



Introduction
Diabetic nephropathy (DN) is the most frequent microvascular complication of diabetes mellitus, which can cause chronic kidney damage leading to glomerulopathy [1, 2]. DN is mainly characterized by progressive proteinuria and renal function decline, which eventually progresses to end-stage renal disease [3]. DN has multiple morphological changes, including podocyte apoptosis, thylakoid cell hypertrophy and proliferation, and epithelial mesenchymal transition (EMT) of renal tubular cells, etc. [4–6]. Among them, tubular cell EMT is the most familiar cause of renal function impairment [7]. Susceptibility factors for DN include disorders of metabolic mechanisms, genetics, hyperglycemia, inflammatory responses, hemodynamic alterations, and oxidative stress, etc. [8, 9]. Research showed that there is a “legacy effect” in diabetic patients, and epigenetic factors play a key role in this effect.
The methylation modification (m6A) occurring on the nitrogen atom at position 6 of adenine is the most familiar methylation modification in eukaryotic mRNA [10]. METTL3 is the core component of the m6A methyltransferase complex and is responsible for catalyzing m6A modifications in RNA [11]. METTL3 can play key regulatory roles at the post-transcriptional levels, including precursor mRNA splicing, export of mature mRNA, and mRNA stability regulation, thus affecting multiple biological processes [12]. In type 2 diabetes, glucose can regulate METTL3-mediated m6A modifications [13]. Recent studies also testified that METTL3-mediated m6A modification of TIMP2 can enhance podocyte injury in DN [14]; METTL3 can attenuate renal injury and interstitial fibrosis by enhancing NSD2 stability [15]. However, studies on the pathogenesis of METTL3 in DN are far from adequate. Further exploration of the genes that METTL3 may modify in DN has significant implications for DN therapy.
Renal fibrosis is a typical pathophysiological change that accompanies the progression of DN disease and is the main pathway leading to chronic kidney disease to end-stage renal failure [16]. And cellular EMT induced by renal tubular epithelial cell injury is thought to be a key factor in renal fibrosis [17]. The Wnt/β-catenin pathway is a key pathway regulating EMT in renal tubular epithelial cells [18]. Therefore, suppression of Wnt/β-catenin pathway attenuates EMT in renal tubular epithelial cells as a key mechanism to alleviate tubulointerstitial fibrosis in DN. WISP1 has been reported to be a downstream molecule of the Wnt/β-catenin pathway [19]. WISP1 can affect the growth and development of cells by influencing their survival, proliferation, migration, and differentiation, resulting in an inflammatory response [20, 21]. WISP1 can affect the proliferation and differentiation of kidney cells by affecting Wnt/β-catenin, which can lead to kidney disease [19]. However, the role of WISP1 in DN is not clear, and whether METTL3 can influence DN processes through modifying WISP1 in m6A-dependent manner has not been reported.
In our study, we first confirmed the change of WISP1 expression in DN model mice. We also verified the effects of WISP1 and METTL3 silencing or overexpression on the proliferation, migration, EMT and fibrosis of high glucose (HG)-induced HK2 cells or HK2 cells. Further, we explored the influence of METTL3 on WISP1 m6A modification and whether METTL3 could accelerate the DN process by changing WISP1 m6A modification. Through our current study, we can conclusively demonstrate that METTL3-mediated WISP1 might be the molecular target for DN therapy, which also provides a m6A modification pattern in DN development.
Results
WISP1 was upregulated in renal tissues of DN model mice and HG-induced HK2 cells
To explore possible key genes in DN development, we first constructed DN mouse model with 40mg/kg streptozotocin. The 24 h urinary albumin of DN mouse was significantly higher than that of control group (1A). That indicated successful diabetic nephropathy modeling. And qRT-PCR data denoted that the level of WISP1 in the DN group was notably increased relative to that in the control group (Figure 1B). Then HK2 cells were induced by HG. And the data revealed that the level of WISP1 in the HG group was obviously elevated compared with that in the NG group (Figure 1C). The expression of WISP1 in renal tissues and HK cells were also to be confirmed at protein levels (Figure 1D). Thus, this result presented that the upregulation of WISP1 might be associated with DN. The proportion (%) of m6A in total RNA was calculated. This data confirmed that the total levels of m6A methylation were higher in DN group than those in control group (Figure 1E). Similar results were also found in cell model. The proportion (%) of m6A in total RNA was increased significantly in HG group (Figure 1F).
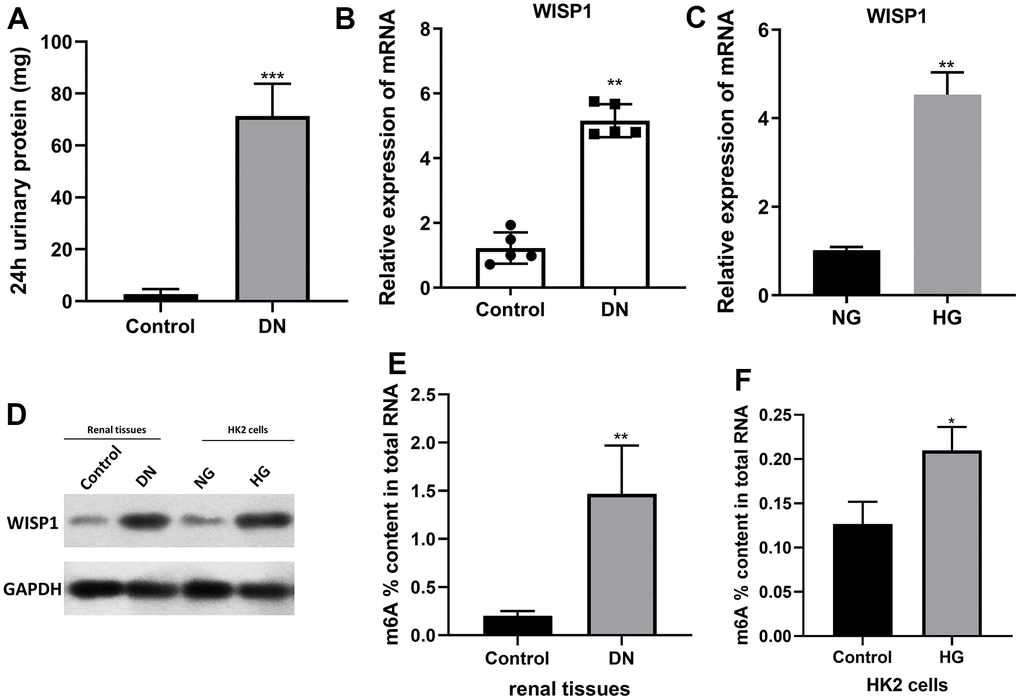
Figure 1. WISP1 expression was highly expressed in DN model mice and HG-treated HK2 cells. DN model mice were first established using streptozotocin. (A) 24-h urinary protein. (B) qRT-PCR analysis of WISP1 expression in kidney tissues which were removed from normal and DN mice (n=5). (C) QRT-PCR analysis of WISP1 expression in HG-treated HK2 cells. (D) The protein level expression of WISP1 in renal tissues and HK2 cells. (E) The total m6A RNA methylation context of total RNA between control and DN group. (F) The total m6A RNA methylation context of total RNA between control and HG group. *P < 0.05, **P < 0.01.
