



Introduction
Oral submucous fibrosis (OSF) is predominantly prevalent in the areas of South and Southeast Asia, with a malignant transformation rate into oral squamous cell carcinoma (OSCC) ranging up to 4-13% [1–3]. Recognized as an areca nut-associated oral potentially malignant disorder (OPMD), OSF is characterized by a gradual reduction in mouth opening, largely due to the dysregulation the extracellular matrix (ECM) synthesis and degradation [4]. Despite various approaches being applied to alleviate symptoms [5–8], none have proven curative for OSF. Hence, deeper understanding in the pathogenesis of OSF is necessary to developing more effective therapies and preventing its malignant transformation.
Myofibroblasts, key ECM-secreting cells, play a major role in wound healing and pathological fibrosis. These cells can escape from apoptosis, leading to continued tissue remodeling and fibrosis through increased production of mediators like pro-inflammatory cytokines and transforming growth factor-β (TGF-β), as well as excessive secretion of ECM components such as collagens [9–11]. In OSF, higher expression of myofibroblast markers has been reported [12]. Therefore, approaches to suppress the persistent activation of myofibroblasts offers a promising strategy to resolve pathological fibrosis.
Emerging studies have revealed the role of epigenetic regulation by non-coding RNAs (ncRNAs) in modulating the activation of myofibroblasts [13]. The interplay between long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) has gained great attention for its impact on the development of pathological fibrosis. Mechanically, lncRNAs can function as competing endogenous RNAs (ceRNAs), acting as miRNA sponges to disrupt the interaction of miRNAs and their target mRNAs [14]. For instance, LINC00084 has been demonstrated to mediate myofibroblast activation in fibrotic buccal mucosa fibroblasts. The upregulation of this lncRNA increases the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 by sponging miR-204 [15]. Another study showed that the arecoline-induced lncRNA H19 binds miR-29b, thereby impeding miR-29b from interacting with type I collagen (COL1A1) and inhibiting several myofibroblast phenotypes [16]. However, the specific functions of dysregulated lncRNAs in the progression of OSF remain inadequately explored.
Myocardial infarction-associated transcript (MIAT), an intergenic lncRNA initially identified as a risk locus for myocardial infarction [17], has since been found to be overexpressed in various cancers, where it regulates multiple biological processes such as cell cycle, invasion, metastasis, and drug resistance [18]. In oral cancer tissues, MIAT is upregulated and associated with poor prognosis [19]. MIAT has also been implicated in several fibrotic diseases, including heart failure [20], renal fibrosis [21], and chronic pancreatitis [22]. Notably, MIAT knockdown has been shown to inhibit TGF-β-stimulated myofibroblast formation in mouse fibroblasts [21]. However, the role of MIAT in OSF development, particularly in influencing myofibroblast transdifferentiation, remains unclear. Given MIAT’s pro-fibrotic role, elucidating its precise molecular interactions and pathways is crucial. This study, therefore, aims to investigate MIAT’s involvement in myofibroblast activation and OSF progression, as well as its ceRNA network, addressing a critical gap in our understanding of OSF pathogenesis.
Results
MIAT expression increases in the fibrotic buccal tissues and its derived primary fibroblasts
To identify key dysregulated lncRNAs involved in OSF progression, we initially established a cohort comprising 25 patients with OSF and 25 healthy individuals. RNA-sequencing analysis showed that MIAT was aberrantly upregulated in the fibrotic buccal tissues from OSF patients (OSF; n=2) compared to that of normal tissues from healthy individuals (N; n=2; Figure 1A). Results from qRT-PCR confirmed overexpression of MIAT in OSF specimens (n=25) and fBMFs (n=5) compared to normal tissues (N; n=25) and fibroblasts derived from non-fibrotic buccal mucosa (nBMFs; n=5), respectively (Figure 1B, 1C). These findings suggest that dysregulated MIAT expression in fibrotic tissues may be associated with myofibroblasts activation. To further validate the association between MIAT expression and myofibroblasts activation, we expanded our cohort to include 45 OSF patients and assessed the expression of pro-fibrotic markers in their fibrotic buccal tissues, including alpha-smooth muscle actin (α-SMA; encoded by ACTA2), collagen type 1 alpha 1 (COL1A1), and fibronectin (FN1). As expected, results from qRT-PCR revealed that the expression of MIAT was positively correlated with ACAT2 (Figure 1D), COL1A1 (Figure 1E), and FN1 (Figure 1F). It has been known that myofibroblast acquires more contractile phenotype by increased actin stress fibers, so α-SMA can be used as a differentiated myofibroblast marker [23]. During differentiation, myofibroblasts display increased expression and secretion of FN and collagen [24]. As such, we postulated that the expression of MIAT may be involved in the regulation of myofibroblast activation and contribute to the development of OSF.
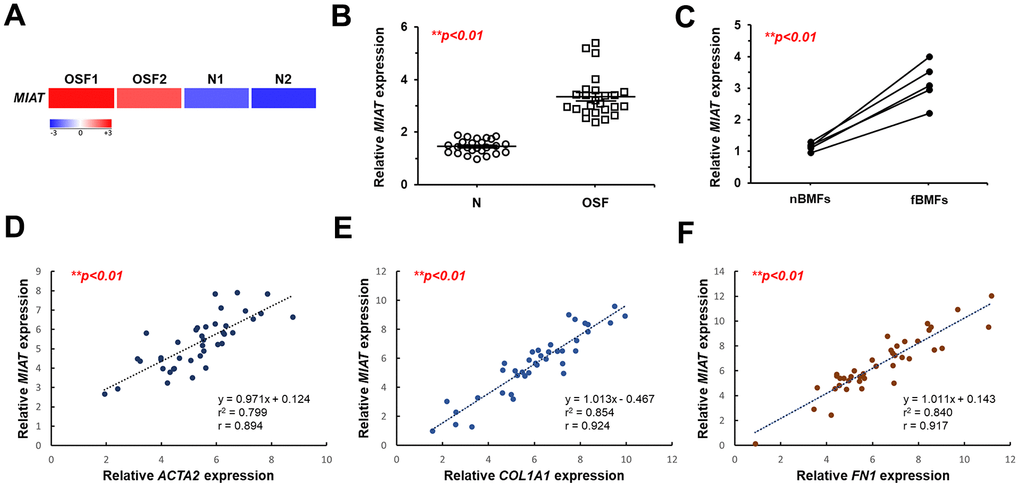
Figure 1. MIAT is upregulated in fibrotic buccal tissues and primary fibrotic buccal mucosa fibroblasts from patients with OSF. (A) RNA-sequencing analysis showed that MIAT was an up-regulated differentially expressed gene (fold change ≥ 2.0; p < 0.01) in fibrotic tissue samples (OSF) compared to normal tissues (N) from patients with OSF (n=2) and healthy individuals (n=2). (B) The relative expression of MIAT in samples of normal (N; n=25) and fibrotic (OSF; n=25) tissue was assessed by qRT-PCR analysis. Data are mean ± S.D. (C) The relative expression of MIAT was further assessed in primary normal buccal mucosa fibroblasts (nBMFs; n=5) and fibrotic buccal mucosa fibroblasts (fBMFs; n=5) by qRT-PCR, with differences between groups analyzed using the paired Student’s t-test. (D–F) A significant positive correlation was observed between the expression of MIAT and fibrotic markers, including ACTA2 (encoding α-SMA; D), COL1A1 (E), and FN1 (F) in samples of fibrotic tissue (n=40).
Silencing MIAT reduces the myofibroblastic properties of fBMFs
To test our presumption, we used a lentiviral vector-mediated short hairpin (sh) RNA targeting MIAT to knock down the expression of MIAT in fBMFs (Figure 2A) and examined the effect of MIAT on myofibroblast phenotypes. First, a collagen gel contraction assay was conducted to assess the contractile forces generated by myofibroblasts, which propagated throughout the collagen matrix and resulted in decreased matrix size. As shown in Figure 2B, the downregulation of MIAT markedly attenuated the collagen gel contractility of fBMFs. Another feature of myofibroblasts is that they proliferate and migrate to the damaged site during wound healing. By using Transwell migration and scratch (wound healing) assays, we demonstrated that suppression of MIAT significantly inhibited the cell migration (Figure 2C) and wound-healing ability (Figure 2D) of fBMFs.
