



Introduction
Breast cancer stands as a pervasive malignancy, impacting women globally and yielding a substantial number of yearly diagnoses. Despite significant advancements in adjuvant therapies, resulting in an admirable 90% 5-year survival rate, breast cancer remains the second most prevalent cause of mortality in women, attributable to its invasive and metastatic proclivities [1–4]. On a global scale, breast cancer prevails as a highly prevalent malignancy, prompting a significant annual diagnostic burden. Notwithstanding advancements in adjuvant therapies that have contributed to a noteworthy 90% 5-year survival rate, the invasive and metastatic characteristics of breast cancer underscore its persistence as the second most common cause of mortality among women [5, 6]. Conventional prognostic scoring systems encompass an array of patient factors, including age, tumor characteristics, grading, and staging, to facilitate diagnosis. Although TNM staging plays a significant role in cancer staging [7, 8], some studies have pointed out that immune markers are more accurate and even better than TNM staging for prognosis [9]. Also, the TNM system may ignore biological heterogeneity within the tumor [10]. Early diagnostic markers hold substantial promise in positively impacting the quality of life and survival duration for individuals grappling with breast cancer.
Glucose deprivation in hypercells with upregulated SLC7A11 gives rise to disulfidptosis, an intriguing mechanism of cell death marked by an anomalous accumulation of intracellular disulfidptosis [11]. This unique mode of cellular demise not only orchestrates the demise of cancer cells but also relies on the vulnerability of the actin cytoskeleton to bond stress induced by disulfidptosis [12]. Thus, GLUT inhibitors cause glucose depletion and disulfidptosis in SLC7A11-high tumor cells [13]. Despite its profound significance, explorations into disulfidptosis within tumors are in their nascent stages, and initial findings are currently underreported.
Long non-coding RNAs (lncRNAs), distinguished by their lack of protein-coding capability and ranging in size from 200 nt to 100 kb, have emerged as crucial players in cancer diagnosis, straddling roles as both oncogenic factors and cancer suppressors [14–17]. The compelling association between lncRNA expression levels and breast cancer subtype classification positions these molecules as promising biomarkers with transformative potential [18–22]. However, to bolster diagnostic precision, a thorough exploration of the involvement of disulfidptosis-associated lncRNA (DAL) in the prognosis and tumor microenvironment of BRCA is indispensable.
In our comprehensive investigation, we meticulously screened eight disulfidptosis-associated lncRNAs (DALs), constructing a model grounded in the TCGA-BRCA cohort. Our analytical focus extended to unraveling the intricate interplay between DALs and critical facets such as immunotherapy response, the tumor microenvironment, and drug sensitivity. Our primary objective was to accentuate the positive significance of DALs in prognostic predictions for BRCA patients, presenting a valuable tool that not only enhances diagnostic accuracy but also aids in the meticulous selection of tailored treatments. This nuanced approach underscores the potential translational impact of our findings in advancing precision medicine within the realm of breast cancer therapeutics.
Materials and Methods
The procurement and compilation of sample data
A comprehensive dataset, encompassing 1097 instances of breast cancer (BRCA), was acquired from the TCGA database (https://portal.gdc.cancer.gov/). This dataset amalgamated gene expression profiles with relevant clinical particulars for each specimen, encompassing survival status, age, gender, survival duration, and TNM stage. In the initial preprocessing of transcriptomic data, a conversion from FPKM (Fragments Per Kilobase Million) to TPM (Transcripts per million) was executed [23]. Following this, the processed expression values underwent a logarithmic transformation with a base of 2. This transformation facilitated the amalgamation of genes and their corresponding expression values, leading to the creation of comprehensive gene expression profiles for each sample.
Concerning clinical information, a streamlined process of downloading and merging relevant datasets sufficed for the subsequent analysis. This meticulous approach ensures the robust integration of gene expression data and clinical parameters, laying a solid foundation for the in-depth exploration of breast cancer within this cohort.
Development and validation of the model
The dataset sourced from the TCGA database underwent a meticulous division into two sets, maintaining a balanced 1:1 ratio, to fulfill distinct roles in the analytical process. The training set assumed the pivotal role of model construction, while the validation set functioned as a critical evaluator of the model’s performance. To pinpoint noteworthy lncRNAs, a univariate Cox regression analysis was executed using the “glmnet” package in R. Subsequently, a LASSO regression approach, incorporating a tenfold cross-validation strategy, was employed to pinpoint the optimal point of minimal error [24]. This stringent methodology facilitated the discernment of pivotal genes and their corresponding parameters via a multivariate Cox regression analysis, leading to the revelation of eight differentially expressed long non-coding RNAs (DALs). These DALs formed the basis for constructing essential models, wherein each patient’s risk score was meticulously calculated. This score was computed by summing the product of the expression level of each long non-coding RNA (lncRNA) and its respective coefficient, offering a quantitative assessment of risk for each participant in the cohort.
Correlation analysis of DAL with different clinical features
To scrutinize the correlation between DALs and diverse clinical characteristics, a comprehensive exploration was undertaken. Utilizing the “Limma” package within the R software, a rigorous examination of differential gene expression was executed on the training dataset. The primary objective of this analysis was to discern variations in the expression levels of DALs associated with clinical indicators and risks. For a nuanced portrayal of the diversity in DAL expression across distinct clusters, we harnessed the capabilities of the “pheatmap” package in R, thereby generating heatmap representations. These depictions offer a visual elucidation of the disparate expression patterns of DALs concerning various clinicopathological features. Such visual aids serve to illuminate the intricate relationship between DAL expression and diverse clinical characteristics.
Model equations
To scrutinize the correlation between differentially expressed long non-coding RNAs (DALs) and various clinical characteristics, we conducted a thorough analysis of gene expression differences within the training dataset, utilizing the “Limma” package in R. The overarching goal of this analysis was to discern discrepancies in the expression levels of DALs associated with clinical indicators and risks. For a graphical representation of the nuanced variations in DAL expression across distinct clusters, we leveraged the capabilities of the “pheatmap” package in R, facilitating the generation of heatmap depictions. These visual representations vividly illustrate the distinct patterns of DAL expression in correlation with clinicopathological features. These visual aids constitute a valuable method for extracting insights into the intricate connection between the expression of differentially expressed DALs and diverse clinical characteristics.
Development of an independent prognostic model and construction of a nomogram
To establish the independent prognostic relevance of the risk score, we performed both univariate and multivariate Cox regression analyses. These assessments aimed to assess the association between the risk score and patient survival outcomes, concurrently accounting for other clinicopathological variables as covariates. Furthermore, employing the ‘rms’ package in R, we crafted histograms to visually illustrate the distribution of risk scores and clinicopathological variables. These graphical representations provided valuable insights into the interrelation between risk scores and various clinical characteristics. Subsequently, based on the results of the prognostic model, we devised a nomogram. Functioning as a graphical predictive instrument, this nomogram amalgamates the risk score with other pertinent clinical variables to estimate personalized survival probabilities for individuals within the TCGA-BRCA cohort.
Investigation of enriched biological functions
To glean insights into the functional annotation and enrichment pathways linked to the eight differentially expressed genes associated with DALs in BRCA, a comprehensive functional enrichment analysis was undertaken. For Gene Ontology (GO) analysis, the ClusterProfiler package in R was employed. Furthermore, GSVA analysis, utilizing the “c2.cp.kegg.v7.4.symbols.gmt” package from MSigDB and the “GSVA” package in R, was conducted to discern pathway disparities among distinct clusters. The outcomes of these analyses were visually presented using the ‘heatmap’ package in R. In addition, Gene Set Enrichment Analysis (GSEA) was carried out, employing the “c2.cp.kegg.Hs.symbols.gmt” package, to delve deeper into enriched pathways associated with the differentially expressed genes. These analytical endeavors were geared towards unraveling the biological functions and pathways potentially influenced by the identified DALs in the context of BRCA.
Immunological scrutiny of risk attributes
To examine the immunological attributes associated with risk assessment, a suite of robust algorithms, including EPIC, MCPCOUNTER, CIBERSORT, CIBERSORT-ABS, TIMER, XCELL, and QUANTISEQ, was deployed for the analysis of immune infiltration scores. Subsequently, using the “limma” package, we performed Spearman’s correlation analyses aimed at elucidating the relationship between the proportion of immune cells and risk assessment. Additionally, the CIBERSORT algorithm was utilized to categorize BRCA patients based on their immune cell characteristics.
In order to assess the enrichment of genetic features associated with the cancer immune cycle and treatment, we performed GSVA analyses on high-risk cohorts using the “clusterProfiler” package [25]. Finally, the “ggcor” R package facilitated the establishment of connections between risk scores and genetic traits. These comprehensive immunological analyses were performed with the overarching objective of offering profound insights into the immune characteristics intricately linked with the risk levels identified in our study.
Drug sensitivity
The Genomics of Drug Sensitivity in Cancer (GDSC) repository, available at https://www.cancerrxgene.org/ (accessed on 22 March 2023) [26], serves as a valuable database enabling the categorization of BRCA patients into low- and high-risk groups based on their half-maximal inhibitory concentration (IC50). This categorization offers crucial insights into the varying sensitivities of distinct patient populations to diverse treatments. To further scrutinize the response of these patient groups to specific therapeutic interventions, the R package “pRRophetic” proves instrumental. This package empowers the prediction of drug sensitivity grounded in genomic profiles, thereby enabling the personalized selection of treatments and the assessment of treatment responses in individuals affected by BRCA.
Somatic mutations
For the analysis of mutation information within BRCA samples, the TCGA-BRCA mutation database (https://portal.gdc.cancer.gov/, accessed on 22 March 2023) served as the primary data source. Subsequently, the Maftools program [27] was employed for the comprehensive analysis and visualization of mutation profiles. In our investigation, particular emphasis was placed on evaluating the risk of breast cancer through a comparative examination of mutational load, commonly referred to as the tumor mutation burden score. The TMB score was calculated by multiplying the mutation-to-covered bases ratio by 10^6, following the methodology outlined in the investigation by Chalmers and collaborators [28]. This approach enabled the quantification of the overall mutational burden, facilitating an assessment of its potential association with the risk of breast cancer within the analyzed samples. We also used the “limma” package to analyze the relationship between mapping tumor stem cells and risk scores.
Statistical analysis
All bioinformatics analyses were conducted leveraging the capabilities of R software version 4.2.2 and Strawberry Perl version 5.30.0. Spearman‘s correlation analysis method was employed to scrutinize the relationship between immune cell infiltration and risk scores. To compare overall survival (OS) outcomes between the high-risk and low-risk groups, Kaplan-Meier (KM) survival curves, complemented by log-rank tests, were utilized. Statistical significance was established at p-values < 0.05, with the control of false discovery rates (FDR) set at < 0.05.
Data availability statement
The datasets utilized in our investigation are accessible via the TCGA repository (http://cancergenome.nih.gov/). The unprocessed data can be obtained from jianguoyun at the following link: https://www.jianguoyun.com/p/DXU0NlwQo47JCxiEw4MFIAA.
Results
Identification of lncRNAs correlated with disulfidptosis-induced cellular demise
Figure 1 presents a synopsis of the pivotal steps undertaken in this investigation. In the initial phase, an exhaustive analysis was performed on the TCGA-BRCA dataset to sift through protein-coding genes, leading to the discovery of 13,162 lncRNAs. Subsequent to this, co-expression analysis was executed between ten established genes linked to disulfidptosis and the lncRNAs associated with disulfidptosis. The aim was to discern those manifesting noteworthy co-expression relationships with disulfidptosis genes (Figure 2A). Moreover, the expression levels of these lncRNAs were extracted, and their affirmative and adverse regulatory associations were ascertained. Univariate Cox analysis was utilized to identify lncRNAs linked to elevated and diminished risk (Figure 2B). This analysis facilitated the identification of lncRNAs significantly linked to the risk levels. Following this, the LASSO algorithm was employed to choose distinctive lncRNAs, utilizing LASSO regression and observing their regression validation trends to pinpoint the juncture with the lowest cross-validation error (Figure 2C, 2D). Using a multivariate Cox proportional hazard regression model, eight disulfidptosis-related lncRNAs (MIR4435-2HG, AL139035.1, AL451085.2, AP001160.1, NRAV, AC015922.2, YTHDF3-AS1, TP53TG1) were chosen along with their corresponding regression coefficients for model construction. Furthermore, a comprehensive investigation into the correlation between the identified lncRNAs and the genes associated with disulfidptosis was undertaken (Figure 2E). The results unveiled a strong correlation between these long non-coding RNAs associated with disulfidptosis (DALs) and the genes implicated in disulfidptosis (Figure 2F).