WISP1 overexpression promoted the proliferation, migration, EMT, and renal fibrosis of HG-induced HK2 cells
On the basis of the above results, we ulteriorly verified the roles of WISP1 in HG-induced HK2 cells through a series of experiments. We first adopted qRT-PCR to evaluate the transfection effect of WISP1-silenced HG-induced HK2 cells and WISP1-overexpressed HK2 cells. As expected, in HG-induced HK2 cells, the level of WISP1 in the silencing group was prominently degraded relative to that in the NC group (Figure 2A). And in HK2 cells, the level of WISP1 in the overexpression group was memorably boosted relative to that in the vector group (Figure 2B). This implies that WISP1 has been successfully silenced in HG-induced HK2 cells or overexpressed in HK2 cells. Then the result of CCK-8 displayed that the proliferation of HG-treated HK2 cells was notably decreased in sh-WISP1 group in comparison with NC group, and the proliferation of HK2 cells was prominently enhanced in OE-WISP1 group compared to vector group (Figure 2C). Correspondingly, the result of wound healing exhibited that the migration ability of HG-treated HK2 cells dramatically decreased after WISP1 silencing, and upregulation of WISP1 observably promoted the migration ability of HK2 cells (Figure 2D). Furthermore, to investigate the relationship between EMT process and WISP1, we applied western blot to monitor the expression levels of EMT associated proteins. The results manifested that WISP1 silencing markedly upregulated E-cadherin, and downregulated a-SMA and Fibronectin in HG-treated HK2 cells, and WISP1 overexpression group had the opposite effects on the expression of these proteins in HK2 cells (Figure 2E). It is well known that EMT is typically characterized by suppression of E-cadherin, facilitation of Fibronectin and a-SMA expressions. So, we revealed that WISP1 overexpression could induce EMT of HK2 cells. Cell fibrosis is typically characterized by encouragement of Wnt1a, c-MYC and β-catenin expressions. Western blot results further disclosed that WISP1 silencing observably decreased c-MYC, Wnt1a, and β-catenin expressions in HG-treated HK2 cells, and WISP1 overexpression notably induced cell fibrosis in HK2 cells (Figure 2F). In short, we demonstrated that WISP1 could induce the development of DN.
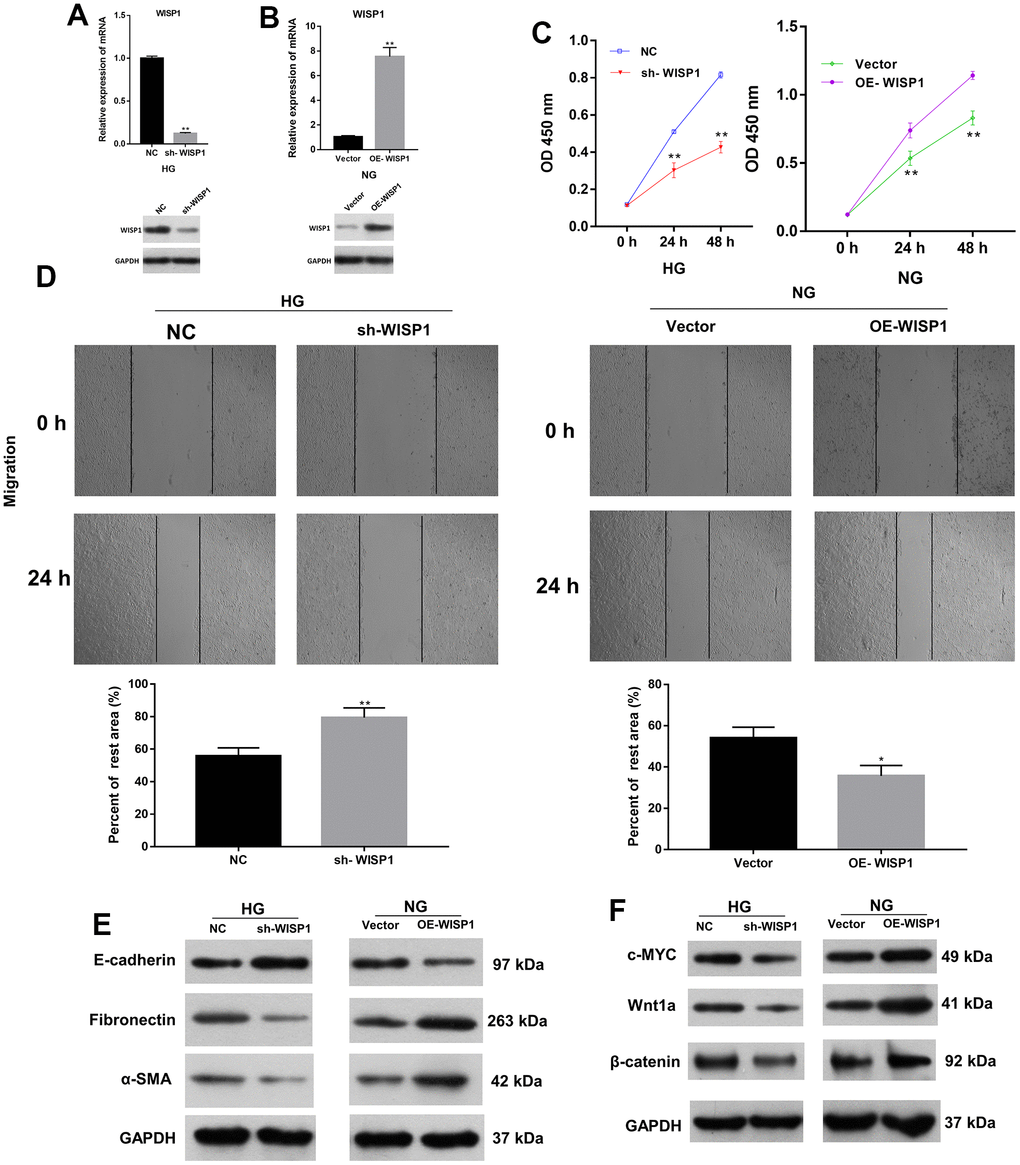
Figure 2. The impacts of WISP1 on the proliferation, migration, EMT, and renal fibrosis of HG-treated HK2 cells. (A) After HG treatment, the expression of WISP1 was confirmed using qRT-PCR and western blot in HK2 cells transfected with NC or sh-WISP1. (B) QRT-PCR and western blot analysis of WISP1 in HK2 cells transfected with OE-WISP1 or Vector. (C) CCK-8 assay for the assessment of cell proliferation in HG-induced HK2 cells with WISP1 silencing and HK2 cells with WISP1 overexpression. (D) Wound healing assay was conducted to identify the migration ability of the WISP1-silenced HG-induced HK2 cells and WISP1-overexpressed HK2 cells. (E) Western blot indicated the expression changes of E-cadherin, Fibronectin and a-SMA in WISP1-silenced HG-induced HK2 cells and WISP1-overexpressed HK2 cells. (F) The expressions of c-MYC, Wnt1 and β-catenin were monitored by applying western blot in each group. *P < 0.05, **P < 0.01.