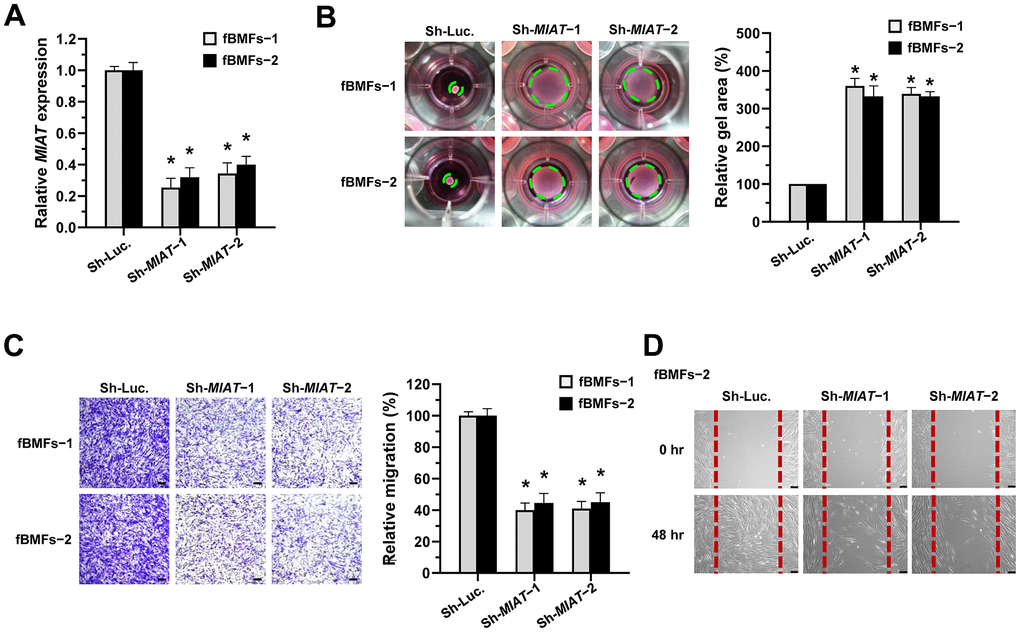
Figure 2. Knockdown of MIAT suppresses the myofibroblastic properties. (A–D) Primary fBMFs (obtained from two patients with OSF; fBMFs−1 and −2) were transfected with lentiviruses expressing non-targeting ShRNA (Sh-Luc.) and Sh-MIAT (Sh-MIAT−1 and −2). The MIAT knockdown efficiency was assessed using qRT-PCR analysis (A). The cells (fBMFs−1 and −2) were then cultured in collagen gel for additional 48 hours, and the gel area after cell contraction was measured (B). Cells (fBMFs−1 and −2) were cultured in Transwell system for an additional 24 hours, and their migration ability was quantified (C). Data are presented as mean ± SD (n=3); *p < 0.05 vs. Sh-Luc. (A–C). Confluent monolayers of fBMFs−2 were scratched and cultured for 48 hours, and the wound closure was assessed. Scale bar, 50 μm (D).
Loss of MIAT inhibits the arecoline-induced myofibroblast transformation of nBMFs
We previously demonstrated that arecoline treatment can effectively induce nBMFs to acquire myofibroblastic properties [15, 25–27]. Thus, to understand whether MIAT is involved in the transformation of fibroblasts to myofibroblasts, we assessed the expression of MIAT in nBMFs after treatment with 0, 5, 10, and 20 μg/mL arecoline for 24 hours. RNA sequencing analysis showed that MIAT levels were up-regulated in nBMFs treated with 10 and 20 μg/mL arecoline (Figure 3A). Also, results of qPCR analysis demonstrated that arecoline gradually increased MIAT expression in a dose-dependent manner (Figure 3B). Based on our previous findings [15, 25–27], in this study, nBMFs were cultured in medium containing 20 μg/mL arecoline for 24 hours. This treatment induced an increase in α-SMA expression (Figure 3D and Supplementary Figure 1), contractility (Figure 3E), and cell migration (Figure 3F), collectively confirming the acquisition of myofibroblast-like properties. As expected, the silencing of MIAT in the arecoline-treated nBMFs (Figure 3C) successfully attenuated the expression of α-SMA (Figure 3D), and significantly reduced both collagen gel contractility (Figure 3E) and Transwell migration capacity (Figure 3F). These results implied that downregulation of MIAT may prevent myofibroblast transformation and potentially mitigate the progression of OSF.
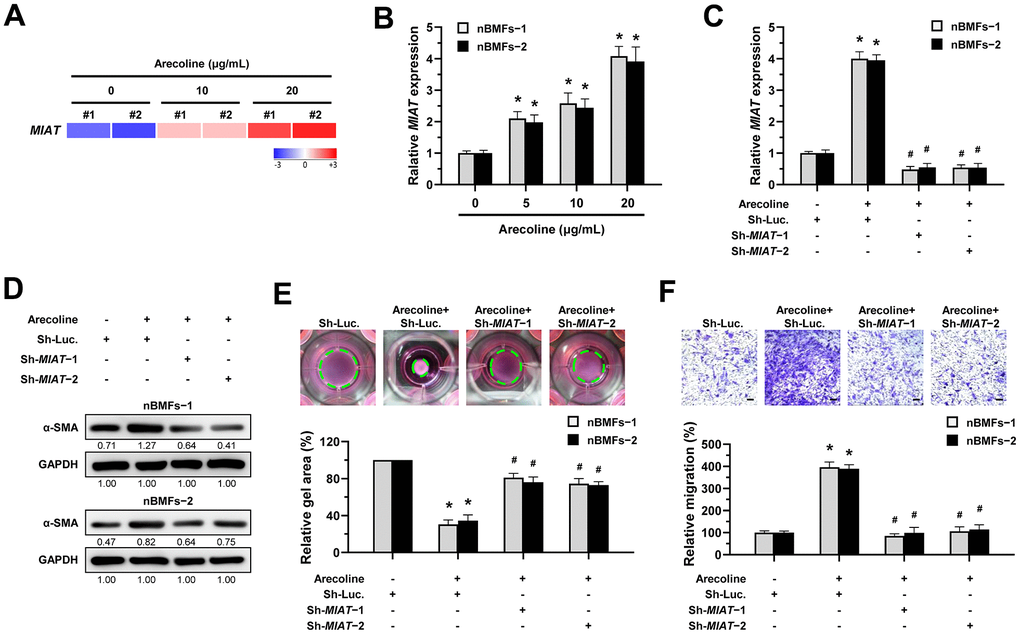
Figure 3. Knockdown of MIAT impairs the arecoline-induced myofibroblastic transformation in BMFs. (A) Primary nBMFs (obtained from two healthy individuals; nBMFs−1 and −2) were cultured with arecoline (0, 10, and 20 μg/mL) for 24 hours, followed by RNA-sequencing analysis to determine the levels of MIAT (p < 0.01). (B) Normal BMFs (−1 and −2) were cultured with arecoline (0, 5, 10, and 20 μg/mL) for 24 hours, followed by qRT-PCR analysis to determine the MIAT expression. (C–F) Normal BMFs (−1 and −2) were transfected with lentiviruses expressing non-targeting ShRNA (Sh-Luc.) and Sh-MIAT (Sh-MIAT−1 and −2). After 48 hours, the cells were cultured with or without arecoline (20 μg/mL) for 24 hours for the induction of myofibroblasts transdifferentiation. The expression of MIAT in each group was assessed using qRT-PCR analysis. Data are presented as mean ± SD (n=3); *p < 0.05 vs. Sh-Luc.; #p < 0.05 vs. Sh-Luc. with arecoline treatment (C). The protein expression of α-SMA in each group was determined using Western blotting analysis (D). Cells (nBMFs−1 and −2) were cultured in collagen gel for an additional 48 hours, and the resulting gel area after cell contraction was measured (E). Cells (nBMFs−1 and −2) were cultured in Transwell system for an additional 24 hours, and their migration ability was quantified (F). Data are presented as mean ± SD (n=3); *p < 0.05 vs. Sh-Luc.; #p < 0.05 vs. Sh-Luc. with arecoline treatment; Scale bar, 50 μm (E, F).
MIAT promotes myofibroblastic properties by acting as a sponge of miR-342-3p
Recent studies focused on the biological roles of lncRNAs as miRNA sponges. Due to the miRNA-mediated post-transcriptional suppression of targeted mRNA occurs in the cytoplasm, determining the subcellular localization of MIAT is precedence. Herein, we showed that MIAT was preferentially located in the cytoplasm of fBMFs (Figure 4A). By using bioinformatics prediction tools, several putative miRNA binding sites contained in MIAT sequences were identified, including miR-342-3p. Since a recent study has demonstrated the interplay between MIAT and miR-342-3p in retinal pericytes [28] and miR-342-3p was downregulated in OSF tissues using RNA sequencing analysis (Figure 4B), we then investigated whether miR-342-3p mediated the fibrosis effect of MIAT. Bioinformatics analysis predicted that MIAT 3’UTR sequence had a potential binding region for miR-342-3p (Figure 4C). To confirm the significance of miR-342-3p in OSF, the qRT-PCR analysis was conducted to evaluate its expression in clinical specimens. As expected, miR-342-3p was significantly decreased in OSF tissues (Figure 4D). Notably, we also found a significantly negative correlation between MIAT expression and miR-342-3p levels in OSF tissues (r = − 0.931, p<0.05, Figure 4E). Results demonstrated that the miR-342-3p mimics evidently lowered the activity in wt-MIAT group but had no effect in mut-MIAT group (Figure 4F). Furthermore, we showed fBMFs with miR-342-3p mimics exhibited lower myofibroblast features, including collagen gel contraction, Transwell migration and wound healing abilities (Figure 4G–4I).
