



Introduction
The mechanism of tumorigenesis is complex, and the same gene has different mechanisms of action in different tumors [1, 2]. Pan-cancer expression analysis can effectively evaluate the mechanism of action of the same gene in different tumors, the predictive value of prognosis and survival of cancer patients and the effect of gene action from the molecular mechanism. Meanwhile, it can better help researchers in different specialties to clarify the overall role of genes in tumorigenesis. GEO and TCGA databases have a large number of functional genome data settings, and mainly focus on the role of these genes in different tumors, which is helpful for our relevant pan-cancer analysis [3–5].
One of the primary goals of cancer research is not only to understand tumor biology but also to improve patient outcomes through therapeutic interventions. Immunotherapies, including neoadjuvant and adjuvant treatments, represent a significant advancement in cancer therapy [6, 7]. However, the efficacy of these treatments often depends on the specific molecular and genetic landscape of the tumor. Understanding the expression of key genes, such as those involved in immune response or cell cycle regulation, across different cancers could help identify patients who would benefit most from immunotherapy. Pan-cancer analysis, by revealing gene expression patterns and immune infiltration across cancer types, plays a crucial role in informing the use of these therapies [8].
The CCNE1 gene undergoes a series of processes such as replication, transcription, and translation to form CCNE1 protein in vivo. The protein can act as a regulatory sub-unit of cyclin-dependent kinases and is important for maintaining genome integrity and the coordination of cell cycle processes [9]. Since the over-expression of the gene was found to be associated with cancer in 1990, a series of studies have shown that the protein encoded by the transcription of the gene can play different roles in multiple phases of the cell cycle [10–13]. Given its importance, CCNE1 was selected as the focus of this study to better understand its function in cancer biology. In subsequent studies, various cancer types have been confirmed to be closely related to CCNE1 over-expression, and the possible mechanism of action has also been analyzed [14–17].
In this study, we performed a pan-cancer analysis for the association of CCNE1 expression with clinicopathological features of cancer. Moreover, several databases including TCGA, GEPIA, HPA, TIMER and STRING databases were applied to explore the association of CCNE1 with prognosis, immune infiltration, and genetic mutation of tumor. This study’s innovative approach of integrating multiple datasets provides new insights into the potential clinical applications of CCNE1 as a prognostic biomarker and its role in immune modulation across different cancers.
Materials and Methods
Expression of CCNE1 in pan-cancer
TIMER2 (tumor immune estimation resource) is an integrated gene data platform that can consistently analyze gene expression differences. Additionally, TIMER2 is widely used in cancer immunology research for its ability to estimate the abundance of six major immune cell types (such as CD4+ T cells, CD8+ T cells, and macrophages) using RNA-seq expression data from The Cancer Genome Atlas (TCGA). Its versatility and ease of use make it a powerful platform for studying the tumor immune microenvironment and its role in cancer progression and therapy responses [18]. TIMER2 was selected because of its wide application in gene expression differences and tumor immunology research, particularly for its reliability in estimating the gene expression differences and immune microenvironment, which was critical for our analysis. CCNE1 was input into “GENE_DE” section that can evaluate the expression of CCNE1 in different types, in the TCGA project. Statistical analysis of the association was performed via the test of purity-adjusted partial Spearman’s correlation. Results are shown via heat-maps and scatter plots. GEPIA2 (gene expression profiling interactive analysis) is an interactive web server to analyze expression of RNA which is derived from the Genotype Tissue Expression (GTEx) program and TCGA [19].
In our research, we obtained CCNE1 expression by using this dataset and analyzed them via GEPIA2’s “Expression Analysis-Box Plots” section, with the DESeq2 package used for normalization and differential expression analysis. Specifically, the parameters were set with a log2FC cutoff of 1 and an adjusted p-value threshold of 0.01. In addition, the limma package was used for differential gene expression analysis in paired tumor and normal tissue comparisons. The limma package is applied to log-transformed data, where it uses empirical Bayes moderation to calculate moderated t-statistics for differential expression. We followed the platform’s default settings for the linear model fitting and moderated t-statistic computation.
Association of CCNE1 with overall survival and disease-free survival in cancer
Overall survival (OS) and disease-free survival (DFS) for all tumors in TCGA were obtained using the “Survival Map” module of GEPIA2. We set high (50%) and low (50%) cut-off to divide high and low cohorts of gene expression. Survival data were visualized with log-rank P-values, 95% confidence intervals, and hazard ratios. The expression of CCNE1 in TCGA was obtained using the GEPIA2 “Pathological Stage Plot” program. Using “similarity for gene detection” module of GEPIA2, the top 100 targeted genes which are associated with CCNE1 can be identified.
CCNE1 expression levels were stratified into high and low expression groups. The stratification was based on the median expression value of CCNE1 across the entire dataset. Samples with CCNE1 expression above the median were classified as the high expression group, while those below the median were classified as the low expression group. GEPIA performs survival analysis based on gene expression levels. GEPIA uses log-rank test, sometimes called the Mantel-Cox test, for the hypothesis evaluation. The cox proportional hazard ratio and the 95% confidence interval information can also be included in the survival plot.
Association of CCNE1 with relapse-free survival in cancer
The Kaplan-Meier plotter that can evaluate genetic impact on prognosis of different cancers was used in our study. The relationship between the CCNE1 gene and progression-free survival (PFS), OS, and first progression in different cancers from TCGA and GEO databases was analyzed using the Kaplan-Meier plotter. Setting “autoselect best cut-off”, the breast, ovary, lung, stomach and liver of the five tumors were divided into two groups. A log p-value of < 0.05 was considered statistically significant.
Expression of CCNE1 protein in cancer
UALCAN provided the protein analysis option through the TCGA and CPTAC datasets [20]. The CPTAC program of UALCAN can be used to obtain the CCNE1 protein expression in different cancers.
Analysis of CCNE1 gene alteration and mutation
C-BioPortal is a complex site, which can visualize, and analyze the cancer genomic [21]. The frequency of CCNE1 gene alteration, mutation type and alteration of copy number in tumors can be seen through “Cancer Types Summary” section of the cBioPortal in TCGA. Setting “TCGA Pan Cancer Atlas Study”, we can query the genetic alteration signature of the CCNE 1 gene via the “Quick select” section. Additionally, we used the “Comparison” section, analyzed data on the relationship among CCNE1 genetic alterations, OS, PFS, and DFS. The results are shown as log-rank P-values.
Immune infiltration analysis of CCNE1 in cancer
Connection between CCNE1 and immune infiltration of cancer was explored using TIMER database. “GENE_CORR” section can be used to analyze the association between gene expression and immune infiltration. Statistical analysis of the association was performed via the test of purity-adjusted partial Spearman’s correlation. Results are shown via heat-maps and scatter plots.
Enrichment analysis of CCNE1 associated genes
STRING can predict protein-protein interactions [22]. The top 50 lists of CCNE1 binding-proteins, which set the required minimum interaction score to “low confidence”, the meaning of the network edge to “evidence”, and display as “no more than 50 interactors”, via STRING site. DAVID (database for annotation, visualization, and integrated discovery) is a functional annotation tool that investigates to clarify gene function [23]. The intersection analysis assesses CCNE1 binding and interacting genes. The genes functional annotation chart was analyzed by the DAVID tool.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Results
CCNE1 gene expression
By comparing the expression of CCNE1, we investigated expression of CCNE1 among cancer types in TCGA by TIMER2. CCNE1 expression in the tumor tissues including BLCA, BRCA, CHOL, COAD, ESCA, HNSC, KICH, KIRC, KIRP, LIHC, LUAD, LUSC, READ, STAD, THCA, UCEC (P < 0.001) and CESC (P < 0.01) was higher than the corresponding normal tissues (Figure 1A). Analyzing the differences in our target gene expression, we used the GTEx dataset and used normal tissues as controls. CCNE1 expression level was upregulated in ACC, BLCA, BRCA, CESC, CHOL, COAD, DLBC, ESCA, HNSC, LIHC, LUAD, LUSC, OV, PAAD, READ, SARC, STAD, THYM, UCEC and UCS, (P < 0.01, Figure 1B). We observed an association between CCNE1 expression and tumor pathological stages (Figure 1C).