Increase of METTL3 expression memorably strengthened proliferation, EMT progression and fibrosis of HG-induced HK2 cells
Besides, we further investigated the roles of METTL3 in EMT and cell fibrosis progression of renal tubular epithelial cells. METTL3 was silenced in HG-treated HK2 cells and overexpressed in HK2 cells, respectively. Based on the qRT-PCR and western blot results, METTL3 was prominently downregulated in sh-METTL3 group, and upregulated in OE-METTL3 group (Figure 3A, 3B). Then we assessed the cell proliferation, and the results presented that the proliferation ability of HG-induced HK2 cells was memorably weakened in sh-METTL3 group relative to that in NC group, and the proliferation ability of HK2 cells was significantly enhanced in OE-METTL3 group relative to that in vector group (Figure 3C). Besides, wound healing results manifested that METTL3 silencing observably attenuated the migration of HG-treated HK2 cells, and overexpression of METTL markedly increased the migration of HK2 cells (Figure 3D). Meanwhile, western blot analysis of EMT and fibrosis marker proteins suggested that the upregulation of METTL3 worsened EMT and induced cell fibrosis in HG-induced HK2 cells, and downregulation of METTL3 had the opposite effect with METTL3 overexpression in HK2 cells (Figure 3E, 3F). Overall, these data indicated that METTL3 could accelerate cell proliferation, migration, EMT and fibrosis of HG-induced HK2 cells.
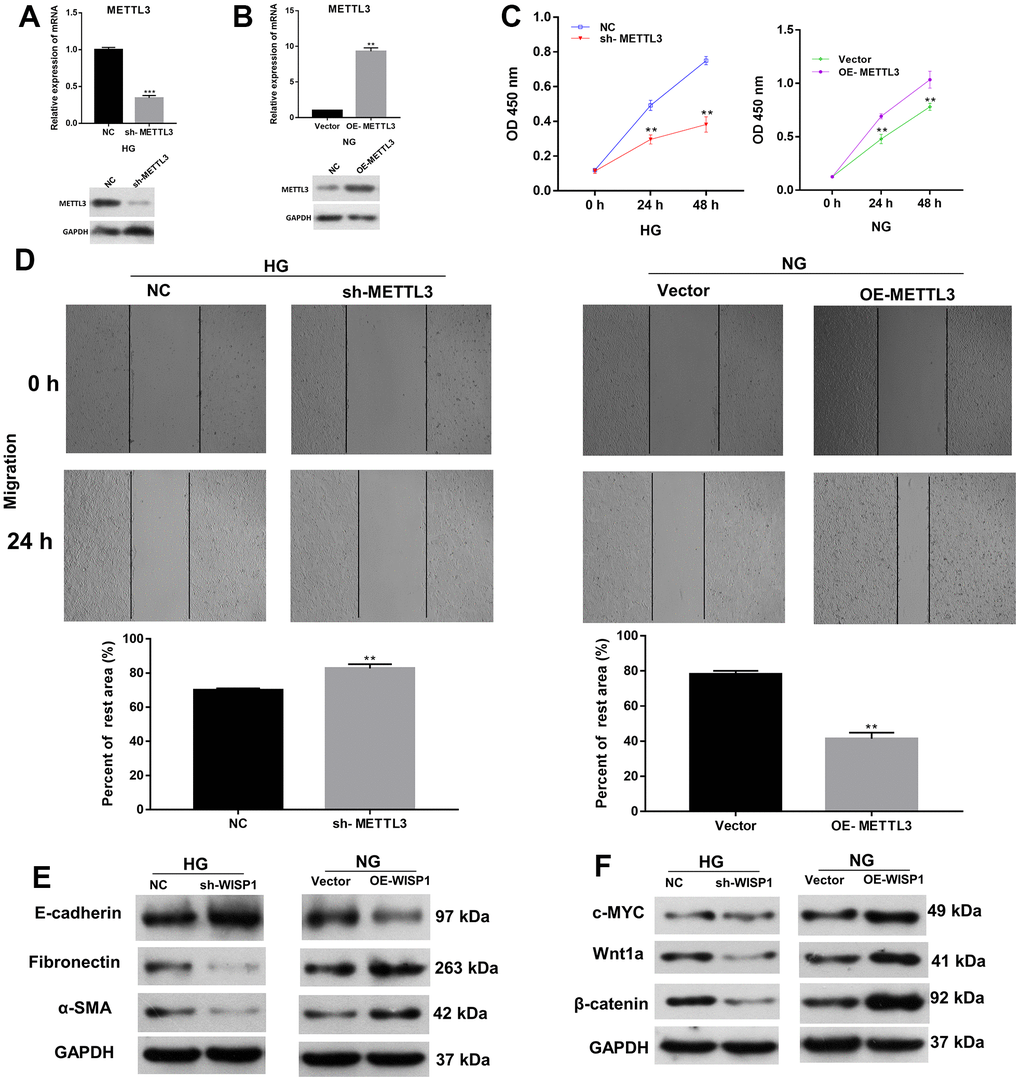
Figure 3. Increase of METTL3 expression memorably strengthened EMT progression and fibrosis of HG-induced HK2 cells. Sh-METTL3 or NC were transfected into HG-treated HK2 cells, and OE-METTL3 or vector were transfected into HK2 cells. (A) QRT-PCR and western blot analysis of METTL3 expression in METTL3-silenced HK2 cells after HG induction. (B) QRT-PCR and western blot analysis of METTL3 expression in METTL3-overexpressed HK2 cells. (C) CCK-8 assay demonstrated the change of cell proliferation. (D) The cell migration was credited through the application of wound healing assay. (E) The changes of E-cadherin, Fibronectin and a-SMA expressions were identified using western blot. (F) Western blot assay for the detection of c-MYC, Wnt1 and β-catenin expressions. **P < 0.01, ***P < 0.001.
Overexpression of WISP1 reversed the effects of METTL3 silencing on proliferation, EMT and fibrosis in HG-induced HK2 cells
