



Introduction
The p53 gene encodes a transcription factor that regulates apoptosis and metabolism and is mutated in the majority of human cancers [1,2]. The p53 protein functions as a tetramer with various protein domains mediating oligomerization, DNA binding and transcriptional transactivation. Drosophila contains a single p53 gene with a structure similar to humans [3-6] including two promoters, and the major protein products are of similar size: 393 amino acid residues for the human protein, Hp53, and 385 amino acid residues for the Drosophila protein, Dmp53 (Drosophila protein diagrammed in Figure 1A). The central DNA binding domain of Dmp53 protein shows partial sequence conservation with Hp53 [3]. The other domains of Dmp53 show less obvious sequence similarity to Hp53, but appear conserved in function. Similar to the N-terminal transcriptional activation domain of Hp53, the N-terminus of Dmp53 contains a high proportion of acidic residues, and Dmp53 has been shown to bind to conserved p53 response elements and activate transcription [3]. The C-terminus of Hp53 contains a basic region (9/26 residues) that can bind either DNA or RNA, and the C-terminus of Dmp53 is also relatively basic (6/24 residues). Finally, the oligomerization domain is located in the C-terminal portion of Hp53, and the corresponding region of Dmp53 contains a conserved critical Gly "hinge" residue, and appears active in oligimerization based on yeast two hybrid assays. The p53 message is expressed at very low levels in adult tissues, with some enrichment indicated for the eye, malphigian tubule (similar to mammalian kidney), and female germ cells [7,8].
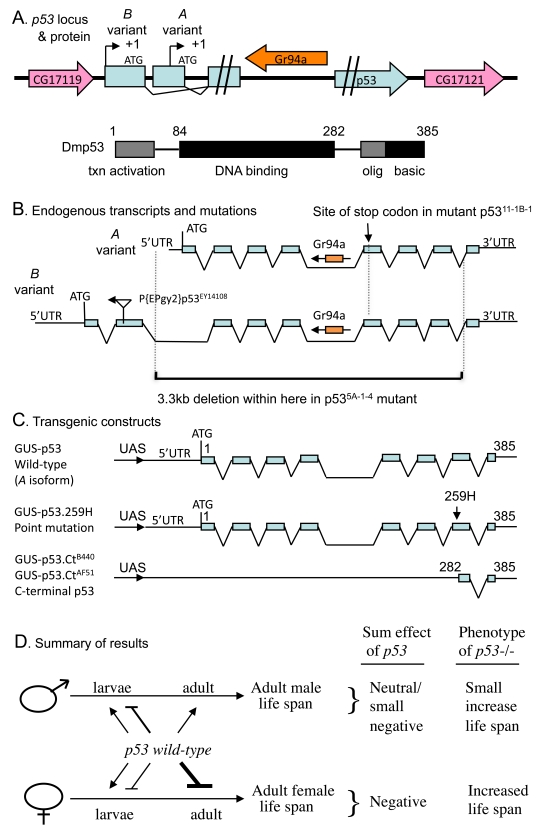
Figure 1. Summaryof Drosophila p53 locus, mutations, transgenes and life span effects. (A) Diagram of p53
locus and major protein product Dmp53. The p53 gene is indicated in
blue, including the two promoters, indicated by black arrows. The internal
intron/exon structure of p53 is omitted here for clarity, but is
shown below in (B). The pink arrows in indicate the genes that
flank p53 on the 5' and 3' side, genes CG17119 and CG17121,
respectively. The orange arrow indicates the gustatory receptor gene Gr94a,
located in the p53 intron. The 385 aa Dmp53 protein is diagrammed
using black and gray boxes, including the N-terminal transcriptional
activation domain, the central DNA binding domain, and the C-terminal
oligomerization domain and basic region. (B) Diagram of endogenous p53
transcripts and mutations. The intron/exon structure of the A and B variant
transcripts is indicated. The Gr94a gene is indicated in orange with an
arrow indicating orientation. The location of insertion of the P element P{EPgy2}p53EY14108
in the second exon of the B isoform is indicated by a triangle, with an
arrow indicating the orientation of the insert. The lower black bracket
indicates the breakpoints of the 3.3kb deletion in the p53[5A-1-4]
mutation. (C) Diagram of transgenic p53 constructs. (D)
Summary of p53 effects on adult life span. The effect on adult life
span of p53 wild type (A variant) over-expression during
larval development and in adults is diagrammed: Bars represent negative
effects of p53 wild-type on adult life span, while arrows represent
positive effects on adult life span; thickness of the lines indicates
relative strength of the effect. "Sum effect of p53" is the expected
summation of effects of p53 on adult life span, which is consistent
with the life span phenotype of p53 null mutation (p53-/-),
as indicated.
Mutant forms of p53 lacking function of a particular domain can have powerful dose-dependent effects that are often dependent upon the presence of wild-type p53 [3,9-11]. For example, specific truncated forms of mouse p53 can cause enhanced cancer resistance and accelerated aging phenotypes, generally interpreted as a state of p53 hyperactivation [12]. Based on studies in mammals it has been suggested that p53 may exhibit antagonistic pleiotropy between life-cycle stages, in that it favors normal development, fecundity and cancer resistance in young animals, but may promote aging in old animals [9,13-15]. Recently p53 gene activity was found to limit the life span of C. elegans hermaphrodites, and this effect was dependent upon the activity of the insulin/IGF1-like signaling (IIS) transcription factor gene Daf-16/FOXO [16]. In Drosophila, several dominant p53 mutations and transgenes have been characterized, that generally appear to antagonize p53 activity [3]. Nervous-tissue expression of one of these dominant p53 transgenes (p53 point mutation 259H) was found to inhibit IIS and extend life span in females [17,18]. However it remains unclear if and how p53 might normally affect the life span of Drosophila males and females. Here the wild-type form of p53, as well as mutant forms, were assayed for effects on Drosophila life span, in both male and female flies.
