


Introduction
Mitochondrial dysfunction and endoplasmic reticulum (ER) stress following ischemia/reperfusion (I/R) injury contribute to the pathogenic processes underlying cardiac damage [1, 2]. The I/R injury-induced excessive production of reactive oxygen species (ROS) within mitochondria activates the apoptotic pathway, leading to cardiomyocyte death and tissue injury [3, 4]. In addition, oxidative stress, produced by mitochondrial ROS accumulation, causes unfolding of proteins within the ER, resulting in ER stress (ERS) [5, 6]. As the heart is highly susceptible to I/R injury, the prevention of ischemic or reperfusion-induced events has become an important aspect of the modern cardiac health care system.
The association between ER and mitochondria—the ER–mitochondrial interface—is a highly dynamic structure that transports calcium, lipids, ROS, and small proteins between mitochondria and ER, as well as coordinates several intracellular processes and signaling pathways [7–9]. Studies by our group [10, 11] and others [12, 13] report that the ER–mitochondrial interface functions in several post-I/R events, including cardiomyocyte death, inflammation response, fibrosis, and angiogenesis [12, 13]. For example, the prolonged influx of misfolded or unfolded proteins into the ER activates ERS-related signaling cascades to clear these abnormal proteins [14, 15]. Moreover, ERS induces apoptosis via the intrinsic pathway involving mitochondria, further contributing to the I/R damage in myocardial tissues [18–21]. Bax-induced hyper-permeabilization of the mitochondrial outer membrane releases pro-apoptotic factors, such as cytochrome c, from the mitochondria into the cytosol to trigger apoptosis [16]. In addition, mitochondrial fission, triggered by Drp1 recruitment from the cytoplasm to the surface of mitochondria, is involved in mitochondrial outer membrane remodeling and mitochondrial apoptotic pathway activation [17–19]. Interestingly, Drp1-induced mitochondrial fission is controlled by ER, which recruits Drp1 to potential division sites [20].
Sarcoplasmic reticulum calcium ATPase 2° (SERCA2a) regulates the functions of ER and mitochondria [21]. Increased SERCA2a expression reduces the calcium levels in mitochondria via calcium reflux into the ER [21]. Decreased intracellular calcium effectively restores the expression and activity of anti-oxidative factors, thus suppressing oxidative stress [21]. Restoration of normal redox biology reduces the levels of oxidized proteins or unfolded peptides, ameliorating the ERS in cardiomyocytes [22, 23]. These findings were first reported in hyperglycemia-treated cardiomyocytes but not in the context of cardiac I/R injury.
The Hippo pathway, found both in Drosophila and mammals, contains a core kinase cassette comprising mammalian STE20-like protein kinases 1 and 2 (MST1/2; homologs of Hpo in D. melanogaster); large tumor suppressor kinases 1 and 2 (LATS1/2; homologs of Wts); and their respective adaptor proteins, Salvador 1 (SAV1; a homolog of Sav), MOB kinase activators 1A and 1B (MOBKL1A/1B; homologs of Mats), and the downstream effectors of the Hippo signaling pathway-transcriptional coactivators YAP and TAZ (the two homologs of Yorkie; YAP, encoded by YAP1; TAZ, also known as WWTR1) [24, 25]. YAP directly targets several mitochondria-associated genes including Drp1, JNK, ERK, and AMPK [25]. Yu et al. [26] demonstrated that overexpressed YAP in transgenic mice with septic cardiomyopathy attenuated lipopolysaccharide (LPS)-induced myocardial injury and cardiac dysfunction by inhibiting mitochondrial fission in a MAPK–ERK pathway-dependent manner. Ma et al. [27] reported that the YAP–Hippo pathway attenuated the hypoxia-induced suppression of OPA1-related mitochondrial fusion both in vivo and in vitro. In the present study, we investigated whether YAP functions as the upstream transcriptional modulator of SERCA2a and affects the mitochondrial performance and ERS.
Results
Overexpression of YAP or SERCA2a attenuates I/R-induced cardiomyocyte apoptosis
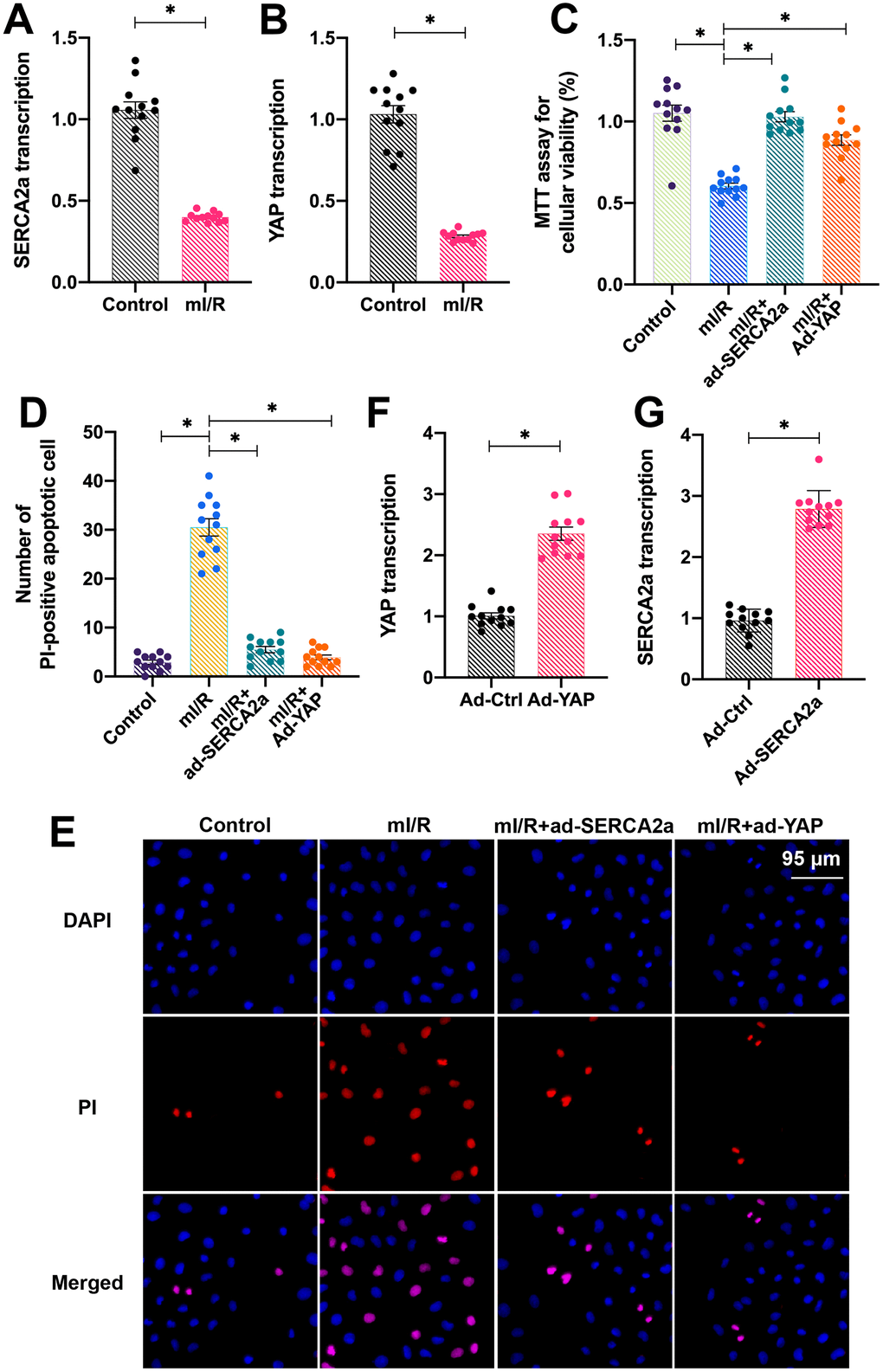
To study the role of overexpressed YAP and SERCA2a in protecting cardiomyocytes against I/R injury, isolated cardiomyocytes were cultured under hypoxic conditions for 2 h and subsequently reoxygenated for 2 h to establish an in vitro mimicked I/R injury (mI/R) model. Next, the total RNA was isolated and the endogenous mRNA levels of YAP and SERCA2a were determined. As shown in Figure 1A, 1B, compared with the control group, the mRNA levels of YAP and SERCA2a were downregulated in response to mI/R injury. To understand the role of SERCA2a and YAP in the setting of cardiac I/R injury, recombinant adenoviruses overexpressing SERCA2a (ad-SERCA2a) and YAP (ad-YAP) were transfected into cardiomyocytes before mI/R injury. Next, the cardiomyocyte viability and apoptotic rate were measured. As shown in Figure 1C, compared with the control group, the overexpression of SERCA2a or YAP decreased the mI/R injury-induced apoptosis. In addition, propidium iodide (PI) staining demonstrated a reduced number of apoptotic cardiomyocytes after transfection with either ad-SERCA2a or ad-YAP (Figure 1D, 1E). The overexpression efficiency was confirmed by quantitative polymerase chain reaction (qPCR) (Figure 1F, 1G). Altogether, our results indicated that overexpression of YAP or SERCA2a attenuated the mI/R injury-induced cardiomyocyte apoptosis.