Moreover, we further verified the moderating relationship between WISP1 and METTL3. And MeRIP-quantitative real-time PCR exhibited that WISP1 m6A modification was notably decreased after METTL3 silencing (Figure 4A). Then we discovered that METTL3 silencing also could notably downregulate WISP1 in HG-induced HK2 cells (Figure 4B). Next, we also confirmed the effects of WISP1 and METTL3 on cell-related functions by the rescue experiment. And HG-treated HK2 cells were co-transfected with sh-METTL3 and WISP1 overexpression plasmid. CCK-8 results displayed that silencing of METTL3 suppressed proliferation of HG-treated HK2 cells, while upregulation of WISP1 dramatically reversed the inhibition effect of METTL3 silencing on cell proliferation (Figure 4C). Similarly, the trend of cell migration was consistent with that of cell proliferation from CCK-8 results (Figure 4D). Besides, western blot results revealed that silencing of METTL3 dramatically upregulated E-cadherin, and downregulated a-SMA and Fibronectin in HG-treated HK2 cells, which also could be remarkably reversed by WISP1 overexpression (Figure 4E). Meanwhile, we testified that the downregulation of c-MYC, Wnt1a, and β-catenin mediated by METTL3 silencing also could be prominently restored by WISP1 overexpression in HG-treated HK2 cells (Figure 4F). Thus, we proved that METTL3 silencing could reduce WISP1 m6A modification, and silencing of METTL3 also could inhibit the degradation process of HG-treated HK2 cells by downregulating WISP1.
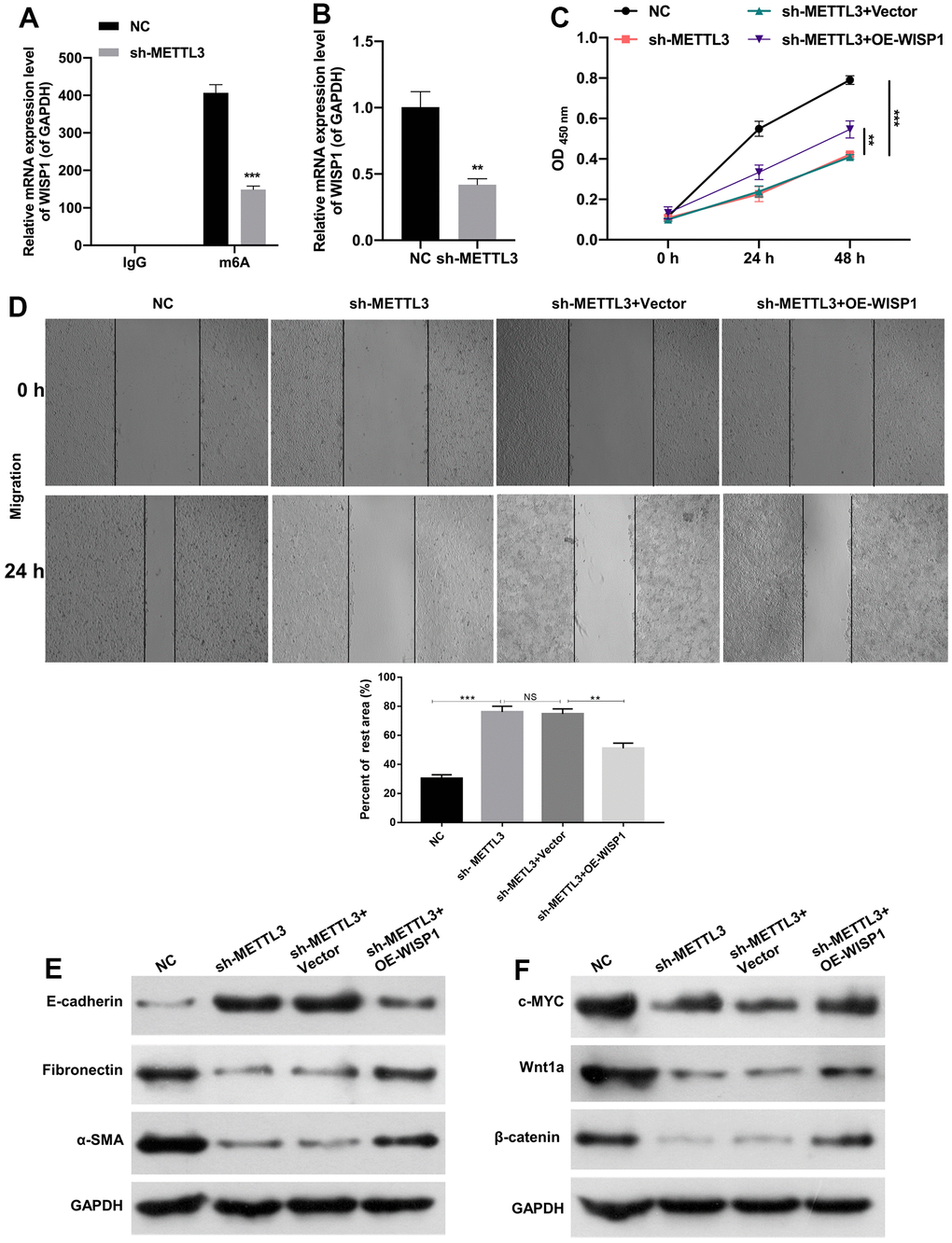
Figure 4. METTL3 silencing prevented EMT progression and fibrosis of HG-induced HK2 cells by decreasing WISP1 with m6A modification. (A) MeRIP-qPCR assay exhibited the change in WISP1 enrichment in HG-treated HK2 cells after METTL3 silencing. (B) WISP1 expression was examined via qRT-PCR in HG-treated HK2 cells which were transfected with METTL3 sh-RNAs. (C) HG-treated HK2 cells were transfected with sh-METTL3 or/and OE-WISP1, and the cell proliferation was tested by CCK-8 assay. (D) Wound healing assay showed the change of cell migration in the processed HK2 cells after HG treatment. (E) E-cadherin, Fibronectin and a-SMA expression levels were assessed by applying western blot. (F) Western blot was adopted to monitor the changes of c-MYC, Wnt1 and β-catenin expressions. **P < 0.01, ***P < 0.001.
Discussion
DN is one of the major microvascular complications of diabetes, accounting for 30% to 40% of patients with diabetes [22]. DN has become the primary cause of end-stage renal disease worldwide and the principal cause of death in patients with diabetes [23]. DN is typically characterized by excessive deposition of extracellular matrix in the glomerulus and tubulointerstitium, ultimately causing glomerulosclerosis and tubulointerstitial fibrosis [24]. Study testified that glomerular injury is an early lesion of DN, and tubulointerstitial injury is secondary to glomerular injury [25]. Tubulointerstitial fibrosis usually occurs early in the course of diabetic kidney injury, which is associated with decreased renal function [26, 27]. Besides, the conversion of renal tubular epithelial cells to mesenchymal cells is one of the vital mechanisms of tubular interstitial fibrosis [28, 29]. Therefore, further investigation of renal tubular epithelial cell EMT and renal fibrosis is of crucial importance for DN therapy.