| Strain # | Genotype | Group (notes) |
| 2 | w[1118] ; + ; Df(3R)Exel6193, P{XP-U}Exel6193 /TM6B, Tb (BL7672) | - (Chromosomal Def uncovers p53) |
| 3 | y[1] w[1118] ; + ; p53[5A-1-4] (BL6815) | - (deletion of p53 gene) |
| 4 | y[1] w[1118] ; + ; p53[11-1B-1] (BL6816) | M (pt mutant) |
| 5 | w[1118] ; p53[1] / TM6B, Tb | M (pt mutant) |
| 6 | w[1118] ; + ; + | M (the same pt mutant as line 4) |
| 7 | Oregon R ( + ; + ; +) | + |
| 8 | y[1] w[67c23]; P{EPgy2}p53[EY14108] (BL 20906) | + |
| 9 | w ; P{Switch}Actin 255B | (GeneSwitch Act-GS-255B driver) |
| 16 | y[1]w[1118]; P{w[+mC]=UAS-p53.Ex}3/T(2;3)TSTL, CyO:TM6B, Tb | (UAS-p53 wild type) |
| 17 | w ; P{w[+mC]=GUS-p53}2.1 | (UAS-p53 wild type - CDM26) |
| 18 | w; P{w[+mC]=GUS-p53.Ct}AF51 | (C-terminal p53 - AF51) |
| 19 | w[1118]; +; P{w[+mC]=GUS-p53.Ct}B440/TM6B, Tb | (C-terminal p53 - B440) |
| 20 | w[1118]; P{w[+mC]=GUS-p53.259H} | (p53 point mutation - 259H) |
Results
Transgenic manipulation of p53 in adult flies
Drosophila p53 transgenes were assayed for effects on life span both in adults and during larval development (see below). The conditional transgenic system Geneswitch [19-21] was used to over-express both wild-type and mutant forms of p53. With the Geneswitch system transgene expression is triggered by feeding flies (or larvae) the drug RU486/Mifepristone. A Geneswitch driver strain called Act-GS-255B was used (Table 1, strain 9), where the tissue-general actin5C promoter drives expression of the Geneswitch transcription factor. In the presence of RU486, the Act- GS-255B driver produces expression of UAS-containing target constructs in all the tissues of either larvae or adults [19,22]: detailed characterization of the system using UAS-GFP reporter constructs demonstrates that the Act-GS-255B driver produces abundant transgene expression throughout all of the tissues of both adult flies and larvae, for both male and female animals, with slightly less (but still abundant) expression in adult males relative to females [22]. All of the flies examined in this study are the progeny of a cross; for example "16-9" flies are the progeny of a cross of males of strain 16 (containing the UAS-p53 wild-type transgene) with females of strain 9 (containing the Act-GS-255B Geneswitch driver) to generate progeny containing both constructs (strains summarized in Table 1); in all cases crosses are indicated with the male parent genotype first, and the female parent genotype second. The RU486 drug itself had no significant effect on male or female life span when administered to adults (Figure 2A; statistical analyses summarized in Supplementary Table 1). When wild-type p53 was over-expressed specifically in adult flies, it had a negative effect (-16%) on mean life span in females (cross 16-9: 95% bootstrap CI for the ratio of the means [-21.11 - 11.61], log-rank p-value = 2.21 x10-6), and a positive effect (+6%) on mean life span in males (cross 16-9: 95% bootstrap CI [2.36 - 10.37], log-rank p-value = 6.97 x10-3) (Figure 2B; Supplementary Table 1). Slightly larger changes were observed for median life spans (Supplementary Table 1), and similar results were obtained with multiple independent transgenic insertions of p53 wild-type (data not shown). In contrast, adult-specific over-expression of the dominant mutantp53 (point mutation p53-259H) transgene did not have a negative effect on female life span, and instead female life span tended to be increased (cross 20-9: +7%, 95% bootstrap CI [4.09 - 9.72], log-rank p-value = 4.05 x10-8) (Supplementary Figure 1B; Supplementary Table 1) [22], and similar results were obtained with p53 dominant mutant transgene p53-Ct[B440] (Supplementary Figure 1C; Supplementary Table 1). Because these Drosophila p53 dominant mutation transgenes are generally expected to antagonize the activity of wild-type p53, the data are consistent with wild-type p53 having a negative effect on adult female life span. The negative effect on life span of wild-type p53 over-expression in adult females and the lack of negative effect with dominant mutant p53 transgenes was also confirmed using the FLP-out conditional system [23] to cause transgene over-expression (data not shown). Taken together, these data indicate that in adult flies, p53 inhibits life span in females and favors life span in males.
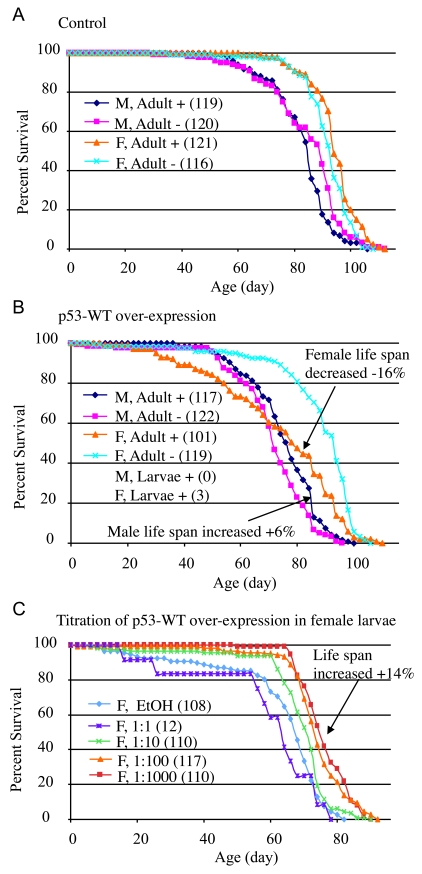
Figure 2. Conditional over-expression of wild-type p53 trans-genes using Geneswitch system.
All flies were the progeny of either Oregon R
control (A) or p53-WT transgenic strain (B, C) crossed
to the tissue-general Geneswitch driver Act-GS-255B. The flies were
cultured in the presence and absence of drug, as larvae or adults, as
indicated: M = males, F = females, + indicates culture in presence of
drug, - indicates culture in absence of drug. The number of flies in each
group are indicated in parentheses. (A, B) Blue diamonds
indicate male adults plus drug, pink squares indicate male adults minus
drug, orange triangles indicate female adult plus drug, turquoise x
indicates female adults minus drug. (A) Control flies, progeny of
Oregon R wild-type and Act-GS-255B. (B) p53 wild-type
transgene over-expression. Note male larvae plus drug produced no adult
flies, whereas female larvae plus drug produced only three escapers. (C)
Titration of p53 wild-type over-expression during female larval
development and effect on subsequent adult life span. EtOH indicates the
ethanol solvent for the drug alone (vector control, indi-cated with light
blue diamonds). Repeats of the titration experiments, including data for
males are presented in Supplementary Figure 1.