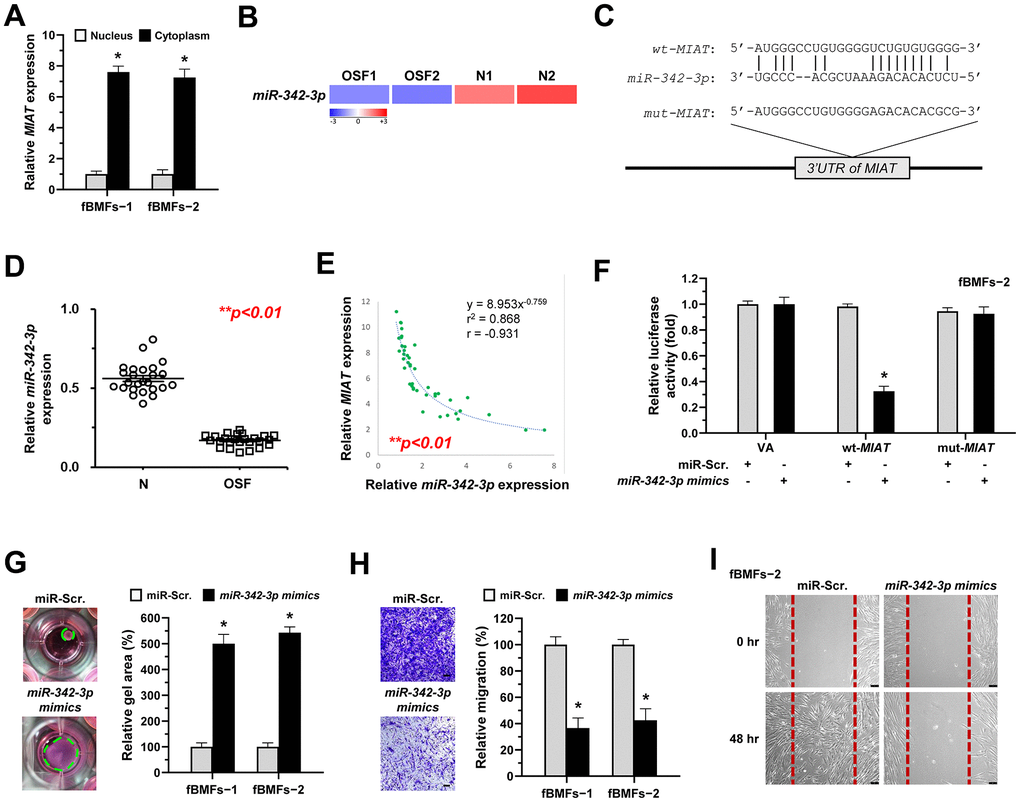
Figure 4. MiR-342-3p negatively correlates to MIAT expression in OSF tissues and acts as an anti-fibrotic miRNA in OSF. (A) RNA of cytoplasmic and nuclear fractions from primary fBMFs (−1 and −2) were analyzed by qRT-PCR to determine the subcellular localization of MIAT. Data are presented as mean ± SD (n=3); *p < 0.05 vs. Nucleus. (B) RNA-sequencing analysis showed that miR-342-3p was a down-regulated differentially expressed gene (fold change ≤ -2.0; p < 0.01) in samples of fibrotic tissues (OSF; n=2) compared to normal tissues (N; n=2). (C) An illustration of the predicted pairing region between miR-342-3p and MIAT 3’UTR, discovered using the miRanda database, and the 3’ UTR regions of full-length (wt-MIAT) and mutated MIAT (mut-MIAT) complementarity to the seed site of miR-342-3p, predicted by TargetScan in silico browser. (D) The relative expression of miR-342-3p in normal (N; n=25) and fibrotic (OSF; n=25) tissues was assessed by qRT-PCR analysis. Data are presented as mean ± SD. (E) A significant negative correlation was observed between MIAT and miR-342-3p expression fibrotic tissue samples (OSF; n=45). (F) Fibrotic BMFs (−1) were co-transfected with either miR-Scramble (miR-Src.) or miR-342-3p mimics, along with the indicated pmirGLO-based constructs shown in (C). Luciferase reporter activity was measured 24 hours post-transfection. Data are presented as mean ± SD (n = 3); *p < 0.05 vs. wt-MIAT with miR-Src. (G–I) Fibrotic BMFs (−1 and −2) expressing Sh-Luc or Sh-MIAT were transfected with either miR-Src or miR-342-3p inhibitor for 24 hours. The cells (fBMFs−1 and −2) were cultured in collagen gel for an additional 48 hours, and the resulting gel area after cell contraction was measured (G). Cells (fBMFs−1 and −2) were cultured in Transwell system for an additional 24 hours, and their migration ability was quantified (H). Data are presented as mean ± SD (n=3); *p < 0.05 vs. miR-Scr. (G, H). Confluent monolayers of fBMF−2 were scratched and cultured for an additional 48 hours, and the wound closure was assessed (I). Scale bar, 50 μm.
Given that miR-342-3p is negatively correlated with MIAT and that adding this miRNA to fBMFs inhibited myofibroblast differentiation, it is crucial to investigate whether MIAT directly sponges miR-342-3p in its exertion of fibrotic trait. Since the evasion of apoptosis by myofibroblasts is a hallmark of fibrotic disorders, understanding this interaction is essential [10]. Thus, we measured the percentage of apoptotic cells in fBMFs infected with MIAT shRNA along with miR-342-3p inhibitor. Our results showed that transfection of miR-342-3p inhibitor into fBMFs successfully avoided cell apoptosis induced by silencing of MIAT (Figure 5A). Similarly, miR-342-3p inhibitor counteracted the effect of silencing of MIAT on collagen gel contraction (Figure 5B), protein expression of α-SMA and COL1A1 (Figure 5C and Supplementary Figure 2), and cell migration (Figure 5D). Taken together, these data indicate that MIAT functions as a miR-342-3p sponge to promote myofibroblast properties in fBMFs.
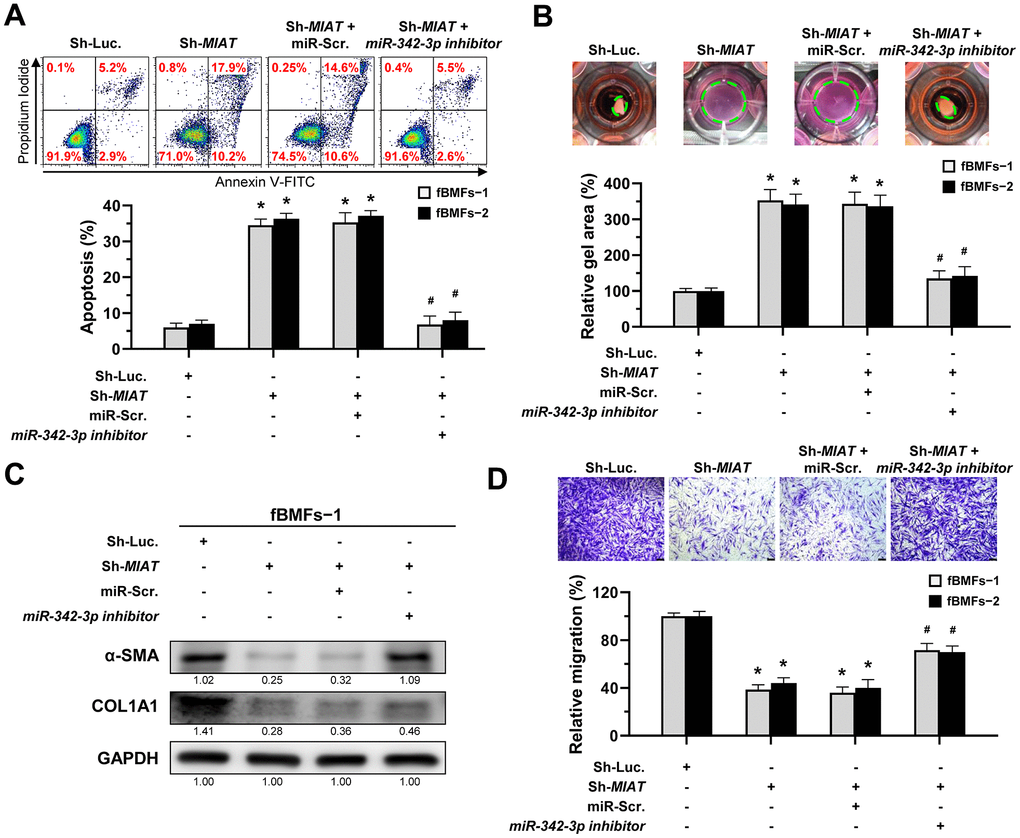
Figure 5. The silencing of MIAT induces apoptosis and inhibits myofibroblastic properties by targeting miR-342-3p. (A–D) Fibrotic BMFs (fBMFs−1 and −2) expressing the Sh-Luc. or Sh-MIAT were transfected with either miR-Scramble (miR-Src.) or miR-342-3p inhibitor for 24 hours. Cell apoptosis (annexin V+ or annexin V+/PI+) was assessed using flow cytometry (A). Cells (fBMFs−1 and −2) were cultured in collagen gel for an additional 48 hours, followed by the measurement of the gel area after cell contraction (B). Data are presented as mean ± SD (n=3); *p < 0.05 vs. Sh-Luc.; #p < 0.05 vs. Sh-MIAT with miR-Src. (A, B). The protein expression of α-SMA and COL1A1 in fBMFs−1 was analyzed using Western blotting (C). Cells (fBMFs−1 and −2) were cultured in Transwell system for an additional 24 hours, and their migration ability was quantified. Data are presented as mean ± SD (n=3); *p < 0.05 vs. Sh-Luc.; #p < 0.05 vs. Sh-MIAT with miR-Src.; Scale bar, 50 μm (D).