The severity of chronic kidney disease (CKD) is positively correlated with the degree of kidney fibrosis, causing end-stage renal disease [30]. The Wnt/β-catenin pathway plays a key role in organ development, tissue homeostasis and injury repair [31]. This pathway is relatively silent in normal adult kidneys, but can be reactivated in uremic model animals and in patients with CKD [32]. Research demonstrated that the Wnt/β-catenin pathway can mediate myofibroblast activation, proliferation and EMT in renal tubular epithelial cells by affecting the expression of related target genes such as fibronectin and E-calcineurin, leading to renal fibrosis [18]. And WISP1 is a key downstream molecule of the Wnt/β-catenin pathway [19]. WISP1 has been reported to be associated with pathological processes such as inflammation, injury repair, and tumor development [20, 21, 33]. Additionally, WISP1 expression can be increased in renal injury models, which may promote the development of renal fibrosis and accelerate the process of CKD [34]. While the role of WISP1 in DN is not fully understood. In our study, we further proved that WISP1 was highly expressed in renal tissues of DN model mice. Meanwhile, we verified that WISP1 silencing could weaken the proliferation, migration, and fibrosis of HG-induced HK2 cells, and WISP1 overexpression could enhance the proliferation, migration, and fibrosis of HK2 cells.
EMT is an evolutionary conserved biological process of epithelial to mesenchymal cell transformation [35]. The EMT phenotypic state is highly dynamic and dependent on cell type and environment [36]. Therefore, the definition of EMT should be evaluated in the context of cellular characteristics and multiple molecular markers, including α-SMA, E-cadherin and fibronectin [37, 38]. In our study, we also revealed that WISP1 silencing can prevent EMT of HG-treated HK2 cells, and WISP1 overexpression can accelerate EMT of HK2 cells through the detection of related molecules.
M6A methylation modification is the most widespread RNA epigenetic modification in eukaryotes [39]. M6A modification is a dynamically reversible post-transcriptional modification that is most abundant in mRNAs and non-coding RNAs [40]. And m6A plays a key role in gene expression and cell fate regulation [41]. In recent years, several studies have confirmed the essential role of m6A modification in DN [42–44]. METTL3, as a major methyltransferase for m6A methylation, has a key role in cell proliferation, differentiation, metastasis and other cellular biological processes [45, 46]. However, only two papers have reported the role and mechanism of METTL3 in DN [14, 15]. In our study, we further confirmed that METTL3 silencing could suppress the proliferation, EMT, migration, and fibrosis of HG-treated HK2 cells, METTL3 overexpression could induce the proliferation, EMT, migration, and fibrosis of HK2 cells. Besides, we first discovered that METTL3 silencing could reduce WISP1 m6A modification, suggesting METTL3 could mediate m6A modification of WISP1 mRNA. More importantly, we uncovered that METTL3 silencing could prevent DN process by reducing WISP1 mediated by m6A modification.