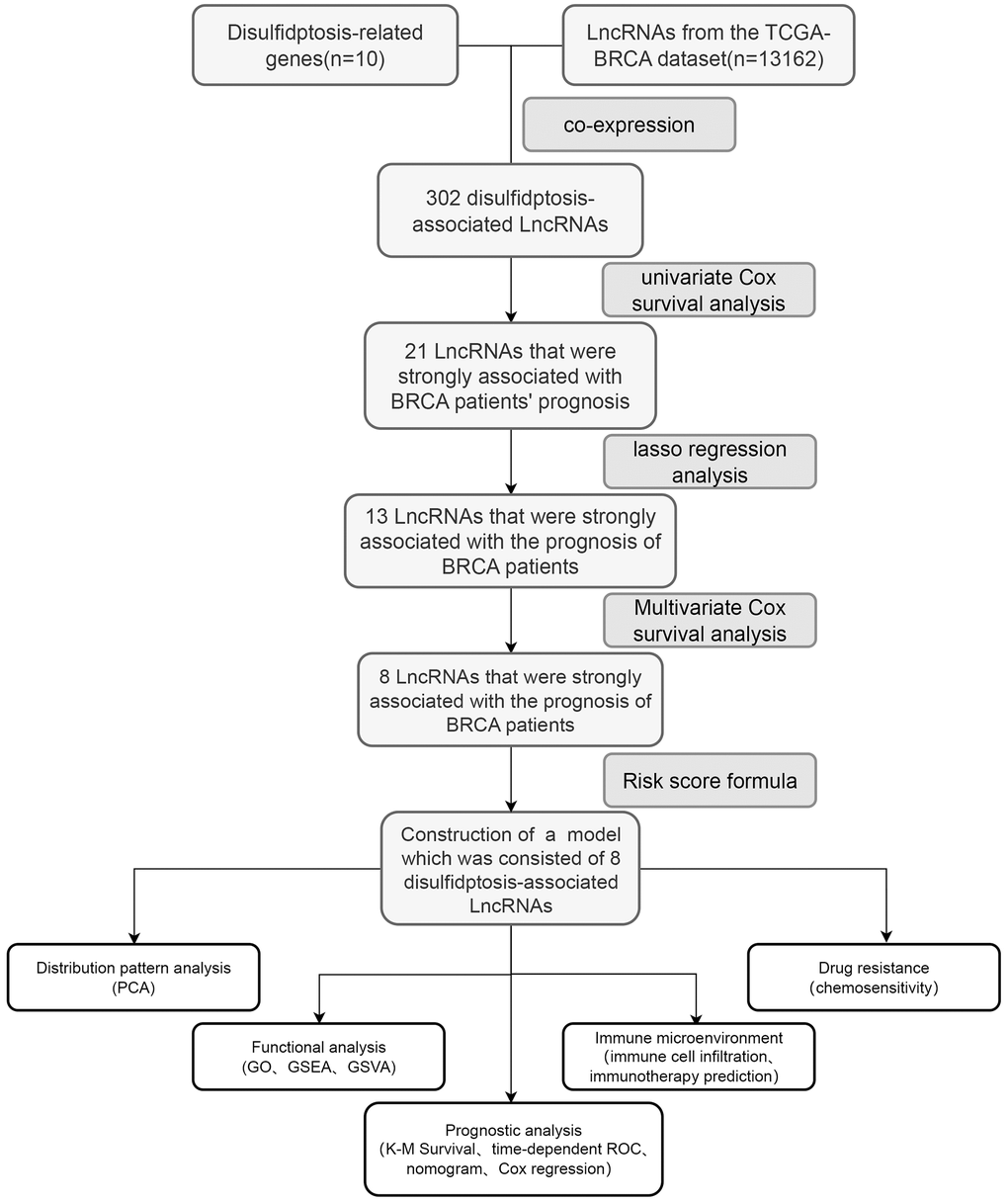
Figure 1. The accompanying schematic diagram presents a thorough and all-encompassing summary of the primary cognitive processes conducted throughout the course of this study.
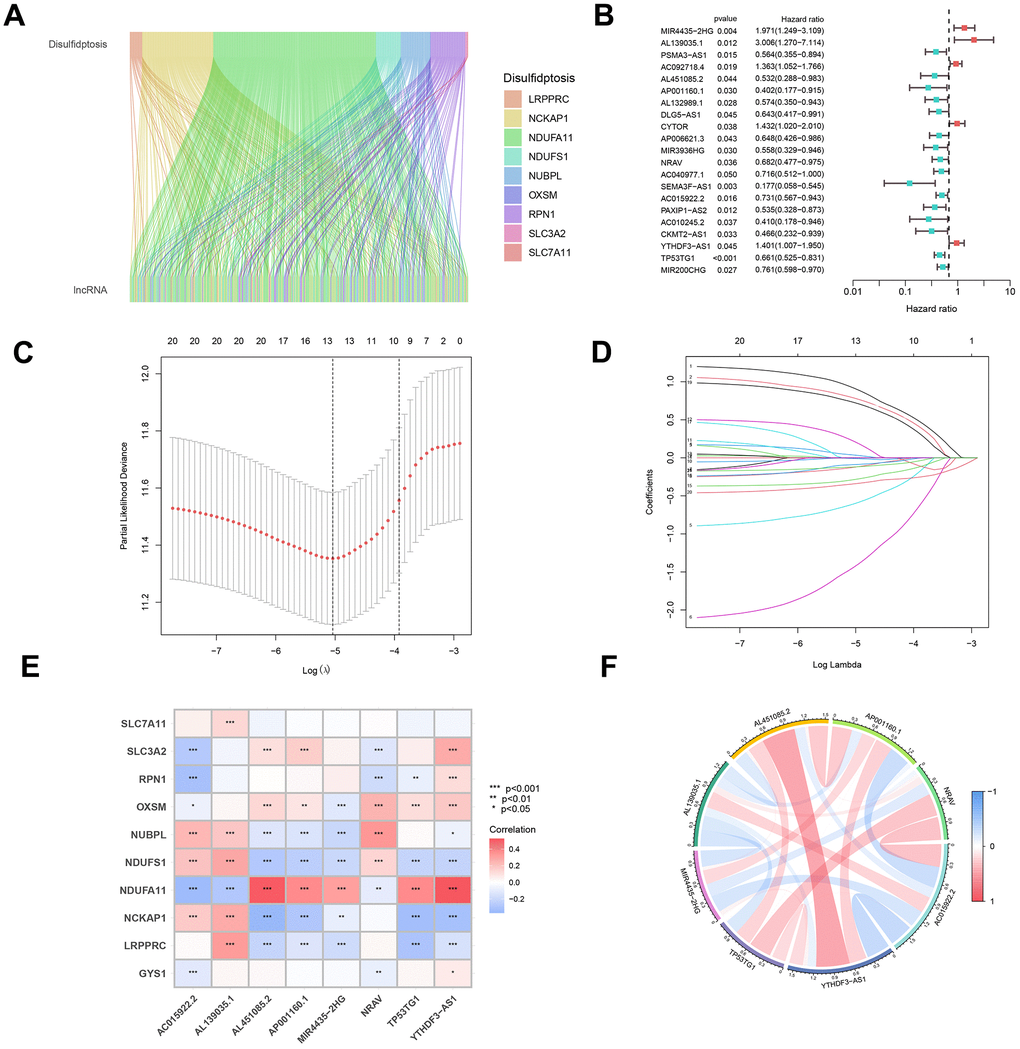
Figure 2. Construction and evaluation of a predictive model for disease onset. (A) A co-expression analysis was executed to explore the intricate interplay between genes associated with disulfidptosis and pivotal lncRNAs, forming the cornerstone of our model. (B) Univariate Cox analysis was employed to pinpoint differentially expressed lncRNAs and assess their correlation with high- and low-risk cohorts. (C) Utilizing the LASSO algorithm, integrated with 10-fold cross-validation, we identified the most significant lncRNAs linked to disulfidptosis. (D) The coefficients obtained from the LASSO algorithm were scrutinized to establish the foundation for our predictive disease model. (E) A correlation heatmap was generated to delve deeper into the intricate relationships between the selected lncRNAs and disulfidptosis-related genes. (F) Subsequent analysis of the interconnections among the chosen lncRNAs provided valuable insights into the underlying mechanisms of disulfidptosis.
Development and validation of the prognostic model
To formulate a resilient prognostic model, the cohort was stratified into two cohorts: the Training group and the Test group, ensuring an equitable distribution of samples in each. The Test group functioned as a validation set to appraise the model’s precision. The expression levels of the eight lncRNAs associated with disulfidptosis were multiplied by their respective regression coefficients for each sample. The resultant values were then aggregated to compute a risk score predicated on the lncRNA expression levels. Following this, samples were arranged in ascending order based on their risk scores, and the median score was employed to categorize them into high-risk and low-risk groups. This methodology was independently applied to all samples, encompassing both the Training and Test groups (Figure 3A–3C).
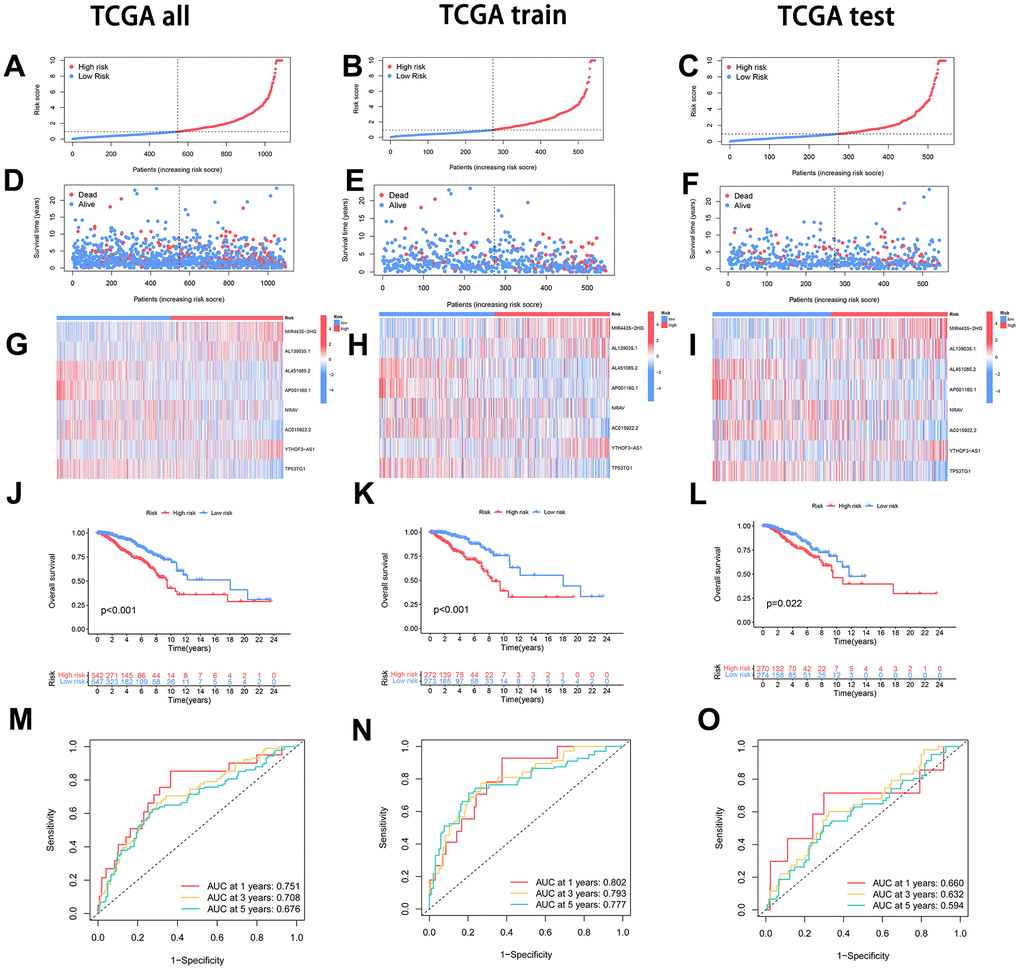
Figure 3. Model construction and evaluation of disease predictive value. (A–C) The dataset, comprising the overall, training, and testing sets, underwent stratification based on the risk score. Following this, samples were delineated into high-risk (depicted in red) and low-risk (depicted in blue) groups, utilizing the median risk score as the threshold. (D–F) The relationship between the risk score and both survival time and patient status was examined across the entire dataset, training set, and testing set. (G–I) Examination of the expression profiles of each Disulfidptosis-Associated LncRNA (DAL) was conducted, comparing high-risk and low-risk groups within the entire dataset, training set, and testing set. (J–L) Survival curves were juxtaposed between the high-risk and low-risk groups in the entire dataset, training set, and testing set to elucidate differences in survival outcomes. (M–O) Time-related Receiver Operating Characteristic (ROC) curve analysis was executed to appraise the predictive performance of the model across the entire dataset, training set, and testing set.