MiR-342-3p mitigates myofibroblast activation by suppressing SOX6
Several studies have shown that SRY-box transcription factor 6 (SOX6) may be a target of miR-342-3p in renal or cardiac injuries [29, 30] and implicate in renal fibrosis [29, 31], so we sought to examine whether SOX6 plays a role for the anti-fibrosis effect of miR-342-3p during oral fibrogenesis. By using RNA sequencing analysis, we showed that SOX6 was upregulated in OSF tissues compared to normal specimens (Figure 6A). We also predicted a potential miR-342-3p binding region on the SOX6 3’UTR sequence via miRDB bioinformatic analysis (Figure 6B), and the results from a luciferase assay showed that the luciferase activity was significantly reduced in wild-type SOX6 in nBMFs (wt-SOX6), while there was no change in mutated SOX6 (mut-SOX6, Figure 6C). Besides, the expression of SOX6 in fBMFs was downregulated after transfection of miR-342-3p mimics (Figure 6D and Supplementary Figure 3). Additionally, we showed that overexpression of SOX6 in nBMFs (Figure 6E and Supplementary Figure 4) elicited multiple myofibroblast phenotypes, including higher collagen gel contractility (Figure 6F), Transwell migration (Figure 6G) and wound healing (Figure 6H) capacities. These results supported that miR-342-3p suppresses myofibroblastic properties by targeting SOX6.
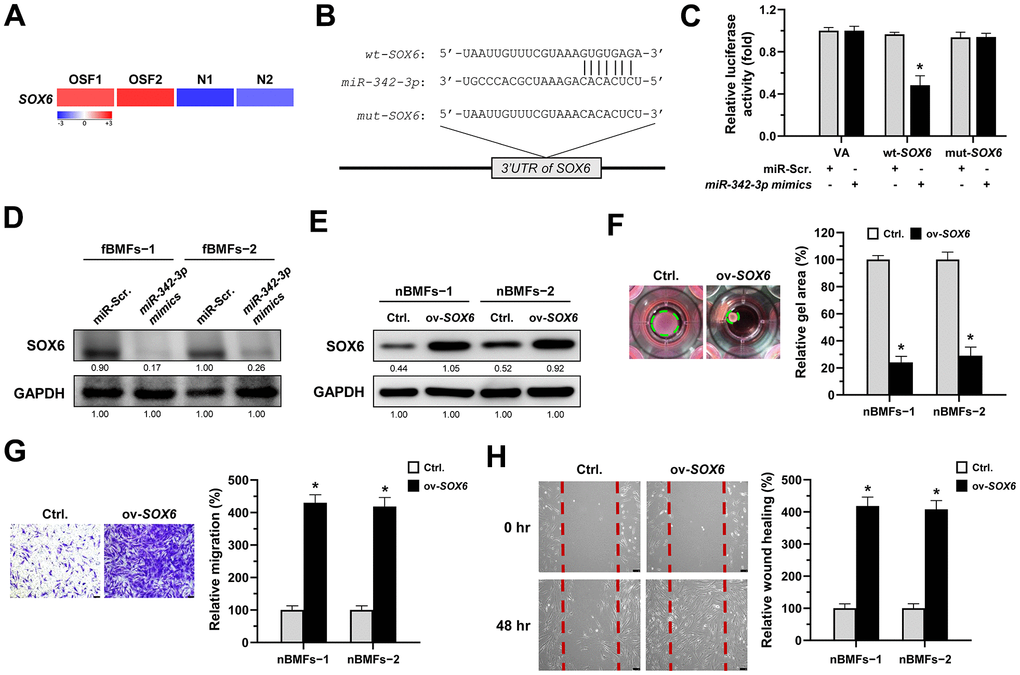
Figure 6. SOX6 is a target of miR-342-3p. (A) RNA-sequencing analysis showed that SOX6 was an up-regulated differentially expressed gene (fold change ≥ 2.0; p < 0.01) in fibrotic tissues samples (OSF; n=2) compared to normal tissues samples (N; n=2). (B) An illustration of the predicted pairing region between miR-342-3p and SOX6 3’UTR were discovered using the miRDB database, and the 3’ UTR regions of full-length (wt-SOX6) and mutated SOX6 (mut-SOX6) complementarity to the seed site of miR-342-3p, predicted by the TargetScan in silico browser. (C) Fibrotic BMFs (−1) were co-transfected with either miR-Scramble (miR-Src.) or miR-342-3p mimics, along with the indicated pmirGLO-based constructs shown in (B). Luciferase reporter activity was measured 24 hours post-transfection. Data are presented as mean ± SD (n = 3); *p < 0.05 vs. wt-SOX6 with miR-Src. (D) Fibrotic BMFs (−1 and −2) were transfected with either miR-Src or miR-342-3p inhibitor for 24 hours, followed by Western blotting analysis to determine the protein expression of SOX6. (E–H) Normal BMFs (−1 and −2) were transfected with lentiviruses expressing control vector (Ctrl.) or SOX6 (ov-SOX6). The overexpression efficiency of SOX6 was assessed by Western blotting analysis (E). Cells (nBMFs−1 and −2) were cultured in collagen gel for an additional 48 hours, and the resulting gel area after cell contraction was measured (F). Cells (nBMFs−1 and −2) were cultured in Transwell system for an additional 24 hours, and their migration ability was quantified (G). Confluent monolayers of cells (nBMFs−1 and −2) were scratched and cultured for an additional 48 hours, and the wound closure was assessed (H). Data are presented as mean ± SD (n=3); *p < 0.05 vs. Ctrl. (F–H). Scale bar, 50 μm.
MIAT promotes myofibroblastic properties through the miR-342-3p/SOX6 axis
According to the above findings that miR-342-3p can bind to MIAT and SOX6 mRNA, we speculated that MIAT may serve as a ceRNA to regulate the miR-342-3p/SOX6 axis during OSF progression. To verify this hypothesis, we assessed the relationship between MIAT and SOX6 and found they were positively correlated (Figure 7A). Furthermore, our results demonstrated that overexpression of SOX6 blocked the effect of MIAT silencing on apoptosis (Figure 7B), collagen gel contraction (Figure 7C), Transwell migration (Figure 7D) and the expression of fibrosis markers (Figure 7E and Supplementary Figure 5). Altogether, our data indicate that upregulation of MIAT in nBMFs following chronic exposure to arecoline may interfere the miR-342-3p-mediated suppression of SOX6, resulting in persistent activation of fBMFs and development of OSF (Figure 8).
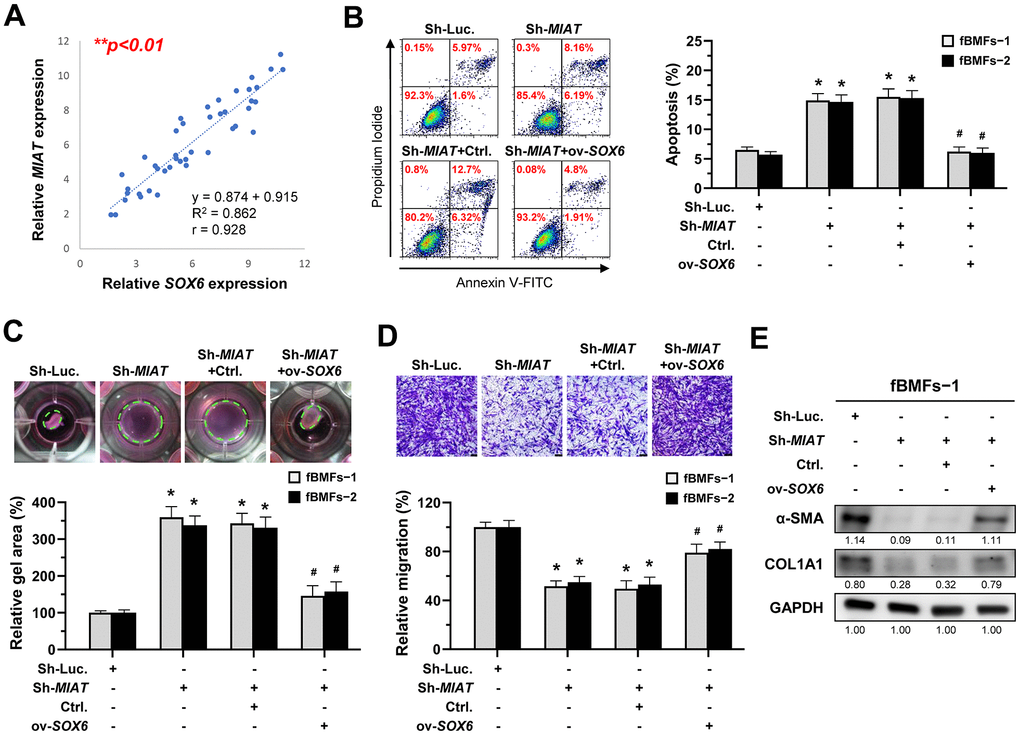
Figure 7. MIAT increases the myofibroblastic properties by positively regulating SOX6. (A) A significant positive correlation between the expression of MIAT and SOX6 in fibrotic tissue sample (OSF; n=43). (B–E) Fibrotic BMFs (−1 and −2) were co-transfected with lentiviruses expressing the following constructs in the indicated combinations: non-targeting ShRNA (Sh-Luc.), Sh-MIAT, control vector (Ctrl.), and SOX6 (ov-SOX6). Cell apoptosis (annexin V+ or annexin V+/PI+) was determined using flow cytometry (B). Cells (fBMFs−1 and −2) were cultured in collagen gel for an additional 48 hours. The resulting gel area after cell contraction was measured (C). Cells were cultured in Transwell system for an additional 24 hours, and their migration ability was assessed. Scale bar, 50 μm (D). Data are presented as mean ± SD (n=3); *p < 0.05 vs. Sh-Luc.; #p < 0.05 vs. Sh-MIAT with Ctrl. (B–D). The protein expression of α-SMA and COL1A1 in fBMFs−1 was analyzed using Western blotting (E).