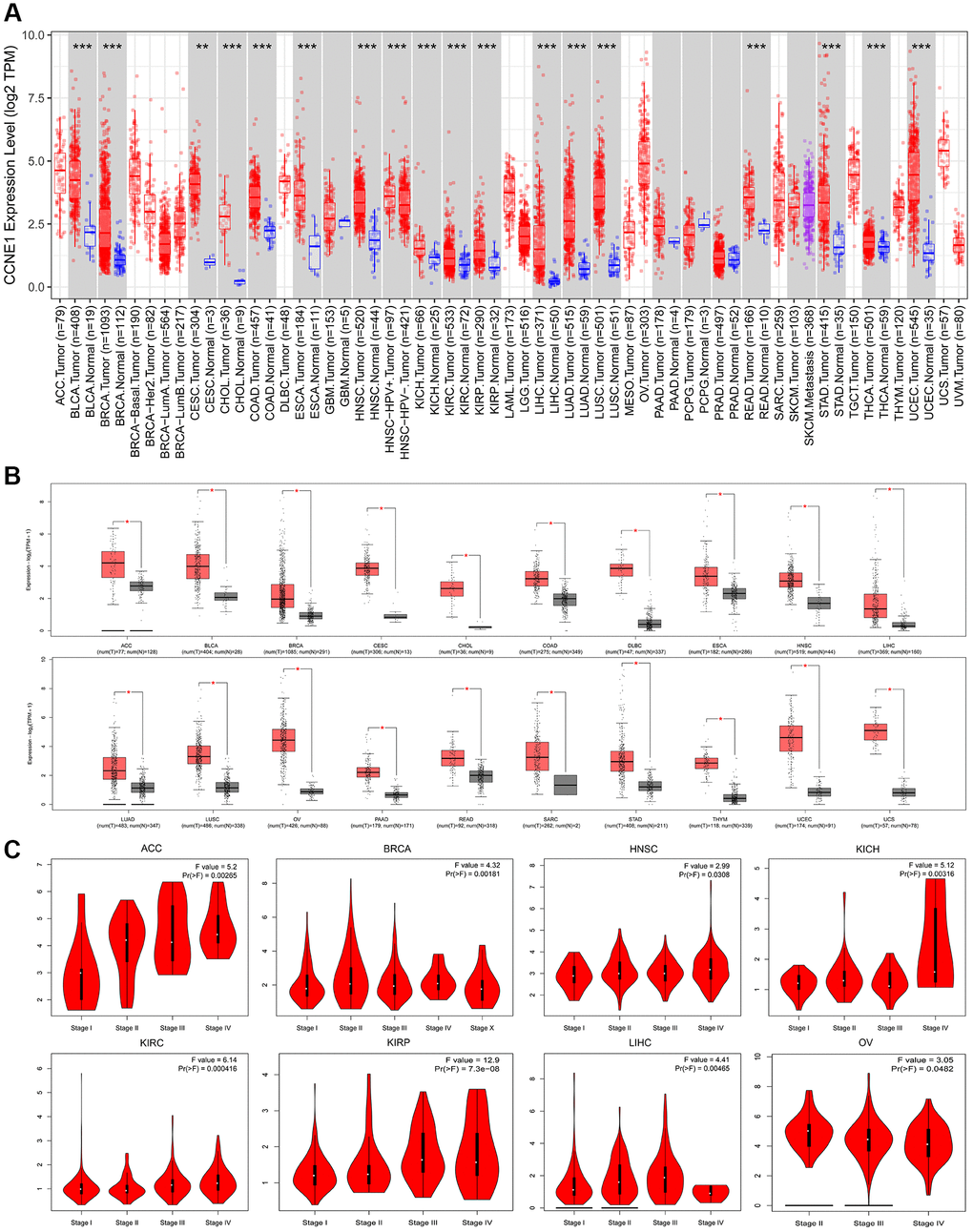
Figure 1. Expression of CCNE1 in different tumors and pathological stages. (A) The expression of CCNE1 in pan-cancer. The expression of CCNE1 in tumor tissue is indicated in red, while the expression of CCNE1 in normal tissue is shown in blue. (B) The expression status of CCNE1 in normal tissue and cancer tissue. The expression of CCNE1 in tumor tissue is indicated in red, while the expression of CCNE1 in normal tissue is shown in grey. (C) Correlation of CCNE1 with pathological stages in multiple cancers.
CCNE1 survival analysis
According to the expression level of CCNE1, the tumor cases were divided into high and low groups. To investigate the correlation between CCNE1 expression and tumor prognosis in different types of tumors, we used TCGA and GEPIA databases for analysis. High CCNE1 gene expression was associated with poor prognosis of OS in cancers, including ACC, BRCA, KIRC, KIRP, LGG, LIHC, LUAD, MESO (Figure 2A). In addition, high gene performance of CCNE1 showed a strong correlation with poor DFS in cancer types, such as BRCA, KIRP, LIHC, LGG, PRAD, MESO, SKCM, THCA and UCEC (Figure 2B). To explore the association between CCNE1 and tumor prognosis, we performed further analysis via Kaplan-Meier plotters tool. High CCNE1 gene expression was linked with poor OS in BRCA, KIRC, KIRP, LIHC, LUAD, OV, PAAD, SARC and UCEC (P < 0.01) (Figure 3A). The results of DFS were shown that a strong correlation between h highly expressed CCNE1 and poor prognosis in BRCA, KIRP, LIHC, LUAD, PAAD, SARC, TGCT, THCA and UCEC (P < 0.01) (Figure 3B).
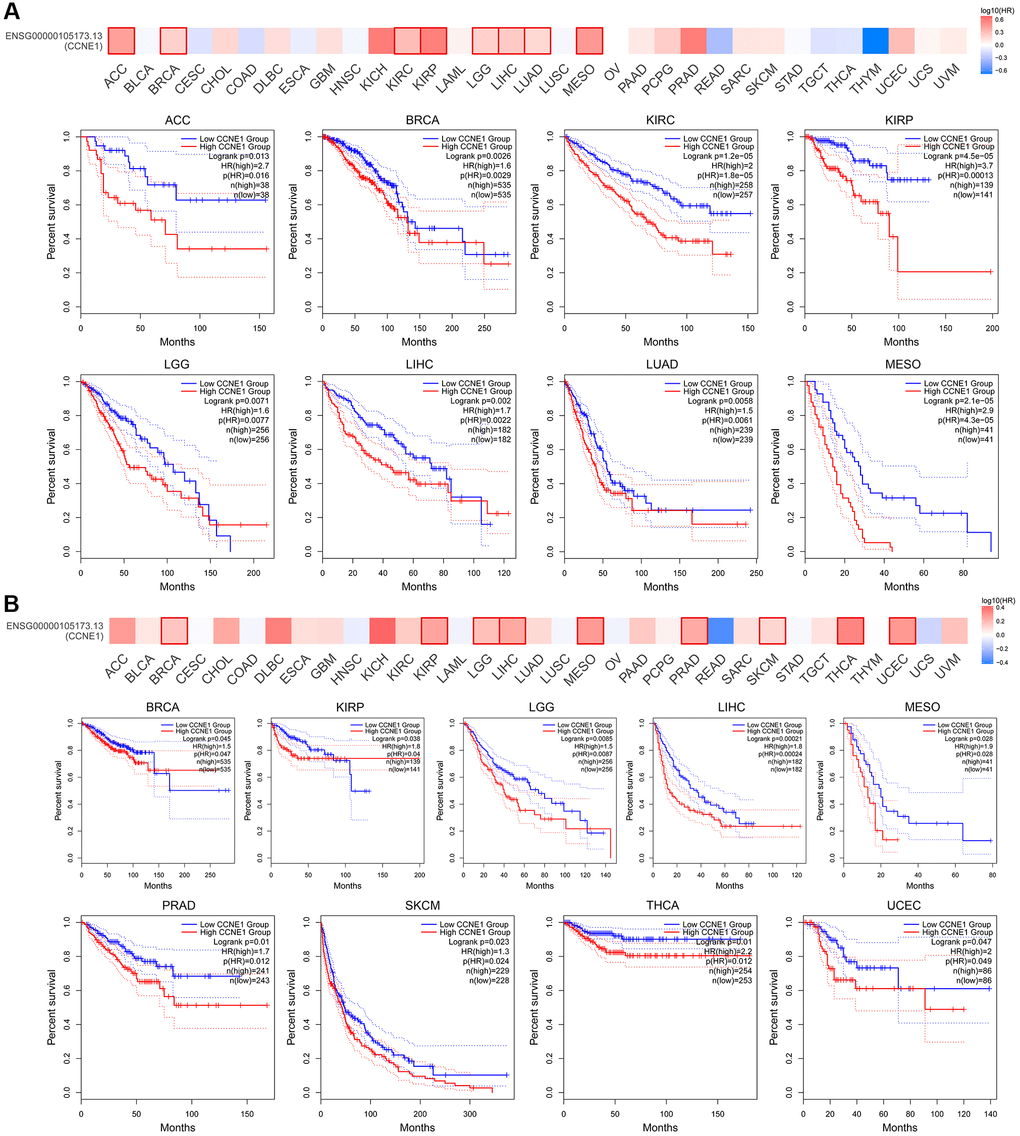
Figure 2. Connection of CCNE1 with (A) overall survival and (B) disease-free survival in cancer. Tumors with higher CCNE1 expression are indicated in red, while those with lower expression are shown in blue.