Transgenic manipulation of p53 during development
A strikingly different set of results was obtained when Drosophila p53 transgenes were expressed specifically during larval development. When administered only during larval development, the drug RU486 itself had no effect on subsequent adult female life span, and a small negative effect on subsequent adult male life span (~-4%; Supplementary Table 1). Over-expression of wild-type p53 at high levels during larval development was toxic to both males and females, in that no male adults were produced, and only three female adults (escapers) were obtained (Figure 2B). Intriguingly, the three female escapers had unusually long life spans: 86 days, 92 days, and 96 days, respectively. To determine if this apparent life span increase was significant, and to investigate the developmental effects of wild-type p53 over-expression in greater detail, the over-expression was modulated by titration of the RU486/Mifepristone drug, in replicated experiments. Titration of wild-type p53 over-expression during development again indicated toxicity at high levels of expression, with greater toxicity evident for males (Supplementary Table 2). Strikingly, at lower levels of induction, wild-type p53 produced both female and male adults with increased mean and maximal life span (Figure 2C; Supplementary Figure 1E-F; Supplementary Table 2; female: +14%, 95% bootstrap CI [9.29 - 19.27]; log-rank p-value ≈ 0; male: +15%, 95% bootstrap CI [10.54 - 19.30]; log-rank p-value = 4.97 x 10-7). These data demonstrate that high-level expression of p53 can be toxic during development, whereas moderate over-expression of p53 during development can cause increased life span in the resulting male and female adults. Consistent with this conclusion, expression of the dominant mutant transgenes during development tended to decrease the life span of the resultant male and female adults (Supplementary Figure 1A-D, Supplementary Table 1).
Effect of mutations in the endogenous p53 gene
To confirm the effects of p53 on Drosophila life span, flies were examined that had a deletion or mutation of the endogenous p53 gene (mutations diagrammed in Figure 1B; strains listed in Table 1) [24]. Multiple trans-heterozygous p53 wild-type and mutant allele combinations were assayed for life span simultaneously as a control for genetic background effects and environmental effects (the "L" cohort, data summarized in Supplementary Table 3, 4). This was done using two p53 wild-type strains (called the "+" group; strains 6 and 7), two strains containing p53 null mutation (called the "-" group; strains 2 and 3), and three strains containing p53 dominant mutations (called the "M" group; strains 4, 5 and 8), and crossing each strain to each of the others in a "round-robin" approach. In this way each of the various p53 genotypes (+/+, -/-, +/-, +/M, -/M, M/M) represents the average of multiple specific genetic backgrounds. This approach avoids the potential complication of identifying p53 effects that might be specific to only one particular genetic background, such as would be created by using a backcrossing strategy.
In flies with mutations of the endogenous p53 gene, the effect on life span should be the sum of the effects of p53 at various life-cycle stages, both positive and negative (diagrammed in Figure 1D); and indeed, p53 mutations were found to have a significant effect on life span in both sexes (ANOVA, p < 0.0001; Supplementary Table 5): Null mutation (-/-) of the p53 gene increased mean female life span by +13% (95% bootstrap CI [9.00 -17.28]; log-rank p-value ≈ 0) relative to wild-type (+/+) controls (Figure 3A; Supplementary Figure 2A; Supplementary Table 4). In the heterozygous p53 mutant genotype (-/+) average female life span was also increased relative to wild-type controls by +11% (95% bootstrap CI [8.41 - 13.59]; log-rank p-value ≈ 0). In male flies null mutation (-/-) of the p53 gene increased mean life span by +12% (95% bootstrap CI [4.92-14.50]; log-rank p-value ≈ 0), whereas the effect of heterozygous mutation was smaller, yielding mean life span increases of +5.5% (95% bootstrap CI [2.15 - 7.53]; log-rank p-value ≈ 0) (Figure 3B; Supplementary Figure 2B; Supplementary Table 4). However, as seen below (Figure 4A, Supplementary Figure 4), the life span increases in p53 mutant males were not consistently observed when crosses were done in the opposite direction, and therefore may not be biologically significant. Similar effects of p53 null (-/-) and heterozygous (+/-) genotypes were obtained when the experiments were repeated using different culture conditions (richer food source and presence of mates) that yield shorter overall life spans (the "W" cohort; Supplementary Figure 3; Supplementary Table 6, Supplementary Table 7). Taken together, these data with endogenous p53 gene mutations support the conclusion that, in sum, p53 limits the life span of female flies, with smaller and more variable effects in male flies.
Several Drosophila p53 dominant mutations (M) were examined and found to have complex effects on adult life span, depending upon the particular allele, and whether or not a wild-type copy of p53 was present in the background (Figure 3; Supplementary Figure 2, Supplementary Figure 3). Some of the variability in life span across genotypes is expected to result from differences in genetic background. Indeed, the complexity of p53 dominant mutations and their interactions with genetic background has recently been reviewed [25]. Strikingly, when the data for the various p53 genotypes in the L cohort were grouped to control for genetic background effects, the dominant mutations tended to increase life span in females (+/M, -/M, M/M), and to decrease life span in males (+/M, M/M) (Figure 3; Supplementary Figure 2; Supplementary Table 4). Since the Drosophila p53 dominant mutations are generally expected to antagonize wild type p53 function, the increased life span of +/M females relative to wild type (+/+) is consistent with the results obtained above suggesting that, in sum, p53 limits the life span of females. However, for the M/M genotype flies, a wild-type copy of the entire p53 gene is not present, and these genotypes produced the greatest increase in life span in females and the greatest decrease in life span in males. Therefore, these data suggest that the mutant forms of p53 may have sexually antagonistic effects on Drosophila life span that are not necessarily dependent upon the presence of a wild-type p53. Strikingly, these effects of dominant mutations on life span were highly dependent upon environment, since in the W cohort the dominant mutations tended to decrease life span in both males and females (Supplementary Figure 3; Supplementary Table 7). It will be of interest in the future to determine what is the mechanism for these opposite effects of dominant p53 mutations in males versus fe-males, and to determine if the dramatic gene-by-environ-ment effect of p53 dominant mutations in females is due to the presence of mates, the richer food source, or both.
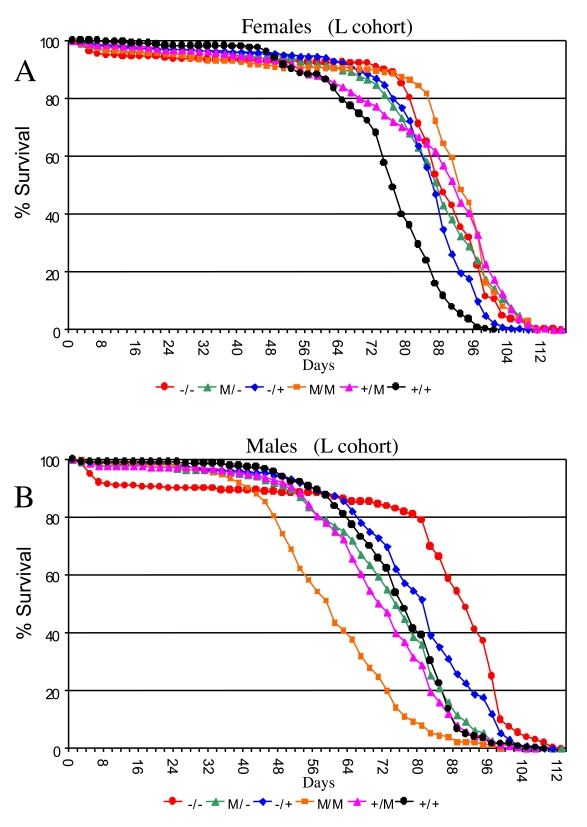
Figure 3. Effect of p53 mutations on life span. Cumulative survival
curves for L cohort. A key of p53 genotypes is presented below the graphs.