In summary, METTL3 plays a key role in human kidney proximal tubular epithelial cell proliferation, EMT and fibrosis, and knockdown of METTL3 could impair EMT and fibrosis in HG-treated HK2 cells. Molecularly, METTL3 silencing could reduce WISP1 transcript level by attenuating WISP1 mRNA m6 methylation modification. And METTL3 silencing also could alleviate DN progression by reducing WISP1 with m6A modification. Therefore, targeted inhibition of METTL3/WISP1 axis is expected to be a key molecular target for DN intervention.
Materials and Methods
Experimental animals
A total of 10 specific Pathogen Free (SPF) grade C57/BL6 mice (male, 8 weeks old, weight 20-25 g) were purchased from the Animal center of Sun Yat-Sen University. Feeding conditions:12 h light day and night, 22-24° C, 40%-70% humidity, well ventilated, ad libitum diet. Animal experiments were performed under the institutional guidelines of the Animal Care and Use Committee of University of Chinese Academy of Sciences Shenzhen Hospital.
Construction of DN model mice
Ten mice were randomly divided into control group and DN model group, with 5 mice in each group. The mice in the model group were fed with high-fat diet for 4 weeks. Subsequently, the diabetic mouse model was established by multiple small-dose intraperitoneal injections of streptozotocin (40 mg/kg for 5 d), and the mice in the control group were injected intraperitoneally with an equal amount of saline. Monitoring of blood glucose concentration was started 72 h after the completion of injection, and the modeling of diabetes was considered successful if the random blood glucose concentration was >16.7 mmol/L. Thereafter, blood glucose and weight were measured regularly once a week, and a 24-h urine protein level >30 mg after 4 weeks indicated successful diabetic nephropathy modeling.
Cell culture
Human kidney proximal tubular epithelial cells (HK2) were from BeNa Culture Collection Company (Beijing, China). After resuscitation, HK2 cells were centrifuged to collect cells. then HK2 cells were grown in DMEM (Aidenbach; Germany) containing 10% fetal bovine serum (FBS, Gibco, USA) at 37° C with 5% CO2. The DMEM medium containing 5.6 mM glucose was set as normal-glucose (NG) group, while the medium containing 30 mM glucose were set as high-glucose (HG) group.
Cell treatment
Negative control (NC), WISP1 shRNAs (sh-WISP1), METTL3 shRNAs (sh-METTL3), vector, WISP1 overexpression plasmid (OE-WISP1), METTL3 overexpression plasmid (OE-METTL3) were provided by Integrated Biotech Solutions (Shanghai, China). HK2 cells at logarithmic growth stage were seeded in 96-well plates at 3×105 cells/well and treated with HG for 48 h. Then HG-treated HK2 cells were transfected with shRNAs or NC, HK2 cells were transfected with the overexpression plasmids or vector for 48 h using Lipofectamine 3000 (Invitrogen, USA) based on the instructions.
Quantitative real-time PCR (qRT-PCR)
Total RNA from the ground kidney tissue or the treated HK2 cells was extracted using Trizol Reagent. After purity and concentration were assessed, the RNA was reverse transcribed to acquire cDNA using PrimeScript™ RT reagent kits (Takara, Dalian, China). Then qRT-PCR was conducted with 2×SYBR Green qPCR Master Mix (Applied Biosystems, USA). The relative levels of METTL3 and WISP1 were calculated by 2-ΔΔCt method.
CCK-8
The processed HK2 cells were inoculated in 96-well plates with 5×103 cells/well and incubated at 37° C. At 0, 24 and 48 h, each well was added with 10 μL CCK-8 solution (Dojindo, Kumamoto, Japan) and fostered in the incubator for 1h. And the absorbance value at 450 nm was carried out by microplate reader.
Wound healing
The processed HK2 cells (5×104 cells) were evenly inoculated in 6-well plates. When the cells showed adnate growth, they were scratched. Then the detached cells were washed off with PBS and the adherent cells were added with serum-free culture medium. After 24 h of continued incubation, the width of the scratch was observed and recorded under a light microscope.
Western blot
The total protein of HK2 cells was extracted using RIPA lysis buffer (Beyotime, Shanghai, China) with 1% PMSF (Beyotime). Then the protein concentration was monitored through BCA kit (Beyotime). 40 μg of protein was subjected to SDS-PAGE gel electrophoresis and transmembrane with 0.22 μm PVDF membrane. After blocking, the membranes were processed with the diluted primary antibodies (Abcam, Cambridge, UK) at 4° C overnight and secondary antibody (Abcam) for 2 h. After treating with Pierce™ ECL substrate (Thermo Fisher Scientific, USA), the blots were colored with an ECL system (Thermo Fisher Scientific).
MeRIP-quantitative real-time PCR
20 μg of total RNA was processed with DNAase I and the RNA free of DNA contamination was digested with RNA fragmentation reagent at 90° C for 30 s. After termination of the reaction, the fragmented RNA was re-extracted by ethanol sedimentation. Subsequently, 2 μg of m6A antibody was co-incubated with 40 μl ProteinA/G magnetic beads in IPP buffer for 1 h. The fragmented RNA was added to the above system and incubation was continued at 4° C for 4 h. After washing the magnetic beads 4 times with IPP buffer, the samples were added to Trizol to extract RNA from the magnetic beads. Finally, qRT-PCR analysis was conducted.
m6A RNA methylation assay
The total amount of m6A in total RNA was measured using the m6A RNA Methylation Assay Kit (Abcam), following the manufacturer manual. 200 ng total RNA from renal tissues or cells was used.
Statistical analysis
All data were processed using SPSS 23.0 software (SPSS Inc., USA), and the measurement data were signified as mean ±SD. One-way ANOVA was adopted for comparison between multiple groups, and t-test was adopted for comparison between two groups. And the difference was defined as statistically significant at P < 0.05.