The correlation analysis between the risk score and survival status revealed a positive association, indicating that with an elevation in the risk score, there was a corresponding increase in the number of fatalities (Figure 3D–3F). Further examination of the expression patterns of the lncRNAs in the high-risk and low-risk groups, encompassing all samples, the Training group, and the Test group, disclosed that MIR4435-2HG, AL139035.1, and YTHDF3-AS1 lncRNAs exhibited an upregulated expression with a rising risk score in the high-risk group. In contrast, AL451085.2, AP001160.1, NRAV, AC015922.2, and TP53TG1 lncRNAs displayed a downregulated expression (Figure 3G–3I).
To evaluate the accuracy of the prognostic model, survival curves were plotted, and receiver operating characteristic (ROC) analysis was conducted at different time points. Consistently, the results demonstrated significantly lower survival rates in the high-risk group compared to the low-risk group at various time points, with statistical significance at P < 0.05. The constructed model effectively differentiated between the high-risk and low-risk groups (Figure 3J–3L). Furthermore, the ROC curves at different time points indicated that a larger area under the curve corresponded to higher predictive accuracy of the constructed model (Figure 3M–3O).
Principal component analysis (PCA) for gene expression
To delve further into gene expression patterns, we performed principal component analysis (PCA) on four distinct categories: all genes, genes associated with disulfidptosis, long non-coding RNAs (lncRNAs) linked to disulfidptosis, and the eight lncRNAs employed in formulating the prognostic model. The PCA analysis unveiled discernible separation among these groups based on their expression profiles. Notably, upon comparing the PCA plots of disulfidptosis-associated genes, all genes, the model lncRNAs, and disulfidptosis-associated lncRNAs, it became apparent that the model lncRNAs exhibited a more pronounced differentiation between the low and high-risk groups. This observation provides compelling evidence that the lncRNAs incorporated into the prognostic model possess the capability to effectively discriminate between low and high-risk sample groups (Figure 4A–4D).
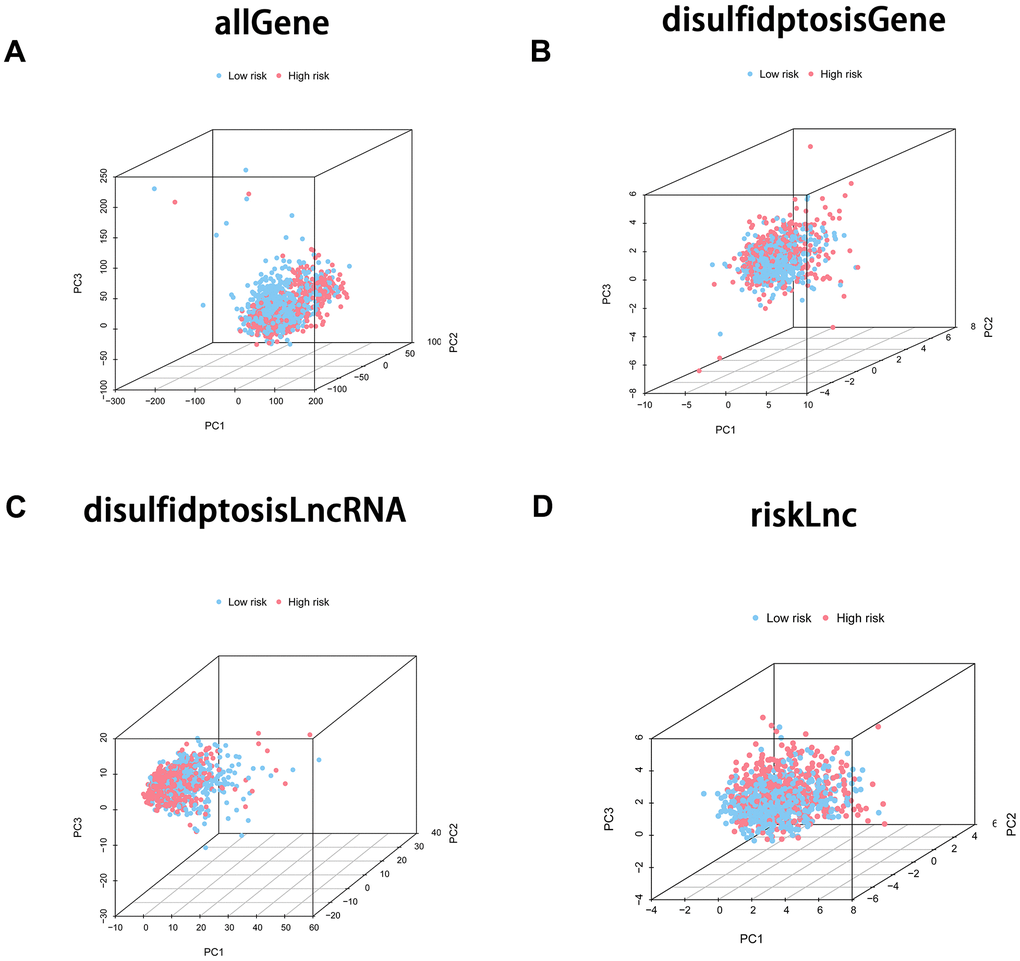
Figure 4. Principal component analysis (PCA) was employed to scrutinize the differentiation between the high-risk and low-risk groups employing diverse gene sets. The four PCA plots depict the sample distribution within the corresponding risk groups based on distinct gene sets: (A) incorporating all genes, (B) comprising disulfidptosis-associated genes, (C) encompassing disulfidptosis-associated lncRNAs, and (D) involving model lncRNAs. The PCA analysis sought to appraise the unique clustering patterns and the potential discriminatory efficacy of these gene sets in distinguishing high-risk from low-risk groups.
Examination of the correlation between DAL and clinical characteristics
To investigate the association between the low and high-risk groups and diverse clinical features, we constructed heatmaps for a visual representation of the relationships. These heatmaps offered a comprehensive overview of the correlation between risk scores and clinical characteristics, encompassing tumor grade, gender, age, T, M, and N (Figure 5A). Moreover, we performed analyses to distinguish the distribution disparities of clinical features, including tumor grade, gender, age, T, M, and N, between the low and high-risk groups. (Figure 5B–5H). These analytical methodologies yielded valuable insights into potential connections between Disulfidptosis-Associated LncRNAs (DALs) and various clinical characteristics, thereby illuminating the potential clinical implications of our findings.
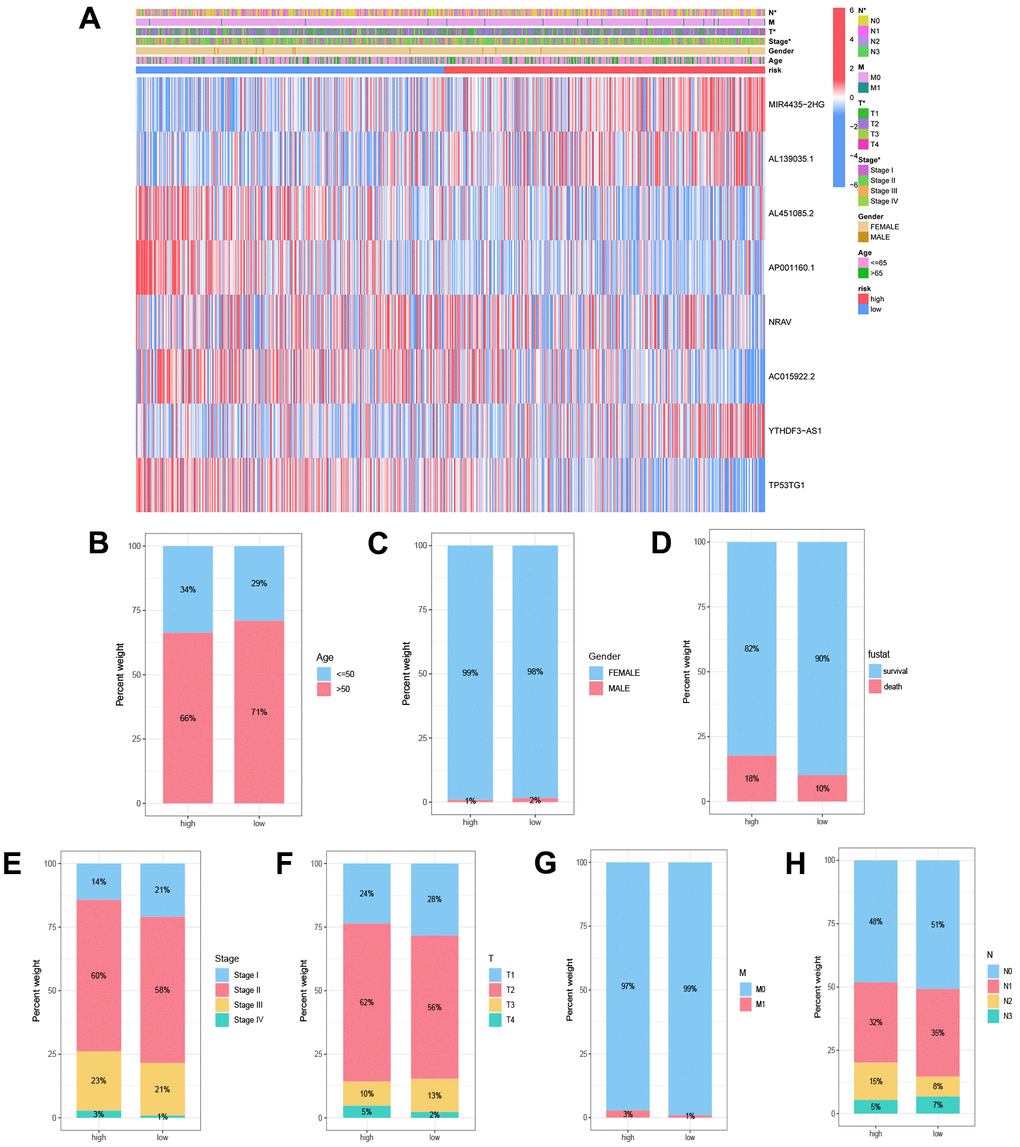
Figure 5. Correlation analysis of disulfidptosis-associated lncRNAs (DALs) with diverse clinical characteristics. (A) Heatmap illustrating the correlation between DALs and risk levels, along with various clinical attributes. (B) Age, (C) gender, (D) survival status, (E) staging, (F) T, (G) N, and (H) M distinctions between the high and low-risk cohorts.
Subgroup analysis of a disulfidptosis-associated lncRNA model in predicting clinical outcomes
To assess the prognostic implications of the DAL model within distinct clinical subgroups, a rigorous survival analysis was undertaken. The low and high-risk groups were stratified based on gender (male and female), age (≤50 and >50 years), clinical stage (I-II and III-IV), and TNM (T1-2 and T3-4, N0-1 and N2-3, M0 and M1). The findings consistently underscored that individuals categorized in the high-risk group experienced markedly diminished survival durations in comparison to their counterparts in the low-risk category across all clinical subgroups (p < 0.05) (Figure 6A–6L). These results affirm the resilience and widespread applicability of the established long non-coding RNA (lncRNA) model associated with disulfidptosis in forecasting clinical outcomes among varied patient cohorts.
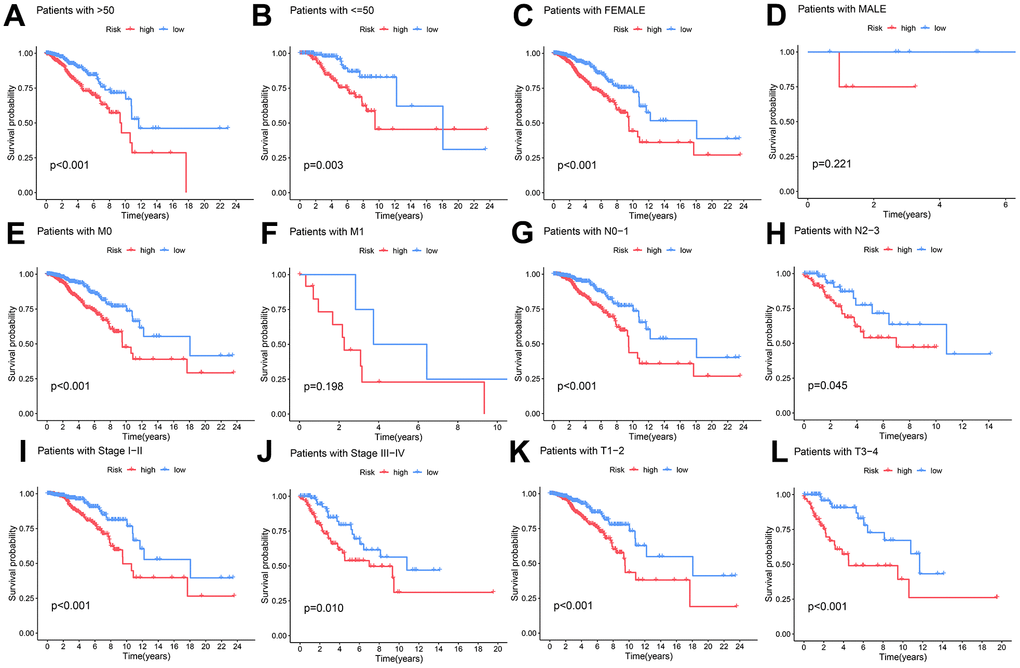
Figure 6. The capacity of the DAL model to forecast survival variations in high and low-risk groups among distinct clinical subgroups. (A, B) age (>50 and ≤50 years), (C, D) gender, (E, F) M stage, (G, H) N stage, (I, J) clinical stage, and (K, L) T stage.