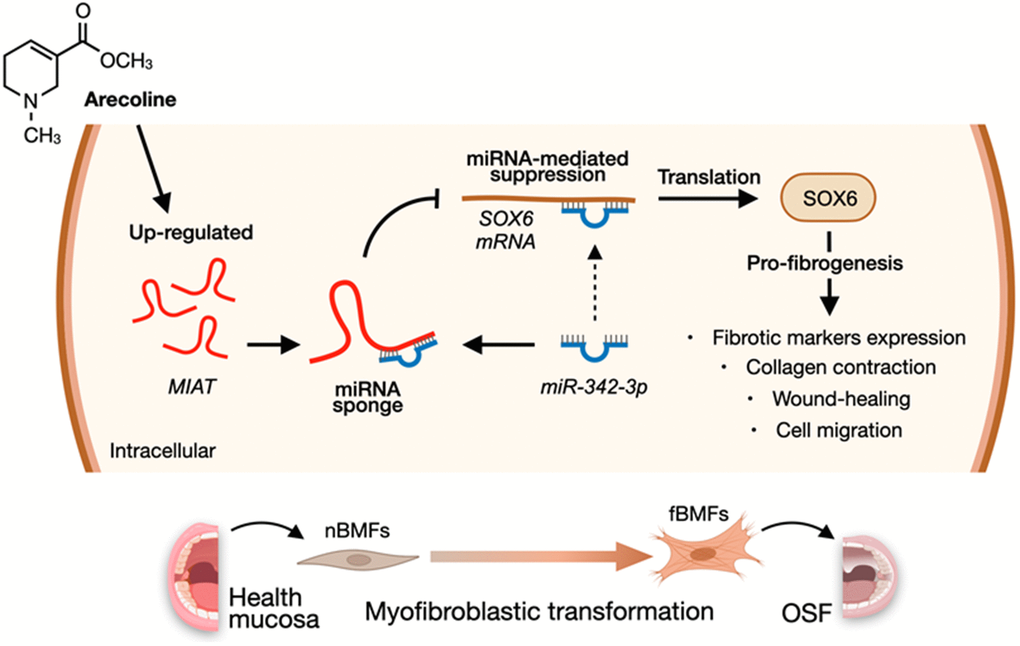
Figure 8. A diagram illustrates the mechanism of MIAT in the development of OSF. Upregulation of MIAT in nBMFs following chronic exposure to arecoline may interfere the miR-342-3p-mediated suppression of SOX6, resulting in persistent activation of fBMFs and development of OSF.
Discussion
MIAT, also termed as Gomafu or LINC00066, is located at 22q12.1 with a length of 30,051 bp and was first identified in 2000 [32]. Ishii et al. later found MIAT is located within a susceptible locus for myocardial infarction (MI), leading them to name this novel gene as MIAT [17]. Beyond its role in cardiovascular diseases [33–35], MIAT dysregulation has been associated with a variety of disorders, such as impaired neuronal function [36] and glioblastoma [37] and various fibrosis diseases. For instance, MIAT has been shown to modulate cardiac fibrosis by serving as a ceRNA and negatively regulating multiple miRNAs, such as miR-29a-3p [38], miR-24 [39], or miR-214-3p [40]. Moreover, TGF-β1-induced MIAT contributes to the activation of pancreatic stellate cells in chronic pancreatitis and proximal tubule epithelial cells in renal interstitial fibrosis by targeting miR-216a-3p/COX-2 [22] or miR-145/EIF5A2 axes [41], respectively. In this study, we showed that MIAT was upregulated in OSF specimens, and the silencing of MIAT reduced myofibroblast phenotypes and activation. Our results further demonstrated that the arecoline-stimulated MIAT conferred myofibroblast transdifferentiation of nBMFs by acting as a ceRNA for miR-342-3p, thereby alleviating its repression on SOX6.
Aberrant expression of miR-342 has been found in various diseases, such as diabetic nephropathy [29, 42] or cardiomyopathy [43]. Unlike most miRNAs where only one guide strand is loaded into RNA-induced silencing complex (RISC) while the other strand is destroyed rapidly, both miR-342-3p and miR-342-5p are excised from the same stem-loop precursor miRNA and become mature miRNAs [44]. It has been revealed that miR-342-3p acts as a tumor suppressor in oral cancer by inhibiting LIM and SH3 protein 1 [45], and our results suggested that dysregulation of miR-342-3p also participated in the development of precancerous OSF. Several studies have demonstrated the involvement of miR-342 in numerous fibrosis diseases by influencing myofibroblast activation. For example, miR-342-3p was found to regulate hepatic stellate cell (HSC) activation by affecting the Zbtb7a-mediated TGF-β signaling in a model of Echinococcus multilocularis infected liver fibrosis [46]. Another study showed that miR-342 directly interacted with Sp1 and inhibited its downstream TGF-β1/Smad signaling, leading to the inhibition of HSCs activation [47]. In renal fibrosis, miR-342-5p has been revealed to target Ptch1 and inhibit its transcription factor FoxO3, leading to autophagy in the TGF-β1-stimulated TCMK-1 (mouse kidney) cells [48]. MiR-342-3p also has been demonstrated to increase cell proliferation and inhibit apoptosis of renal mesangial cells by reducing SOX6 expression [29]. In line with this finding, we showed that miR-342-3p exhibited anti-fibrosis properties in fBMFs, possibly through direct suppression of SOX6.
SOX6 is a transcription factor that was discovered in 1990s [49, 50] and belongs to the SOXD subfamily along with SOX5 and SOX13 [51]. In humans, SOX6 and SOX5 are located in paralogous chromosomal regions on 11p15.3–15.2 and 12p12.1, respectively. Although SOX6 do not harbor any transactivation or transrepression domains, it has been found to bind to various proteins, cofactors, and miRNAs to regulate the transcription and functions of multiple genes [52–54]. Several studies suggested that differential expression of SOX6 may account for numerous diseases, such as cardiomyopathy [54, 55]. In fact, emerging evidence demonstrated that SOX6 serves as a target for a number of miRNAs in renal fibrosis such as miR-342-3p [29], miR-19b [56], and miR-185-5p [31]. Notably, these studies showed that miRNA-mediated sponging of SOX6 promotes cell apoptosis and reduces the expression of fibrosis markers like fibronectin and type I collagen [29, 31]. Our results are consistent with these findings, showing that SOX6 suppresses apoptosis in myofibroblasts, thereby increasing fibrosis markers. Furthermore, we demonstrated that manipulation of SOX6 affected myofibroblast marker (α-SMA) expression and phenotypes, which provided direct evidence that upregulation of SOX6 leads to oral fibrogenesis through modulation of myofibroblast transdifferentiation.
A limitation of this study is the inconsistency in sample sizes between the experiments analyzing gene expression correlations (e.g. Figure 1D–1F; n=40) and those comparing gene expression differences between OSF and normal (N) groups (e.g. Figure 1B; n=25). To minimize potential confounding variables, we utilized samples from patients with OSF and healthy individuals recruited during the same period (n=25 per group) when comparing the expression differences of target genes. To enhance the statistical significance of our results, we continued to recruit OSF patients, expanding the sample size from 25 to 45. Our association analysis has indicated that the expression of MIAT strongly correlated to each gene of interest (Figures 1D–1F, 4E, 7A), suggesting that similar findings could be obtained from the smaller sample size (n=25). However, it is necessary to continue expanding both the number and diversity of patients, such as genetic background and clinical stage of disease progression. This would be beneficial to strengthen the evidence base of our study and to further elucidate the role of MIAT in OSF pathogenesis.
In conclusion, our findings revealed that the aberrantly overexpressed MIAT in OSF tissues may be due to chronic stimulation of arecoline, resulting in transdifferentiation of nBMFs via titrating the inhibitory effect of miR-342-3p on SOX6 expression. These results not only offered insight into how upregulated MIAT led to OSF, but also demonstrated that targeting this MIAT/miR-342-3p/SOX6 pathway may be a promising treatment direction.
Materials and Methods
Tissue preparation
45 samples of fibrotic buccal mucosa tissues (OSF) were obtained from surgical resection of OSF patients who did not receive any preoperative treatment at the Department of Dentistry, Chung Shan Medical University Hospital; and 25 samples of normal buccal mucosa tissues (N) were obtained from the surgical removal of impacted third molars of healthy individuals. All samples were obtained with the written informed consent of patients. For RNA-sequencing and qRT-PCR analysis, the obtained samples were immediately stored at -80° C before use. All ethical regulations and operations were complied with Institutional Review Board of Chung Shan Medical University Hospital. The histopathological identification of OSF samples was verified by two pathologists independently. Tissue specimens from 25 OSF patients and 25 healthy individuals, initially recruited during the same period, were used to analyze the statistical differences in the expression levels of various genes between the OSF and normal groups. Specimens from all recruited OSF patients (n = 45, including the initial 25 and an additional 20 subsequently recruited) were used to analyze the correlations among the expression levels of various genes of interest. Due to the limited availability of clinical tissue samples, the actual number of samples used in the experiments ranged from 40 to 45 (specific sample sizes are indicated in the respective figure legends).
