, 38]. On the contrary, PDCD10 was reported as a tumor suppressor in glioblastoma cells [18]. These contradictory results hint that function of PDCD10 is tissue- and disease-specific in different tumors. As shown in our previous results, PDCD10 participates in the crosstalk between glioblastoma cells and microglia/macrophages and promotes tumor growth by CXCL2-CXCR2 signaling in vivo [25], which suggests that CXCR2 may play a critical role in aggressive behaviors mediated by PDCD10 in tumors. In this study, we report for the first time that PDCD10 can promote cellular proliferation, migration, and invasion of PAs by regulating the CXCR2-AKT/ERK signaling axis.
EMT is a cellular process that cells lose their epithelial features and acquire mesenchymal phenotype. It is well established that EMT is associated with tumor initiation, tumor cell migration, tumor stemness, metastasis, and treatment resistance in various cancer types [39]. Some studies have reported that EMT plays a key role in tumor progression of PAs [40, 41]. For example, Li et al. found that high CCNB1 expression resulted in cavernous sinus invasion of PAs through promoting EMT process [40]. PDCD10 was also reported to regulate EMT in hepatocellular carcinoma and ovarian cancer [17, 21]. Thus, it can be speculated that PDCD10 might contribute to tumor progression in PAs by inducing EMT. In our study, we observed that PDCD10 silencing increased epithelial marker expression (E-cadherin), but reduced the mesenchymal marker (N-cadherin and VIM), while overexpression of PDCD10 reduced E-cadherin expression but increased the expression of N-cadherin and VIM in PA cells. These results suggest that PDCD10 acts as regulators of EMT in PAs.
As a seven-transmembrane (7TM) protein, the C-terminus of CXCR2 is related to phosphorylation and internalization of the receptor, while the N-terminal dominant and extracellular loops determine the binding with ligands [42]. The ligands of CXCR2 include CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8, among which CXCL1, CXCL2, CXCL3, CXCL,5, and CXCL7 bind CXCR2 only [43]. After binding of these cognate chemokine ligands to CXCR2 on the cell surface, the associated G-protein is activated and dissociates, which in turn leads to activation of multiple downstream signaling cascades including the phosphatidylinositol-3 kinase (PI3K)/Akt and RAS-Raf-MEK1/2-ERK1/2 signaling pathway [42].
To date, CXCR2 has been extensively studied in various types of neoplasms. In lung adenocarcinoma, up-regulation of CXCR2 in tumor cells promotes invasion and metastasis, which results in a poor prognosis [44]. In addition, high expression of CXCR2 has also been observed in PAs, medullary carcinomas of the thyroid, and pheochromocytomas [23]. CXCR2 was also reported to be associated with EMT and resistance of tumor cells to chemotherapy [45, 46]. Besides, CXCR2 was up-regulated in cancer stem cells (CSCs) and promotes the growth and migration of CSCs [47, 48]. In our study, we observed that PDCD10 silencing and overexpression significantly inhibited and increased protein expression level of CXCR2 together with phosphorylation levels of AKT and ERK1/2 in Att-20 and TtT/GF cells, respectively. In PAs, aberrant activation of AKT and ERK pathways was reported to lead to abnormal hormone production and neoplastic growth [49, 50]. Interestingly, activation of CXCR2 reversed the down-regulation of ERK1/2 and AKT phosphorylation level and suppression of proliferation, migration, and invasion induced by knockdown of PDCD10. Thus, we assumed that CXCR2 is involved in aggressive behaviors of tumor cells mediated by PDCD10 in PAs. Proteomic analysis revealed that PDCD10 may be involved in vesicle trafficking of membrane proteins [16, 32]. Consistent with this, several recent studies reported that PDCD10 was involved in regulation of exocytosis and AQP2 membrane targeting [51, 52]. Thus, we speculate that CXCR2 expressed as a membrane protein, maybe stabilized by PDCD10 and its interactors via regulating protein trafficking. However, the precise mechanism of how PDCD10 and CXCR2 cooperate in the cellular context requires further exploration in the future.
In vivo, PDCD10 silencing significantly reduced the sizes and weights of PA cell-derived tumors in xenograft models. In addition, the expression levels of Ki-67, which represents a level of tumor proliferation, together with CXCR2 were decreased in the PDCD10-shRNA group. These results further verify the pro-oncogenic effect of PDCD10 in PAs and imply the therapeutic potential of targeting PDCD10. Although temozolomide has been included in the guidelines of the Endocrine Society and European Society of Endocrinology, long-term use of it could result in an increase of toxicity including the risk of leukemia and bone marrow suppression [53–56]. Besides, the efficacy and safety of existing targeted therapies (e.g., RTK inhibitors) for PAs still need further investigation [57]. Thus, PDCD10 could be a new promising target for the design of drugs and treatment of PAs.