Males are indicated with solid symbols and females are indicated with open
symbols. (A) Females. (B) Males.
Controls for maternal effects and X chromosome effects
In an effort to control for possible maternal effects and X chromosome effects, several life span assays were repeated with the crosses done in both directions simultaneously, i.e., varying which strain serves as mother or father for the cross (Supplementary Figure 4). An increase in life span of p53 null mutant (-/-) flies relative to wild-type (+/+) controls was obtained in female progeny regardless of cross direction (Supplementary Figure 4; Supplementary Table 8), thereby ruling out a primary effect of maternal genotype. In males a consistent change in life span was not observed, in that although the null mutants exhibited slight differences in life span compared to controls, the direction of change differed depending on the direction of the cross. Furthermore, while the survival curves of many of the reverse cross pairs differed from one another in both sexes (log-rank test, data not shown), in females there was strong concordance and highly significant results from comparisons of survival curves in both cross directions and relative to both controls, while this was not the case for males (Supplementary Table 8). These results demonstrate that the increased life span in females due to p53 mutation cannot be simply due to maternal or X chromosome effects, and in conjunction with the above findings, these data again suggest that p53 preferentially limits the life span of female flies.
Sex-specific effects p53 on fly stress resistance
Drosophila p53 is required for normal resistance of larval cells and tissues to certain kinds of stress, for example, ionizing radiation and UV toxicity [26,27], and third-instar larvae that are null for p53 exhibit decreased survival when challenged with 4,000 Rads of ionizing radiation [28]. To determine if p53 genotype might have sex-specific effects on stress resistance in adult flies, male and female flies that were either wild-type or mutant for p53 were subjected to two types of life-shortening stress, ionizing radiation and 100% oxygen atmosphere, in replicated experiments (Figure 4, Supplementary Table 9). Treatment with 90,000 Rads of gamma-irradiation on day 10 of adult age reduced adult life spans by half, and p53 mutant female flies were again found to have greater mean life span than wild-type controls (+/-: +18%, 95% bootstrap CI [13.13 - 23.36]; log-rank p-value = 0; -/-: +13%, 95% bootstrap CI [9.09 - 16.71]; log-rank p-value = 2.98 x10-4). In contrast, p53 mutations were found to slightly reduce the survival of female flies subject to 100% oxygen atmosphere (-/+: not significantly different than wild-type; -/-: -4%, 95% bootstrap CI [-5.06 - -3.34]; log-rank p-value = 1.28 x10-13). In males, p53 null mutants subject to ionizing radiation had significantly reduced mean life span, whereas heterozygotes fared slightly better than wild-type (+/-: +4%, 95% bootstrap CI [1.80 - 6.00]; log-rank p-value = 2.02 x10-7; -/-: -19%, 95% bootstrap CI [-20.68 - -17.06]; log-rank p-value ≈ 0). As with females, p53 gene mutations tended to reduce male survival in response to a 100% oxygen environment (+/-: -4%, 95% bootstrap CI [-4.38 - -3.05]; log-rank p-value = 4.44 x10-16; -/-: -15%, 95% bootstrap CI [-16.13 - -14.10]; log-rank p-value ≈ 0). Therefore, wild-type p53 tended to favor the survival of both sexes under 100% oxygen stress conditions, yet was detrimental to female life span in flies subject to ionizing radiation. Therefore the results for adults subject to ionizing radiation were similar to those observed during normal aging: normal p53 function increased survival of males and decreased survival of females. The fact that p53 favored the survival of both sexes under the more severe life-shortening condition of 100% oxygen stress may be indicative of a threshold effect on survival that is sex-specific.
Discussion
In these experiments a combination of genetic and transgenic approaches were used to study how p53 affects the life span of male and female Drosophila. The conditional transgenic system Geneswitch was employed to produce tissue-general expression of p53, either during development or specifically in adults. Detailed characterization of the Geneswitch driver strain ("Actin-GS-255B") using GFP reporter constructs demonstrated that the system yields truly tissue-general expression during larval development, as well as tissue-general expression in both male and female adults [22]. The data indicate that Drosophila p53 has effects on adult life span that are antagonistically pleiotropic between developmental stages and sexes (summarized in Figure 1A). One advance of the present study is that life span effects were identified using transgenes encoding the full length, wild-type form of Drosophila p53 protein, as well as ones encoding mutant forms. In adults, wild-type p53 over-expression limited life span in females and favored life span in males. In contrast, during development, p53 over-expression acted in a dose-dependent manner to either reduce or increase the subsequent longevity of both male and female adults: high level expression during development was detrimental, whereas moderate over-expression produced increased life span. The dominant mutation transgenes generally produced the opposite effect of wild type p53 transgenes, in both males and females. This indicates that the opposing effects of p53 transgenes on male and female life span cannot be simply due to some cryptic difference in the efficiency of transgene expression in males versus females, or to some differential toxicity of the encoded proteins in males versus females.
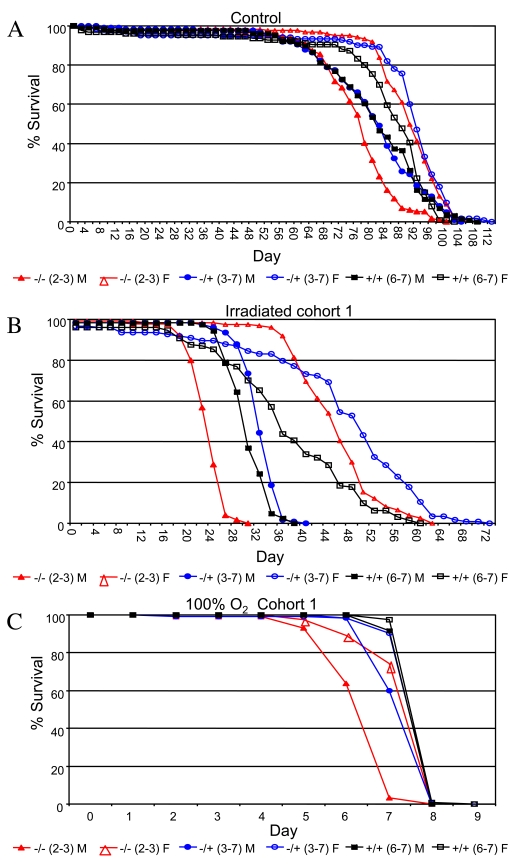
Figure 4. Survival curves for the indicated genotypes under stress conditions.
(A) Ionizing radiation. (B) 100% oxygen survival. A key of p53 genotypes
is presented below the graphs. Males are indicated with solid symbols and females
are indicated with open symbols. Survival curves for replicate experiments (cohort 2)
are presented in Supplementary Figure 5. Survival statistics for these and replicate
experiments are summarized in Supplementary Table 9.