Cell culture
Fibrotic buccal mucosa fibroblasts (fBMFs) and normal BMFs (nBMFs) were extracted from buccal mucosa tissues of OSF patients and healthy individuals, respectively. The details of primary cell isolation and culture were previously described [16]. In brief, buccal mucosa tissues obtained from surgery were incubated in Hanks’ Balanced Salt Solution (HBSS) at 4° C and transferred to the laboratory immediately for further processing. After trypsinization, the tissues were cultured in DMEM medium (with 10% fetal bovine serum [FBS], and 1% penicillin-streptomycin cocktail) and plated into 25-T flasks for 14 days. The cells which are spindle-shaped and migrated out of the tissues were defined as buccal fibroblasts. All cells were continuously passaged and used for subsequent experiments between the 3rd and 8th passages and were negative for mycoplasma contamination verified using short tandem repeat (STR) DNA profiling. Arecoline was purchased from Sigma (St. Louis, MO, USA). Arecoline was dissolved in phosphate-buffered saline (PBS) as a stock solution stored at -20° C before use. When nBMFs reached a designated density, cells were treated with arecoline at a series concentration (0-20 μg/mL) for 24 hours.
RNA-sequencing analysis
Total RNA was extracted from the collected buccal tissue samples and primary BMF cells using Trizol reagent as directed by the manufacturer (Invitrogen, Carlsbad, CA, USA). RNA quality and quantity were evaluated using NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA). All procedures of RNA library preparation, sequencing, and transcriptome discrepancies analysis were performed at Genomics Inc. The detail was described in our previous studies [57]. To verify the RNA-sequencing results, the selected differentially expressed gene expressions were further confirmed by qRT-PCR analysis.
QRT-PCR analysis
The methods of RNA extraction, quality control and quantitation are described above. For MIAT subcellular localization detection, the nuclear and cytoplasmic RNA from fBMFs were isolated using a PARIS™ Kit (Thermo Fisher Scientific). cDNA was prepared using The Superscript III first-strand synthesis system (Invitrogen) to reverse-transcribe RNA. ABI StepOne™ Real-Time PCR Systems (Applied Biosystems, Waltham, MA, USA) were used for PCR experiments with the resultant cDNAs. The specific primer sequences are listed as follows (5’-3’): MIAT, TATTTGCAGGGGGTGCTCTG (forward), GGGCAGGGGGTCTAACTCTA (reverse); α-SMA, AGCACATGGAAAAGATCTGGCACC (forward), TTTTCTCCCGGTTGGCCTTG (reverse); COL1A1, GATTCCCTGGACCTAAAGGTGC (forward), AGCCTCTCCATCTTTGCCAGCA (reverse); FN1, ACTGCGAGAGTAAACCTGAAGC (forward), GCGGTTTGCGATGGTACAGCT (reverse); miR-342-3p, GTGCTATCTGTGATTGAGGGA (forward), CGGGTGCGATTTCTGTG (reverse); SOX6, GTCGCTTAATGTGTGGCTCG (forward), TGTTCTTCCTGCCCTGACATT (reverse); GAPDH, GTGGCTGGCTCAGAAAAAGG (forward), GGGGAGATTCAGTGTGGTGG (reverse); U6, CTCGCTTCGGCAGCACA (forward), AACGCTTCACGAATTTGCGT (reverse). Relative expression levels were normalized using GAPDH as the internal control for total and cytoplasmic RNA, while U6 served as the internal control for nuclear RNA and miRNA normalization, and the 2-ΔCt method was applied.
Knockdown of MIAT
A lentiviral pLKO-shRNA-expressing vector was purchased from the National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). Lentivirus particle production was performed in 293T cells by co-transfecting the pLKO-shRNA-expressing vectors with packaging and envelope plasmids with a ratio of 10:10:1 using Lipofectamine 2000 reagent (Invitrogen). The virus-containing supernatant was collected after 48 post-transfection and filtered through a 0.45-μm PVDF filter. Fibrotic BMFs were cultured with the filtered virus-containing supernatant supplemented with 8 μg/mL polybrene (Merck, Darmstadt, Germany) for 48 hours. The lentivirus-infected cells were selected by puromycin. Quantitative RT-PCR analysis was performed to verify the knockdown efficiency. The sequences for Sh-MIAT are listed below: Sh-MIAT-1, AAAAGCAGTCCAGGGTCTATTTATTGGATCCAATAAATAGACCCTGGACTGC; Sh-MIAT-2, AAAAGGGTTTGAACCTTTAGGATTTGGATCCAAATCCTAAAGGTTCAAACCC. A pLKO-luciferase-expressing vector was used as the non-targeting control (Sh-Luc.).
Collagen gel contraction assay
A total of 0.5 mL of the collagen solution (2 mg collagen/mL) was mixed with 1 × 105 cells and added into each well of a 24−well plate. After gel polymerization by 30 minutes of incubation at 37° C, 0.5 mL DMEM medium was added to cover the polymerized gels entirely. Then the gels were scraped out and plated into another 24−well plate for 48 hours of incubation at 37° C. Images of the contracted collagen gels were captured and quantified using ImageJ software (NIH). The area of the contracted gel was measured and compared to the initial area to calculate the percentage of contraction. A decrease in relative gel area represents an increase in the gel contraction ability of cells.
Transwell migration assay
A total of 0.4 mL of a serum-free DMEM containing 1 × 105 cells was added into the upper 8-μm pore insert, then the lower chamber was filled with 0.6 mL DMEM containing 10% FBS. After incubation at 37° C for 24 hours, non-migrated cells on the upper side of the insert membrane were removed, and the migrated cells on the lower side were fixed with cold-100% methanol for 30 minutes at room temperature. The fixed cells were stained with 0.1% crystal violet solution for 20 minutes. Migrated cells were visualized by capturing images using a microscope. For the evaluation of migrated cells, 0.4 mL of 30% acetic acid was added to each insert and incubated for 15 minutes to dissolve the crystal violet stain. The absorbance value at 570 nm of crystal violet stain from each insert was measured using a microplate reader (Infinite M200 Pro, Tecan Group Ltd., Männedorf, Germany) and quantified according to the manufacturer’s protocols.
Wound healing assay
After cell confluence reached 90%, a straight wound was introduced across the center of each well using a 200 μL sterile pipette tip. Each well was washed with PBS to remove any detached cells and debris, and cells were cultured with a serum−free DMEM medium for 48 hours. Images of the wound area were captured at the time points of 0 and 48 hours by microscope.
Western blot analysis
In brief, whole-cell lysate containing 20 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto a polyvinylidene fluoride membrane. After blocking for non-specific sites with 5% bovine serum albumin (BSA), the membrane was incubated with primary antibodies for 16 hours at 4° C, and subsequently incubated with the corresponding HRP-conjugated secondary antibody for 1 hour at room temperature. The signals of antibody binding were developed by ECL substrate (Merck) and then captured using the LAS−1000 plus analyzer (GE Healthcare, Piscataway, NJ, USA). GAPDH was used as an internal reference. All the antibodies were purchased from Cell Signaling Inc. (Danvers, MA, USA).
MiR-342-3p knockdown and overexpression
MiR-342-3p mimics, miR-342-3p inhibitor, and miR-scramble negative control (miR-Scr.) were purchased from Applied Biosystems (Waltham, MA, USA), and a total of 100 nM oligonucleotide sequences were transfected into cells using Lipofectamine 2000 (Invitrogen). After 48 hours of transfection, cells were collected for subsequent experiments.
Luciferase reporter activity assay
The binding of miR-342-3p to MIAT or SOX6 was examined using a luciferase reporter activity assay (Promega, Madison, WI, USA). MIAT and SOX6 3’-UTR with full-length (wild-type, wt) and mutant-type (mut) were synthesized and cloned into pmirGLO vectors according to the manufacturer’s protocols. Luciferase vectors were mixed with miR-342-3p mimics and miR-scramble (miR-Src.), respectively, and the mixtures were then co-transfected into cells using Lipofectamine 2000 reagent. The activity of luciferase reporter was detected using the Luciferase Assay System according to the manufacturer’s instructions. The relative activity of each group was normalized with Renilla luciferase internal control (VA).
Apoptosis assay
Briefly, 2 × 105 cells were incubated with stained with 1.5 μl Annexin V-FITC reagent and 1.5 μl propidium iodide (PI) for 5 minutes at room temperature in the dark. The stained cells were then directly measured using a Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Data were analyzed using FlowJo software v10 (FlowJo LLC, Ashland, OR, USA). The Annexin single-positive and Annexin/PI double-positive populations were considered apoptotic cells.
Overexpression of SOX6
The SOX6 cDNA was cloned into the pCDHI-MCS1-EF1-CopGFP plasmid (System Biosciences, Mountain View, CA, USA). The pCDH and two helper plasmids (packaging and envelope plasmids) were co-transfected into 293T cells using Lipofectamine 2000 reagent to produce lentiviral particles. The conditions for lentivirus production and infection were as described in the Knockdown of MIAT section. Following lentivirus infection, GFP-positive cells were sorted using flow cytometry. Overexpression of SOX6 was confirmed using qRT-PCR and Western blot analysis. An empty pCDHI-MCSI-EF1-COpGFP vector was used as the control group (Ctrl.).
Statistical analysis
All results were plotted as mean ± standard deviation (S.D.). A p-value of 0.05 or less was considered statistically significant. Differences between two groups were analyzed using paired or unpaired Student’s t-test, while differences among multiple groups were analyzed using one-way analysis of variance (ANOVA). The correlation between two genes’ expression in clinical specimens was obtained using Pearson correlation analysis. All analyses were performed using Prism software, version 9.0 (Graph-Pad Inc., La Jolla, CA, USA) and SPSS software, version 21.0 (SPSS Inc., Chicago, IL, USA).