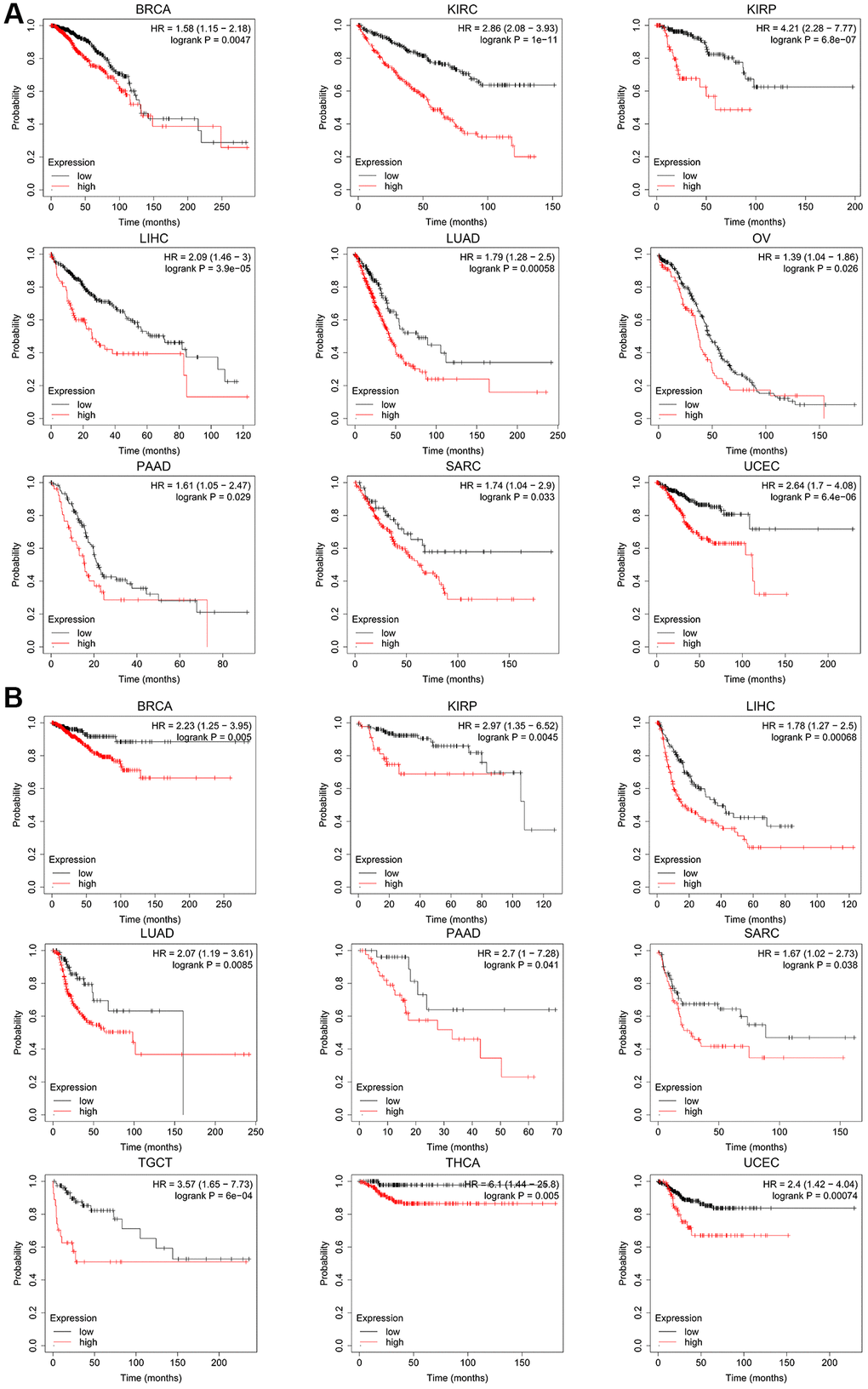
Figure 3. The Kaplan-Meier plotter reflecting CCNE1 expression and (A) overall survival and (B) relapse-free survival. Tumors with higher CCNE1 expression are indicated in red, while those with lower expression are shown in black.
CCNE1 genetic change
In this study, to analyze the genetic pattern of CCNE1, we used different tumor datasets from the TCGA cohort for our study. CCNE1 has the highest genetic change frequency in uterine carcinoma patients (40%) (Figure 4A). And the main type is “amplification”. In UCEC, we found that the prominent types are the “amplification” and “mutation”, accounting for about 5% of the frequency of occurrence. As shown in the results, an interesting phenomenon is that most of the tumors listed in the figure are formed under the influence of CCNE1, and the “amplification”, is the predominant type. In Figure 4B, the analysis of the type, location and number of cases of CCNE1 genetic changes is shown, including missense, truncating, and fusion mutations. Based on the analysis of our data, missense mutations were found to be the predominant type of genetic change in CCNE1, with M336I/T/V changes detected in 1 case of COAD, 1 case of LUAD, and 1 case of STAD (Figure 4B). The 3D structure of CCNE1 protein is shown in Figure 4C. Potential association in different types of cancer was analyzed between CCNE1 changes and prognosis by crude analysis of the change pattern. The clinical survival prognosis value of CCNE1 alterations reflected prognosis in COAD patients with regard to OS (P < 0.05), DSS (P < 0.001), DFS (P < 0.001), but not PFS (P = 0.140) (Figure 4D).
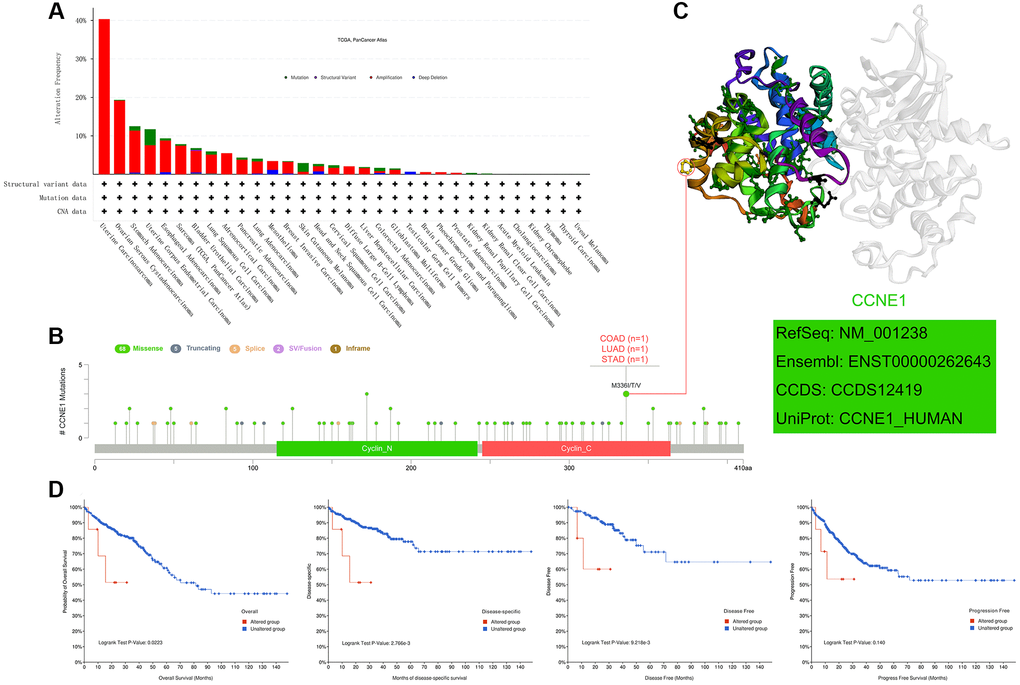
Figure 4. Mutation features of CCNE1 in cancer. (A) Bar chart representing the distribution of different CCNE1 mutation types across various cancers. (B) Lollipop plot highlighting the mutation sites of CCNE1, with higher frequency mutations displayed prominently. (C) 3D structural model of CCNE1, focusing on regions with the highest mutation frequency. (D) Kaplan-Meier curves linking CCNE1 mutation status with survival outcomes (overall survival, disease-specific survival, disease-free survival, and progression-free survival) in COAD.