Independent prognostic analysis of clinical features and the creation of nomograms
To ascertain the autonomous prognostic efficacy of the formulated model, we executed independent prognostic analyses on clinical attributes. Univariate independent prognostic analysis was employed to scrutinize the association between singular clinical characteristics and the temporal and categorical aspects of survival. Additionally, a multivariate independent prognostic analysis was undertaken to appraise the collective impact of various clinical traits on survival metrics, taking into account plausible interactions between these factors. Our observations unveiled age, staging, and risk score as autonomous prognostic determinants, underscoring their potential to exert influence on patient outcomes irrespective of other variables (Figure 7A, 7B).
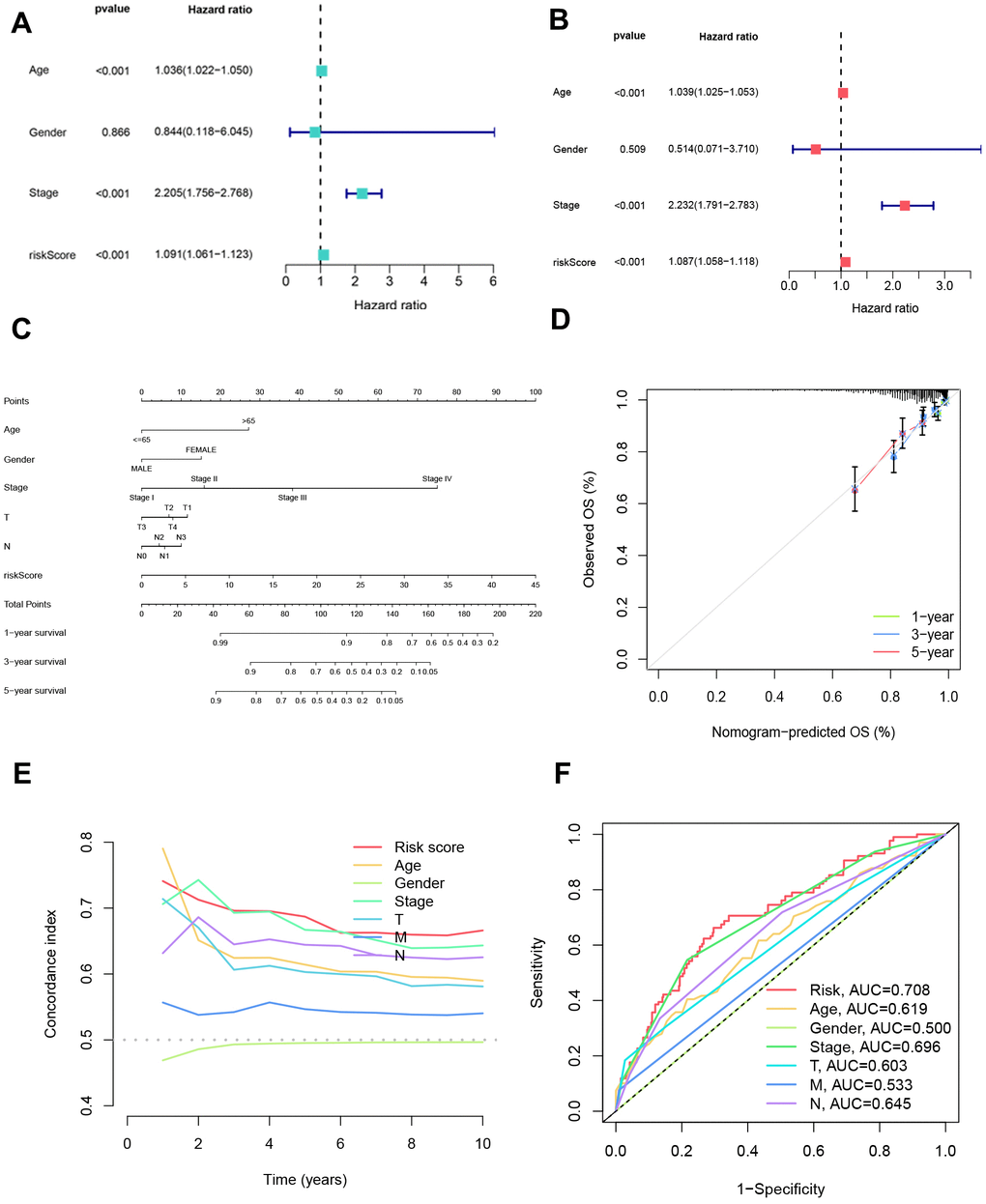
Figure 7. Independent prognostic analysis of clinical characteristics and nomogram creation. (A) Assessment of the independent prognostic impact of each clinical factor individually. (B) Evaluation of the independent prognostic influence of multiple factors for each clinical variable. (C) Nomograms were formulated incorporating each clinical indicator and risk scores to present a visual aid for predicting personalized patient outcomes. (D) Calibration curves were generated to appraise the nomograms’ accuracy, comparing predicted survival probabilities with actual survival rates at 1-year, 3-year, and 5-year intervals. (E) Calculation of the C-index was performed to gauge the discriminatory efficacy of gender, age, stage, risk scores, T category, N category, and M category in forecasting patient outcomes. (F) Receiver operating characteristic (ROC) analysis was undertaken for each clinical indicator to ascertain its predictive performance and capacity to differentiate between distinct outcome groups.
Moreover, a nomogram was formulated, amalgamating diverse clinical attributes to forecast patient survival. The calibration curves for 1, 3, and 5 years demonstrated a close alignment with the gray line segment, denoting a commendable concordance between projected and actualized survival probabilities (Figure 7C, 7D). Remarkably, the C-index value and the area under the ROC curve for the risk score surpassed those of alternative clinical traits, indicating the superior predictive accuracy of the devised model in prognosticating patient survival (Figure 7E, 7F). These results highlight the robustness and superior prognostic ability of the developed model, underscoring its potential clinical utility in predicting patient outcomes.
Enrichment analysis of prognostic markers in breast cancer patients
To unravel the functional implications of the identified prognostic markers, we conducted Gene Ontology (GO) enrichment analysis. The differential genes between the two groups were found to participate in diverse biological processes, encompassing ovulatory cycles and endocrine processes. At the cellular level, these genes were associated with blood particle composition and intermediate fibers, while at the molecular level, they exhibited activities such as peptide chain endonuclease activity and hormone functions (Figure 8A).
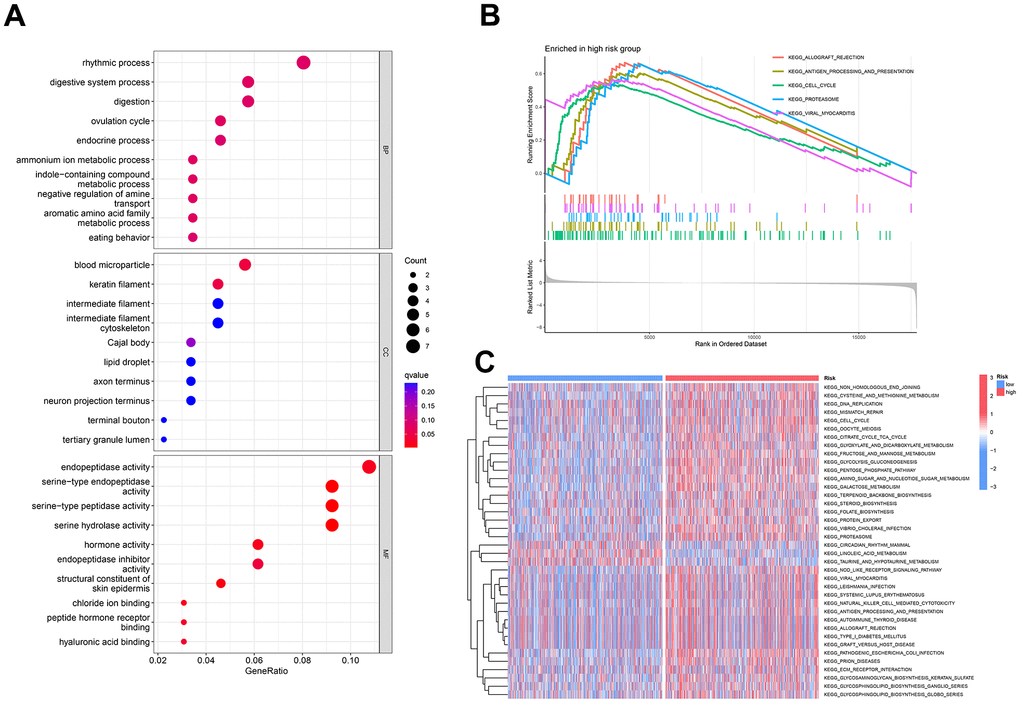
Figure 8. Enrichment analysis of prognostic markers in breast cancer patients. (A) Illustrates the pathway enrichment of differentially expressed genes concerning cellular components, molecular functions, and biological processes. (B) GSEA enrichment analysis reveals five upregulated functional pathways. (C) GSVA is employed to analyze the differential expression of various KEGG pathways between the two risk groups.
Moreover, GSEA highlighted a notable upregulation of pathways associated with immune rejection and antigen processing and presentation within the high-risk group (Figure 8B). For a holistic comprehension of differentially enriched pathways, GSVA enrichment analysis was executed. The outcomes unveiled heightened activity in pathways such as circadian rhythm in mammals, linoleic acid metabolism, and taurine and hypotaurine metabolism within the low-risk group. Conversely, the remaining pathways exhibited augmented activity in the high-risk group (Figure 8C).
These discoveries offer insightful revelations into the potential biological processes and pathways linked to the identified prognostic markers in individuals with breast cancer. The heightened activation of immune-related pathways in the high-risk cohort implies the engagement of immune mechanisms in disease progression. Simultaneously, the divergent activities across pathways between the low and high-risk cohorts underscore potential molecular signatures associated with distinct risk profiles.
Predictive analysis of the correlation between disulfidptosis-related lncRNA model and immune cell infiltration and tumor microenvironment
To probe the interconnection between the disulfidptosis-related lncRNA model and immune cell infiltration, along with the tumor microenvironment, a battery of algorithms, including QUANTISEQ, XCELL, TIMER, EPIC, CIBERSORT, MCPCOUNTER, and CIBERSORT-ABS, was employed. These analyses unveiled both affirmative and adverse correlations between risk scores and specific immune cell types (Figure 9A). CIBERSORT analysis additionally disclosed noteworthy distinctions in the abundance of plasma cells, Macrophages M2, T cells CD4 memory resting, Macrophages M0, T cells CD8, B cells naive, Tregs, T cells CD4 memory activated, Monocytes, and Mast cells resting between the high and low-risk cohorts (Figure 9B). Furthermore, disparities in immune functions, such as Type II IFN Response, were noted between the two risk groups (Figure 9C, 9D).