Supplementary Materials
Author Contributions
C-C. Yu, and C-Y. Ho contributed study concept and critical design, M-Y. Lu, C-Y. Fang, and P-L. Hsieh conducted the experiments. S-C. Chao acquired, analyzed, and interpreted data. Y-W. Liao fulfilled the initial manuscript and Y. Ohiro critically reviewed and revised the final manuscript. All authors were involved in the current study and contributed to this study. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Statement and Consent
This study was performed in accordance with the Declaration of Helsinki and was approved by the Chung Shan Medical University Hospital (IRB approval no. CSMUH No: CS2-22044). All enrolled patients signed informed consent forms.
Funding
This study is supported by grants from the Chung Shan Medical University Hospital (grant number: CSH-2024-C-064) in Taiwan; Taipei Medical University (grant number: TMU112-AE1-B15) in Taiwan; National Science and Technology Council/Ministry of Science and Technology (grant number: MOST 111-2314-B-040-020-MY3) in Taiwan.
References
- 1. Pindborg JJ, Murti PR, Bhonsle RB, Gupta PC, Daftary DK, Mehta FS. Oral submucous fibrosis as a precancerous condition. Scand J Dent Res. 1984; 92:224–9. https://doi.org/10.1111/j.1600-0722.1984.tb00883.x [PubMed]
- 2. Hsue SS, Wang WC, Chen CH, Lin CC, Chen YK, Lin LM. Malignant transformation in 1458 patients with potentially malignant oral mucosal disorders: a follow-up study based in a Taiwanese hospital. J Oral Pathol Med. 2007; 36:25–9. https://doi.org/10.1111/j.1600-0714.2006.00491.x [PubMed]
- 3. Kujan O, Mello FW, Warnakulasuriya S. Malignant transformation of oral submucous fibrosis: A systematic review and meta-analysis. Oral Dis. 2021; 27:1936–46. https://doi.org/10.1111/odi.13727 [PubMed]
- 4. Angadi PV, Rao SS. Areca nut in pathogenesis of oral submucous fibrosis: revisited. Oral Maxillofac Surg. 2011; 15:1–9. https://doi.org/10.1007/s10006-010-0219-8 [PubMed]
- 5. Patil S, Halgatti V, Maheshwari S, Santosh BS. Comparative study of the efficacy of herbal antioxdants oxitard and aloe vera in the treatment of oral submucous fibrosis. J Clin Exp Dent. 2014; 6:e265–70. https://doi.org/10.4317/jced.51424 [PubMed]
- 6. Anuradha A, Patil B, Asha VR. Evaluation of efficacy of aloe vera in the treatment of oral submucous fibrosis - a clinical study. J Oral Pathol Med. 2017; 46:50–5. https://doi.org/10.1111/jop.12463 [PubMed]
- 7. Patil P, Hazarey V, Chaudhari R, Nimbalkar-Patil S. Clinical Efficacy of a Mouth-Exercising Device Adjunct to Local Ointment Intra-Lesional Injections and Surgical Treatment for Oral Submucous Fibrosis: a Randomized Controlled Trial. Asian Pac J Cancer Prev. 2016; 17:1255–9. https://doi.org/10.7314/apjcp.2016.17.3.1255 [PubMed]
- 8. Bande C, Dawane P, Gupta MK, Gawande M, Rode V. Immediate versus delayed aggressive physical therapy following buccal fat pad interposition in oral submucous fibrosis-a prospective study in Central India. Oral Maxillofac Surg. 2016; 20:397–403. https://doi.org/10.1007/s10006-016-0580-3 [PubMed]
- 9. Angadi PV, Kale AD, Hallikerimath S. Evaluation of myofibroblasts in oral submucous fibrosis: correlation with disease severity. J Oral Pathol Med. 2011; 40:208–13. https://doi.org/10.1111/j.1600-0714.2010.00995.x [PubMed]
- 10. Hinz B, Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol. 2020; 16:11–31. https://doi.org/10.1038/s41584-019-0324-5 [PubMed]
- 11. Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. 2013; 229:298–309. https://doi.org/10.1002/path.4104 [PubMed]
- 12. Chang YC, Tsai CH, Lai YL, Yu CC, Chi WY, Li JJ, Chang WW. Arecoline-induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated by ZEB1. J Cell Mol Med. 2014; 18:698–708. https://doi.org/10.1111/jcmm.12219 [PubMed]
- 13. Zhang Y, Luo G, Zhang Y, Zhang M, Zhou J, Gao W, Xuan X, Yang X, Yang D, Tian Z, Ni B, Tang J. Critical effects of long non-coding RNA on fibrosis diseases. Exp Mol Med. 2018; 50:e428. https://doi.org/10.1038/emm.2017.223 [PubMed]
- 14. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011; 43:904–14. https://doi.org/10.1016/j.molcel.2011.08.018 [PubMed]
- 15. Lee YH, Liao YW, Lu MY, Hsieh PL, Yu CC. LINC00084/miR-204/ZEB1 Axis Mediates Myofibroblastic Differentiation Activity in Fibrotic Buccal Mucosa Fibroblasts: Therapeutic Target for Oral Submucous Fibrosis. J Pers Med. 2021; 11:707. https://doi.org/10.3390/jpm11080707 [PubMed]
- 16. Yu CC, Liao YW, Hsieh PL, Chang YC. Targeting lncRNA H19/miR-29b/COL1A1 Axis Impedes Myofibroblast Activities of Precancerous Oral Submucous Fibrosis. Int J Mol Sci. 2021; 22:2216. https://doi.org/10.3390/ijms22042216 [PubMed]
- 17. Ishii N, Ozaki K, Sato H, Mizuno H, Susumu Sa, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, Satoshi Sa, Nakamura Y, Tanaka T. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. 2006; 51:1087–99. https://doi.org/10.1007/s10038-006-0070-9 [PubMed]
- 18. Da CM, Gong CY, Nan W, Zhou KS, Wu ZL, Zhang HH. The role of long non-coding RNA MIAT in cancers. Biomed Pharmacother. 2020; 129:110359. https://doi.org/10.1016/j.biopha.2020.110359 [PubMed]
- 19. Zhong W, Xu Z, Wen S, Xie T, Wang F, Wang Q, Chen J. Long non-coding RNA myocardial infarction associated transcript promotes epithelial-mesenchymal transition and is an independent risk factor for poor prognosis of tongue squamous cell carcinoma. J Oral Pathol Med. 2019; 48:720–7. https://doi.org/10.1111/jop.12892 [PubMed]
- 20. Yang L, Deng J, Ma W, Qiao A, Xu S, Yu Y, Boriboun C, Kang X, Han D, Ernst P, Zhou L, Shi J, Zhang E, et al. Ablation of lncRNA Miat attenuates pathological hypertrophy and heart failure. Theranostics. 2021; 11:7995–8007. https://doi.org/10.7150/thno.50990 [PubMed]
- 21. Bijkerk R, Au YW, Stam W, Duijs JM, Koudijs A, Lievers E, Rabelink TJ, van Zonneveld AJ. Long Non-coding RNAs Rian and Miat Mediate Myofibroblast Formation in Kidney Fibrosis. Front Pharmacol. 2019; 10:215. https://doi.org/10.3389/fphar.2019.00215 [PubMed]
- 22. Liu H, Yu K, Ma P, Xiong L, Wang M, Wang W. Long noncoding RNA myocardial infarction-associated transcript regulated the pancreatic stellate cell activation to promote the fibrosis process of chronic pancreatitis. J Cell Biochem. 2019; 120:9547–55. https://doi.org/10.1002/jcb.28231 [PubMed]
- 23. Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001; 12:2730–41. https://doi.org/10.1091/mbc.12.9.2730 [PubMed]
- 24. Ignotz RA, Massagué J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986; 261:4337–45. [PubMed]
- 25. Fang CY, Chen SH, Huang CC, Liao YW, Chao SC, Yu CC. Fucoidan-Mediated Inhibition of Fibrotic Properties in Oral Submucous Fibrosis via the MEG3/miR-181a/Egr1 Axis. Pharmaceuticals (Basel). 2022; 15:833. https://doi.org/10.3390/ph15070833 [PubMed]
- 26. Yu CC, Yu CH, Chang YC. Aberrant SSEA-4 upregulation mediates myofibroblast activity to promote pre-cancerous oral submucous fibrosis. Sci Rep. 2016; 6:37004. https://doi.org/10.1038/srep37004 [PubMed]
- 27. Yu CC, Tsai CH, Hsu HI, Chang YC. Elevation of S100A4 expression in buccal mucosal fibroblasts by arecoline: involvement in the pathogenesis of oral submucous fibrosis. PLoS One. 2013; 8:e55122. https://doi.org/10.1371/journal.pone.0055122 [PubMed]
- 28. Yu X, Ma X, Lin W, Xu Q, Zhou H, Kuang H. Long noncoding RNA MIAT regulates primary human retinal pericyte pyroptosis by modulating miR-342-3p targeting of CASP1 in diabetic retinopathy. Exp Eye Res. 2021; 202:108300. https://doi.org/10.1016/j.exer.2020.108300 [PubMed]
- 29. Jiang ZH, Tang YZ, Song HN, Yang M, Li B, Ni CL. miRNA-342 suppresses renal interstitial fibrosis in diabetic nephropathy by targeting SOX6. Int J Mol Med. 2020; 45:45–52. https://doi.org/10.3892/ijmm.2019.4388 [PubMed]