CCNE1 protein expression
Through CPTAC, the expression of CCNE1 protein in five types of tumors including BRCA, HNSC, HCC, LUAD, and OV were analyzed. We observed that the expression level of CCNE1 protein was higher in BRCA, HNSC, HCC, LUAD and OV (P < 0.001) (Figure 5).
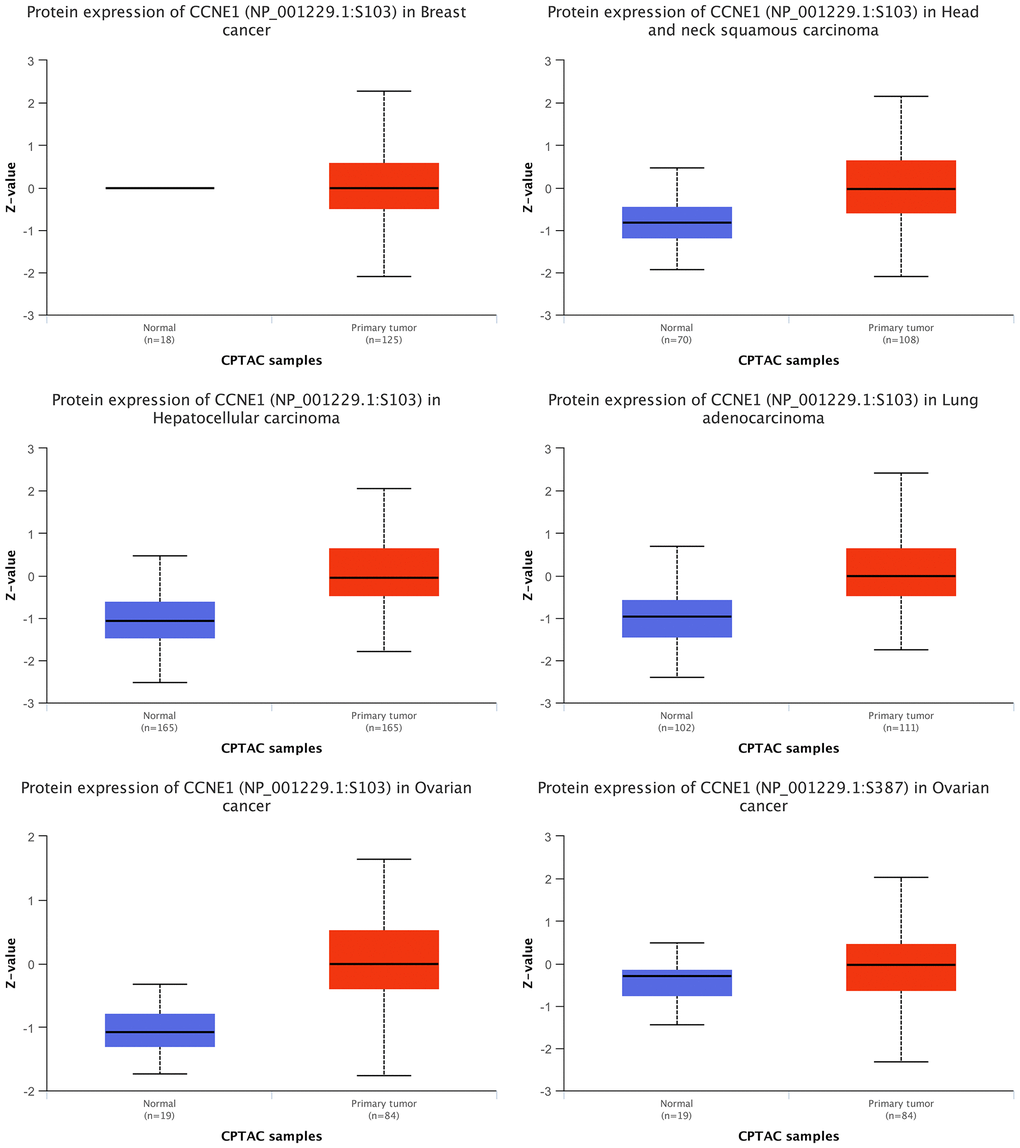
Figure 5. Expression of CCNE1 protein in multiple cancers. The expression of CCNE1 in tumor tissue is indicated in red, while the expression of CCNE1 in normal tissue is shown in blue.
Patients’ immune cell infiltration of CCNE1
To explore the involvement of CCNE1 in immune infiltration and the role of this process in the initiation, progression and metastasis of tumor development, we used TIMER2, EPIC, MCPCOUNTER, CIBERSORT, CIBERSORT-ABS, QUANTISEQ, XCELL, naive_XCELL, central memory_XCELL, and effector memory_XCELL algorithms to analyze the correlation between immune cell infiltration and CCNE1 differential expression in TCGA. CCNE1 expression positively correlated with the cancer-associated immune infiltration level in BRCA, COAD, LUSC, STAD and THYM (Figure 6).
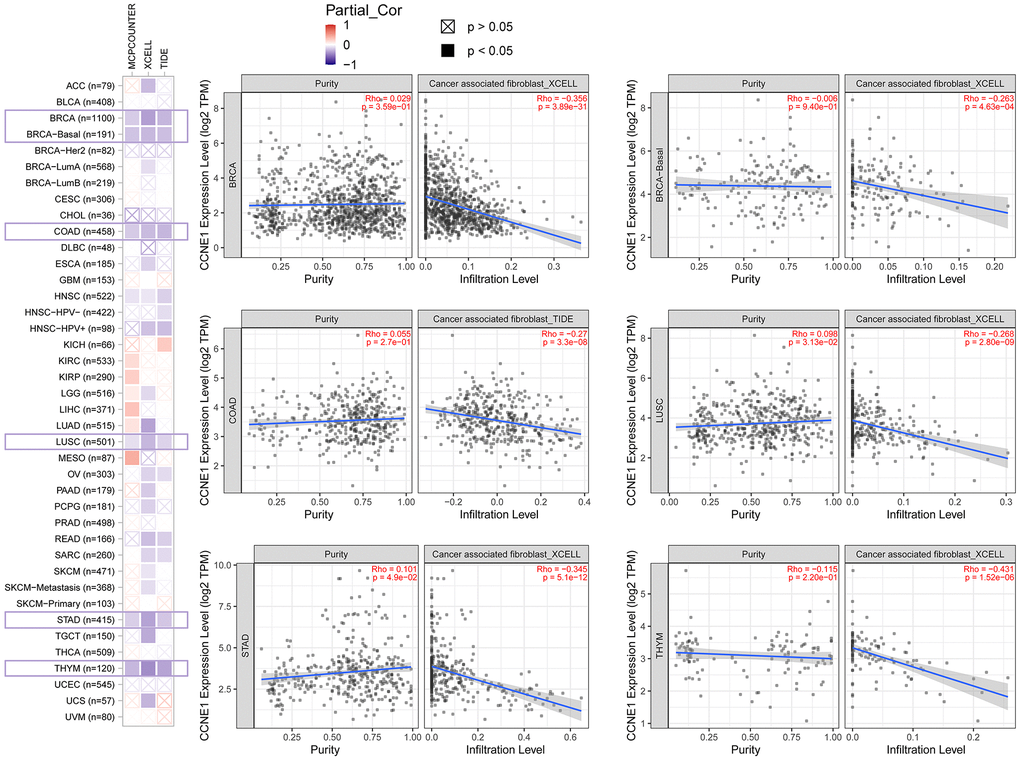
Figure 6. Correlation between CCNE1 expression and immune infiltration of cancer-associated fibroblasts in TCGA. Scatter plot displaying the correlation between CCNE1 mRNA levels and the infiltration levels of cancer-associated fibroblasts across TCGA cancer types. The strength of the correlation is represented by the Pearson correlation coefficient (r), and the p-values indicate the statistical significance of these associations.