SERCA2a is transcriptionally regulated by YAP in cardiomyocytes
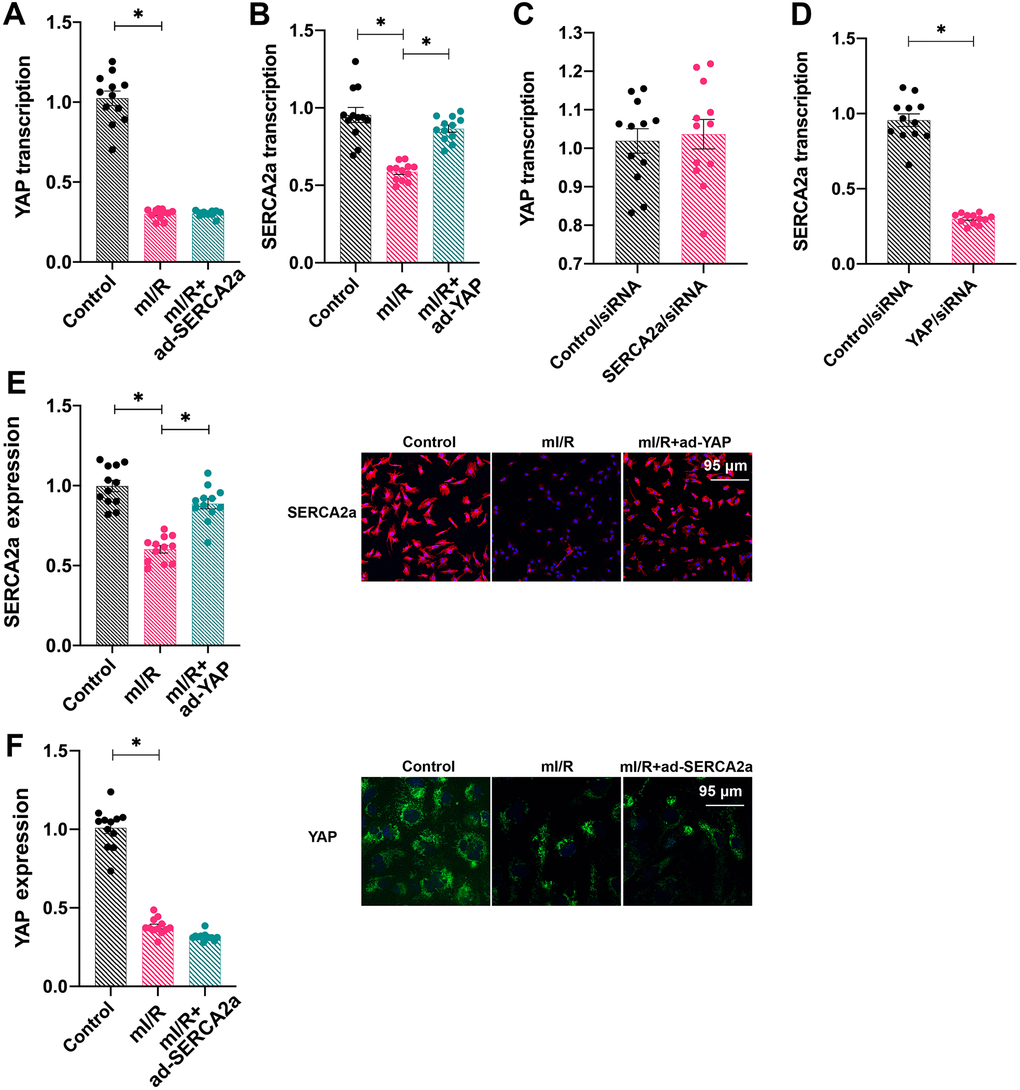
To understand the relationship between SERCA2a and YAP, ad-SERCA2a and ad-YAP were transfected into cardiomyocytes under mI/R setting and their mRNA levels were measured. As shown in Figure 2A, 2B, ad-YAP transfection increased the transcription of SERCA2a, whereas ad-SERCA2a had no effect on YAP transcription, suggesting that YAP overexpression promoted the SERCA2a translation. To confirm this finding, SERCA2a siRNA and YAP siRNA were transfected into cardiomyocytes under normal conditions. The silencing of SERCA2a had no effect on YAP transcription; however, YAP knockdown reduced the transcription of SERCA2a (Figure 2C, 2D). In addition, immunofluorescence assays demonstrated that the protein expression of SERCA2a was downregulated in response to mI/R injury, whereas ad-YAP transfection increased its expression (Figure 2E, 2F). In contrast, ad-SERCA2a overexpression had no marked effect on the protein expression of YAP in mI/R-treated cardiomyocytes (Figure 2E, 2F). Altogether, our results indicated that SERCA2a was transcriptionally regulated by YAP.