Results consistent with the transgenic manipulations were obtained from analysis of the endogenous p53 gene: Null mutation of the endogenous p53 gene increased life span in females, and had smaller, more variable effects on male life span. The effects of p53 on adult fly survival under stress conditions were also sex-biased: wild-type p53 was found to favor the survival of both sexes under 100% oxygen stress conditions, yet to be detrimental to female life span in flies subject to ionizing radiation. In these experiments p53 expression and function is being altered in all of the tissues of the animal simultaneously, and therefore the effects observed are the sum of any possible tissue-specific effects of p53. Indeed our results suggest that the positive and negative effects of p53 on life span observed here with tissue-general alterations are comprised of a mix of both positive and negative tissue-specific effects, that combine to result in the observed opposite effects in males versus females (J.S. and J.T., 2009 Experimental Gerontology, in press).
The data presented here indicate that p53 null mutation increases life span in female flies, with smaller, more variable increases observed for male flies. Helfand and coworkers have previously reported that p53 null mutant male and female flies were sickly, with a shortened life span, however, statistical analysis was not presented [17]. One possibility is that the apparent reduction in life span and vigor previously reported for p53 null flies may have resulted from inbreeding depression in the homozygous mutant flies used in that study. In contrast, in the experiments presented here, multiple trans-heterozygous p53 null mutant genotypes were examined, so as to reduce possible inbreeding effects, and thereby reveal the life span benefit of p53 null mutations. Helfand and coworkers also analyzed the effect on life span of nervous system-specific expression of two p53 dominant mutant transgenes, a C-terminal fragment transgene (p53-Ct), and the point mutant (p53-259H). They found that nervous system expression of p53-Ct throughout both development and adulthood increased female life span by +58%, and increased male life span by +32% [17]. Because the dominant mutations are generally expected to antagonize p53 activity, their results are consistent with our conclusion that, in sum, p53 limits life span in females, with smaller effect in males (summarized in Figure 1D). Using the Elav-Geneswitch driver to restrict expression to the adult nervous system, Helfand and coworkers found that the p53-Ct transgene increased female life span by +18% to +26%, and the p53-259H transgene increased female life span by +11% to +13%, again consistent with our finding that p53 limits the life span of adult females. Indeed, using the tissue-general Act-GS-255B driver to restrict transgene expression to adults, we also found that the p53-Ct and p53-259H transgenes produced an increase in median life span in females (Supplementary Figure 1A-D) [22]. For adult-specific expression in male nervous system, Helfand and coworkers reported life span data for only two assays, both using the p53-Ct transgene: using a high-calorie food condition, male life span was reported to be increased by +13%, whereas using a low-calorie food, male life span was unchanged, and results for normal food were not presented [17]. That result might at first appear to be partly inconsistent with our conclusion that p53 favors life span in adult males, however, there are several possible explanations that might reconcile these results. First, the previous experiment involved the p53-Ct transgene, encoding the p53 C-terminal fragment, and data from mammals suggests that certain dominant p53 mutants are capable of either antagonizing or promoting p53 activity, depending upon the level of expression and the cellular context [11]. Second, the life span increase was observed only under a high-calorie food condition, and our data suggest sex-specific interactions between dominant p53 mutations and diet/environment with regard to life span (Figure 3, Supplementary Figure 2). Under our conditions and using tissue-general expression, we found that adult-specific expression of the dominant mutant p53 transgenes tended to decrease male life span (Supplementary Figure 1, Supplementary Table 1), consistent with our conclusion that p53 normally favors adult male life span. Finally, the effects of tissue-general expression, as tested here, will be the sum of all tissue-specific effects, be they positive or negative. Indeed our results suggest that the positive and negative effects of p53 on life span observed here with tissue-general alterations are comprised of a mix of both positive and negative tissue-specific effects (J.S. and J.T., 2009 Experimental Gerontology, in press), that combine to result in opposite effects in males versus females (summarized in Figure 1D). Therefore, the previous results from the Helfand group (with the possible exception of a single assay of males under a high-calorie food condition), are generally consistent with the results presented here.
One possible mechanism by which p53 might act in adult flies to preferentially limit female life span is by stimulating IIS, since IIS appears to preferentially limit life span in females of Drosophila and other species [29,30]. Studies in mammals provide precedent for crosstalk between p53 and the IIS pathway, including the target transcription factor FOXO, in regulating both aging and cancer [31,32]. Consistent with this idea, life span extension in Drosophila females produced by nervous system-specific expression of the dominant mutant p53-259H transgene was found to correlate with a reduction in IIS signaling [18]. In C. elegans, mutation of the p53 homolog cep-1 increased life span of adult hermaphrodites, and this increase required the function of the IIS target transcription factor gene Daf-16/FOXO [16]. To definitively rule in (or out) a role for IIS in Drosophila p53 life span effects will require future assays in the presence and absence of the Foxo transcription factor.
Another possible mechanism by which p53 might affect life span is by altering proliferation or causing apoptosis in particular cell types. For example, ablation of germ-line cells in adult animals by forced over-expression of the bam gene caused increased life span in males and females [33]. However, while germ line ablation might be attractive as a possible mechanism for the increased life span observed in p53-over-expressing males, it is not consistent with the life span decrease observed in females. Alternatively, over-expression of wild-type p53 specifically in adult diploid cells using an escargot-GAL4 driver caused ablation of most stem cells in the gut, and gut stem cell proliferation appears to be more rapid in females than in males [34]. While this might be attractive as a possible mechanism for the life span decrease observed in p53-over-expressing females, it is not consistent with the life span increase observed in males; indeed other experiments involving disruption of adult diploid cell function caused an equally dramatic decrease in life span in both sexes [35]. It will be of interest in the future to ask if p53 might be affecting life span through highly sex-specific or sexually opposite effects on cell proliferation and survival. Notably, over-expression of strong caspase inhibitors and other apoptosis and senescence regulatory genes in adult flies did not yield increased life span in either sex, and where negative effects on life span were observed, such as with wingless and activated Ras, the negative effects were similar in males and females [22]. Those results tend to suggest that p53 may be acting through some other mechanisms, such as alterations in metabolism or autophagy. Additional possible mechanisms by which p53 might affect life span include sex-specific alterations in behavior, such as food intake, or potentially costly activities such as movement or aggression.