- 30. Wang B, Cao C, Han D, Bai J, Guo J, Guo Q, Li D, Zhang J, Zhang Z, Wang Y, Tang J, Shen D, Zhang J. Dysregulation of miR-342-3p in plasma exosomes derived from convalescent AMI patients and its consequences on cardiac repair. Biomed Pharmacother. 2021; 142:112056. https://doi.org/10.1016/j.biopha.2021.112056 [PubMed]
- 31. Li G, Qin Y, Qin S, Zhou X, Zhao W, Zhang D. Circ_WBSCR17 aggravates inflammatory responses and fibrosis by targeting miR-185-5p/SOX6 regulatory axis in high glucose-induced human kidney tubular cells. Life Sci. 2020; 259:118269. https://doi.org/10.1016/j.lfs.2020.118269 [PubMed]
- 32. Ohnishi Y, Tanaka T, Yamada R, Suematsu K, Minami M, Fujii K, Hoki N, Kodama K, Nagata S, Hayashi T, Kinoshita N, Sato H, Sato H, et al. Identification of 187 single nucleotide polymorphisms (SNPs) among 41 candidate genes for ischemic heart disease in the Japanese population. Hum Genet. 2000; 106:288–92. https://doi.org/10.1007/s004390051039 [PubMed]
- 33. Fasolo F, Jin H, Winski G, Chernogubova E, Pauli J, Winter H, Li DY, Glukha N, Bauer S, Metschl S, Wu Z, Koschinsky ML, Reilly M, et al. Long Noncoding RNA MIAT Controls Advanced Atherosclerotic Lesion Formation and Plaque Destabilization. Circulation. 2021; 144:1567–83. https://doi.org/10.1161/CIRCULATIONAHA.120.052023 [PubMed]
- 34. Vausort M, Wagner DR, Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ Res. 2014; 115:668–77. https://doi.org/10.1161/CIRCRESAHA.115.303836 [PubMed]
- 35. Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, Tao ZF, Song YC, Chen Q, Jiang Q. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015; 116:1143–56. https://doi.org/10.1161/CIRCRESAHA.116.305510 [PubMed]
- 36. Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS, Nayler SP, Nones K, Hu J, Bredy TW, Nakagawa S, Rigo F, Taft RJ, et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry. 2014; 19:486–94. https://doi.org/10.1038/mp.2013.45 [PubMed]
- 37. Zhang XQ, Sun S, Lam KF, Kiang KM, Pu JK, Ho AS, Lui WM, Fung CF, Wong TS, Leung GK. A long non-coding RNA signature in glioblastoma multiforme predicts survival. Neurobiol Dis. 2013; 58:123–31. https://doi.org/10.1016/j.nbd.2013.05.011 [PubMed]
- 38. Zhou J, Zhou Y, Wang CX. LncRNA-MIAT regulates fibrosis in hypertrophic cardiomyopathy (HCM) by mediating the expression of miR-29a-3p. J Cell Biochem. 2019; 120:7265–75; 120:7265–75. https://doi.org/10.1002/jcb.28001 [PubMed]
- 39. Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S, Yang T, Zhan L, Yuan Y, Chu W, Pan Z, Wang Z, Yang B, Lu Y. MIAT Is a Pro-fibrotic Long Non-coding RNA Governing Cardiac Fibrosis in Post-infarct Myocardium. Sci Rep. 2017; 7:42657. https://doi.org/10.1038/srep42657 [PubMed]
- 40. Qi Y, Wu H, Mai C, Lin H, Shen J, Zhang X, Gao Y, Mao Y, Xie X. LncRNA-MIAT-Mediated miR-214-3p Silencing Is Responsible for IL-17 Production and Cardiac Fibrosis in Diabetic Cardiomyopathy. Front Cell Dev Biol. 2020; 8:243. https://doi.org/10.3389/fcell.2020.00243 [PubMed]
- 41. Wang Z, Zhang B, Chen Z, He Y, Ru F, Liu P, Chen X. The long noncoding RNA myocardial infarction-associated transcript modulates the epithelial-mesenchymal transition in renal interstitial fibrosis. Life Sci. 2020; 241:117187. https://doi.org/10.1016/j.lfs.2019.117187 [PubMed]
- 42. Assmann TS, Recamonde-Mendoza M, de Souza BM, Bauer AC, Crispim D. MicroRNAs and diabetic kidney disease: Systematic review and bioinformatic analysis. Mol Cell Endocrinol. 2018; 477:90–102. https://doi.org/10.1016/j.mce.2018.06.005 [PubMed]
- 43. Florian A, Patrascu A, Tremmel R, Rösch S, Sechtem U, Schwab M, Schaeffeler E, Yilmaz A. Identification of Cardiomyopathy-Associated Circulating miRNA Biomarkers in Muscular Dystrophy Female Carriers Using a Complementary Cardiac Imaging and Plasma Profiling Approach. Front Physiol. 2018; 9:1770. https://doi.org/10.3389/fphys.2018.01770 [PubMed]
- 44. Wu H, Ye C, Ramirez D, Manjunath N. Alternative processing of primary microRNA transcripts by Drosha generates 5' end variation of mature microRNA. PLoS One. 2009; 4:e7566. https://doi.org/10.1371/journal.pone.0007566 [PubMed]
- 45. Song X, Jin Y, Yan M, Zhang Y, Chen B. MicroRNA-342-3p functions as a tumor suppressor by targeting LIM and SH3 protein 1 in oral squamous cell carcinoma. Oncol Lett. 2019; 17:688–96. https://doi.org/10.3892/ol.2018.9637 [PubMed]
- 46. Cao S, Wang D, Wu Y, Zhang J, Pu L, Luo X, Zhang X, Sun X, Zheng Y, Wang S, Guo X. mmu-miRNA-342-3p promotes hepatic stellate cell activation and hepatic fibrosis induced by Echinococcus multilocularis infection via targeting Zbtb7a. PLoS Negl Trop Dis. 2023; 17:e0011520. https://doi.org/10.1371/journal.pntd.0011520 [PubMed]
- 47. Shu B, Zhang RZ, Zhou YX, He C, Yang X. METTL3-mediated macrophage exosomal NEAT1 contributes to hepatic fibrosis progression through Sp1/TGF-β1/Smad signaling pathway. Cell Death Discov. 2022; 8:266. https://doi.org/10.1038/s41420-022-01036-y [PubMed]
- 48. Tang S, Wang Y, Xie G, Li W, Chen Y, Liang J, Liu P, Song F, Zhou J. Regulation of Ptch1 by miR-342-5p and FoxO3 Induced Autophagy Involved in Renal Fibrosis. Front Bioeng Biotechnol. 2020; 8:583318. https://doi.org/10.3389/fbioe.2020.583318 [PubMed]
- 49. Roose J, Korver W, Oving E, Wilson A, Wagenaar G, Markman M, Lamers W, Clevers H. High expression of the HMG box factor sox-13 in arterial walls during embryonic development. Nucleic Acids Res. 1998; 26:469–76. https://doi.org/10.1093/nar/26.2.469 [PubMed]
- 50. Connor F, Wright E, Denny P, Koopman P, Ashworth A. The Sry-related HMG box-containing gene Sox6 is expressed in the adult testis and developing nervous system of the mouse. Nucleic Acids Res. 1995; 23:3365–72. https://doi.org/10.1093/nar/23.17.3365 [PubMed]
- 51. Schepers GE, Teasdale RD, Koopman P. Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev Cell. 2002; 3:167–70. https://doi.org/10.1016/s1534-5807(02)00223-x [PubMed]
- 52. Leow SC, Poschmann J, Too PG, Yin J, Joseph R, McFarlane C, Dogra S, Shabbir A, Ingham PW, Prabhakar S, Leow MK, Lee YS, Ng KL, et al. The transcription factor SOX6 contributes to the developmental origins of obesity by promoting adipogenesis. Development. 2016; 143:950–61. https://doi.org/10.1242/dev.131573 [PubMed]
- 53. Yi Z, Cohen-Barak O, Hagiwara N, Kingsley PD, Fuchs DA, Erickson DT, Epner EM, Palis J, Brilliant MH. Sox6 directly silences epsilon globin expression in definitive erythropoiesis. PLoS Genet. 2006; 2:e14. https://doi.org/10.1371/journal.pgen.0020014 [PubMed]
- 54. Li X, Wang J, Jia Z, Cui Q, Zhang C, Wang W, Chen P, Ma K, Zhou C. MiR-499 regulates cell proliferation and apoptosis during late-stage cardiac differentiation via Sox6 and cyclin D1. PLoS One. 2013; 8:e74504. https://doi.org/10.1371/journal.pone.0074504 [PubMed]
- 55. Hagiwara N, Klewer SE, Samson RA, Erickson DT, Lyon MF, Brilliant MH. Sox6 is a candidate gene for p100H myopathy, heart block, and sudden neonatal death. Proc Natl Acad Sci USA. 2000; 97:4180–5. https://doi.org/10.1073/pnas.97.8.4180 [PubMed]
- 56. Xia WP, Chen X, Ru F, He Y, Liu PH, Gan Y, Zhang B, Li Y, Dai GY, Jiang ZX, Chen Z. Knockdown of lncRNA XIST inhibited apoptosis and inflammation in renal fibrosis via microRNA-19b-mediated downregulation of SOX6. Mol Immunol. 2021; 139:87–96. https://doi.org/10.1016/j.molimm.2021.07.012 [PubMed]
- 57. Peng CY, Liao YW, Lu MY, Yang CM, Hsieh PL, Yu CC. Positive Feedback Loop of SNAIL-IL-6 Mediates Myofibroblastic Differentiation Activity in Precancerous Oral Submucous Fibrosis. Cancers (Basel). 2020; 12:1611. https://doi.org/10.3390/cancers12061611 [PubMed]