CCNE1 enrichment data analysis
In this study, the CCNE1 gene molecule was investigated to screen for gene segments related to CCNE1 binding protein and CCNE1 performance, and a series of pathway enrichment studies were performed for this purpose, using the STRING online database. In our study, the network had 51 nodes which means genes and 397 edges that represent the links between binding genes (Figure 7A). We identified the top 100 genes that correlated with CCNE1 expression, via GEPIA online databases. Additionally, the heatmap indicated a positive correlation between CCNE1 top five related genes in cancer (Figure 7B) and the relationship between CCNE1 and these five genes was also shown (Figure 7C). In Venn diagram, we analyzed the interaction and showed five common members including CDK1, CKS1B, E2F1, CKS2, and FOXM1 (Figure 7D). Investigating functional and pathway enrichment analyses of CCNE1, we performed GO and KEGG enrichment analyses via the DAVID 6.8 online tool. GO enrichment analysis data suggested that most of genes are linked to regulation of cell division in BP category, nucleus in CC category, and protein binding in MF category (Figure 7E). KEGG analysis indicated that the CCNE1 and correlated genes and interacted genes were mainly enriched in cell cycle and cellular senescence pathway (Figure 7F).
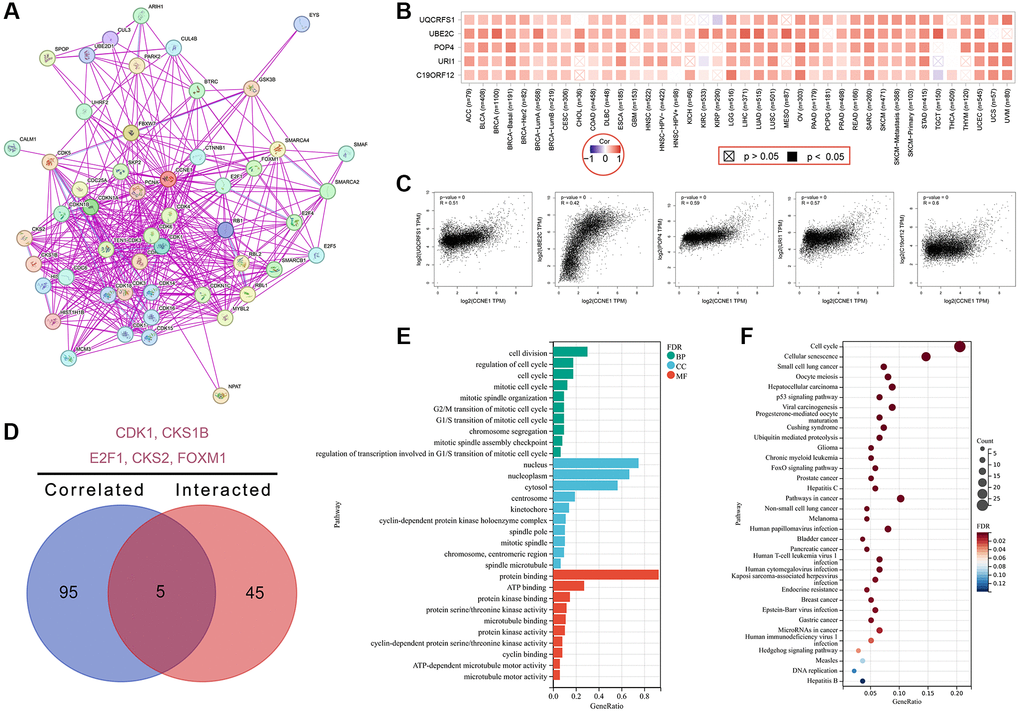
Figure 7. Enrichment analysis for CCNE1 interacted or related genes. (A) Determining interacted genes of CCNE1. (B) Top 5 CCNE1 associated genes in TCGA projects. (C) The corresponding heatmap map for correlation between CCNE1 and top 5 related genes in various cancers. (D) An intersection analysis of CCNE1 interacted and associated genes. (E) GO and (F) KEGG pathway analysis of CCNE1 interacted or related genes.
Discussion
The main role of CCNE 1 encoding cyclin E1 is to promote the transition of the cell cycle from G1 to S phase, thereby facilitating the initiation of DNA synthesis [24]. More and more studies on the role of CCNE1 in tumor tissues have been reported, for example, CCNE1 over-expression in BRCA [25], BLCA [26] and OV [27] tissues are associated with poor tumor survival, although there are also some opposite results [28–30]. Previous studies have reported that 19q12 amplification mutations in CCNE1 are associated with tumor types, especially in uterine tumors, high-grade serous ovarian cancer, and gastroesophageal cancer [31–35]. Moreover, the findings from relevant scholars suggest that such changes enhance genomic instability, genome-wide proliferation, and resistance to cytotoxicity [31–35]. However, whether there is a corresponding common pathway of CCNE1 in its translation, transcription, expression and other processes to produce effects in different tumor pathogenesis is still not clear. After an extensive literature review, there is no unified conclusion on the results of our pan-cancer analysis of CCNE1 [36, 37]. But interestingly, we found that the conserved structure of CCNE1 protein exists in different types of tumor tissues [25, 38, 39], which also implies that CCNE1 may have a similar mechanism in the action of CCNE1 in tumor tissues. Therefore, the CPTAC, GEO, TCGA databases were analyzed in our study to examine the role of CCNE1 in 33 tumors in terms of gene mutation, gene expression and molecular characterization of CCNE1.
Our results suggested that the abnormal expression of CCNE1 occurs frequently in multiple types of tumors. We found that CCNE1 expression level in the tumor tissues of BLCA, BRCA, CHOL, COAD, ESCA, HNSC, KICH, KIRC, KIRP, LIHC, LUAD, READ, STAD, THCA, UCEC, and CESC was higher than the corresponding normal tissues. Functionally, CCNE1 plays a crucial role in driving the transition from G1 to S phase in the cell cycle by forming a complex with CDK2, thereby promoting DNA replication and cell division. This process is critical in maintaining normal cellular homeostasis. However, in cancer, aberrant CCNE1 expression can lead to uncontrolled proliferation [40]. For instance, the overexpression of CCNE1 in ovarian cancer has been shown to promote genomic instability through its interaction with CDK2, which accelerates the cell cycle and impairs DNA repair mechanisms. CCNE1 has been implicated in epithelial-mesenchymal transition (EMT), further promoting cancer cell dissemination [41]. These findings underline the multifaceted role of CCNE1 not only in cell cycle regulation but also in driving aggressive cancer phenotypes through various molecular pathways. From the above results, it is not difficult to conclude that the down-regulation of CCNE1 expression will become the next direction to focus on. Our study found that the abnormal expression of CCNE1 occurred not only in one tumor, but also in a variety of tumor types. For example, in the tumor tissues of BLCA, CESC, and ESCA, CCNE1 expression levels are higher than normal tissues. Furthermore, in the tumor tissue of HNSC, KIRP, and THCA, the results were the same as above. The elevated expression of CCNE1 in various cancers indicates its significant role in tumorigenesis and cancer progression [42]. Clinically, high CCNE1 expression has been correlated with several critical aspects of cancer behavior and patient outcomes. Elevated CCNE1 levels are often associated with increased tumor aggressiveness and invasiveness. This is likely due to its role in driving the cell cycle progression from G1 to S phase, promoting rapid cell division and proliferation. Studies have shown that higher CCNE1 expression can lead to enhanced metastatic potential, making it a marker for aggressive cancer phenotypes. CCNE1 overexpression may influence the response to various cancer treatments. For instance, cancers with high CCNE1 expression may exhibit resistance to certain chemotherapeutic agents that target cell cycle pathways. This resistance can be attributed to the rapid cell cycle progression driven by CCNE1, which might reduce the efficacy of treatments designed to halt cell division. On the other hand, targeting CCNE1 directly or its associated pathways could offer a novel therapeutic approach, potentially improving treatment outcomes for patients with CCNE1-overexpressing tumors. As demonstrated in our study, high CCNE1 expression is associated with poor overall survival in several cancers, including ACC, BRCA, KIRC, KIRP, LGG, LIHC, LUAD, and MESO. This highlights its potential as a prognostic biomarker. Patients with elevated CCNE1 levels may require more aggressive and tailored therapeutic strategies to improve their prognosis. Furthermore, innovative therapeutic approaches focusing on the downregulation of CCNE1 have shown promise in mitigating tumor growth and enhancing treatment efficacy [43].
Among these, RNA interference (RNAi) technologies and small molecule inhibitors specifically targeting CCNE1 activity have emerged as effective strategies [44]. For instance, RNAi approaches, such as siRNA and shRNA, have been shown to successfully silence CCNE1 expression, reducing tumor growth in breast cancer [45]. In addition to RNAi, small molecule inhibitors like Adavosertib, a WEE1 inhibitor, have been explored for their ability to inhibit cyclin E/CDK2 complexes, slowing down cell cycle progression in cancers with CCNE1 amplification [46]. These advancements indicate that targeting CCNE1 pathways can offer substantial therapeutic benefits, particularly for patients with CCNE1-overexpressing tumors where traditional treatments have failed [47].