Activation of the YAP/SERCA2a pathway reduces mitochondrial damage in I/R-treated cardiomyocytes
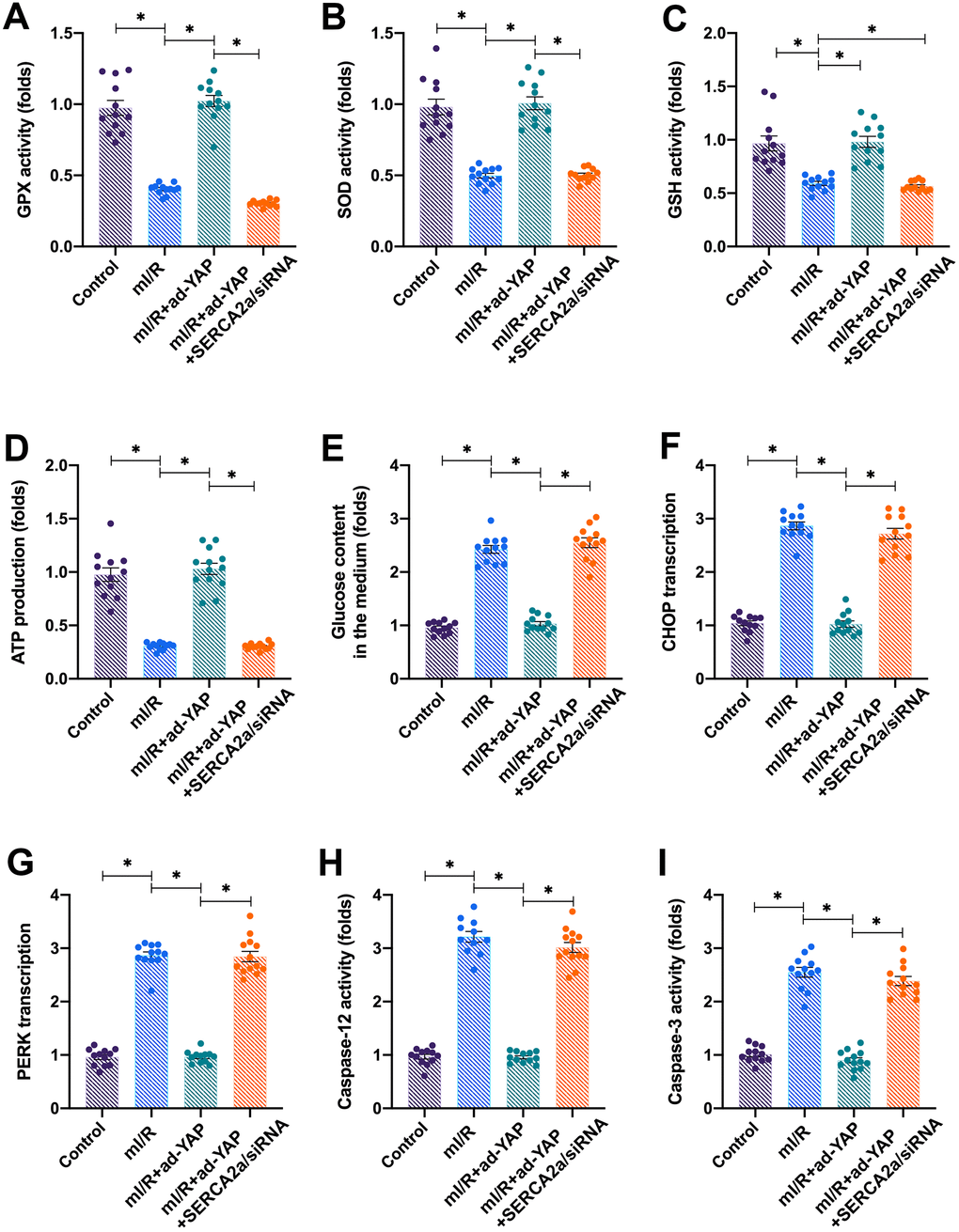
We next investigated how the YAP/SERCA2a pathway protected cardiomyocytes against mI/R injury. Several previous studies have suggested that mitochondria and ER are the two primary targets for reperfusion-induced myocardial injury [16, 21]. Thus, we focused on the alterations in mitochondrial function in response to YAP and/or SERCA2a overexpression. The levels of mitochondrial ROS rapidly increased after mI/R injury (Figure 3A, 3B), indicating oxidative stress. Interestingly, the overexpression of YAP or SERCA2a reduced the levels of mitochondrial ROS (Figure 3A, 3B). In addition, the levels of anti-oxidative factors, such as glutathione (GSH), superoxide dismutase (SOD), and glutathione peroxidase (GPx), which rapidly decreased following mI/R injury, were elevated after overexpression of YAP or SERCA2a (Figure 3C–3E). These results indicated that the mitochondrial redox balance was sustained by SERCA2a and YAP in cardiomyocytes.
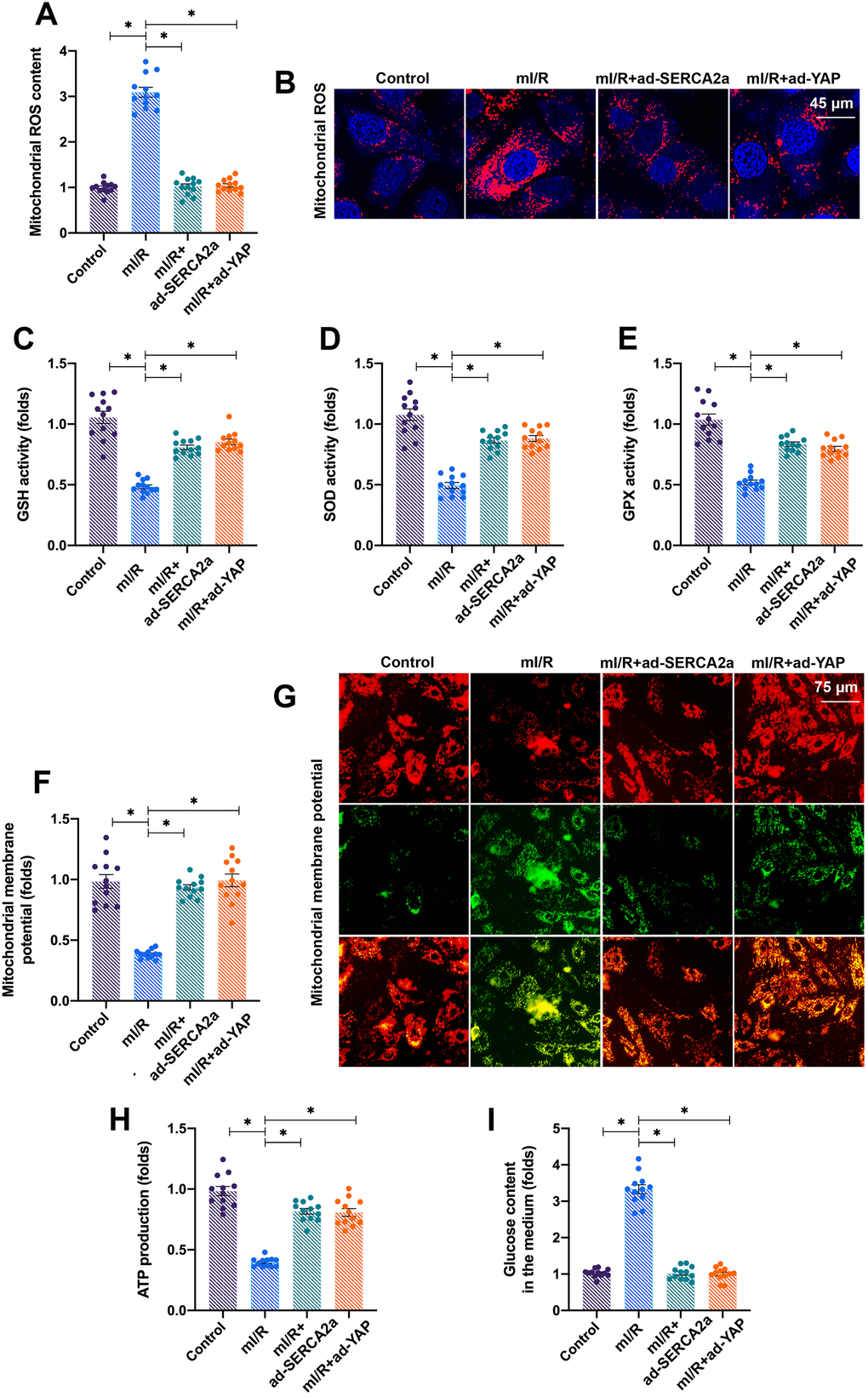
In addition to oxidative stress, we observed that the mI/R injury reduced the mitochondrial membrane potential, which was reversed by YAP or SERCA2a overexpression (Figure 3F, 3G). The mitochondrial membrane potential acts as the driving force for cellular ATP production by controlling glucose consumption. The mI/R injury inhibited ATP production in cardiomyocytes (Figure 3H), followed by an accumulation of glucose in the culture medium (Figure 3I). The overexpression of YAP or SERCA2a enhanced the consumption of glucose, thus sustaining ATP production (Figure 3H, 3I). Altogether, our results showed that YAP and SERCA2a controlled the mitochondrial redox balance and bioenergetics in mI/R-treated cardiomyocytes.