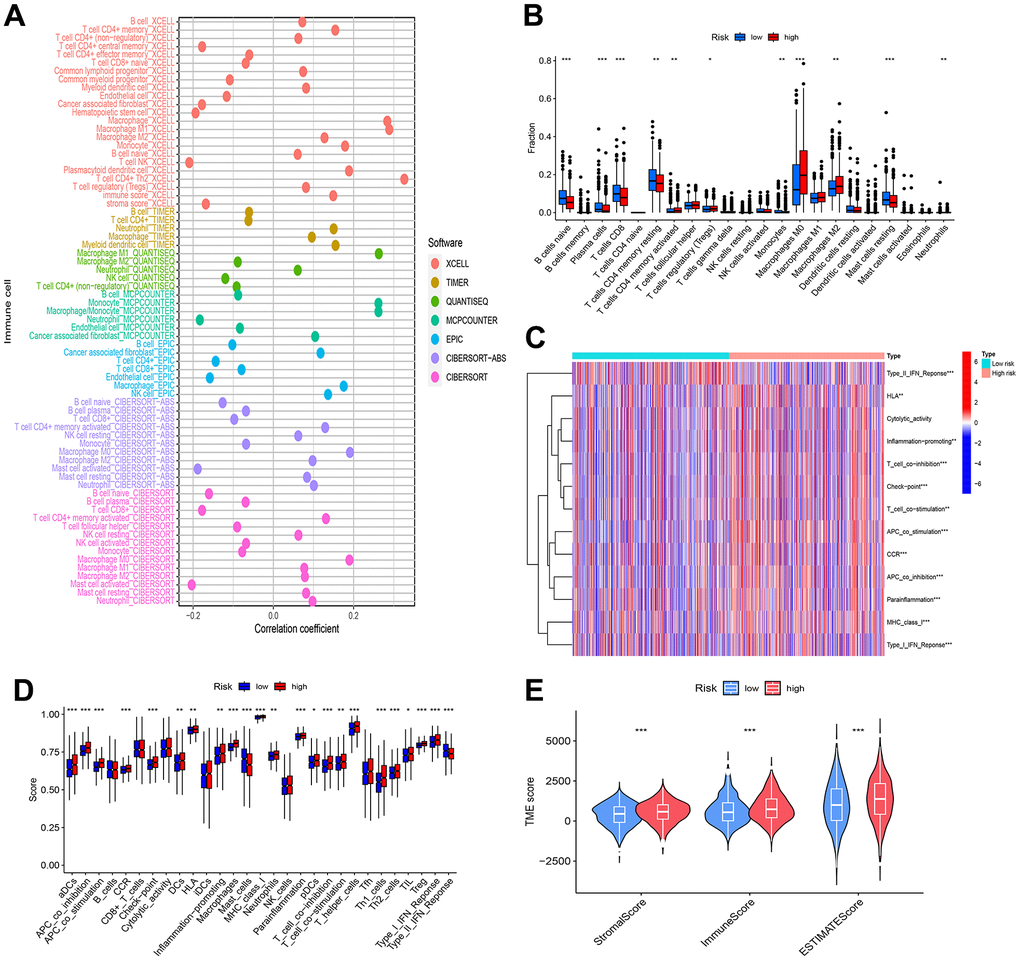
Figure 9. Analysis of immune cell infiltration and tumor microenvironment. (A) Bubble plots were employed for the visualization of immune cell infiltration within the tumor microenvironment. (B) Variances in immune response were evident through differential immune cell infiltration between the high and low-risk groups. (C, D) The high and low-risk groups exhibited significant disparities in immune function, implying modifications in immune activity associated with breast cancer risk. (E) Observable distinctions in tumor microenvironment scores between the high and low-risk groups indicated variations in the composition and characteristics of the tumor microenvironment. Statistical significance was denoted by * (p < 0.05), ** (p < 0.01), and *** (p < 0.001).
In addition, studies of the tumor microenvironment found that both stromal and immune cells were more abundant in the high-risk group than in the low-risk group, which may be related to their poor prognosis (Figure 9E).
While further validation is warranted, these findings suggest that the constructed disulfidptosis-related lncRNA model can offer valuable insights into the relationship between immune cell infiltration, risk scores, and immunotherapy pathways in BRCA.
Disparate analysis of drug sensitivity linked to long non-coding RNAs correlated with Disulfidoptosis-induced mortality
The investigation into disparities in drug sensitivity between cohorts classified as high and low risk represents a potential avenue for refining strategies in breast cancer therapeutics. Our scrutiny disclosed noteworthy distinctions in the IC50 values of 32 pharmaceutical agents among these risk-stratified groups, implying that risk scores could function as predictive benchmarks for discerning drug sensitivity and informing judicious clinical dosing decisions. Notably, BI-2536, Erlotinib, KU-55933, and MG-132 exhibited lower IC50 values in the high-risk group, implying enhanced efficacy of these drugs in treating high-risk breast cancer patients. Conversely, Afuresertib, BMS-345541, GNE-317, Ipatasertib, IWP-2, Leflunomide, Mitoxantrone, and MK-2206 demonstrated relatively higher IC50 values in the high-risk group, suggesting potentially reduced effectiveness and the necessity for higher dosages in high-risk patients. These findings propose the potential utility of risk scores in drug selection and dosage optimization for breast cancer treatment. However, it is crucial to note that further validation is necessary to confirm these results and evaluate the clinical applicability of risk scores in predicting drug sensitivity (Figure 10A–10L).
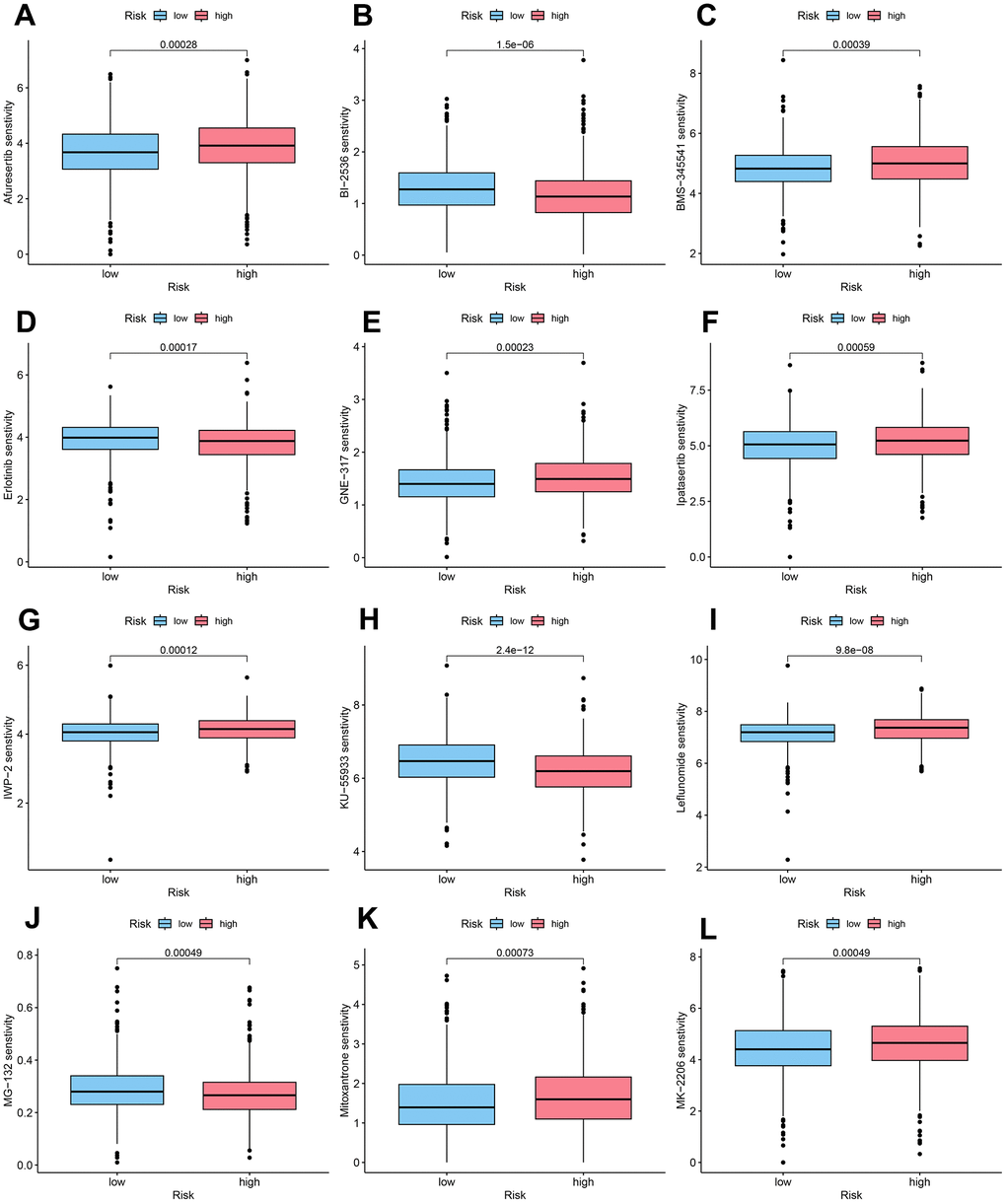
Figure 10. DAL-related drug sensitivity analysis. (A) Afuresertib, (B) BI-2536, (C) BMS-345541, (D) Erlotinib, (E) GNE-317, (F) Ipatasertib, (G) IWP-2, (H) KU-55933, (I) Leflunomide, (J) MG-132, (K) Mitoxantrone, (L) MK-2206.
Comparison of somatic mutations in the low and high-risk groups
We undertook an examination of somatic mutation frequencies in cohorts of breast cancer patients, aiming to discern variances between those deemed low and high risk. Our observations unveiled discernible patterns in the mutation frequencies of pivotal genes. In particular, the incidence of PIK3CA mutations was less frequent in the high-risk category (31%) in contrast to the low-risk subset (38%), while TP53 mutations exhibited a heightened prevalence in the high-risk group (42%) compared to their low-risk counterparts (21%). It is noteworthy that PIK3CA and TP53 emerged as the most frequently mutated genes among individuals with breast cancer (Figure 11A, 11B).
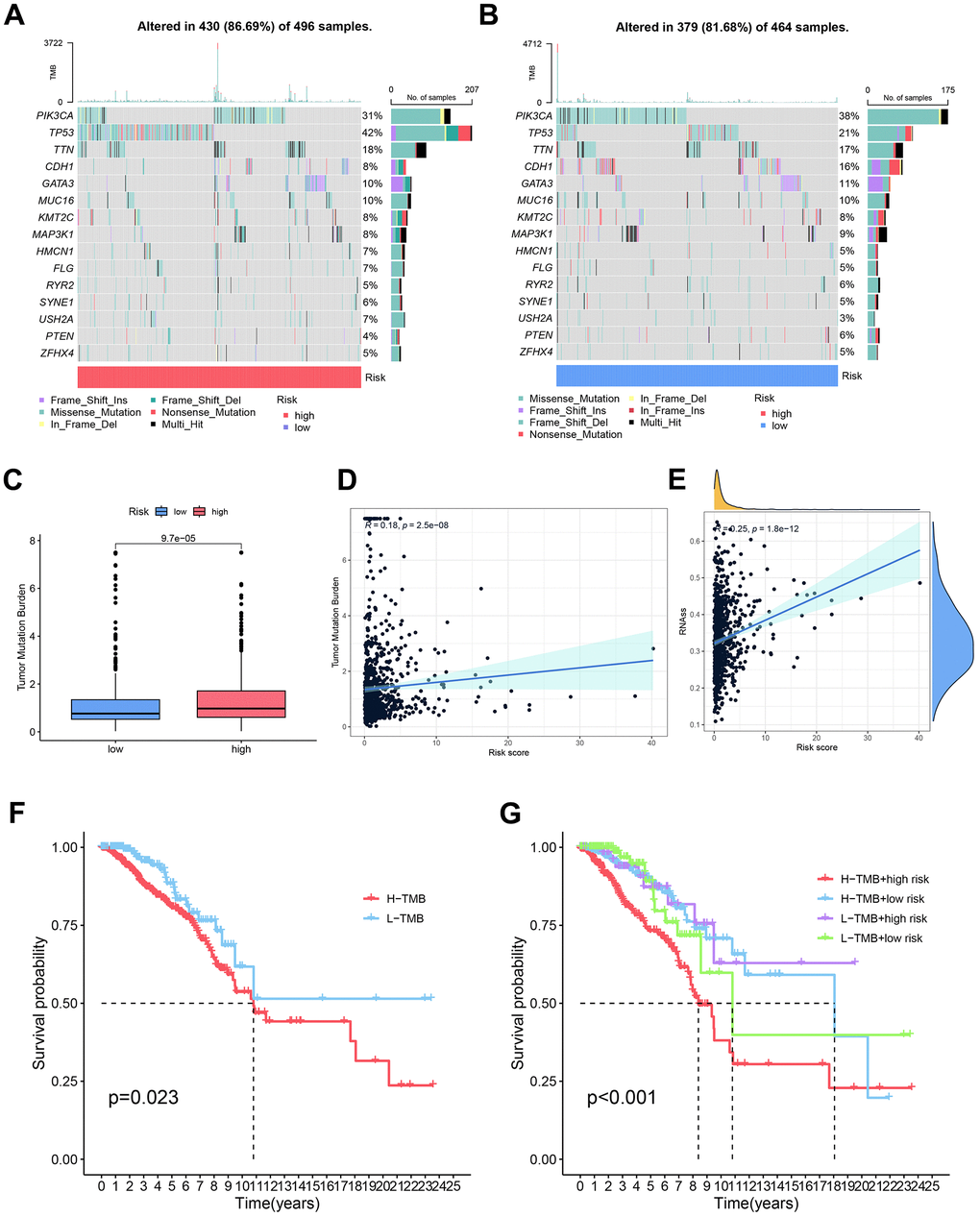
Figure 11. Comparative analysis of somatic mutation rates and influencing factors in high and low-risk groups. (A, B) Comprehensive assessment was undertaken to discern disparities in somatic mutations between the high and low-risk groups. (C) Observable distinctions in tumor mutation burden (TMB) were noted between the high and low-risk groups, signifying variations in the mutational landscape. (D) Investigation into the relationship between TMB and risk scores yielded insights into the connection between mutational burden and breast cancer risk. (E) Examination of the correlation between risk scores and cancer stem cells (CSCs) provided illumination on the potential involvement of CSCs in breast cancer risk. (F) Comparative analysis of survival curves over time between high and low tumor mutation burdens elucidated the impact of mutational burden on patient survival. (G) Survival curves over time were scrutinized for different combinations of tumor mutation burdens and risk scores, emphasizing the synergistic effect of mutational burden and risk scores on patient prognosis.