Despite the high level of CCNE 1 expression in many tumors, different conclusions from previous studies. To explore the association of CCNE 1 with different tumor outcomes, a meta-analysis study found that over expression of CCNE 1 was associated with poor survival in cancer patients [30]. In this research, in this study, there was a statistically significant relationship between a high expression of CCNE 1 and a poor survival prognosis (P < 0.01). The reason may be different data processing methods and new survival information. At the same time, we explored the relationship between OV pathological stage and CCNE1 expression level through the “Pathological Stage Map” module of GEPIA2 (P < 0.01). In addition, we performed a survival analysis of OV by the Kaplan-Meier plotter approach. The results suggested that CCNE1 expression was not statistically significant with the clinical prognosis of DFS and OS. Therefore, the current evidence based on clinical big data can only prove that the over expression of CCNE1 is correlated with OV, and cannot support the clinical prognosis role of OV. Our study also found that the poor prognosis of different tumors was closely associated with the high expression level of CCNE 1, suggesting that the high expression status of CCNE 1 may be a biomarker of clinical prognosis in cancer patients. Meanwhile, it also means that down-regulation of CCNE1 expression may be one of the methods to improve the prognosis of patients. Recent studies have confirmed that PKMYT1 inhibitors may be an effective approach for the treatment of CCNE1-amplified cancers [34].
Tumor microenvironment (TME) is a setting that promotes tumor progression, and tumor cells often use this setting to escape the surveillance of immune function. In addition, TME also plays an important role in reflecting the therapeutic effect and predicting the clinical outcome of tumors. The research results of relevant scholars showed that TME components include tumor-associated mesenchymal stem cells, extracellular matrix, lymphocytes, fibroblasts [48–51]. Tumor immune-infiltrating cells play an important role in immune regulation by transferring from blood to tumor tissue [52]. CCNE1, as a major component of focal adhesions, plays a vital role in the extracellular matrix [53]. Importantly, CCNE 1 may also have significant effects on the immune response and TME. In our research, we analyzed the relationship between immune cell infiltration and differential expression of CCNE1 using CIBERSORT-ABS, XCELL, MCPCOUNTER, QUANTISEQ, EPIC, CIBERSORT central memory_XCELL, effector memory_XCELL algorithms, and TIMER2. In different tumors, high expression of CCNE1 is associated with cancer-associated fibroblasts, especially in BRCA, COAD, LUSC, STAD, THYM. It has been reported that cancer-associated fibroblasts and endothelial cells have been able to exert tumorigenic effects in the TME by secreted various growth factors, cytokines and chemokines and promoting the degradation of the extracellular matrix [54, 55]. However, in our study, there was no obvious association between CCNE1 expression and MSCs, monocytes, or bone marrow-derived suppressor cells, but the link between CCNE1 and TME through cancer-associated fibroblasts may explain the prognostic impact of CCNE1 in different cancers. At the same time, we found no significant correlation between CCNE 1 expression and MSCs, monocytes, or bone marrow-derived suppressor cells. MSCs are regulators of the tumor niche and are involved in tumorigenesis and metastasis. CCNE1 was previously reported to be important in the proliferation and colony formation of MSCs [56, 57]. Our study reveals a significant correlation between CCNE1 expression and increased immune cell infiltration in multiple cancers, including breast, colon, lung, stomach, and thymic cancers. By employing various algorithms, we’ve confirmed that elevated CCNE1 levels enhance immune presence, aligning with its known roles in cell cycle regulation and tumorigenesis. Notably, this correlation suggests CCNE1’s involvement in modulating the tumor microenvironment, which could influence immune cell recruitment and activation, thus presenting potential therapeutic implications for enhancing cancer immunotherapy effectiveness. More analyses on immunotherapy to further reveal the biological properties and roles of CCNE1 in different tumours are needed to provide an important reference for subsequent studies.
Moreover, genetic mutations also play a critical role in the mechanism of action of carcinogenesis [58]. The overexpression of CCNE1 has been found in a variety of tumors, so, we further explored the corresponding mutation characteristics of CCNE1. Kang et al. showed that CCNE 1 gene amplification leads to poor prognosis in cancer patients [59]. CCNE1 gene mutations include amplification, missense mutations, truncating mutations, and fusion mutations. Missense mutations such as M336I/T/V result in amino acid substitutions that may impact the stability and function of the CCNE1 protein. Amplification may lead to overexpression of CCNE1 protein, causing dysregulation of cell cycle control and promoting tumorigenesis. Fusion mutations can produce fusion proteins with novel functions or loss of original functions, affecting cell proliferation and differentiation. Truncating mutations may result in the production of truncated proteins lacking essential functional domains, thereby impairing their normal function. CCNE1 plays a critical role in cell cycle regulation, and its mutations and overexpression are closely associated with the development of various cancers. CCNE1 mutations may lead to cell cycle dysregulation, promoting cancer cell proliferation. A detailed analysis of different mutation types can enhance our understanding of the role of CCNE1 in cancer development and progression, providing a theoretical basis for precision medicine.
Our study showed that CCNE 1 had the highest frequency of changes in uterine cancer patients, at 40%, whose main type is “amplification”. And we also found that in UCEC, the more prominent types are the “amplification” and “mutation”, accounting for about 5% of the frequency of occurrence. Above all, most of the tumors listed in the study are formed under the influence of CCNE1, and the “amplification” is the predominant type. In conclusion, CCNE1 amplification may effectively predict the occurrence of adverse prognostic events in cancer patients, and the control of CCNE1 amplification will become a hot research direction for the control of adverse prognostic events. This suggested that the enriched pathways associated with CCNE 1 could be used as potential therapeutic options for the treatment of cancer patients. Additionally, the analysis found that the amplification of CCNE1 was associated with poor prognosis of patients, so the CCNE1 gene mutation may also be a potential molecular marker for poor prognosis in endometrioma patients.
For the first time, the top 100 proteins were screened by software and finally found CDK1, CKS1B, E2F1, CKS2, and FOXM1 to be associated with CCNE1. Although the different roles of the above proteins in tumor pathogenesis have been reported, little is known about the exact pathogenic mechanism of tumors, which is also the focus of our future studies. Meanwhile, in this study, the CPTAC dataset was used to explore the expression of CCNE1 protein in breast cancer, hepatocellular carcinoma, head and neck squamous cell carcinoma, ovarian cancer, and lung adenocarcinoma.
We used GO enrichment analysis and KEGG pathway enrichment analysis to investigate the differential expression signatures of CCNE 1. The results found that the differentially expressed CCNE 1 was mainly associated with the regulation of cell division in the BP class. Moreover, it is also related with the protein binding regulation of MF class and the nuclear regulation of CC class. Importantly, miR-30c-2-3p has an important role in cancer cell migration, by regulating CCNE 1 gene expression to regulating cytokine expression, invasion and cell proliferation in breast cancer cells. Previous study has shown that miR-30c-2-3p negatively regulates NF-κB signaling and cell cycle progression by downregulating CCNE 1 in breast cancer, resulting in reduced cytokine expression in vitro, cell invasion, and cell proliferation in vitro [60]. In addition, previous studies in gastric cancer showed that CircDENND2A can promote the progression of non-small-cell lung cancer by regulating the CCNE1 signaling pathway [61]. However, there were some limitations existing in our study. The data used for analysis were obtained from online services. More cell-based studies and clinical experiments are needed to confirm our findings and to further explore interactions between relevant molecules, the precise mechanisms involved, and the potential clinical applications of CCNE1 in cancer.
Conclusions
In this study, a series of pan-cancer analyses were performed to determine the relevance of CCNE1 in pan-cancer and its potential predictive value. We found that the expression of CCNE1 is related to clinical prognosis, immune infiltration of tumor cells, and gene mutation. The pan-cancer analysis of CCNE1 can analyze the role and effect of CCNE 1 in tumor development and development from multiple perspectives, and can provide some help for clinical diagnosis, treatment and basic research.
Author Contributions
Yujie Ouyang and Ziyi Wu were co-first authors. Yujie Ouyang, Ziyi Wu, Dilihumaer Aili, Chunhua Yang, Hui Zhang and Tong Wu had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Concept and design: Yujie Ouyang, Ziyi Wu, Hui Zhang, Tong Wu. Acquisition, analysis, or interpretation of data: Yujie Ouyang, Ziyi Wu, Hui Zhang, Tong Wu.