Author Contributions
Conceptualization: Yuanzhen Chen, Ping Li and Dan Song. Methodology: Yuanzhen Chen, Ping Li, Mei Lin and Ying Jiang. Formal analysis: Yuanzhen Chen, Guifang Tan and Lianfang Huang. Writing – original draft preparation: Yuanzhen Chen, Ping Li and Dan Song. Project administration: Yuanzhen Chen, Ping Li and Dan Song. Funding acquisition: Dan Song. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
This study was approved by the Ethics Committee of The University of Chinese Academy of Sciences Shenzhen Hospital (LL-KT-2021143). All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Funding
This work was supported the Guangming District Economic Development Special Fund (2020R01129; 2021R01003).
References
- 1. Samsu N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. Biomed Res Int. 2021; 2021:1497449. https://doi.org/10.1155/2021/1497449 [PubMed]
- 2. Zhu H, Zhong S, Yan H, Wang K, Chen L, Zhou M, Li Y. Resveratrol reverts Streptozotocin-induced diabetic nephropathy. Front Biosci (Landmark Ed). 2020; 25:699–709. https://doi.org/10.2741/4829 [PubMed]
- 3. Sagoo MK, Gnudi L. Diabetic Nephropathy: An Overview. Methods Mol Biol. 2020; 2067:3–7. https://doi.org/10.1007/978-1-4939-9841-8_1 [PubMed]
- 4. Dai H, Liu Q, Liu B. Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. J Diabetes Res. 2017; 2017:2615286. https://doi.org/10.1155/2017/2615286 [PubMed]
- 5. Tang G, Li S, Zhang C, Chen H, Wang N, Feng Y. Clinical efficacies, underlying mechanisms and molecular targets of Chinese medicines for diabetic nephropathy treatment and management. Acta Pharm Sin B. 2021; 11:2749–67. https://doi.org/10.1016/j.apsb.2020.12.020 [PubMed]
- 6. Pan Y, Liu L, Yang H, Chen W, Chen Z, Xu J. Sacubitril/Valsartan Improves Progression of Early Diabetic Nephropathy in Rats Through Inhibition of NLRP3 Inflammasome Pathway. Diabetes Metab Syndr Obes. 2022; 15:2479–88. https://doi.org/10.2147/DMSO.S366518 [PubMed]
- 7. Xu Y, Ouyang C, Lyu D, Lin Z, Zheng W, Xiao F, Xu Z, Ding L. Diabetic nephropathy execrates epithelial-to-mesenchymal transition (EMT) via miR-2467-3p/Twist1 pathway. Biomed Pharmacother. 2020; 125:109920. https://doi.org/10.1016/j.biopha.2020.109920 [PubMed]
- 8. Wei L, Xiao Y, Li L, Xiong X, Han Y, Zhu X, Sun L. The Susceptibility Genes in Diabetic Nephropathy. Kidney Dis (Basel). 2018; 4:226–37. https://doi.org/10.1159/000492633 [PubMed]
- 9. Dronavalli S, Duka I, Bakris GL. The pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol Metab. 2008; 4:444–52. https://doi.org/10.1038/ncpendmet0894 [PubMed]
- 10. Reichel M, Köster T, Staiger D. Marking RNA: m6A writers, readers, and functions in Arabidopsis. J Mol Cell Biol. 2019; 11:899–910. https://doi.org/10.1093/jmcb/mjz085 [PubMed]
- 11. Yu J, Shen L, Liu Y, Ming H, Zhu X, Chu M, Lin J. The m6A methyltransferase METTL3 cooperates with demethylase ALKBH5 to regulate osteogenic differentiation through NF-κB signaling. Mol Cell Biochem. 2020; 463:203–10. https://doi.org/10.1007/s11010-019-03641-5 [PubMed]
- 12. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017; 18:31–42. https://doi.org/10.1038/nrm.2016.132 [PubMed]
- 13. Yang J, Liu J, Zhao S, Tian F. N6-Methyladenosine METTL3 Modulates the Proliferation and Apoptosis of Lens Epithelial Cells in Diabetic Cataract. Mol Ther Nucleic Acids. 2020; 20:111–6. https://doi.org/10.1016/j.omtn.2020.02.002 [PubMed]
- 14. Jiang L, Liu X, Hu X, Gao L, Zeng H, Wang X, Huang Y, Zhu W, Wang J, Wen J, Meng X, Wu Y. METTL3-mediated m6A modification of TIMP2 mRNA promotes podocyte injury in diabetic nephropathy. Mol Ther. 2022; 30:1721–40. https://doi.org/10.1016/j.ymthe.2022.01.002 [PubMed]
- 15. Tang W, Zhao Y, Zhang H, Peng Y, Rui Z. METTL3 enhances NSD2 mRNA stability to reduce renal impairment and interstitial fibrosis in mice with diabetic nephropathy. BMC Nephrol. 2022; 23:124. https://doi.org/10.1186/s12882-022-02753-3 [PubMed]
- 16. Calle P, Hotter G. Macrophage Phenotype and Fibrosis in Diabetic Nephropathy. Int J Mol Sci. 2020; 21:2806. https://doi.org/10.3390/ijms21082806 [PubMed]
- 17. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, Kalluri R. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015; 21:998–1009. https://doi.org/10.1038/nm.3902 [PubMed]
- 18. Feiteng C, Lei C, Deng L, Chaoliang X, Zijie X, Yi S, Minglei S. Relaxin inhibits renal fibrosis and the epithelial-to-mesenchymal transition via the Wnt/β-catenin signaling pathway. Ren Fail. 2022; 44:513–24. https://doi.org/10.1080/0886022X.2022.2044351 [PubMed]