Activation of the YAP/SERCA2a pathway attenuates ERS in I/R-treated cardiomyocytes
Mitochondria are the centers of cellular energy metabolism, and ER acts as the manufactory for protein synthesis [28]. Under physiological conditions, cells employ ERS, a process activated by the accumulation of unfolded proteins, for rapid protein turnover [29]. In the present study, we found that the mI/R injury activated ERS in cardiomyocytes, as evidenced by increased mRNA levels of PREK and CHOP (Figure 4A, 4B). Interestingly, the overexpression of YAP or SERCA2a repressed the ERS in mI/R-treated cardiomyocytes.
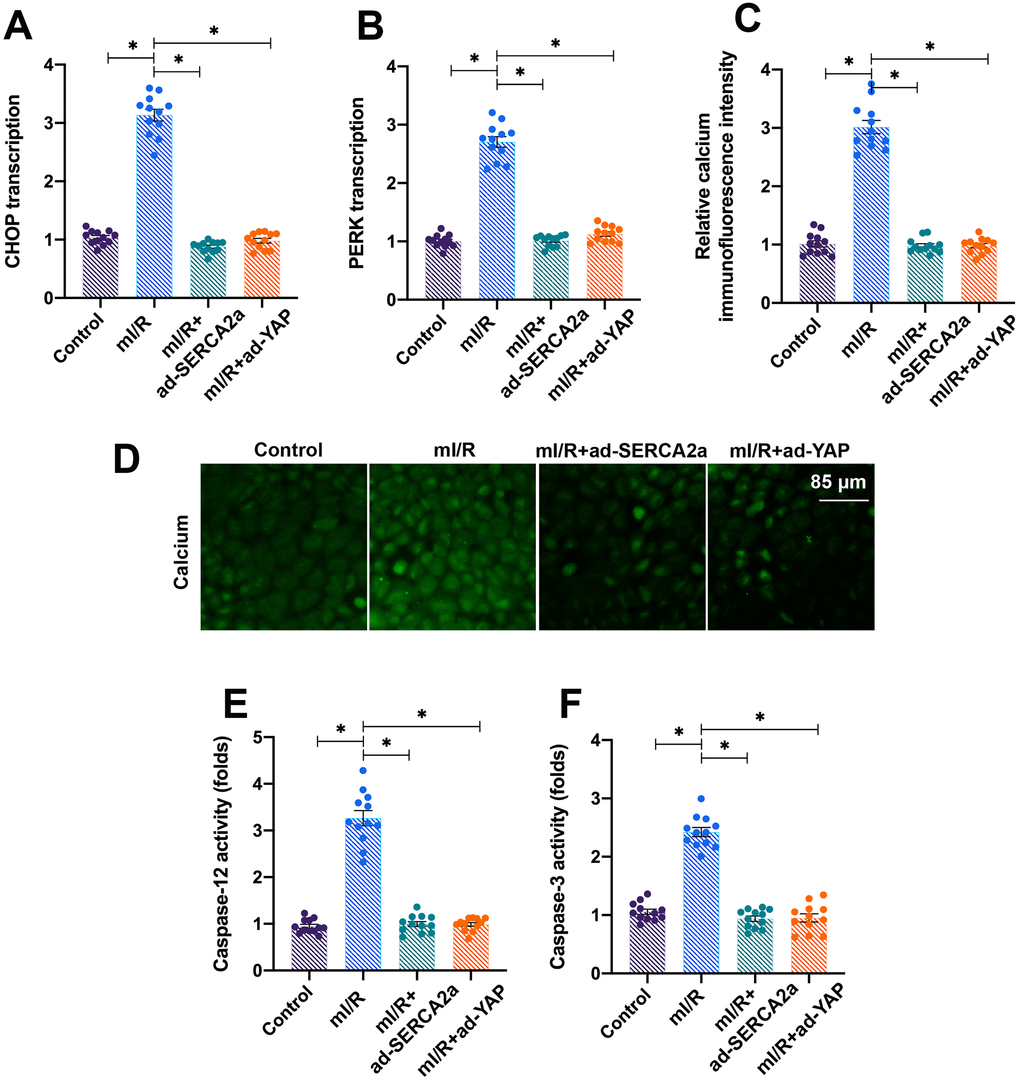
Furthermore, ER maintains the intracellular calcium balance by regulating the activity of calcium channels, such as IP3R and RyR, localized on its surface [22, 30]. In the present study, we found that the levels of intracellular calcium increased after exposure of cardiomyocytes to mI/R injury (Figure 4C, 4D), indicating baseline calcium overload. The activities of apoptotic proteins caspase-12 and caspase-3 increased following mI/R injury, indicating that apoptosis was triggered by ERS (Figure 4E, 4F). Interestingly, the overexpression of YAP or SERCA2a attenuated the calcium overload (Figure 4C, 4D) and reduced the activity of caspase-12 and caspase-3 (Figure 4E, 4F). Altogether, our results demonstrated that the activation of YAP/SERCA2a attenuated ERS in cardiomyocytes.
Knockdown of SERCA2a attenuates YAP-induced protection on I/R-treated cardiomyocytes
To verify whether YAP-induced cardiac protection was dependent on SERCA2a, SERCA2a siRNA and ad-YA were co-transfected into cardiomyocytes. Next, mitochondrial function and ERS were measured. Although the overexpression of YAP attenuated the mI/R-induced mitochondrial oxidative stress (Figure 5A–5C), this effect was suppressed by co-transfection with SERCA2a siRNA. In addition, the overexpression of YAP sustained the mitochondrial ATP production and glucose consumption (Figure 5D, 5E); these effects were negated by SERCA2a siRNA co-transfection. In mI/R-treated cardiomyocytes, YAP overexpression suppressed the mRNA levels of CHOP and PERK, whereas this effect was negated in cardiomyocytes co-transfected with SERCA2a siRNA (Figure 5F, 5G). Similarly, mI/R-induced caspase-12 and caspase-3 activation was suppressed by YAP overexpression, whereas SERCA2a siRNA suppressed the inhibitory action of YAP on caspase-3 and caspase-12 (Figure 5H, 5I). Altogether, our results indicated that YAP protected cardiomyocytes from mitochondrial damage and ER stress following I/R injury in a SERCA2a-dependent manner.