Moreover, the cohort identified as high-risk demonstrated a markedly elevated tumor mutation load in comparison to their low-risk counterparts. Additionally, a discernible positive correlation emerged between the risk score and the tumor mutation load (R=0.18, P=2.5e-08) (Figure 11C, 11D). Tumor stem cells (DNAss and RNAss) are crucial regulators in the advancement of tumors. We also investigated the relationship between stem cell scores, derived from transcriptomic data, and patient risk, revealing a positive correlation between the two (P=1.8e-12, R=0.25) (Figure 11E).
Survival analysis based on tumor mutational load demonstrated a gradual decrease in patient survival over time. Individuals with a diminished mutation load exhibited a more favorable prognosis compared to those harboring elevated mutation burdens, whereas those with concurrent high-risk scores and heightened tumor mutation burden demonstrated the most unfavorable prognosis (P<0.01) (Figure 11F, 11G).
Discussion
Breast carcinoma (BC) stands as the most widespread malignancy impacting women on a global scale [29], constituting roughly 15% of all newly diagnosed cases [30]. Extensive evidence indicates that the effectiveness of conventional approaches such as chemotherapy, surgery, or radiotherapy in treating metastatic breast cancer remains consistently low [31]. Henceforth, the imperative lies in crafting multi-gene composite models capable of precision in prognosticating outcomes for breast cancer patients.
Gan Boyi et al. recently introduced an innovative form of cellular demise termed disulfidptosis, which distinguishes itself from other mechanisms of cell death by relying on the intracellular accumulation of disulfidptosis for binding abnormal disulfidptosis to actin and cytoskeletal proteins [12]. This unique mode of cell death holds potential for effective induction in cancer treatment, thus presenting a promising avenue for targeted cancer therapy. Simultaneously, as cancer research progresses, the significance of lncRNAs in tumorigenesis is increasingly acknowledged. Notably, certain investigations have demonstrated that the abundance of lncRNAs even surpasses that of proteins in diseases like acute granulocytic leukemia and T acute lymphoblastic leukemia, underscoring the pivotal role of lncRNAs in cancer prediction [32].
Building upon the significant implications of disulfidptosis-induced cell death and the pivotal role of lncRNAs in tumor biology, we developed a model comprising eight differentially expressed lncRNAs (DALs) with the objective of identifying their positive prognostic significance in patients with breast cancer (BRCA). Our approach involved employing Cox analysis in conjunction with LASSO regression cross-validation to pinpoint eight DALs (MIR4435-2HG, AL139035.1, AL451085.2, AP001160.1, NRAV, AC015922.2, YTHDF3-AS1, TP53TG1) that exhibited independent predictive power, forming the basis of our model construction.
Among these eight DALs, antecedent data suggests that MIR4435-2HG, AL451085.2, NRAV, AC015922.2, YTHDF3, and TP53TG1 exhibit potential as prognostic indicators in cancer. Conversely, additional scrutiny is warranted to validate the prognostic significance of AL139035.1 and AP001160.1. Remarkably, the aberration in MIR4435-2HG has been linked to an adverse prognosis in breast cancer, potentially amplifying cellular proliferation and invasiveness through its interaction with β-linked proteins [33]. Moreover, our discoveries correspond with prior investigations, affirming that elevated expression levels of AL451085.2 correlate with a propitious prognosis in patients [34].
NRAV, acknowledged as an immune-associated lncRNA and a pivotal mediator of antiviral innate immunity, has been implicated in diverse malignancies, including endometrial and breast cancers, gastric cancer, and glioma, elucidated by interactive molecular pathway analyses [35, 36]. The heightened expression of AC015922.2 has been discerned as a prognostic determinant in populations characterized by elevated risk [37]. In addition, YTHDF3 destabilizes lncRNA GAS5, thereby promoting the progression of colorectal cancer [38]. On the other hand, our investigation revealed TP53TG1 to be a safeguarding element. Previous investigations have demonstrated a significant association between TP53TG1 and lymph node infiltration in breast cancer patients, suggesting its potential oncogenic role in the official cavity subtype of breast cancer [39], possibly due to its characteristic expression in a specific subtype. Overall, our model based on these eight DALs holds promise for early detection and prognostic prediction in BRCA patients, highlighting the potential clinical significance of these lncRNAs in the management of breast cancer.
Upon careful examination of the survival curves, a distinct divergence in the survival rates becomes evident over time between the groups categorized as high and low-risk. This differentiation is further supported by the successful application of principal component analysis, which effectively distinguishes these two risk groups. Moreover, the ROC curves, especially those pertaining to time points 1, 3, and 5, attest to the heightened accuracy of the devised model.
To augment the clinical significance and practicality of our investigation, we formulated and authenticated nomograms encompassing various clinical parameters (each possessing autonomous predictive efficacy, with the exception of gender) and the exceedingly precise risk scores derived from the DAL model. Comparing the C-index and the area under the curve (AUC) for each indicator reveals that the risk scores obtained from our DAL model outperform other existing clinical indicators as better prognostic predictors. This integration of clinical indicators and risk scores through nomograms offers a more comprehensive and robust approach for prognostic assessment in clinical practice.
Moreover, we conducted GO and GSEA to explore the functional characteristics of genes exhibiting differential expression between the low and high-risk groups. Our findings suggest the engagement of these disparately expressed genes in pivotal biological processes, including the ovulatory cycle, endocrine mechanisms, and hormone secretion. As we all know, drugs can act on estrogen receptor (ER) and progesterone receptor (PR) to achieve endocrine therapy for breast cancer, which shows the important role of hormones in breast cancer. The enrichment further illustrates that the prognostic predictor genes we screened play a regulatory role in endocrinology. Meanwhile, we found that the high-risk group was up-regulated in allograft rejection and antigen processing and presentation. Combined with the immune correlation analysis, it is not difficult to see that the high-risk group has higher expression of many immune functions compared with the low-risk group, which to a certain extent confirms why HER-2 positive and triple-negative breast cancers, usually have higher immune cell infiltration and worse prognosis [40]. This suggests the essential role of these genes in regulating and participating in these specific physiological processes.
The outcomes of our investigation propose a close linkage between breast cancer and endocrine regulation. Tumor cells demonstrate proficiency in orchestrating their microenvironment through autocrine and paracrine secretions, thereby fostering their autonomous sustenance, proliferation, and maturation [41]. Given this perspective, we delved more profoundly into the breast cancer tumor microenvironment, frequently denoted as the tumor microenvironment, enshrining the cellular milieu where tumor or cancer stem cells reside, concomitant with the pivotal involvement of immune cells [42]. Our investigation extended to analyzing the immune infiltration patterns of the eight differentially expressed lncRNAs (DALs) in relation to tumor cells. We identified remarkable variances in the composition of immune cells between the high and low-risk cohorts. In the high-risk group, there was a downregulation observed in plasma cells, T cells CD8, Monocytes, Mast cells resting, B cells naïve, and T cells CD4 memory resting. Conversely, T cells CD4 memory activated, Macrophages M2, Neutrophils, Tregs, and Macrophages M0 exhibited an upregulation in the high-risk group. Notably, naive B cells, which constitute the majority of B cells (accounting for 65%) in normal breast tissue [43], exhibited significant downregulation in the high-risk group. This reduction could be ascribed to the transformation of naïve B cells into alternative immune cell types under the influence of myeloid-derived suppressor cells (MDSCs) associated with breast cancer [44, 45]. These converted naive B cells may acquire immunosuppressive properties, thereby suppressing T cell responses. Regarding plasma cells, studies provide conflicting evidence regarding their role in breast cancer prognosis. Some suggest that high expression of plasma cell subgenes correlates with a favorable prognosis, while others propose a promotive role, potentially indicating variations in expression depending on tumor type and subtype [46]. In the realm of invasive ductal carcinoma of the breast, the existence of tumor-infiltrating lymphocytes, notably comprising CD8+ cells, has been recognized as a favorable prognostic indicator, exerting a substantial influence within the tumor microenvironment [47]. Tumor-associated macrophages (TAMs), a subset of monocytes, have been recognized as important monocytes within the breast cancer microenvironment, contributing to cancer metastasis to distant sites [48]. Notably, resting mast cells within fully developed breast tumors have been suggested to impede the progression of breast cancer [49].
In the high-risk cohort, we noted elevated expression levels of activated CD4+ memory T cells in contrast to the low-risk group. This observation implies that CD4+ memory T cells were prompted and stimulated to sustain an immune response [50]. A study undertaken by Olkhanud et al. underscored the function of regulatory B cells (Bregs) in transforming quiescent CD25-, CD4+ T cells into CD25+, Foxp3+, CD4+ regulatory T cells (Tregs) among breast cancer patients [51]. Consequently, the upregulation of Tregs in breast cancer patients may be attributed to this conversion process. The elevation of M0 macrophages in the high-risk cohort can be ascribed to the existence of circulating macrophages within the tumor microenvironment. These macrophages have the potential to modify the tumor microenvironment, consequently fostering tumor progression [52]. Additionally, cancer-associated fibroblasts (CAFs) in invasive breast cancer have been reported to facilitate the induction of monocytes into M2 macrophages [53, 54]. This implies that SAA1 might play a role in the immunosuppressive effects evident in individuals with breast cancer. These results underscore the intricate interplay between immune cells and the tumor microenvironment in breast cancer. The activation and modulation of specific immune cell populations, including CD4+ memory T cells, Tregs, macrophages, and neutrophils, can profoundly impact the advancement and prognosis of breast cancer. A comprehension of these immune interactions offers valuable insights into the mechanisms governing tumor development and may offer guidance for the formulation of immunotherapeutic strategies in breast cancer treatment.
Our findings indicate that both the immune score and the stromal score were higher in the high-risk group compared to the low-risk group, an observation that suggests that both the stromal microenvironment and the immune microenvironment have a strong influence on the high-risk phenotype of breast cancer patients. It is firmly established that the interplay between stromal and epithelial constituents of the breast, facilitated by paracrine interactions, can sustain the growth and progression of breast tumors [55]. The established understanding dictates that the dynamic interplay between the stromal and epithelial constituents of the breast, facilitated through paracrine interactions, possesses the capacity to perpetuate the growth and progression of breast tumors. Significantly, accumulating evidence posits that the intercellular communication dynamics between stromal constituents and triple-negative breast cancer (TNBC) cells may contribute to fostering tumor growth, metastatic extravasation, and the colonization of TNBC tumors [56]. The stromal microenvironment appears to exert significant effects on various aspects of tumor growth and metastasis. These findings emphasize the importance of considering the stromal component in understanding breast cancer biology and its clinical implications. Targeting the stromal-epithelial interactions and modulating the tumor microenvironment may hold potential for therapeutic interventions aimed at restraining tumor growth and metastasis among breast cancer patients at high risk.
Breast cancer is generally not associated with a high tumor mutational burden (TMB). However, our findings indicate a trend of increasing TMB with higher risk scores, and there are variations in TMB among different breast cancer subtypes [57]. In both cohorts, PIK3CA and TP53 emerged as the most frequently mutated genes. Remarkably, the low-risk cohort displayed a heightened incidence of PIK3CA mutations compared to the high-risk cohort. PIK3CA mutations are renowned for instigating the PI3K-AKT-mTOR pathway, a pivotal regulator of cellular growth and survival frequently implicated in breast cancer [58]. The diminished occurrence of PIK3CA mutations in the high-risk demographic could imply that these mutations are primarily prevalent in the initial stages of breast carcinoma, such as ductal carcinoma in situ [59]. Alternatively, this observation might signify the pre-existence of an activated PI3K-AKT-mTOR pathway in high-risk patients, thereby contributing to their heightened risk. Conversely, TP53 demonstrated an augmented mutation rate in the high-risk cohort in contrast to the low-risk group, suggesting a plausible involvement of TP53 in the progression of breast cancer. TP53 mutations are linked to genomic instability and therapy resistance, and the heightened frequency of TP53 mutations in the high-risk category could contribute to their unfavorable prognosis [60]. These discoveries underscore the significance of discerning distinct genetic modifications, particularly PIK3CA and TP53 mutations, within varied risk cohorts of breast cancer patients. The presence of these mutations may have implications for prognosis, therapeutic response, and potential targeted treatment strategies. Further investigations into the molecular mechanisms driven by these mutations could provide valuable insights for personalized management of breast cancer patients.