In these experiments Drosophila p53 was also found to have sex-specific effects on survival under stress conditions. Wild-type p53 favored the survival of both sexes under 100% oxygen stress, yet was detrimental to female life span in flies subject to ionizing radiation. This may be indicative of a threshold effect on survival that is sex-specific. Mechanistically the ability of p53 to either favor survival or mortality may be related to p53's ability to regulate both repair and apoptotic pathways [1,36-38], and perhaps the functional connection between p53 and FOXO in response to oxidative stress [25]. In line with our findings, C. elegans hermaphrodites that are long-lived due to p53 (cep-1) mutation did not demonstrate increased resistance to oxidative (or UV) stress [16], however resistance to gamma irradiation was not examined. Strikingly, in C. elegans hermaphrodites, p53 has recently been found to increase life span in response to mild mitochondrial stress, and to decrease life span in response to severe mitochondrial stress, consistent with a threshold effect on survival [39] ; however effects in males have not been reported. In mice, reduced p53 function results in resistance to lethality caused by moderate gamma irradiation and increased sensitivity to severe irradiation [40,41], again suggestive of a threshold effect, however any potential sex-bias has not been reported. Finally, long-lived female Drosophila that over-expressed dominant-mutant p53 in neurons exhibited increased resistance to the oxidative stressor paraquat [17]; however effects in males were not reported. Taken together the data are consistent with a model in whichp53 has a threshold effect on survival under stress, and the threshold for the transition from favorable to detrimental depends upon the type of stress and the sex of the animal. Such a threshold model is consistent with extensive data from mammals and model systems demonstrating that p53 can either favor oxidative stress resistance and cell survival, or favor oxidative stress and cell death, depending upon the cellular and environmental context, and the degree of activation of p53 [38]. In mammals, physiological levels of p53 activity appear to maintain normal cellular redox status, through sustained expression of antioxidant genes (e.g., Sesn1&2, GPX1, AIF) and metabolic genes (e.g., SCO2, PGM, TIGAR). In contrast, hypo-physiological levels of p53 activity can suppress expression of antioxidant genes (e.g., Sesn1&2, GPX1) and cause increased oxidative stress. Similarly, hyper-physiological levels of p53 activity can induce pro-oxidant and apoptosis-promoting genes (e.g., NQO1, POX, BAX, PUMA, p66shc), and/or cause an imbalance in expression of antioxidant genes (e.g., MnSOD, PIG12, ALDH4, GPX), and again cause increased oxidative stress [38].
Antagonistic pleiotropy of gene function between younger and older animals is generally accepted as one of the most likely genetic mechanisms underlying aging [42]; however, specific genes exhibiting such pleiotropy have generally not been identified. One notable exception is data from mammals that suggests p53 exhibits antagonistic pleiotropy between developmental stages. At young ages p53 favors fecundity and favors survival by acting as a tumor suppressor, yet at late ages it may limit survival by promoting cell senescence, or through other mechanisms [13,43]. Increasing evidence suggests that genes can also exhibit antagonistic plieotropy of function between the sexes, affecting a variety of traits including reproductive fitness and life span [30,44-47]. The data presented here suggest that Drosophila p53 exhibits a combination of both developmental stage-specific and sex-specific antagonistic pleiotropy with regard to life span. If this result were to translate to humans, it would have implications for human aging related diseases such as cancer. Consistent with our results using flies, the effects of human p53 and p53-interacting genes such as MDM2 on cancer incidence and longevity are often sex-biased [48], and p53 has recently been implicated in regulating mammalian maternal fecundity [49]. Moreover, during mouse development, p53 null mutations cause a high frequency of neural tube defects and lethality that preferentially affects female embryos [50,51], and interestingly, this sex difference appears to result from the number of X chromosomes rather than the presence or absence of the Y [52]. The sex-specific effects ofp53 may be related to recent observations that in humans the X-chromosome dosage-compensation gene MOF can regulate p53 [53]; and notably the MOF gene is conserved and also X-linked in flies. Taken together the data support a sexual antagonistic pleiotropy model in which p53 function may be maintained by positive selection for fecundity and/or survival benefit during development, in young animals, and under certain stress conditions, despite acting at another stage of the life cycle and in the other sex to limit adult life span (summarized in Figure 1D).
Methods
Drosophila culture. Drosophila culture and life span assays were performed as previously described [19]. Briefly, crosses were conducted in 250 ml urine-specimen bottles (Genessee Scientific) containing 35 ml of medium. Adult flies were maintained in narrow polystyrene vials (Genesee Scientific) containing 5 ml medium. Drosophila culture media contained cornmeal, agar, dextrose, yeast, and propionic acid to inhibit bacterial growth and tegosept to inhibit fungal growth [54]; except for the W cohort which were cultured on an older recipe containing molasses rather than dextrose (food recipes summarized in Supplementary Table 10). Flies were maintained at 25oC and on a 12:12 dark/light cycle, and were removed to room temperature for less than 1 hour every 2 days to provide fresh medium and remove and enumerate dead flies. To estimate life expectancy, single-sex mortality vials were established, with ~25 flies per vial (sample sizes were occasionally reduced due to rare escapers) and 5 or 10 replicate vials (depending on the experiment) per sex for every cohort. The L cohort deletion experiment used 10 replicate vials per sex, the reverse-cross experiments used 5 vials per sex, the stress experiments used 5 vials per sex, the Geneswitch experiments used 5 vials per sex, and the drug-titration experiments used 5 vials per sex. Note that for each line in the W cohort ~125 flies were maintained at ~25 flies per vial with mates.
Drosophila strains . All Drosophila strains and genotypes are listed in Table 1, and several mutants and transgenes are diagrammed in Figure 1. Wild-type (A-isoform) and dominant-mutant p53 transgene stocks were obtained from Michael Brodsky [3] and Bloomington Drosophila Stock Center. P{UAS-p53.Ex}, p53 wild-type. P{GUS-p53.Ct}AF51, C-terminal fragment AA285-385, chromosome 2. P{GUS-p53.Ct}B440, C-terminal fragment AA285-385, chromosome 3. P{GUS-p53.259H}, AA substitution, chromosome 3. The p53 mutant strains were obtained from Kent Golic and Bloomington Drosophila Stock Center [55]. Df(3R)slo3 is deletion of entire p53 gene ("-"). Df(3R)Exel, P{XP-U}Exel is deletion of entire p53 gene ("-"). p53[5A-1-4] is 3.3kb internal deletion ("-"), and it's structure was confirmed by PCR amplification and sequencing (diagrammed in Figure 1B). p53[11-1B-1] is a point mutation that introduces a stop codon at nucleotide residue 211, and is predicted to yield a 70AA truncated protein ("M"). P{EPgy2}p53[EY14108] is a P element insert mutation obtained from Bloomington Drosophila Stock Center (BL 20906), and the insertion was mapped to the first exon of the p53 B-variant using inverse PCR (diagrammed in Figure 1B) [56]. Because the p53[EY14108] mutation is predicted to produce an altered complement of p53 protein isoforms, it is grouped here with the dominant mutants ("M").