Author Contributions
Jingdian Liu and Junwen Wang contributes equally in this work. Ting Lei, Kai Shu: project design, manuscript revision. Junwen Wang and Jingdian Liu: experiments validation, statistical analysis, preliminary manuscript written. Weidong Tian, Yu Xu, Ran Li, Kai Zhao, Chao You, Hongquan Niu, Huaqiu Zhang: clinical data collection and evaluation, statistical analysis. Yuan Zhu, Joerg Walter Bartsch: manuscript revision, proof-reading.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical Statement and Consent
The ethics committee at Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology approved this study and written informed consent was obtained from all participants or their family members.
Funding
This work was supported by National Natural Science Foundation of China (grant no. 81702478 and 81602204).
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence.
References
- 1. Daly AF, Tichomirowa MA, Beckers A. The epidemiology and genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab. 2009; 23:543–54. https://doi.org/10.1016/j.beem.2009.05.008 [PubMed]
- 2. Larysz D, Blamek S, Rudnik A. Clinical aspects of molecular biology of pituitary adenomas. Folia Neuropathol. 2012; 50:110–7. [PubMed]
- 3. Raverot G, Castinetti F, Jouanneau E, Morange I, Figarella-Branger D, Dufour H, Trouillas J, Brue T. Pituitary carcinomas and aggressive pituitary tumours: merits and pitfalls of temozolomide treatment. Clin Endocrinol (Oxf). 2012; 76:769–75. https://doi.org/10.1111/j.1365-2265.2012.04381.x [PubMed]
- 4. Langlois F, McCartney S, Fleseriu M. Recent Progress in the Medical Therapy of Pituitary Tumors. Endocrinol Metab (Seoul). 2017; 32:162–70. https://doi.org/10.3803/EnM.2017.32.2.162 [PubMed]
- 5. Losa M, Mortini P, Barzaghi R, Ribotto P, Terreni MR, Marzoli SB, Pieralli S, Giovanelli M. Early results of surgery in patients with nonfunctioning pituitary adenoma and analysis of the risk of tumor recurrence. J Neurosurg. 2008; 108:525–32. https://doi.org/10.3171/JNS/2008/108/3/0525 [PubMed]
- 6. Santoro A, Minniti G, Ruggeri A, Esposito V, Jaffrain-Rea ML, Delfini R. Biochemical remission and recurrence rate of secreting pituitary adenomas after transsphenoidal adenomectomy: long-term endocrinologic follow-up results. Surg Neurol. 2007; 68:513–8. https://doi.org/10.1016/j.surneu.2007.05.057 [PubMed]
- 7. Elbelt U, Schlaffer SM, Buchfelder M, Knappe UJ, Vila G, Micko A, Deutschbein T, Unger N, Lammert A, Topuzoglu-Müller T, Bojunga J, Droste M, Johanssen S, et al. Efficacy of Temozolomide Therapy in Patients With Aggressive Pituitary Adenomas and Carcinomas-A German Survey. J Clin Endocrinol Metab. 2020; 105:dgz211. https://doi.org/10.1210/clinem/dgz211 [PubMed]
- 8. Losa M, Bogazzi F, Cannavo S, Ceccato F, Curtò L, De Marinis L, Iacovazzo D, Lombardi G, Mantovani G, Mazza E, Minniti G, Nizzoli M, Reni M, Scaroni C. Temozolomide therapy in patients with aggressive pituitary adenomas or carcinomas. J Neurooncol. 2016; 126:519–25. https://doi.org/10.1007/s11060-015-1991-y [PubMed]
- 9. Petit N, Blécon A, Denier C, Tournier-Lasserve E. Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development. Gene Expr Patterns. 2006; 6:495–503. https://doi.org/10.1016/j.modgep.2005.11.001 [PubMed]
- 10. Busch CR, Heath DD, Hubberstey A. Sensitive genetic biomarkers for determining apoptosis in the brown bullhead (Ameiurus nebulosus). Gene. 2004; 329:1–10. https://doi.org/10.1016/j.gene.2004.01.004 [PubMed]
- 11. Wang Y, Liu H, Zhang Y, Ma D. cDNA cloning and expression of an apoptosis-related gene, humanTFAR15 gene. Sci China C Life Sci. 1999; 42:323–9. https://doi.org/10.1007/BF03183610 [PubMed]