Elevated tumor mutation burden (TMB) and augmented risk scores were linked to diminished patient survival in breast cancer, establishing their potential as pivotal prognostic indicators. The identified association between high-risk breast cancer cohorts and heightened TMB suggests a potential utility of immunotherapeutic modalities, encompassing immune checkpoint inhibitors, for these patients. Strategically targeting the immune milieu in high-risk cases holds promise for augmenting treatment efficacy. Furthermore, the positive correlation between risk scores and stem cell abundance intimates a potentially higher prevalence of cancer stem cells in high-risk breast cancer patients. Cancer stem cells, acknowledged for their implication in cancer advancement, resistance to therapies, and adverse prognoses, accentuate the intricacies of the disease. This finding further emphasizes the importance of considering risk scores in personalized treatment decision-making for breast cancer patients, as it could help identify patients who may require more aggressive treatment approaches or novel therapies targeting cancer stem cells [61].
It has been suggested that leflunomide can inhibit mitochondrial fission in triple-negative breast cancer, and has also been shown to inhibit breast cancer growth in combination with doxorubicin [62, 63]. The difference in sensitivity of different drugs in high and low-risk groups provides doctors with clinical recommendations for more refined treatment, but more research is needed to further confirm the use of specific drugs.
In summary, the results suggest that risk scores hold valuable information for predicting drug sensitivity, tumor mutation load, stem cell content, and patient prognosis in breast cancer. Incorporating risk scores into clinical decision-making could assist in tailoring treatment strategies to individual patients. However, additional validation and prospective studies are necessary to confirm these findings and assess their clinical utility in a broader patient population.
Despite the clinical significance of our study in providing accurate prognostic predictions for BRCA patients and informing treatment decisions, there are several notable limitations that should be acknowledged. Firstly, individual variations among BRCA patients may have an impact on the expression patterns of the eight identified DALs, and it is challenging to completely eliminate these confounding factors. Future studies could consider incorporating additional patient-specific factors to enhance the precision of prognostic predictions. Due to the lack of comprehensive lncRNA information in other datasets such as GEO and ICGC, our study mainly utilized random group validation within the TCGA dataset. Despite diligent attempts to corroborate our results with external datasets, the persistent absence of consistent and precise lncRNA data across diverse platforms and cohorts poses an ongoing challenge. Subsequent studies ought to aspire towards the inclusion of larger, more diverse datasets to authenticate and broaden the scope of our findings. At the same time, this study lacked experimental validation of the constructed genes, and more experimental studies are still needed to exemplify the prediction of DAL for the difference in prognostic level of breast cancer. Notwithstanding these constraints, our inquiry imparts valuable insights into the prognostic relevance of the identified DALs in BRCA patients. The prospects for future research lie in the refinement of data collection and analysis methodologies to surmount these limitations, thereby amplifying the clinical utility of our discoveries.
Conclusions
In summary, our research underscores the prospective significance of lncRNAs associated with disulfidptosis in serving as prognostic indicators and therapeutic targets for breast cancer. Integrating eight identified DALs as prognostic benchmarks can enhance the accuracy of prognosis prediction for BRCA patients. This insight offers hope for clinicians to devise personalized treatment strategies (including chemotherapy and immunotherapy) based on an understanding of the immune environment and unique subtypes of breast cancer. Further follow-up studies and clinical validation are needed to thoroughly ascertain the effectiveness of their clinical assessment.
Author Contributions
CJ, SZ, LJ, ZC, HC, GY and SH conceived the study. CJ, SZ, LJ, ZC, HC, GY and SH drafted the manuscript. CJ, SZ, LJ, JH and HQC performed the literature search and collected the data. CJ, SZ, LJ, ZC, XZ, JT, XG, HC, GY and SH analyzed and visualized the data. CJ, SZ, LJ, ZC, XZ, JT, XG, HC, GY and SH completed in vitro experiments. CJ, SZ, LJ, ZC, HQC, JH, XZ, JT, XG, HC, GY and SH helped with the final revision of this manuscript. All authors reviewed and approved the final manuscript.
Acknowledgments
All authors acknowledge the contributions from the TCGA project.
Conflicts of Interest
The authors affirm that this research was performed without any existing or potential conflicts of interest in terms of commercial or financial relationships.
Ethical Statement
The data used in this study originated from public databases and there are no ethically related issues.
Funding
This study was granted by the Doctoral Startup Fund of the Affiliated Hospital of Southwest Medical University (No. 19025), Innovation and Entrepreneurship Training Program of Southwest Medical University (No. 2023393), and the Science and Technology Strategic Cooperation Programs of Luzhou Municipal People’s Government and Southwest Medical University (No.2023LZXNYDJ033).
References
- 1. Qiu Y, Liu Y, Li WH, Zhang HQ, Tian XX, Fang WG. P2Y2 receptor promotes the migration and invasion of breast cancer cells via EMT-related genes Snail and E-cadherin. Oncol Rep. 2018; 39:138–50. https://doi.org/10.3892/or.2017.6081 [PubMed]
- 2. Maurizi A, Ciocca M, Giuliani C, Di Carlo I, Teti A. Role of Neural (N)-Cadherin in Breast Cancer Cell Stemness and Dormancy in the Bone Microenvironment. Cancers (Basel). 2022; 14:1317. https://doi.org/10.3390/cancers14051317 [PubMed]
- 3. Jiang C, Zhang S, Jiang L, Chen Z, Chen H, Huang J, Tang J, Luo X, Yang G, Liu J, Chi H. Precision unveiled: Synergistic genomic landscapes in breast cancer-Integrating single-cell analysis and decoding drug toxicity for elite prognostication and tailored therapeutics. Environ Toxicol. 2024; 39:3448–72. https://doi.org/10.1002/tox.24205 [PubMed]
- 4. Huang G, Zhou J, Chen J, Liu G. Identification of pyroptosis related subtypes and tumor microenvironment infiltration characteristics in breast cancer. Sci Rep. 2022; 12:10640. https://doi.org/10.1038/s41598-022-14897-1 [PubMed]
- 5. Yahalom G, Weiss D, Novikov I, Bevers TB, Radvanyi LG, Liu M, Piura B, Iacobelli S, Sandri MT, Cassano E, Allweis TM, Bitterman A, Engelman P, et al. An Antibody-based Blood Test Utilizing a Panel of Biomarkers as a New Method for Improved Breast Cancer Diagnosis. Biomark Cancer. 2013; 5:71–80. https://doi.org/10.4137/BIC.S13236 [PubMed]
- 6. Huang G, Xiao S, Jiang Z, Zhou X, Chen L, Long L, Zhang S, Xu K, Chen J, Jiang B. Machine learning immune-related gene based on KLRB1 model for predicting the prognosis and immune cell infiltration of breast cancer. Front Endocrinol (Lausanne). 2023; 14:1185799. https://doi.org/10.3389/fendo.2023.1185799 [PubMed]
- 7. Tellez-Gabriel M, Knutsen E, Perander M. Current Status of Circulating Tumor Cells, Circulating Tumor DNA, and Exosomes in Breast Cancer Liquid Biopsies. Int J Mol Sci. 2020; 21:9457. https://doi.org/10.3390/ijms21249457 [PubMed]
- 8. McVeigh TP, Kerin MJ. Clinical use of the Oncotype DX genomic test to guide treatment decisions for patients with invasive breast cancer. Breast Cancer (Dove Med Press). 2017; 9:393–400. https://doi.org/10.2147/BCTT.S109847 [PubMed]
- 9. Bisheshar SK, De Ruiter EJ, Devriese LA, Willems SM. The prognostic role of NK cells and their ligands in squamous cell carcinoma of the head and neck: a systematic review and meta-analysis. Oncoimmunology. 2020; 9:1747345. https://doi.org/10.1080/2162402X.2020.1747345 [PubMed]
- 10. Shan Z, Wu W, Yan X, Yang Y, Luo D, Liu Q, Li X, Goel A, Ma Y. A novel epithelial-mesenchymal transition molecular signature predicts the oncological outcomes in colorectal cancer. J Cell Mol Med. 2021; 25:3194–204. https://doi.org/10.1111/jcmm.16387 [PubMed]
- 11. Chi H, Huang J, Yan Y, Jiang C, Zhang S, Chen H, Jiang L, Zhang J, Zhang Q, Yang G, Tian G. Unraveling the role of disulfidptosis-related LncRNAs in colon cancer: a prognostic indicator for immunotherapy response, chemotherapy sensitivity, and insights into cell death mechanisms. Front Mol Biosci. 2023; 10:1254232. https://doi.org/10.3389/fmolb.2023.1254232 [PubMed]
- 12. Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, Olszewski K, Horbath A, Chen X, Lei G, Mao C, Wu S, Zhuang L, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023; 25:404–14. https://doi.org/10.1038/s41556-023-01091-2 [PubMed]
- 13. Tang J, Peng X, Xiao D, Liu S, Tao Y, Shu L. Disulfidptosis-related signature predicts prognosis and characterizes the immune microenvironment in hepatocellular carcinoma. Cancer Cell Int. 2024; 24:19. https://doi.org/10.1186/s12935-023-03188-y [PubMed]
- 14. Hur K, Kim SH, Kim JM. Potential Implications of Long Noncoding RNAs in Autoimmune Diseases. Immune Netw. 2019; 19:e4. https://doi.org/10.4110/in.2019.19.e4 [PubMed]
- 15. Chi H, Peng G, Wang R, Yang F, Xie X, Zhang J, Xu K, Gu T, Yang X, Tian G. Cuprotosis Programmed-Cell-Death-Related lncRNA Signature Predicts Prognosis and Immune Landscape in PAAD Patients. Cells. 2022; 11:3436. https://doi.org/10.3390/cells11213436 [PubMed]
- 16. Huang J, Liu M, Chen H, Zhang J, Xie X, Jiang L, Zhang S, Jiang C, Zhang J, Zhang Q, Yang G, Chi H, Tian G. Elucidating the Influence of MPT-driven necrosis-linked LncRNAs on immunotherapy outcomes, sensitivity to chemotherapy, and mechanisms of cell death in clear cell renal carcinoma. Front Oncol. 2023; 13:1276715. https://doi.org/10.3389/fonc.2023.1276715 [PubMed]
- 17. Huang G, Cao H, Liu G, Chen J. Role of androgen receptor signaling pathway-related lncRNAs in the prognosis and immune infiltration of breast cancer. Sci Rep. 2022; 12:20631. https://doi.org/10.1038/s41598-022-25231-0 [PubMed]
- 18. Amorim M, Salta S, Henrique R, Jerónimo C. Decoding the usefulness of non-coding RNAs as breast cancer markers. J Transl Med. 2016; 14:265. https://doi.org/10.1186/s12967-016-1025-3 [PubMed]
- 19. Huang X, Chi H, Gou S, Guo X, Li L, Peng G, Zhang J, Xu J, Nian S, Yuan Q. An Aggrephagy-Related LncRNA Signature for the Prognosis of Pancreatic Adenocarcinoma. Genes (Basel). 2023; 14:124. https://doi.org/10.3390/genes14010124 [PubMed]
- 20. Tan M, Huang G, Chen J, Yi J, Liu X, Liao N, Hu Y, Zhou W, Guo Q. Construction and validation of an eight pyroptosis-related lncRNA risk model for breast cancer. Am J Transl Res. 2022; 14:2779–800. [PubMed]
- 21. Wang YQ, Huang G, Chen J, Cao H, Xu WT. LncRNA SNHG6 promotes breast cancer progression and epithelial-mesenchymal transition via miR-543/LAMC1 axis. Breast Cancer Res Treat. 2021; 188:1–14. https://doi.org/10.1007/s10549-021-06190-y [PubMed]