Discussion
According to the ACCF/AHA/SCAI guidelines, early revascularization is required to control myocardial injury, limb injury, and complications during organ transplantation. However, optimal clinical outcomes have not been reported after the restoration of blood flow to the ischemic tissue [31]. Post-ischemic damage or I/R injury significantly compromises the clinical benefits of revascularization strategies [32]. Previous studies have reported the involvement of mitochondria and ER in the progression of cardiac I/R injury [33, 34]. Damaged mitochondria fail to supply sufficient ATP required for cardiomyocyte contraction, leading to myocardial depression [35, 36]. Impaired ER function or ERS activates the members of the caspase family, triggering apoptosis of cardiomyocytes [37, 38]. Therefore, understanding the link between mitochondrial damage and ERS during cardiac I/R injury is vital for the design and development of novel drugs or therapeutic approaches for patients undergoing revascularization. In this study, we suggest that the YAP/SERCA2a signaling pathway functions upstream of mitochondrial damage and ERS in I/R-challenged cardiomyocytes. Exposure to I/R injury downregulated the expression of YAP and SERCA2a, and reduced cardiomyocyte viability. Overexpression of YAP transcriptionally upregulated SERCA2a expression and sustained mitochondrial function and ER homoeostasis. These findings identified the YAP/SERCA2a axis to simultaneously protect mitochondria and ER in I/R-treated cardiomyocytes. Therefore, developing therapeutic approaches that target the YAP/SERCA2a pathway would clinically benefit patients with cardiac I/R injury.
The hearts of mammals, including humans, have a poor regenerative capacity. Cardiovascular disorders in most patients advance to heart failure [39], ultimately leading to death. Inactivation of YAP in the myocardium of adult mice improved cell survival and cardiac function after myocardial infarction [40]. Moreover, a study reported that YAP activation reversed mitophagy and promoted mitochondrial fission, both induced by oxidative stress, to attenuate cardiac dysfunction after I/R injury [26]. Similarly, phosphorylation of YAP activated the Hippo pathway in mice with diabetic cardiomyopathy, increasing cardiomyocyte contractility and reducing cardiac injury by coordinating mitophagy and apoptosis [41]. In mice with septic cardiomyopathy, inhibition of YAP augmented cardiomyocyte damage by altering the mitochondrial quality control system under inflammatory stress [26]. Several drugs are used to target the YAP–Hippo signaling pathway in different kinds of cancers [42, 43]; however, few medicines are available for cardiovascular disorders. We report YAP as a transcriptional factor that promotes the expression of SERCA2a in I/R-treated cardiomyocytes [44]. Previous studies have described a direct role of YAP in regulating cardiomyocyte viability. Our study identifies the downstream effectors of YAP and reveals how YAP indirectly acts on them. More studies are required to explore how YAP binds to the promoter of SERCA2a and control its mRNA levels.
SERCA2a is an ATP-dependent protein kinase that utilizes mitochondrial ATP to maintain calcium homeostasis during the cardiac cycle. It controls the transport of cytoplasmic calcium to the ER and thus prevents ERS. We have previously reported that both the expression and activity of SERCA2a were downregulated in cardiomyocytes following I/R injury [45]. In the present study, we report that SERCA2a is transcriptionally regulated by YAP, such that reduced levels of YAP cause SERCA2a downregulation in I/R-treated cardiomyocytes. In addition, a recent study has reported that HAX-1 regulates post-transcriptional modifications, including oxidation and degradation, of SERCA2a in cardiomyocytes [46]. SERCA2a downregulation or inactivation is closely associated with myocardial infarction [47], atrial arrhythmias [48], hypertension [49], cardiac aging [50], heart failure [51], and myocardial hypertrophy [52]. We show that SERCA2a sustains mitochondrial function and ER homeostasis by promoting mitochondrial metabolism, maintaining intracellular calcium homeostasis and preventing ERS.
In conclusion, cardiac I/R injury promotes YAP downregulation and SERCA2a inactivation. Activation of the YAP/SERCA2a pathway sustains cardiomyocyte viability and function by attenuating I/R injury-induced mitochondrial damage and ERS. These findings highlight that the dysregulated YAP/SERCA2a pathway could be involved in the pathology underlying cardiac I/R injury in a mitochondria and ER-dependent manner.
Materials and Methods
Isolation and primary culture of mouse cardiomyocytes
Cardiomyocytes were isolated from 1- to 3-day old C57BL/6 mice according to the procedures described in our previous studies [23, 53]. In brief, after the heart tissue was dissected and washed, it was finely minced and the chunks were placed in 0.25% trypsin. The pooled cell suspension was centrifuged and resuspended in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The resuspension was cultured in a tissue culture flask for 90 min at 37°C to allow the fibroblasts to attach to the flask bottom [54]. The non-adherent and weakly attached cells, mainly cardiomyocytes, were removed and seeded into culture plates. Next, 5-bromo-2’-deoxyuridine (5-BrdU, 10 nM; #B5002; Sigma, Saint Louis, USA) was added to the culture medium to remove fibroblasts. The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. After 48 h, cardiomyocytes that adhered to the culture dish were used for subsequent experiments [55]. Cells were deprived of serum and placed in an anoxic chamber for 12 h in a humidified atmosphere comprising 5% CO2 and 95% N2.
Protein extraction and western blotting
Total proteins were extracted from cardiac tissue using the T-PER extraction reagent (Thermo Scientific) [56]. All cell extracts were prepared using lysis buffer that included the following: 50 mM Tris (pH 7.5), 120 mmol/L NaCl, 6 mmol/L EGTA, 1 mmol/L EDTA, 20 mmol/L NaF, 1 mmol/L sodium pyrophosphate, 30 mmol/L 4-nitrophenyl phosphate, 0.1% Nonidet P-40, 1 mmol/L benzamidine, and a protease inhibitor mixture (Roche). The extracted proteins were resolved on 10% SDS PAGE and electroblotted onto PVDF membranes (Bio-Rad). After blocking the membranes in TBST (Tris-buffered saline with Tween 20) containing 5% [w/v] non-fat dry milk for 1 h at room temperature, these were washed thrice [57], and incubated overnight with primary antibodies diluted at optimal concentrations in 5% non-fat milk solution. The membranes were washed and incubated with a donkey anti-rabbit or anti-mouse IRDye-conjugated IgG (Li-Cor Odyssey) secondary antibody (dilution 1:3000) for 1 h [58]. The blots were scanned, and the images were displayed in grayscale. The intensity of the protein bands was quantified using an image processing program (Li-Cor Odyssey).
Construction of adenoviruses
To construct adenoviral vectors overexpressing SERCA2a and YAP genes, the DNA sequences encoding for these genes were PCR amplified using high-fidelity Pfu polymerase (Agilent Technologies). PCR products were sequenced and cloned into the vector AdTrack-CMV (Agilent Technologies) [59]. Next, complete genomes of recombinant adenoviruses were constructed by homologous recombination between the recombinant AdTrack-CMV and the Ad-Easy vectors in Escherichia coli [60]. The adenoviruses were packaged in HEK293 cells and purified by CsCl2 density gradient ultracentrifugation. Adenovirus genomic DNA was purified using the NucleoSpin virus kit (Macherey-Nagel), and adenoviral titers were determined using the Adeno-X qPCR titration kit (632252, Clontech).
siRNA-induced gene knockdown
Cardiomyocytes were transfected with SERCA2a siRNA or non-targeting siRNA (negative control #1 siRNA, Thermo Fisher Scientific) using Lipofectamine RNAiMAX reagent (13778150, Thermo Fisher Scientific) according to the manufacturer’s instructions [61]. The silencing of SERCA2a miRNA was confirmed through qPCR [62].