- 12. Hwang J, Pallas DC. STRIPAK complexes: structure, biological function, and involvement in human diseases. Int J Biochem Cell Biol. 2014; 47:118–48. https://doi.org/10.1016/j.biocel.2013.11.021 [PubMed]
- 13. Madsen CD, Hooper S, Tozluoglu M, Bruckbauer A, Fletcher G, Erler JT, Bates PA, Thompson B, Sahai E. STRIPAK components determine mode of cancer cell migration and metastasis. Nat Cell Biol. 2015; 17:68–80. https://doi.org/10.1038/ncb3083 [PubMed]
- 14. Ma X, Zhao H, Shan J, Long F, Chen Y, Chen Y, Zhang Y, Han X, Ma D. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007; 18:1965–78. https://doi.org/10.1091/mbc.e06-07-0608 [PubMed]
- 15. Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, et al, and Société Française de Neurochirurgie. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005; 76:42–51. https://doi.org/10.1086/426952 [PubMed]
- 16. Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL, Wu CC. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res. 2007; 6:4343–55. https://doi.org/10.1021/pr0704276 [PubMed]
- 17. Fan L, Lei H, Zhang S, Peng Y, Fu C, Shu G, Yin G. Non-canonical signaling pathway of SNAI2 induces EMT in ovarian cancer cells by suppressing miR-222-3p transcription and upregulating PDCD10. Theranostics. 2020; 10:5895–913. https://doi.org/10.7150/thno.43198 [PubMed]
- 18. Wan X, Saban DV, Kim SN, Weng Y, Dammann P, Keyvani K, Sure U, Zhu Y. PDCD10-Deficiency Promotes Malignant Behaviors and Tumor Growth via Triggering EphB4 Kinase Activity in Glioblastoma. Front Oncol. 2020; 10:1377. https://doi.org/10.3389/fonc.2020.01377 [PubMed]
- 19. Yang D, Wang JJ, Li JS, Xu QY. miR-103 Functions as a Tumor Suppressor by Directly Targeting Programmed Cell Death 10 in NSCLC. Oncol Res. 2018; 26:519–28. https://doi.org/10.3727/096504017X15000757094686 [PubMed]
- 20. Tan P, Ye Y, He L, Xie J, Jing J, Ma G, Pan H, Han L, Han W, Zhou Y. TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PLoS Biol. 2018; 16:e3000051. https://doi.org/10.1371/journal.pbio.3000051 [PubMed]
- 21. Sun B, Zhong FJ, Xu C, Li YM, Zhao YR, Cao MM, Yang LY. Programmed cell death 10 promotes metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma via PP2Ac-mediated YAP activation. Cell Death Dis. 2021; 12:849. https://doi.org/10.1038/s41419-021-04139-z [PubMed]
- 22. Le Y, Zhou Y, Iribarren P, Wang J. Chemokines and chemokine receptors: their manifold roles in homeostasis and disease. Cell Mol Immunol. 2004; 1:95–104. [PubMed]
- 23. Tecimer T, Dlott J, Chuntharapai A, Martin AW, Peiper SC. Expression of the chemokine receptor CXCR2 in normal and neoplastic neuroendocrine cells. Arch Pathol Lab Med. 2000; 124:520–5. https://doi.org/10.5858/2000-124-0520-EOTCRC [PubMed]
- 24. Hertzer KM, Donald GW, Hines OJ. CXCR2: a target for pancreatic cancer treatment? Expert Opin Ther Targets. 2013; 17:667–80. https://doi.org/10.1517/14728222.2013.772137 [PubMed]
- 25. Zhang Q, Wang J, Yao X, Wu S, Tian W, Gan C, Wan X, You C, Hu F, Zhang S, Zhang H, Zhao K, Shu K, Lei T. Programmed Cell Death 10 Mediated CXCL2-CXCR2 Signaling in Regulating Tumor-Associated Microglia/Macrophages Recruitment in Glioblastoma. Front Immunol. 2021; 12:637053. https://doi.org/10.3389/fimmu.2021.637053 [PubMed]
- 26. Knosp E, Steiner E, Kitz K, Matula C. Pituitary adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery. 1993; 33:610–7. https://doi.org/10.1227/00006123-199310000-00008 [PubMed]
- 27. Buonassisi V, Sato G, Cohen AI. Hormone-producing cultures of adrenal and pituitary tumor origin. Proc Natl Acad Sci USA. 1962; 48:1184–90. https://doi.org/10.1073/pnas.48.7.1184 [PubMed]