- 22. Li Y, Jiang B, Wu X, Huang Q, Chen W, Zhu H, Qu X, Xie L, Ma X, Huang G. Long non-coding RNA MIAT is estrogen-responsive and promotes estrogen-induced proliferation in ER-positive breast cancer cells. Biochem Biophys Res Commun. 2018; 503:45–50. https://doi.org/10.1016/j.bbrc.2018.05.146 [PubMed]
- 23. Yao Z, Zhu G, Too J, Duan M, Wang Z. Feature Selection of OMIC Data by Ensemble Swarm Intelligence Based Approaches. Front Genet. 2022; 12:793629. https://doi.org/10.3389/fgene.2021.793629 [PubMed]
- 24. Yao Z, Li F, Xie W, Chen J, Wu J, Zhan Y, Wu X, Wang Z, Zhang G. DeepSF-4mC: A deep learning model for predicting DNA cytosine 4mC methylation sites leveraging sequence features. Comput Biol Med. 2024; 171:108166. https://doi.org/10.1016/j.compbiomed.2024.108166 [PubMed]
- 25. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. https://doi.org/10.1186/1471-2105-14-7 [PubMed]
- 26. Geeleher P, Cox NJ, Huang RS. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014; 15:R47. https://doi.org/10.1186/gb-2014-15-3-r47 [PubMed]
- 27. Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018; 28:1747–56. https://doi.org/10.1101/gr.239244.118 [PubMed]
- 28. Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, Schuetze S, Alva A, Siddiqui J, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017; 548:297–303. https://doi.org/10.1038/nature23306 [PubMed]
- 29. Shi W, Jin X, Wang Y, Zhang Q, Yang L. High serum exosomal long non-coding RNA DANCR expression confers poor prognosis in patients with breast cancer. J Clin Lab Anal. 2022; 36:e24186. https://doi.org/10.1002/jcla.24186 [PubMed]
- 30. Cui Y, Xu Y, Li Y, Sun Y, Hu J, Jia J, Li X. Antibody Drug Conjugates of Near-Infrared Photoimmunotherapy (NIR-PIT) in Breast Cancers. Technol Cancer Res Treat. 2023; 22:15330338221145992. https://doi.org/10.1177/15330338221145992 [PubMed]
- 31. Peng XC, Yang L, Yang LP, Mao YQ, Yang HS, Liu JY, Zhang DM, Chen LJ, Wei YQ. Efficient inhibition of murine breast cancer growth and metastasis by gene transferred mouse survivin Thr34-->Ala mutant. J Exp Clin Cancer Res. 2008; 27:46. https://doi.org/10.1186/1756-9966-27-46 [PubMed]
- 32. Wang J, Zhang X, Chen W, Li J, Liu C. CRlncRNA: a manually curated database of cancer-related long non-coding RNAs with experimental proof of functions on clinicopathological and molecular features. BMC Med Genomics. 2018 (Suppl 6); 11:114. https://doi.org/10.1186/s12920-018-0430-2 [PubMed]
- 33. Bárcenas-López DA, Núñez-Enríquez JC, Hidalgo-Miranda A, Beltrán-Anaya FO, May-Hau DI, Jiménez-Hernández E, Bekker-Méndez VC, Flores-Lujano J, Medina-Sansón A, Tamez-Gómez EL, López-García VH, Lara-Ramos JR, Núñez-Villegas NN, et al. Transcriptome Analysis Identifies LINC00152 as a Biomarker of Early Relapse and Mortality in Acute Lymphoblastic Leukemia. Genes (Basel). 2020; 11:302. https://doi.org/10.3390/genes11030302 [PubMed]
- 34. Gao L, Li Q. Identification of Novel Pyroptosis-Related lncRNAs Associated with the Prognosis of Breast Cancer Through Interactive Analysis. Cancer Manag Res. 2021; 13:7175–86. https://doi.org/10.2147/CMAR.S325710 [PubMed]
- 35. Gao C, Zhou G, Cheng M, Feng L, Cao P, Zhou G. Identification of senescence-associated long non-coding RNAs to predict prognosis and immune microenvironment in patients with hepatocellular carcinoma. Front Genet. 2022; 13:956094. https://doi.org/10.3389/fgene.2022.956094 [PubMed]
- 36. Wu X, Deng Z, Liao X, Ruan X, Qu N, Pang L, Shi X, Qin S, Jiang H. Establishment of Prognostic Signatures of N6-Methyladenosine-Related lncRNAs and Their Potential Functions in Hepatocellular Carcinoma Patients. Front Oncol. 2022; 12:865917. https://doi.org/10.3389/fonc.2022.865917 [PubMed]
- 37. Jia H, Hao S, Cao M, Wang L, Bai H, Shui W, Yang X. m6A-Related lncRNAs Are Potential Prognostic Biomarkers of Cervical Cancer and Affect Immune Infiltration. Dis Markers. 2022; 2022:8700372. https://doi.org/10.1155/2022/8700372 [PubMed]
- 38. Hu Y, Tang J, Xu F, Chen J, Zeng Z, Han S, Wang F, Wang D, Huang M, Zhao Y, Huang Y, Zhuo W, Zhao G. A reciprocal feedback between N6-methyladenosine reader YTHDF3 and lncRNA DICER1-AS1 promotes glycolysis of pancreatic cancer through inhibiting maturation of miR-5586-5p. J Exp Clin Cancer Res. 2022; 41:69. https://doi.org/10.1186/s13046-022-02285-6 [PubMed]
- 39. Motalebzadeh J, Eskandari E. Comprehensive analysis of DRAIC and TP53TG1 in breast cancer luminal subtypes through the construction of lncRNAs regulatory model. Breast Cancer. 2022; 29:1050–66. https://doi.org/10.1007/s12282-022-01385-7 [PubMed]
- 40. Mehani B, Asanigari S, Chung HJ, Dazelle K, Singh A, Hannenhalli S, Aldape K. Immune cell gene expression signatures in diffuse glioma are associated with IDH mutation status, patient outcome and malignant cell state, and highlight the importance of specific cell subsets in glioma biology. Acta Neuropathol Commun. 2022; 10:19. https://doi.org/10.1186/s40478-022-01323-w [PubMed]
- 41. Zhang H, Duan J, Wu O. The expression of macrophage migration inhibitory factor in the non-small cell lung cancer. Saudi J Biol Sci. 2020; 27:1527–32. https://doi.org/10.1016/j.sjbs.2020.04.027 [PubMed]
- 42. Wu X, Bai Z. Multi-omics analysis of m6A modification-related patterns based on m6A regulators and tumor microenvironment infiltration in lung adenocarcinoma. Sci Rep. 2021; 11:20921. https://doi.org/10.1038/s41598-021-00272-z [PubMed]
- 43. Feng X, Zhang T, Chou J, Liu L, Miller LD, Sullivan CA, Browne JD. Comprehensive gene cluster analysis of head and neck squamous cell carcinoma TCGA RNA-seq data defines B cell immunity-related genes as a robust survival predictor. Head Neck. 2022; 44:443–52. https://doi.org/10.1002/hed.26944 [PubMed]
- 44. Lee-Chang C, Rashidi A, Miska J, Zhang P, Pituch KC, Hou D, Xiao T, Fischietti M, Kang SJ, Appin CL, Horbinski C, Platanias LC, Lopez-Rosas A, et al. Myeloid-Derived Suppressive Cells Promote B cell-Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol Res. 2019; 7:1928–43. https://doi.org/10.1158/2326-6066.CIR-19-0240 [PubMed]
- 45. Xiong J, Chi H, Yang G, Zhao S, Zhang J, Tran LJ, Xia Z, Yang F, Tian G. Revolutionizing anti-tumor therapy: unleashing the potential of B cell-derived exosomes. Front Immunol. 2023; 14:1188760. https://doi.org/10.3389/fimmu.2023.1188760 [PubMed]
- 46. Yeong JCT, Lim JC, Lee B, Li H, Chia N, Ong CCH, Lye WK, Putti TC, Dent R, Lim E, Thike AA, Tan PH, Iqbal J. High Densities of Tumor-Associated Plasma Cells Predict Improved Prognosis in Triple Negative Breast Cancer. Front Immunol. 2018; 9:1209. https://doi.org/10.3389/fimmu.2018.01209 [PubMed]
- 47. Biylgi O, Karagöz B, Türken O, Gültepe M, Özgün A, Tunçel T, Emirzeoğlu L, Çelik S, Müftüoğlu T, Gökhan Kandemir E. CD4+CD25(high), CD8+CD28- cells and thyroid autoantibodies in breast cancer patients. Cent Eur J Immunol. 2014; 39:338–44. https://doi.org/10.5114/ceji.2014.45945 [PubMed]
- 48. Li Z, Wang D, Wang W, Chen X, Tang A, Hou P, Li M, Zheng J, Bai J. Macrophages-stimulated PRMT1-mediated EZH2 methylation promotes breast cancer metastasis. Biochem Biophys Res Commun. 2020; 533:679–84. https://doi.org/10.1016/j.bbrc.2020.10.037 [PubMed]
- 49. He L, Zhu Z, Chen S, Wang Y, Gu H. Mammary tumor growth and metastasis are reduced in c-Kit mutant Sash mice. Cancer Med. 2016; 5:1292–7. https://doi.org/10.1002/cam4.696 [PubMed]
- 50. Li Q, Yang H, Wang P, Liu X, Lv K, Ye M. XGBoost-based and tumor-immune characterized gene signature for the prediction of metastatic status in breast cancer. J Transl Med. 2022; 20:177. https://doi.org/10.1186/s12967-022-03369-9 [PubMed]
- 51. Chen T, Song D, Min Z, Wang X, Gu Y, Wei B, Yao J, Chen K, Jiang Z, Xie H, Zhou L, Zheng S. Perioperative dynamic alterations in peripheral regulatory T and B cells in patients with hepatocellular carcinoma. J Transl Med. 2012; 10:14. https://doi.org/10.1186/1479-5876-10-14 [PubMed]
- 52. Zhang SC, Hu ZQ, Long JH, Zhu GM, Wang Y, Jia Y, Zhou J, Ouyang Y, Zeng Z. Clinical Implications of Tumor-Infiltrating Immune Cells in Breast Cancer. J Cancer. 2019; 10:6175–84. https://doi.org/10.7150/jca.35901 [PubMed]
- 53. Zhai X, Chen X, Wan Z, Ge M, Ding Y, Gu J, Hua J, Guo D, Tan M, Xu D. Identification of the novel therapeutic targets and biomarkers associated of prostate cancer with cancer-associated fibroblasts (CAFs). Front Oncol. 2023; 13:1136835. https://doi.org/10.3389/fonc.2023.1136835 [PubMed]
- 54. Niu X, Yin L, Yang X, Yang Y, Gu Y, Sun Y, Yang M, Wang Y, Zhang Q, Ji H. Serum amyloid A 1 induces suppressive neutrophils through the Toll-like receptor 2-mediated signaling pathway to promote progression of breast cancer. Cancer Sci. 2022; 113:1140–53. https://doi.org/10.1111/cas.15287 [PubMed]
- 55. Quinn AL, Burak WE
Jr , Brueggemeier RW. Effects of matrix components on aromatase activity in breast stromal cells in culture. J Steroid Biochem Mol Biol. 1999; 70:249–56. https://doi.org/10.1016/s0960-0760(99)00113-2 [PubMed] - 56. Zhou J, Wang XH, Zhao YX, Chen C, Xu XY, Sun Q, Wu HY, Chen M, Sang JF, Su L, Tang XQ, Shi XB, Zhang Y, et al. Cancer-Associated Fibroblasts Correlate with Tumor-Associated Macrophages Infiltration and Lymphatic Metastasis in Triple Negative Breast Cancer Patients. J Cancer. 2018; 9:4635–41. https://doi.org/10.7150/jca.28583 [PubMed]
- 57. Ribeiro R, Carvalho MJ, Goncalves J, Moreira JN. Immunotherapy in triple-negative breast cancer: Insights into tumor immune landscape and therapeutic opportunities. Front Mol Biosci. 2022; 9:903065. https://doi.org/10.3389/fmolb.2022.903065 [PubMed]
- 58. Pang Y, Bai G, Zhao J, Wei X, Li R, Li J, Hu S, Peng L, Liu P, Mao H. The BRD4 inhibitor JQ1 suppresses tumor growth by reducing c-Myc expression in endometrial cancer. J Transl Med. 2022; 20:336. https://doi.org/10.1186/s12967-022-03545-x [PubMed]
- 59. Schleifman EB, Desai R, Spoerke JM, Xiao Y, Wong C, Abbas I, O’Brien C, Patel R, Sumiyoshi T, Fu L, Tam RN, Koeppen H, Wilson TR, et al. Targeted biomarker profiling of matched primary and metastatic estrogen receptor positive breast cancers. PLoS One. 2014; 9:e88401. https://doi.org/10.1371/journal.pone.0088401 [PubMed]
- 60. Yabe M, Omarbekova AZ, Hsu M, May H, Arcila ME, Liu Y, Dogan A, Brunner AM, Nardi V, Hasserjian RP, Klimek VM. TP53 Combined Phenotype Score Is Associated with the Clinical Outcome of TP53-Mutated Myelodysplastic Syndromes. Cancers (Basel). 2021; 13:5502. https://doi.org/10.3390/cancers13215502 [PubMed]
- 61. Qian W, Kong X, Zhang T, Wang D, Song J, Li Y, Li X, Geng H, Min J, Kong Q, Liu J, Liu Z, Wang D, et al. Cigarette smoke stimulates the stemness of renal cancer stem cells via Sonic Hedgehog pathway. Oncogenesis. 2018; 7:24. https://doi.org/10.1038/s41389-018-0029-7 [PubMed]
- 62. Jones A, Thornton C. Mitochondrial dynamics in the neonatal brain - a potential target following injury? Biosci Rep. 2022; 42:BSR20211696. https://doi.org/10.1042/BSR20211696 [PubMed]
- 63. Hanson K, Robinson SD, Al-Yousuf K, Hendry AE, Sexton DW, Sherwood V, Wheeler GN. The anti-rheumatic drug, leflunomide, synergizes with MEK inhibition to suppress melanoma growth. Oncotarget. 2017; 9:3815–29. https://doi.org/10.18632/oncotarget.23378 [PubMed]