Immunostaining
Cells were seeded onto collagen type I-coated cell culture chamber slides, fixed with 4% paraformaldehyde, and permeabilized using 0.3% Triton-X 100 [63]. JC-1 probe (Beyotime, China, Cat. No:C2006) was used to detect mitochondrial membrane potential. The cells were incubated with primary antibodies followed by incubation with Alexa Fluor secondary antibodies (1:1000). Coverslips were mounted onto slides using the ProLong Gold Antifade Mountant containing DAPI (Life Technologies) [64], and images were acquired with an Olympus IX73 microscope. Appropriate negative controls were used to perform background correction. Five fields/muscle section or culture slide were randomly counted, and all quantifications were performed by investigators blinded to the treatment [65].
TUNEL assay
The samples were fixed with 4% paraformaldehyde and analyzed for apoptosis by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay [66]. The assay was performed using the ApopTag peroxidase in situ apoptosis detection kit (S7100; EMD Millipore, CA, USA) according to the manufacturer’s instructions [67].
Measurement of intracellular calcium
Cardiomyocytes were transfected with siRNA against SERCA2a and after 48 h, approximately 2 × 106 cells were infected with adenovirus. After incubation with serum-free M199 for 4 h, intracellular calcium was detected using the Fluo-8 calcium flux assay kit (Abcam, ab112129) according to the manufacturer’s instructions [68]. In brief, the cells were loaded with calcium-sensitive probe Fluo-8 in Hanks’ buffer with 20 mM Hepes (HHBS) for 30 min at 37°C [69]. The cells were washed with HHBS twice. Afterward, the cells were excited at 490 nm and Ca2+-bound Fluo-8 emission was recorded at 525 nm.
Statistical analysis
Data were statistically analyzed using Student’s t test or one-way analysis of variance (ANOVA), followed by Newman–Keuls test. Quantitative data are expressed as mean ± standard error of mean (SEM). Non-quantitative data are representative of at least three independent experiments. A P-value less than 0.05 was considered significant.
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work is funded by Natural Science Foundation of Guangdong Province of China (2018A030313067), Shunde Hospital of Southern Medical University Scientific Research Start Plan Project (Key Project: SRSP2019004), Foshan Self-financing Technology Project (2018AB002083) and Key Specialist Department Training Project of Foshan City, Guangdong Province of China (FSPY3-2015034).
References
- 1. Abukar Y, Ramchandra R, Hood SG, McKinley MJ, Booth LC, Yao ST, May CN. Increased cardiac sympathetic nerve activity in ovine heart failure is reduced by lesion of the area postrema, but not lamina terminalis. Basic Res Cardiol. 2018; 113:35. https://doi.org/10.1007/s00395-018-0695-9 [PubMed]
- 2. Aluja D, Inserte J, Penela P, Ramos P, Ribas C, Iñiguez MÁ, Mayor F
Jr , Garcia-Dorado D. Calpains mediate isoproterenol-induced hypertrophy through modulation of GRK2. Basic Res Cardiol. 2019; 114:21. https://doi.org/10.1007/s00395-019-0730-5 [PubMed] - 3. Lee BW, Ghode P, Ong DS. Redox regulation of cell state and fate. Redox Biol. 2019; 25:101056. https://doi.org/10.1016/j.redox.2018.11.014 [PubMed]
- 4. Li D, Wang X, Huang Q, Li S, Zhou Y, Li Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-κB pathway in vivo and in vitro. Redox Biol. 2018; 15:62–73. https://doi.org/10.1016/j.redox.2017.11.023 [PubMed]
- 5. Schmidt HM, Kelley EE, Straub AC. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019; 21:101072. https://doi.org/10.1016/j.redox.2018.101072 [PubMed]
- 6. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2017; 113:5. https://doi.org/10.1007/s00395-017-0663-9 [PubMed]
- 7. Wang J, He W, Tsai PJ, Chen PH, Ye M, Guo J, Su Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020; 19:72. https://doi.org/10.1186/s12944-020-01210-0 [PubMed]
- 8. Santin Y, Fazal L, Sainte-Marie Y, Sicard P, Maggiorani D, Tortosa F, Yücel YY, Teyssedre L, Rouquette J, Marcellin M, Vindis C, Shih JC, Lairez O, et al. Mitochondrial 4-HNE derived from MAO-a promotes mitoCa2+ overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020; 27:1907–23. https://doi.org/10.1038/s41418-019-0470-y [PubMed]
- 9. Moltedo O, Remondelli P, Amodio G. The mitochondria-endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Front Cell Dev Biol. 2019; 7:172. https://doi.org/10.3389/fcell.2019.00172 [PubMed]
- 10. Zhou H, Wang S, Hu S, Chen Y, Ren J. ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Front Physiol. 2018; 9:755. https://doi.org/10.3389/fphys.2018.00755 [PubMed]
- 11. Zhou H, Toan S. Pathological roles of mitochondrial oxidative stress and mitochondrial dynamics in cardiac microvascular ischemia/reperfusion injury. Biomolecules. 2020; 10:85. https://doi.org/10.3390/biom10010085 [PubMed]
- 12. Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res. 2018; 64. https://doi.org/10.1111/jpi.12471 [PubMed]
- 13. Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017; 63:e12413. https://doi.org/10.1111/jpi.12413 [PubMed]
- 14. Filadi R, Pizzo P. ER-mitochondria tethering and ca2+ crosstalk: the IP3R team takes the field. Cell Calcium. 2019; 84:102101. https://doi.org/10.1016/j.ceca.2019.102101 [PubMed]
- 15. Kovuru N, Raghuwanshi S, Sharma DS, Dahariya S, Pallepati A, Gutti RK. Endoplasmic reticulum stress induced apoptosis and caspase activation is mediated through mitochondria during megakaryocyte differentiation. Mitochondrion. 2020; 50:115–20. https://doi.org/10.1016/j.mito.2019.10.009 [PubMed]
- 16. Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma S, Zhu H, Ren J, Zhou H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018; 14:576–87. https://doi.org/10.1016/j.redox.2017.11.004 [PubMed]
- 17. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018; 15:335–46. https://doi.org/10.1016/j.redox.2017.12.019 [PubMed]
- 18. Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S, Ren J, Chen Y. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting mff-required mitochondrial fission by CK2α. Basic Res Cardiol. 2018; 113:23. https://doi.org/10.1007/s00395-018-0682-1 [PubMed]
- 19. Zhou H, Shi C, Hu S, Zhu H, Ren J, Chen Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. 2018; 21:599–615. https://doi.org/10.1007/s10456-018-9611-z [PubMed]