- 28. Inoue K, Matsumoto H, Koyama C, Shibata K, Nakazato Y, Ito A. Establishment of a folliculo-stellate-like cell line from a murine thyrotropic pituitary tumor. Endocrinology. 1992; 131:3110–6. https://doi.org/10.1210/endo.131.6.1446645 [PubMed]
- 29. Wang J, Voellger B, Benzel J, Schlomann U, Nimsky C, Bartsch JW, Carl B. Metalloproteinases ADAM12 and MMP-14 are associated with cavernous sinus invasion in pituitary adenomas. Int J Cancer. 2016; 139:1327–39. https://doi.org/10.1002/ijc.30173 [PubMed]
- 30. Raverot G, Dantony E, Beauvy J, Vasiljevic A, Mikolasek S, Borson-Chazot F, Jouanneau E, Roy P, Trouillas J. Risk of Recurrence in Pituitary Neuroendocrine Tumors: A Prospective Study Using a Five-Tiered Classification. J Clin Endocrinol Metab. 2017; 102:3368–74. https://doi.org/10.1210/jc.2017-00773 [PubMed]
- 31. Fidalgo M, Guerrero A, Fraile M, Iglesias C, Pombo CM, Zalvide J. Adaptor protein cerebral cavernous malformation 3 (CCM3) mediates phosphorylation of the cytoskeletal proteins ezrin/radixin/moesin by mammalian Ste20-4 to protect cells from oxidative stress. J Biol Chem. 2012; 287:11556–65. https://doi.org/10.1074/jbc.M111.320259 [PubMed]
- 32. Baxter SS, Dibble CF, Byrd WC, Carlson J, Mack CR, Saldarriaga I, Bencharit S. Role of cytoskeletal proteins in cerebral cavernous malformation signaling pathways: a proteomic analysis. Mol Biosyst. 2014; 10:1881–9. https://doi.org/10.1039/c3mb70199a [PubMed]
- 33. Louvi A, Nishimura S, Günel M. Ccm3, a gene associated with cerebral cavernous malformations, is required for neuronal migration. Development. 2014; 141:1404–15. https://doi.org/10.1242/dev.093526 [PubMed]
- 34. Lauenborg B, Kopp K, Krejsgaard T, Eriksen KW, Geisler C, Dabelsteen S, Gniadecki R, Zhang Q, Wasik MA, Woetmann A, Odum N. Programmed cell death-10 enhances proliferation and protects malignant T cells from apoptosis. APMIS. 2010; 118:719–28. https://doi.org/10.1111/j.1600-0463.2010.02669.x [PubMed]
- 35. De Marco C, Zoppoli P, Rinaldo N, Morganella S, Morello M, Zuccalà V, Carriero MV, Malanga D, Chirillo R, Bruni P, Malzoni C, Di Vizio D, Venturella R, et al. Genome-wide analysis of copy number alterations led to the characterisation of PDCD10 as oncogene in ovarian cancer. Transl Oncol. 2021; 14:101013. https://doi.org/10.1016/j.tranon.2021.101013 [PubMed]
- 36. Feng ZM, Qiu J, Chen XW, Liao RX, Liao XY, Zhang LP, Chen X, Li Y, Chen ZT, Sun JG. Essential role of miR-200c in regulating self-renewal of breast cancer stem cells and their counterparts of mammary epithelium. BMC Cancer. 2015; 15:645. https://doi.org/10.1186/s12885-015-1655-5 [PubMed]
- 37. Dong C, Fan B, Ren Z, Liu B, Wang Y. CircSMARCA5 Facilitates the Progression of Prostate Cancer Through miR-432/PDCD10 Axis. Cancer Biother Radiopharm. 2021; 36:70–83. https://doi.org/10.1089/cbr.2019.3490 [PubMed]
- 38. Wu K, Mu XY, Jiang JT, Tan MY, Wang RJ, Zhou WJ, Wang X, He YY, Li MQ, Liu ZH. miRNA-26a-5p and miR-26b-5p inhibit the proliferation of bladder cancer cells by regulating PDCD10. Oncol Rep. 2018; 40:3523–32. https://doi.org/10.3892/or.2018.6734 [PubMed]
- 39. Pastushenko I, Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019; 29:212–26. https://doi.org/10.1016/j.tcb.2018.12.001 [PubMed]
- 40. Li B, Cheng J, Wang H, Zhao S, Zhu H, Li C, Zhang Y, Zhao P. CCNB1 affects cavernous sinus invasion in pituitary adenomas through the epithelial-mesenchymal transition. J Transl Med. 2019; 17:336. https://doi.org/10.1186/s12967-019-2088-8 [PubMed]
- 41. Jia W, Zhu J, Martin TA, Jiang A, Sanders AJ, Jiang WG. Epithelial-mesenchymal Transition (EMT) Markers in Human Pituitary Adenomas Indicate a Clinical Course. Anticancer Res. 2015; 35:2635–43. [PubMed]