Acknowledgments
The authors would like to thank all of the co-investigators and colleagues who made this study possible.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
All clinical information and data of this research were obtained from public databases - TCGA and CPTAC, cBioPortal datasets. Therefore, approval from the ethics committee is not required for this study.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities of Central South University (2023ZZTS0026) and Hunan Province Natural Science Foundation (Grant No. 2021JJ30944). The study funders/sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
References
- 1. Jiang LJ, Guo SB, Zhou ZH, Li ZY, Zhou FJ, Yu CP, Li M, Huang WJ, Liu ZW, Tian XP. Snai2-mediated upregulation of NADSYN1 promotes bladder cancer progression by interacting with PHB. Clin Transl Med. 2024; 14:e1555. https://doi.org/10.1002/ctm2.1555 [PubMed]
- 2. Guo SB, Feng XZ, Huang WJ, Zhou ZZ, Tian XP. Global research hotspots, development trends and prospect discoveries of phase separation in cancer: a decade-long informatics investigation. Biomark Res. 2024; 12:39. https://doi.org/10.1186/s40364-024-00587-9 [PubMed]
- 3. Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015; 19:A68–77. https://doi.org/10.5114/wo.2014.47136 [PubMed]
- 4. Blum A, Wang P, Zenklusen JC. SnapShot: TCGA-Analyzed Tumors. Cell. 2018; 173:530. https://doi.org/10.1016/j.cell.2018.03.059 [PubMed]
- 5. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013; 41:D991–5. https://doi.org/10.1093/nar/gks1193 [PubMed]
- 6. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020; 20:651–68. https://doi.org/10.1038/s41577-020-0306-5 [PubMed]
- 7. van de Donk NWC, Usmani SZ, Yong K. CAR T-cell therapy for multiple myeloma: state of the art and prospects. Lancet Haematol. 2021; 8:e446–61. https://doi.org/10.1016/S2352-3026(21)00057-0 [PubMed]
- 8. Wu Z, Xu N, Li G, Yang W, Zhang C, Zhong H, Wu G, Chen F, Li D. Multi-omics analysis of the oncogenic role of optic atrophy 1 in human cancer. Aging (Albany NY). 2023; 15:12982–97. https://doi.org/10.18632/aging.205214 [PubMed]
- 9. Sonntag R, Giebeler N, Nevzorova YA, Bangen JM, Fahrenkamp D, Lambertz D, Haas U, Hu W, Gassler N, Cubero FJ, Müller-Newen G, Abdallah AT, Weiskirchen R, et al. Cyclin E1 and cyclin-dependent kinase 2 are critical for initiation, but not for progression of hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2018; 115:9282–7. https://doi.org/10.1073/pnas.1807155115 [PubMed]
- 10. Pardee AB. G1 events and regulation of cell proliferation. Science. 1989; 246:603–8. https://doi.org/10.1126/science.2683075 [PubMed]
- 11. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009; 9:153–66. https://doi.org/10.1038/nrc2602 [PubMed]
- 12. Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009; 9:785–97. https://doi.org/10.1038/nrc2696 [PubMed]
- 13. Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992; 257:1689–94. https://doi.org/10.1126/science.1388288 [PubMed]
- 14. Guerrero-Zotano Á, Belli S, Zielinski C, Gil-Gil M, Fernandez-Serra A, Ruiz-Borrego M, Ciruelos Gil EM, Pascual J, Muñoz-Mateu M, Bermejo B, Margeli Vila M, Antón A, Murillo L, et al. CCNE1 and PLK1 Mediate Resistance to Palbociclib in HR+/HER2- Metastatic Breast Cancer. Clin Cancer Res. 2023; 29:1557–68. https://doi.org/10.1158/1078-0432.CCR-22-2206 [PubMed]
- 15. Zheng X, Chen L, Liu W, Zhao S, Yan Y, Zhao J, Tian W, Wang Y. CCNE1 is a predictive and immunotherapeutic indicator in various cancers including UCEC: a pan-cancer analysis. Hereditas. 2023; 160:13. https://doi.org/10.1186/s41065-023-00273-0 [PubMed]
- 16. Xu H, George E, Kinose Y, Kim H, Shah JB, Peake JD, Ferman B, Medvedev S, Murtha T, Barger CJ, Devins KM, D'Andrea K, Wubbenhorst B, et al. CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models. Cell Rep Med. 2021; 2:100394. https://doi.org/10.1016/j.xcrm.2021.100394 [PubMed]
- 17. Gorski JW, Ueland FR, Kolesar JM. CCNE1 Amplification as a Predictive Biomarker of Chemotherapy Resistance in Epithelial Ovarian Cancer. Diagnostics (Basel). 2020; 10:279. https://doi.org/10.3390/diagnostics10050279 [PubMed]
- 18. Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017; 77:e108–10. https://doi.org/10.1158/0008-5472.CAN-17-0307 [PubMed]
- 19. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017; 45:W98–102. https://doi.org/10.1093/nar/gkx247 [PubMed]
- 20. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVS, Varambally S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017; 19:649–58. https://doi.org/10.1016/j.neo.2017.05.002 [PubMed]
- 21. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088 [PubMed]
- 22. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019; 47:D607–13. https://doi.org/10.1093/nar/gky1131 [PubMed]
- 23. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57. https://doi.org/10.1038/nprot.2008.211 [PubMed]
- 24. Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005; 24:2776–86. https://doi.org/10.1038/sj.onc.1208613 [PubMed]
- 25. Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, DeMichele A, Harbeck N, André F, Bayar MA, Michiels S, Zhang Z, Giorgetti C, et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol. 2019; 37:1169–78. https://doi.org/10.1200/JCO.18.00925 [PubMed]
- 26. Song BN, Kim SK, Chu IS. Bioinformatic identification of prognostic signature defined by copy number alteration and expression of CCNE1 in non-muscle invasive bladder cancer. Exp Mol Med. 2017; 49:e282. https://doi.org/10.1038/emm.2016.120 [PubMed]
- 27. Petersen S, Wilson AJ, Hirst J, Roby KF, Fadare O, Crispens MA, Beeghly-Fadiel A, Khabele D. CCNE1 and BRD4 co-amplification in high-grade serous ovarian cancer is associated with poor clinical outcomes. Gynecol Oncol. 2020; 157:405–10. https://doi.org/10.1016/j.ygyno.2020.01.038 [PubMed]
- 28. Shariat SF, Ashfaq R, Sagalowsky AI, Lotan Y. Correlation of cyclin D1 and E1 expression with bladder cancer presence, invasion, progression, and metastasis. Hum Pathol. 2006; 37:1568–76. https://doi.org/10.1016/j.humpath.2006.05.017 [PubMed]
- 29. Pils D, Bachmayr-Heyda A, Auer K, Svoboda M, Auner V, Hager G, Obermayr E, Reiner A, Reinthaller A, Speiser P, Braicu I, Sehouli J, Lambrechts S, et al. Cyclin E1 (CCNE1) as independent positive prognostic factor in advanced stage serous ovarian cancer patients - a study of the OVCAD consortium. Eur J Cancer. 2014; 50:99–110. https://doi.org/10.1016/j.ejca.2013.09.011 [PubMed]
- 30. Zhao H, Wang J, Zhang Y, Yuan M, Yang S, Li L, Yang H. Prognostic Values of CCNE1 Amplification and Overexpression in Cancer Patients: A Systematic Review and Meta-analysis. J Cancer. 2018; 9:2397–407. https://doi.org/10.7150/jca.24179 [PubMed]
- 31. Yuan J, Hu Z, Mahal BA, Zhao SD, Kensler KH, Pi J, Hu X, Zhang Y, Wang Y, Jiang J, Li C, Zhong X, Montone KT, et al. Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers. Cancer Cell. 2018; 34:549–60.e9. https://doi.org/10.1016/j.ccell.2018.08.019 [PubMed]
- 32. Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al, and Australian Ovarian Cancer Study Group. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015; 521:489–94. https://doi.org/10.1038/nature14410 [PubMed]