- 20. Wang B, Xiao X, Huang F, Liu R. Syntaxin-17-dependent mitochondrial dynamics is essential for protection against oxidative-stress-induced apoptosis. Antioxidants (Basel). 2019; 8:522. https://doi.org/10.3390/antiox8110522 [PubMed]
- 21. Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T, Ma Q, Han T, Zhang Y, Tian F, Chen Y. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic Biol Med. 2016; 95:278–92. https://doi.org/10.1016/j.freeradbiomed.2016.03.035 [PubMed]
- 22. Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D, Zhou H, Chen Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones. 2018; 23:101–13. https://doi.org/10.1007/s12192-017-0827-4 [PubMed]
- 23. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018; 25:1080–93. https://doi.org/10.1038/s41418-018-0086-7 [PubMed]
- 24. Dobrokhotov O, Samsonov M, Sokabe M, Hirata H. Mechanoregulation and pathology of YAP/TAZ via hippo and non-hippo mechanisms. Clin Transl Med. 2018; 7:23. https://doi.org/10.1186/s40169-018-0202-9 [PubMed]
- 25. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014; 94:1287–312. https://doi.org/10.1152/physrev.00005.2014 [PubMed]
- 26. Yu W, Mei X, Zhang Q, Zhang H, Zhang T, Zou C. Yap overexpression attenuates septic cardiomyopathy by inhibiting DRP1-related mitochondrial fission and activating the ERK signaling pathway. J Recept Signal Transduct Res. 2019; 39:175–86. https://doi.org/10.1080/10799893.2019.1641822 [PubMed]
- 27. Ma S, Dong Z. Melatonin attenuates cardiac reperfusion stress by improving OPA1-related mitochondrial fusion in a yap-hippo pathway-dependent manner. J Cardiovasc Pharmacol. 2019; 73:27–39. https://doi.org/10.1097/FJC.0000000000000626 [PubMed]
- 28. Li X, Lee Y, Kang Y, Dai B, Perez MR, Pratt M, Koay EJ, Kim M, Brekken RA, Fleming JB. Hypoxia-induced autophagy of stellate cells inhibits expression and secretion of lumican into microenvironment of pancreatic ductal adenocarcinoma. Cell Death Differ. 2019; 26:382–93. https://doi.org/10.1038/s41418-018-0207-3 [PubMed]
- 29. Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019; 26:199–212. https://doi.org/10.1038/s41418-018-0246-9 [PubMed]
- 30. Zhou H, Wang J, Zhu P, Hu S, Ren J. Ripk3 regulates cardiac microvascular reperfusion injury: the role of IP3R-dependent calcium overload, XO-mediated oxidative stress and f-action/filopodia-based cellular migration. Cell Signal. 2018; 45:12–22. https://doi.org/10.1016/j.cellsig.2018.01.020 [PubMed]
- 31. van Duinen V, Zhu D, Ramakers C, van Zonneveld AJ, Vulto P, Hankemeier T. Perfused 3D angiogenic sprouting in a high-throughput in vitro platform. Angiogenesis. 2019; 22:157–65. https://doi.org/10.1007/s10456-018-9647-0 [PubMed]
- 32. Yang J, Liu X, Fu Y, Song Y. Recent advances of microneedles for biomedical applications: drug delivery and beyond. Acta Pharm Sin B. 2019; 9:469–83. https://doi.org/10.1016/j.apsb.2019.03.007 [PubMed]
- 33. Beckendorf J, van den Hoogenhof MM, Backs J. Physiological and unappreciated roles of CaMKII in the heart. Basic Res Cardiol. 2018; 113:29. https://doi.org/10.1007/s00395-018-0688-8 [PubMed]
- 34. Bøtker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femminò S, García-Dorado D, et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. 2018; 113:39. https://doi.org/10.1007/s00395-018-0696-8 [PubMed]
- 35. Coverstone ED, Bach RG, Chen L, Bierut LJ, Li AY, Lenzini PA, O’Neill HC, Spertus JA, Sucharov CC, Stitzel JA, Schilling JD, Cresci S. A novel genetic marker of decreased inflammation and improved survival after acute myocardial infarction. Basic Res Cardiol. 2018; 113:38. https://doi.org/10.1007/s00395-018-0697-7 [PubMed]
- 36. Kanwar MK, Yu J, Zhou J. Phytomelatonin: recent advances and future prospects. J Pineal Res. 2018; 65:e12526. https://doi.org/10.1111/jpi.12526 [PubMed]
- 37. Ranhotra HS. The estrogen-related receptors in metabolism and cancer: newer insights. J Recept Signal Transduct Res. 2018; 38:95–100. https://doi.org/10.1080/10799893.2018.1456552 [PubMed]
- 38. Takahashi K, Fukushima K, Otagaki S, Ishimoto K, Minami K, Fukushima N, Honoki K, Tsujiuchi T. Effects of LPA1 and LPA6 on the regulation of colony formation activity in colon cancer cells treated with anticancer drugs. J Recept Signal Transduct Res. 2018; 38:71–75. https://doi.org/10.1080/10799893.2018.1426608 [PubMed]
- 39. Thieltges KM, Avramovic D, Piscitelli CL, Markovic-Mueller S, Binz HK, Ballmer-Hofer K. Characterization of a drug-targetable allosteric site regulating vascular endothelial growth factor signaling. Angiogenesis. 2018; 21:533–43. https://doi.org/10.1007/s10456-018-9606-9 [PubMed]
- 40. Meliambro K, Campbell KN. Hippo pathway deficiency and recovery from heart failure after myocardial infarction: potential implications for kidney disease. Kidney Int. 2018; 93:290–92. https://doi.org/10.1016/j.kint.2017.12.001 [PubMed]
- 41. Yang Y, Wang H, Ma Z, Hu W, Sun D. Understanding the role of mammalian sterile 20-like kinase 1 (MST1) in cardiovascular disorders. J Mol Cell Cardiol. 2018; 114:141–49. https://doi.org/10.1016/j.yjmcc.2017.11.010 [PubMed]
- 42. Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu S, Zhu P, Wang W, Zhou H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/f-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018; 14:59–71. https://doi.org/10.1016/j.redox.2017.08.013 [PubMed]
- 43. Stewart IE, Lukka PB, Liu J, Meibohm B, Gonzalez-Juarrero M, Braunstein MS, Lee RE, Hickey AJ. Development and characterization of a dry powder formulation for anti-tuberculosis drug spectinamide 1599. Pharm Res. 2019; 36:136. https://doi.org/10.1007/s11095-019-2666-8 [PubMed]
- 44. Zhang HF, Wang YL, Tan YZ, Wang HJ, Tao P, Zhou P. Enhancement of cardiac lymphangiogenesis by transplantation of CD34+ VEGFR-3+ endothelial progenitor cells and sustained release of VEGF-C. Basic Res Cardiol. 2019; 114:43. https://doi.org/10.1007/s00395-019-0752-z [PubMed]
- 45. Yellon DM, He Z, Khambata R, Ahluwalia A, Davidson SM. The GTN patch: a simple and effective new approach to cardioprotection? Basic Res Cardiol. 2018; 113:20. https://doi.org/10.1007/s00395-018-0681-2 [PubMed]
- 46. Bidwell PA, Liu GS, Nagarajan N, Lam CK, Haghighi K, Gardner G, Cai WF, Zhao W, Mugge L, Vafiadaki E, Sanoudou D, Rubinstein J, Lebeche D, et al. HAX-1 regulates SERCA2a oxidation and degradation. J Mol Cell Cardiol. 2018; 114:220–33. https://doi.org/10.1016/j.yjmcc.2017.11.014 [PubMed]