Geneswitch conditional gene expression system . Geneswitch strains and protocols are as previously described [19-21]. The strain Act-GS-255B [19,22] contains two inserts on the second chromosome of a construct in which the actin5C promoter drives expression of the Geneswitch coding region. RU486 (Mifepristone, Sigma) was fed to adult flies or developing larvae by adjusting the food to ~160ug/ml final concentration. A stock solution of 3.2mg/ml of RU486 was prepared by dissolving drug in ethanol (100%). Control food received ethanol solvent alone. In certain experiments RU486 concentrations were titrated as indicated. All ages are expressed as days from eclosion at 25oC. To generate flies containing both the Act-GS-255B driver and the UAS-transgenes, virgins from the Act-GS-255B strain were crossed to males from each transgenic strain and the Oregon R wild-type strain as a control. Certain crosses were done in the opposite direction, as indicated in the "reverse cross" experiments. The life span assay result for p53-259H transgene over-expression in adult flies using Act-GS-255B driver has been previously published [22], and is included here with additional statistical analysis for comparison purposes (Supplementary Table 1).
Statistical analyses . Initial cohort size was taken to be the number of flies in the vials at the beginning of the second two-day interval. Deaths during the first interval after transfer were considered to be due to injury during collection and therefore were excluded from the calculations. Survivorship was scored every other day and final cohort size was taken as summed deaths. The effect ofp53 deletion, mutation, and over-expression on Drosophila life span was assayed in multiple trials for several lines. Life span summary statistics for each of the experiments (data pooled across replicate vials) and detailed statistical analyses are presented in the Supplementary Materials (Supplementary Table 1-9). A non-parametric log-rank test was employed to compare the survival functions betweenp53 deficient or over-expression genotypes and controls [57]. To further assess the effect of p53 on mean, median, and "maximal lifespan" (defined operationally here as the 90th percentile of life span), 95% double bootstrap-t confidence intervals for the ratio of the means (or ratio of the percentiles) of the experimental and control samples were computed using a custom Fortran script. Mixed effects models were fit to data from each sex separately to ascertain the effects of mutation type (M) and genotype (G) (fixed main effects) on life expectancy, with replicate vials (R) treated as a random effect using the nlme package in R. Mixed-effects models allow for a flexible representation of the covariance structure due to the grouping of the data and enabled the variation induced in the survival response by replicate vials to be characterized. As appropriate, the models were y = μ + M + R(M) + ε (where M = +/+, +/-, etc and G = 6-7, 2-6, etc was treated as an "inner" grouping) and y = μ + G + R(G) + ε, where ε indicates the within vial error variance. Post-hoc Tukey tests were performed to assess significant differences among means after correcting for multiple testing. Analyses were performed using the R statistical environment [58], unless otherwise noted.
Supplementary Materials
Supplementary Materials
Acknowledgments
We thank Michelle Arbeitman and Heidi Scrable for helpful comments. This work was supported by a Senior Scholar Award from the Ellison Medical Foundation to JT, and by grants from the Department of Health and Human Services to ST (GM067243) and to JT (AG011833), and by a pilot project award to JT from the USC ADRC (1P50 AG05142). ST is a Royal Society-Wolfson Research Merit Award holder.
Conflicts of Interest
The authors of this manuscript have no conflicts of interest to declare.
References
- 1. Soussi T p53 alterations in human cancer: more questions than answers. Oncogene. 2007; 26: 2145 -2156. [PubMed] .
- 2. Yaswen P and Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell. 2007; 128: 233 -234. [PubMed] .
- 3. Brodsky MH , Nordstrom W , Tsang G , Kwan E , Rubin GM and Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000; 101: 103 -113. [PubMed] .
- 4. Ollmann M , Young LM , Di Como CJ , Karim F , Belvin M , Roberts S , Whittaker K , Demsky M , Fisher WW and Buchman A. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell. 2000; 101: 91 -101. [PubMed] .
- 5. Akdemir F , Christich A , Sogame N , Chapo J and Abrams JM. p53 directs focused genomic responses in Drosophila. Oncogene. 2007; 26: 5184 -5193. [PubMed] .
- 6. Bourdon JC , Fernandes K , Murray-Zmijewski F , Liu G , Diot A , Xirodimas DP , Saville MK and Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005; 19: 2122 -2137. [PubMed] .
- 7. Chintapalli VR , Wang J and Dow JA. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007; 39: 715 -720. [PubMed] .
- 8. Jin S , Martinek S , Joo WS , Wortman JR , Mirkovic N , Sali A , Yandell MD , Pavletich NP , Young MW and Levine AJ. Identification and characterization of a p53 homologue in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2000; 97: 7301 -7306. [PubMed] .
- 9. Sharpless NE and DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004; 113: 160 -168. [PubMed] .
- 10. Campisi J Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005; 120: 513 -522. [PubMed] .
- 11. Moore L , Lu X , Ghebranious N , Tyner S and Donehower LA. Aging-associated truncated form of p53 interacts with wild-type p53 and alters p53 stability, localization, and activity. Mech Ageing Dev. 2007; 128: 717 -730. [PubMed] .
- 12. Gatza C , Hinkel G , Moore L , Dumble M and Donehower LA. Burlington, MA Elsevier 149 -171. p53 and mouse aging models. In: Masoro EJ, Austad SN, eds. Handbook of the Biology of Aging. 2006; .
- 13. Kang HJ , Feng Z , Sun Y , Atwal G , Murphy ME , Rebbeck TR , Rosenwaks Z , Levine AJ and Hu W. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc Natl Acad Sci U S A. 2009; 106: 9761 -9766. [PubMed] .
- 14. Campisi J Cancer and ageing: rival demons. Nat Rev Cancer. 2003; 3: 339 -349. [PubMed] .
- 15. Aranda-Anzaldo A and Dent MA. Reassessing the role of p53 in cancer and ageing from an evolutionary perspective. Mech Ageing Dev. 2007; 128: 293 -302. [PubMed] .
- 16. Arum O and Johnson TE. Reduced expression of the Caenorhabditis elegans p53 ortholog cep-1 results in increased longevity. J Gerontol A Biol Sci Med Sci. 2007; 62: 951 -959. [PubMed] .
- 17. Bauer JH , Poon PC , Glatt-Deeley H , Abrams JM and Helfand SL. Neuronal expression of p53 dominant-negative proteins in adult Drosophila melanogaster extends life span. Curr Biol. 2005; 15: 2063 -2068. [PubMed] .
- 18. Bauer JH , Chang C , Morris SN , Hozier S , Andersen S , Waitzman JS and Helfand SL. Expression of dominant-negative Dmp53 in the adult fly brain inhibits insulin signaling. Proc Natl Acad Sci U S A. 2007; 104: 13355 -61330. [PubMed] .
- 19. Ford D , Hoe N , Landis GN , Tozer K , Luu A , Bhole D , Badrinath A and Tower J. Alteration of Drosophila life span using conditional, tissue-specific expression of transgenes triggered by doxycycline or RU486/Mifepristone. Exp Gerontol. 2007; 42: 483 -497. [PubMed] .