- 42. Cheng Y, Ma XL, Wei YQ, Wei XW. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev Cancer. 2019; 1871:289–312. https://doi.org/10.1016/j.bbcan.2019.01.005 [PubMed]
- 43. Liu K, Wu L, Yuan S, Wu M, Xu Y, Sun Q, Li S, Zhao S, Hua T, Liu ZJ. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature. 2020; 585:135–40. https://doi.org/10.1038/s41586-020-2492-5 [PubMed]
- 44. Saintigny P, Massarelli E, Lin S, Ahn YH, Chen Y, Goswami S, Erez B, O’Reilly MS, Liu D, Lee JJ, Zhang L, Ping Y, Behrens C, et al. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 2013; 73:571–82. https://doi.org/10.1158/0008-5472.CAN-12-0263 [PubMed]
- 45. Xu H, Lin F, Wang Z, Yang L, Meng J, Ou Z, Shao Z, Di G, Yang G. CXCR2 promotes breast cancer metastasis and chemoresistance via suppression of AKT1 and activation of COX2. Cancer Lett. 2018; 412:69–80. https://doi.org/10.1016/j.canlet.2017.09.030 [PubMed]
- 46. Sharma B, Nawandar DM, Nannuru KC, Varney ML, Singh RK. Targeting CXCR2 enhances chemotherapeutic response, inhibits mammary tumor growth, angiogenesis, and lung metastasis. Mol Cancer Ther. 2013; 12:799–808. https://doi.org/10.1158/1535-7163.MCT-12-0529 [PubMed]
- 47. Infanger DW, Cho Y, Lopez BS, Mohanan S, Liu SC, Gursel D, Boockvar JA, Fischbach C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013; 73:7079–89. https://doi.org/10.1158/0008-5472.CAN-13-1355 [PubMed]
- 48. Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS One. 2008; 3:e3077. https://doi.org/10.1371/journal.pone.0003077 [PubMed]
- 49. Monsalves E, Juraschka K, Tateno T, Agnihotri S, Asa SL, Ezzat S, Zadeh G. The PI3K/AKT/mTOR pathway in the pathophysiology and treatment of pituitary adenomas. Endocr Relat Cancer. 2014; 21:R331–44. https://doi.org/10.1530/ERC-14-0188 [PubMed]
- 50. Suojun Z, Feng W, Dongsheng G, Ting L. Targeting Raf/MEK/ERK pathway in pituitary adenomas. Eur J Cancer. 2012; 48:389–95. https://doi.org/10.1016/j.ejca.2011.11.002 [PubMed]
- 51. Jenny Zhou H, Qin L, Zhang H, Tang W, Ji W, He Y, Liang X, Wang Z, Yuan Q, Vortmeyer A, Toomre D, Fuh G, Yan M, et al. Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med. 2016; 22:1033–42. https://doi.org/10.1038/nm.4169 [PubMed]
- 52. Wang R, Wu ST, Yang X, Qian Y, Choi JP, Gao R, Song S, Wang Y, Zhuang T, Wong JJ, Zhang Y, Han Z, Lu HA, et al. Pdcd10-Stk24/25 complex controls kidney water reabsorption by regulating Aqp2 membrane targeting. JCI Insight. 2021; 6:e142838. https://doi.org/10.1172/jci.insight.142838 [PubMed]
- 53. Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, Trouillas J, Dekkers OM, and European Society of Endocrinology. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. 2018; 178:G1–24. https://doi.org/10.1530/EJE-17-0796 [PubMed]
- 54. Kourelis TV, Buckner JC, Gangat N, Patnaik MM. Temozolomide induced bone marrow Suppression--A single institution outcome analysis and review of the literature. Am J Hematol. 2015; 90:E183–4. https://doi.org/10.1002/ajh.24066 [PubMed]
- 55. Momota H, Narita Y, Miyakita Y, Shibui S. Secondary hematological malignancies associated with temozolomide in patients with glioma. Neuro Oncol. 2013; 15:1445–50. https://doi.org/10.1093/neuonc/not036 [PubMed]
- 56. Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, Wass JA, and Endocrine Society. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011; 96:273–88. https://doi.org/10.1210/jc.2010-1692 [PubMed]
- 57. Lin AL, Donoghue MTA, Wardlaw SL, Yang TJ, Bodei L, Tabar V, Geer EB. Approach to the Treatment of a Patient with an Aggressive Pituitary Tumor. J Clin Endocrinol Metab. 2020; 105:3807–20. https://doi.org/10.1210/clinem/dgaa649 [PubMed]