- 47. Jiao L, Li M, Shao Y, Zhang Y, Gong M, Yang X, Wang Y, Tan Z, Sun L, Xuan L, Yu Q, Li Y, Gao Y, et al. lncRNA-ZFAS1 induces mitochondria-mediated apoptosis by causing cytosolic ca2+ overload in myocardial infarction mice model. Cell Death Dis. 2019; 10:942. https://doi.org/10.1038/s41419-019-2136-6 [PubMed]
- 48. Liu B, Lou Q, Smith H, Velez-Cortes F, Dillmann WH, Knollmann BC, Armoundas AA, Györke S. Conditional up-regulation of SERCA2a exacerbates RyR2-dependent ventricular and atrial arrhythmias. Int J Mol Sci. 2020; 21:2535. https://doi.org/10.3390/ijms21072535 [PubMed]
- 49. Hadipour-Lakmehsari S, Driouchi A, Lee SH, Kuzmanov U, Callaghan NI, Heximer SP, Simmons CA, Yip CM, Gramolini AO. Nanoscale reorganization of sarcoplasmic reticulum in pressure-overload cardiac hypertrophy visualized by dSTORM. Sci Rep. 2019; 9:7867. https://doi.org/10.1038/s41598-019-44331-y [PubMed]
- 50. Roh JD, Hobson R, Chaudhari V, Quintero P, Yeri A, Benson M, Xiao C, Zlotoff D, Bezzerides V, Houstis N, Platt C, Damilano F, Lindman BR, et al. Activin type II receptor signaling in cardiac aging and heart failure. Sci Transl Med. 2019; 11:eaau8680. https://doi.org/10.1126/scitranslmed.aau8680 [PubMed]
- 51. Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, Chepurko V, Yang DK, Kwak TH, Eom SH, Park ZY, Yoo YJ, Kim DH, et al. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum ca 2+-ATPase in heart failure. Circ Res. 2019; 124:e63–80. https://doi.org/10.1161/CIRCRESAHA.118.313865 [PubMed]
- 52. Oh JG, Watanabe S, Lee A, Gorski PA, Lee P, Jeong D, Liang L, Liang Y, Baccarini A, Sahoo S, Brown BD, Hajjar RJ, Kho C. miR-146a suppresses SUMO1 expression and induces cardiac dysfunction in maladaptive hypertrophy. Circ Res. 2018; 123:673–85. https://doi.org/10.1161/CIRCRESAHA.118.312751 [PubMed]
- 53. Zhou H, Toan S, Zhu P, Wang J, Ren J, Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res Cardiol. 2020; 115:11. https://doi.org/10.1007/s00395-019-0773-7 [PubMed]
- 54. Zhou H, Li N, Yuan Y, Jin YG, Guo H, Deng W, Tang QZ. Activating transcription factor 3 in cardiovascular diseases: a potential therapeutic target. Basic Res Cardiol. 2018; 113:37. https://doi.org/10.1007/s00395-018-0698-6 [PubMed]
- 55. Zhang H, Jin B, Faber JE. Mouse models of alzheimer’s disease cause rarefaction of pial collaterals and increased severity of ischemic stroke. Angiogenesis. 2019; 22:263–79. https://doi.org/10.1007/s10456-018-9655-0 [PubMed]
- 56. Zhu Y, Wang P, Zhang L, Bai G, Yang C, Wang Y, He J, Zhang Z, Zhu G, Zou D. Superhero rictor promotes cellular differentiation of mouse embryonic stem cells. Cell Death Differ. 2019; 26:958–68. https://doi.org/10.1038/s41418-018-0177-5 [PubMed]
- 57. Trieb M, Kornej J, Knuplez E, Hindricks G, Thiele H, Sommer P, Scharnagl H, Dagres N, Dinov B, Bollmann A, Husser D, Marsche G, Buettner P. Atrial fibrillation is associated with alterations in HDL function, metabolism, and particle number. Basic Res Cardiol. 2019; 114:27. https://doi.org/10.1007/s00395-019-0735-0 [PubMed]
- 58. Zarfati M, Avivi I, Brenner B, Katz T, Aharon A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis. 2019; 22:185–96. https://doi.org/10.1007/s10456-018-9649-y [PubMed]
- 59. Zhang Y, Zou X, Qian W, Weng X, Zhang L, Zhang L, Wang S, Cao X, Ma L, Wei G, Wu Y, Hou Z. Enhanced PAPSS2/VCAN sulfation axis is essential for snail-mediated breast cancer cell migration and metastasis. Cell Death Differ. 2019; 26:565–79. https://doi.org/10.1038/s41418-018-0147-y [PubMed]
- 60. Trindade F, Vitorino R, Leite-Moreira A, Falcão-Pires I. Pericardial fluid: an underrated molecular library of heart conditions and a potential vehicle for cardiac therapy. Basic Res Cardiol. 2019; 114:10. https://doi.org/10.1007/s00395-019-0716-3 [PubMed]
- 61. Wolint P, Bopp A, Woloszyk A, Tian Y, Evrova O, Hilbe M, Giovanoli P, Calcagni M, Hoerstrup SP, Buschmann J, Emmert MY. Cellular self-assembly into 3D microtissues enhances the angiogenic activity and functional neovascularization capacity of human cardiopoietic stem cells. Angiogenesis. 2019; 22:37–52. https://doi.org/10.1007/s10456-018-9635-4 [PubMed]
- 62. Zhang N, Zhang H, Liu Y, Su P, Zhang J, Wang X, Sun M, Chen B, Zhao W, Wang L, Wang H, Moran MS, Haffty BG, Yang Q. SREBP1, targeted by miR-18a-5p, modulates epithelial-mesenchymal transition in breast cancer via forming a co-repressor complex with snail and HDAC1/2. Cell Death Differ. 2019; 26:843–59. https://doi.org/10.1038/s41418-018-0158-8 [PubMed]
- 63. Zhang M, Zhang Y, Xu E, Mohibi S, de Anda DM, Jiang Y, Zhang J, Chen X. Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ. 2018; 25:1118–30. https://doi.org/10.1038/s41418-017-0029-8 [PubMed]
- 64. Vargas LA, Velasquez FC, Alvarez BV. Retraction note to: compensatory role of the NBCn1 sodium/bicarbonate cotransporter on ca2+-induced mitochondrial swelling in hypertrophic hearts. Basic Res Cardiol. 2018; 113:21. https://doi.org/10.1007/s00395-018-0680-3 [PubMed]
- 65. Walraven M, Homs MY, van der Veldt AA, Dekker H, Koldenhof J, Honeywell R, Barendrecht A, Sebastian SA, Parr N, Koekman AC, Voest EE, Roest M, Korporaal SJ, Verheul HM. Platelet function is disturbed by the angiogenesis inhibitors sunitinib and sorafenib, but unaffected by bevacizumab. Angiogenesis. 2018; 21:325–34. https://doi.org/10.1007/s10456-018-9598-5 [PubMed]
- 66. Xia Q, Zhang Y, Li Z, Hou X, Feng N. Red blood cell membrane-camouflaged nanoparticles: a novel drug delivery system for antitumor application. Acta Pharm Sin B. 2019; 9:675–89. https://doi.org/10.1016/j.apsb.2019.01.011 [PubMed]
- 67. Vargas LA, Velasquez FC, Alvarez BV. Editorial expression of concern to: compensatory role of the NBCn1 sodium/bicarbonate cotransporter on ca2+-induced mitochondrial swelling in hypertrophic hearts. Basic Res Cardiol. 2018; 113:17. https://doi.org/10.1007/s00395-018-0676-z [PubMed]
- 68. Wider J, Undyala VV, Whittaker P, Woods J, Chen X, Przyklenk K. Remote ischemic preconditioning fails to reduce infarct size in the zucker fatty rat model of type-2 diabetes: role of defective humoral communication. Basic Res Cardiol. 2018; 113:16. https://doi.org/10.1007/s00395-018-0674-1 [PubMed]
- 69. Venugopal S, Kao C, Chandna R, Sulochana KN, Subramanian V, Chen M, Kini RM, Ge R. Angio-3, a 10-residue peptide derived from human plasminogen kringle 3, suppresses tumor growth in mice via impeding both angiogenesis and vascular permeability. Angiogenesis. 2018; 21:653–65. https://doi.org/10.1007/s10456-018-9616-7 [PubMed]