- 20. Osterwalder T , Yoon KS , White BH and Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci U S A. 2001; 98: 12596 -12601. [PubMed] .
- 21. Roman G , Endo K , Zong L and Davis RL. P[Switch], a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2001; 98: 12602 -12607. [PubMed] .
- 22. Shen J , Curtis C , Tavaré S and Tower J. A screen of apoptosis and senescence regulatory genes for life span effects when over-expressed in Drosophila. Aging. 2009; 1: 191 -211. .
- 23. Sun J and Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol Cell Biol. 1999; 19: 216 -228. [PubMed] .
- 24. Rong YS , Titen SW , Xie HB , Golic MM , Bastiani M , Bandyopadhyay P , Olivera BM , Brodsky M , Rubin GM and Golic KG. Targeted mutagenesis by homologous recombination in D.melanogaster. Genes Dev. 2002; 16: 1568 -1581. [PubMed] .
- 25. van der Horst A and Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007; 8: 440 -450. [PubMed] .
- 26. Brodsky MH , Weinert BT , Tsang G , Rong YS , McGinnis NM , Golic KG , Rio DC and Rubin GM. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol. 2004; 24: 1219 -1231. [PubMed] .
- 27. Jassim OW , Fink JL and Cagan RL. Dmp53 protects the Drosophila retina during a developmentally regulated DNA damage response. Embo J. 2003; 22: 5622 -5632. [PubMed] .
- 28. Sogame N , Kim M and Abrams JM. Drosophila p53 preserves genomic stability by regulating cell death. Proc Natl Acad Sci U S A. 2003; 100: 4696 -4701. [PubMed] .
- 29. Toivonen JM and Partridge L. Endocrine regulation of ageing and reproduction in Drosophila. Mol Cell Endocrinol. 2009; 299: 39 -50. [PubMed] .
- 30. Tower J Sex-specific regulation of aging and apoptosis. Mech Ageing Dev. 2006; 127: 705 -718. [PubMed] .
- 31. You H and Mak TW. Crosstalk between p53 and FOXO transcription factors. Cell Cycle. 2005; 4: 37 -38. [PubMed] .
- 32. Maier B , Gluba W , Bernier B , Turner T , Mohammad K , Guise T , Sutherland A , Thorner M and Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004; 18: 306 -319. [PubMed] .
- 33. Flatt T , Min KJ , D'Alterio C , Villa-Cuesta E , Cumbers J , Lehmann R , Jones DL and Tatar M. Drosophila germ-line modulation of insulin signaling and lifespan. Proc Natl Acad Sci U S A. 2008; 105: 6368 -6373. [PubMed] .
- 34. Jiang H , Patel PH , Kohlmaier A , Grenley MO , McEwen DG and Edgar BA. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell. 2009; 137: 1343 -1355. [PubMed] .
- 35. Biteau B , Hochmuth CE and Jasper H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell. 2008; 3: 442 -455. [PubMed] .
- 36. Sablina AA , Budanov AV , Ilyinskaya GV , Agapova LS , Kravchenko JE and Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005; 11: 1306 -1313. [PubMed] .
- 37. Edwards MG , Anderson RM , Yuan M , Kendziorski CM , Weindruch R and Prolla TA. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics. 2007; 8: 80 [PubMed] .
- 38. Liu B , Chen Y and St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008; 44: 1529 -1535. [PubMed] .
- 39. Ventura N , Rea SL , Schiavi A , Torgovnick A , Testi R and Johnson TE. p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell. 2009; 8: 380 -393. [PubMed] .
- 40. Komarova EA , Kondratov RV , Wang K , Christov K , Golovkina TV , Goldblum JR and Gudkov AV. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004; 23: 3265 -3271. [PubMed] .
- 41. Strom E , Sathe S , Komarov PG , Chernova OB , Pavlovska I , Shyshynova I , Bosykh DA , Burdelya LG , Macklis RM , Skaliter R , Komarova EA and Gudkov AV. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006; 2: 474 -479. [PubMed] .
- 42. Hughes KA and Reynolds RM. Evolutionary and mechanistic theories of aging. Annu Rev Entomol. 2005; 50: 421 -445. [PubMed] .
- 43. Rodier F , Campisi J and Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res. 2007; 35: 7475 -7484. [PubMed] .
- 44. Nuzhdin SV , Pasyukova EG , Dilda CL , Zeng ZB and Mackay TF. Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1997; 94: 9734 -9739. [PubMed] .
- 45. Tower J and Arbeitman M. The genetics of gender and life span. J Biol. 2009; 8: 38 [PubMed] .
- 46. Rice WR , Gavrilets S and Friberg U. Sexually antagonistic "zygotic drive" of the sex chromosomes. PLoS Genet. 2008; 4: e1000313 [PubMed] .
- 47. Morrow EH , Stewart AD and Rice WR. Assessing the extent of genome-wide intralocus sexual conflict via experimentally enforced gender-limited selection. J Evol Biol. 2008; 21: 1046 -54. [PubMed] .
- 48. Feng Z , Hu W , Rajagopal G and Levine AJ. The tumor suppressor p53: cancer and aging. Cell Cycle. 2008; 7: 842 -847. [PubMed] .
- 49. Hu W , Feng Z , Atwal GS and Levine AJ. p53: a new player in reproduction. Cell Cycle. 2008; 7: 848 -852. [PubMed] .
- 50. Sah VP , Attardi LD , Mulligan GJ , Williams BO , Bronson RT and Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995; 10: 175 -180. [PubMed] .
- 51. Armstrong JF , Kaufman MH , Harrison DJ and Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995; 5: 931936 .
- 52. Chen X , Watkins R , Delot E , Reliene R , Schiestl RH , Burgoyne PS and Arnold AP. Sex difference in neural tube defects in p53-null mice is caused by differences in the complement of X not Y genes. Dev Neurobiol. 2008; 68: 265 -273. [PubMed] .
- 53. Rea S , Xouri G and Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007; 26: 5385 -5394. [PubMed] .
- 54. Ren C , Webster P , Finkel SE and Tower J. Increased internal and external bacterial load during Drosophila aging without life-span trade-off. Cell Metab. 2007; 6: 144 -152. [PubMed] .
- 55. Xie HB and Golic KG. Gene deletions by ends-in targeting in Drosophila melanogaster. Genetics. 2004; 168: 1477 -1489. [PubMed] .
- 56. Landis GN , Bhole D and Tower J. A search for doxycycline-dependent mutations that increase Drosophila melanogaster life span identifies the VhaSFD, Sugar baby, filamin, fwd and Cct1 genes. Genome Biol. 2003; 4: R8 [PubMed] .
- 57. Miller RG New York John Wiley & Sons Survival Analysis. 1981; .
- 58. RDevelopment CoreTeam Vienna, Austria R Foundation for Statistical Computing R: A language and environment for statistical computing. 2006; .