- 33. Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013; 45:1134–40. https://doi.org/10.1038/ng.2760 [PubMed]
- 34. Gallo D, Young JTF, Fourtounis J, Martino G, Álvarez-Quilón A, Bernier C, Duffy NM, Papp R, Roulston A, Stocco R, Szychowski J, Veloso A, Alam H, et al. CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature. 2022; 604:749–56. https://doi.org/10.1038/s41586-022-04638-9 [PubMed]
- 35. Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, Moore DA, Grönroos E, Rowan A, Dewhurst SM, Demeulemeester J, Dentro SC, Horswell S, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature. 2020; 587:126–32. https://doi.org/10.1038/s41586-020-2698-6 [PubMed]
- 36. Ooi WF, Nargund AM, Lim KJ, Zhang S, Xing M, Mandoli A, Lim JQ, Ho SWT, Guo Y, Yao X, Lin SJ, Nandi T, Xu C, et al. Integrated paired-end enhancer profiling and whole-genome sequencing reveals recurrent CCNE1 and IGF2 enhancer hijacking in primary gastric adenocarcinoma. Gut. 2020; 69:1039–52. https://doi.org/10.1136/gutjnl-2018-317612 [PubMed]
- 37. Li J, Zhou L, Liu Y, Yang L, Jiang D, Li K, Xie S, Wang X, Wang S. Comprehensive Analysis of Cyclin Family Gene Expression in Colon Cancer. Front Oncol. 2021; 11:674394. https://doi.org/10.3389/fonc.2021.674394 [PubMed]
- 38. Amininia S, Hashemi M, Ebrahimi M, Mashhadi MA, Hashemi SM, Taheri M, Ghavami S. Association between CCNE1 polymorphisms and the risk of breast cancer in a sample of southeast Iranian population. Med Oncol. 2014; 31:189. https://doi.org/10.1007/s12032-014-0189-z [PubMed]
- 39. Zhao ZM, Yost SE, Hutchinson KE, Li SM, Yuan YC, Noorbakhsh J, Liu Z, Warden C, Johnson RM, Wu X, Chuang JH, Yuan Y. CCNE1 amplification is associated with poor prognosis in patients with triple negative breast cancer. BMC Cancer. 2019; 19:96. https://doi.org/10.1186/s12885-019-5290-4 [PubMed]
- 40. Zeng J, Hills SA, Ozono E, Diffley JFX. Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell. 2023; 186:528–42.e14. https://doi.org/10.1016/j.cell.2022.12.036 [PubMed]
- 41. Freeman-Cook K, Hoffman RL, Miller N, Almaden J, Chionis J, Zhang Q, Eisele K, Liu C, Zhang C, Huser N, Nguyen L, Costa-Jones C, Niessen S, et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell. 2021; 39:1404–21.e11. https://doi.org/10.1016/j.ccell.2021.08.009 [PubMed]
- 42. Yuan Q, Zheng L, Liao Y, Wu G. Overexpression of CCNE1 confers a poorer prognosis in triple-negative breast cancer identified by bioinformatic analysis. World J Surg Oncol. 2021; 19:86. https://doi.org/10.1186/s12957-021-02200-x [PubMed]
- 43. Zhang C, Zhu Q, Gu J, Chen S, Li Q, Ying L. Down-regulation of CCNE1 expression suppresses cell proliferation and sensitizes gastric carcinoma cells to Cisplatin. Biosci Rep. 2019; 39:BSR20190381. https://doi.org/10.1042/BSR20190381 [PubMed]
- 44. Nakayama K, Rahman MT, Rahman M, Nakamura K, Ishikawa M, Katagiri H, Sato E, Ishibashi T, Iida K, Ishikawa N, Kyo S. CCNE1 amplification is associated with aggressive potential in endometrioid endometrial carcinomas. Int J Oncol. 2016; 48:506–16. https://doi.org/10.3892/ijo.2015.3268 [PubMed]
- 45. Chen X, Low KH, Alexander A, Jiang Y, Karakas C, Hess KR, Carey JPW, Bui TN, Vijayaraghavan S, Evans KW, Yi M, Ellis DC, Cheung KL, et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin Cancer Res. 2018; 24:6594–610. https://doi.org/10.1158/1078-0432.CCR-18-1446 [PubMed]
- 46. Fu S, Yao S, Yuan Y, Previs RA, Elias AD, Carvajal RD, George TJ, Yuan Y, Yu L, Westin SN, Xing Y, Dumbrava EE, Karp DD, et al. Multicenter Phase II Trial of the WEE1 Inhibitor Adavosertib in Refractory Solid Tumors Harboring CCNE1 Amplification. J Clin Oncol. 2023; 41:1725–34. https://doi.org/10.1200/JCO.22.00830 [PubMed]
- 47. Krivega MV, Geens M, Heindryckx B, Santos-Ribeiro S, Tournaye H, Van de Velde H. Cyclin E1 plays a key role in balancing between totipotency and differentiation in human embryonic cells. Mol Hum Reprod. 2015; 21:942–56. https://doi.org/10.1093/molehr/gav053 [PubMed]
- 48. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013; 19:1423–37. https://doi.org/10.1038/nm.3394 [PubMed]
- 49. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015; 348:74–80. https://doi.org/10.1126/science.aaa6204 [PubMed]
- 50. Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. 2017; 16:31. https://doi.org/10.1186/s12943-017-0597-8 [PubMed]
- 51. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016; 16:582–98. https://doi.org/10.1038/nrc.2016.73 [PubMed]
- 52. Fridman WH, Galon J, Dieu-Nosjean MC, Cremer I, Fisson S, Damotte D, Pagès F, Tartour E, Sautès-Fridman C. Immune infiltration in human cancer: prognostic significance and disease control. Curr Top Microbiol Immunol. 2011; 344:1–24. https://doi.org/10.1007/82_2010_46 [PubMed]
- 53. Ghosh T, Varshney A, Kumar P, Kaur M, Kumar V, Shekhar R, Devi R, Priyanka P, Khan MM, Saxena S. MicroRNA-874-mediated inhibition of the major G1/S phase cyclin, CCNE1, is lost in osteosarcomas. J Biol Chem. 2017; 292:21264–81. https://doi.org/10.1074/jbc.M117.808287 [PubMed]
- 54. Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment - Accomplices in tumor malignancy. Cell Immunol. 2019; 343:103729. https://doi.org/10.1016/j.cellimm.2017.12.003 [PubMed]
- 55. Hida K, Maishi N, Annan DA, Hida Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int J Mol Sci. 2018; 19:1272. https://doi.org/10.3390/ijms19051272 [PubMed]
- 56. Liang Y, Zhang D, Li L, Xin T, Zhao Y, Ma R, Du J. Exosomal microRNA-144 from bone marrow-derived mesenchymal stem cells inhibits the progression of non-small cell lung cancer by targeting CCNE1 and CCNE2. Stem Cell Res Ther. 2020; 11:87. https://doi.org/10.1186/s13287-020-1580-7 [PubMed]
- 57. Wu H, Mu X, Liu L, Wu H, Hu X, Chen L, Liu J, Mu Y, Yuan F, Liu W, Zhao Y. Bone marrow mesenchymal stem cells-derived exosomal microRNA-193a reduces cisplatin resistance of non-small cell lung cancer cells via targeting LRRC1. Cell Death Dis. 2020; 11:801. https://doi.org/10.1038/s41419-020-02962-4 [PubMed]
- 58. Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, Mularoni L, Pich O, Bonet J, Kranas H, Gonzalez-Perez A, Lopez-Bigas N. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020; 20:555–72. https://doi.org/10.1038/s41568-020-0290-x [PubMed]
- 59. Kang EY, Weir A, Meagher NS, Farrington K, Nelson GS, Ghatage P, Lee CH, Riggan MJ, Bolithon A, Popovic G, Leung B, Tang K, Lambie N, et al, and AOCS Group. CCNE1 and survival of patients with tubo-ovarian high-grade serous carcinoma: An Ovarian Tumor Tissue Analysis consortium study. Cancer. 2023; 129:697–713. https://doi.org/10.1002/cncr.34582 [PubMed]
- 60. Shukla K, Sharma AK, Ward A, Will R, Hielscher T, Balwierz A, Breunig C, Münstermann E, König R, Keklikoglou I, Wiemann S. MicroRNA-30c-2-3p negatively regulates NF-κB signaling and cell cycle progression through downregulation of TRADD and CCNE1 in breast cancer. Mol Oncol. 2015; 9:1106–19. https://doi.org/10.1016/j.molonc.2015.01.008 [PubMed]
- 61. Zhang Y, Shan C, Chen Y, Sun S, Liu D, Zhang X, Zhang S. CircDENND2A Promotes Non-small Cell Lung Cancer Progression via Regulating MiR-34a/CCNE1 Signaling. Front Genet. 2020; 11:987. https://doi.org/10.3389/fgene.2020.00987 [PubMed]