- 19. Chen YZ, Sun DQ, Zheng Y, Zheng GK, Chen RQ, Lin M, Huang LF, Huang C, Song D, Wu BQ. WISP1 silencing confers protection against epithelial-mesenchymal transition of renal tubular epithelial cells in rats via inactivation of the wnt/β-catenin signaling pathway in uremia. J Cell Physiol. 2019; 234:9673–86. https://doi.org/10.1002/jcp.27654 [PubMed]
- 20. Maiese K. Prospects and Perspectives for WISP1 (CCN4) in Diabetes Mellitus. Curr Neurovasc Res. 2020; 17:327–31. https://doi.org/10.2174/1567202617666200327125257 [PubMed]
- 21. Tao W, Chu C, Zhou W, Huang Z, Zhai K, Fang X, Huang Q, Zhang A, Wang X, Yu X, Huang H, Wu Q, Sloan AE, et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat Commun. 2020; 11:3015. https://doi.org/10.1038/s41467-020-16827-z [PubMed]
- 22. Gallagher H, Suckling RJ. Diabetic nephropathy: where are we on the journey from pathophysiology to treatment? Diabetes Obes Metab. 2016; 18:641–7. https://doi.org/10.1111/dom.12630 [PubMed]
- 23. Nagib AM, Elsayed Matter Y, Gheith OA, Refaie AF, Othman NF, Al-Otaibi T. Diabetic Nephropathy Following Posttransplant Diabetes Mellitus. Exp Clin Transplant. 2019; 17:138–46. https://doi.org/10.6002/ect.2018.0157 [PubMed]
- 24. Kolset SO, Reinholt FP, Jenssen T. Diabetic nephropathy and extracellular matrix. J Histochem Cytochem. 2012; 60:976–86. https://doi.org/10.1369/0022155412465073 [PubMed]
- 25. Cohen-Bucay A, Viswanathan G. Urinary markers of glomerular injury in diabetic nephropathy. Int J Nephrol. 2012; 2012:146987. https://doi.org/10.1155/2012/146987 [PubMed]
- 26. Ina K, Kitamura H, Tatsukawa S, Takayama T, Fujikura Y, Shimada T. Transformation of interstitial fibroblasts and tubulointerstitial fibrosis in diabetic nephropathy. Med Electron Microsc. 2002; 35:87–95. https://doi.org/10.1007/s007950200011 [PubMed]
- 27. Shi Y, Huang C, Zhao Y, Cao Q, Yi H, Chen X, Pollock C. RIPK3 blockade attenuates tubulointerstitial fibrosis in a mouse model of diabetic nephropathy. Sci Rep. 2020; 10:10458. https://doi.org/10.1038/s41598-020-67054-x [PubMed]
- 28. Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011; 121:468–74. https://doi.org/10.1172/jci44595 [PubMed]
- 29. Seccia TM, Caroccia B, Piazza M, Rossi GP. The Key Role of Epithelial to Mesenchymal Transition (EMT) in Hypertensive Kidney Disease. Int J Mol Sci. 2019; 20:3567. https://doi.org/10.3390/ijms20143567 [PubMed]
- 30. Kalantar-Zadeh K, Jafar TH, Nitsch D, Neuen BL, Perkovic V. Chronic kidney disease. Lancet. 2021; 398:786–802. https://doi.org/10.1016/S0140-6736(21)00519-5 [PubMed]
- 31. Bastakoty D, Young PP. Wnt/β-catenin pathway in tissue injury: roles in pathology and therapeutic opportunities for regeneration. FASEB J. 2016; 30:3271–84. https://doi.org/10.1096/fj.201600502R [PubMed]
- 32. Zhou L, Liu Y. Wnt/β-catenin signaling and renin-angiotensin system in chronic kidney disease. Curr Opin Nephrol Hypertens. 2016; 25:100–6. https://doi.org/10.1097/MNH.0000000000000205 [PubMed]
- 33. Maiese K. WISP1: Clinical insights for a proliferative and restorative member of the CCN family. Curr Neurovasc Res. 2014; 11:378–89. https://doi.org/10.2174/1567202611666140912115107 [PubMed]
- 34. Zhong X, Tu YJ, Li Y, Zhang P, Wang W, Chen SS, Li L, Chung AC, Lan HY, Chen HY, Li GS, Wang L. Serum levels of WNT1-inducible signaling pathway protein-1 (WISP-1): a noninvasive biomarker of renal fibrosis in subjects with chronic kidney disease. Am J Transl Res. 2017; 9:2920–32. [PubMed]
- 35. Roy S, Sunkara RR, Parmar MY, Shaikh S, Waghmare SK. EMT imparts cancer stemness and plasticity: new perspectives and therapeutic potential. Front Biosci (Landmark Ed). 2021; 26:238–65. https://doi.org/10.2741/4893 [PubMed]
- 36. Lin YT, Wu KJ. Epigenetic regulation of epithelial-mesenchymal transition: focusing on hypoxia and TGF-β signaling. J Biomed Sci. 2020; 27:39. https://doi.org/10.1186/s12929-020-00632-3 [PubMed]
- 37. Kyung SY, Kim DY, Yoon JY, Son ES, Kim YJ, Park JW, Jeong SH. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol Toxicol. 2018; 19:13. https://doi.org/10.1186/s40360-018-0204-7 [PubMed]
- 38. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019; 20:69–84. https://doi.org/10.1038/s41580-018-0080-4 [PubMed]
- 39. Zhang H, Shi X, Huang T, Zhao X, Chen W, Gu N, Zhang R. Dynamic landscape and evolution of m6A methylation in human. Nucleic Acids Res. 2020; 48:6251–64. https://doi.org/10.1093/nar/gkaa347 [PubMed]
- 40. Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, Yuan W, Kan Q, Sun Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019; 12:121. https://doi.org/10.1186/s13045-019-0805-7 [PubMed]
- 41. Qin Y, Li L, Luo E, Hou J, Yan G, Wang D, Qiao Y, Tang C. Role of m6A RNA methylation in cardiovascular disease (Review). Int J Mol Med. 2020; 46:1958–72. https://doi.org/10.3892/ijmm.2020.4746 [PubMed]
- 42. Lan J, Xu B, Shi X, Pan Q, Tao Q. WTAP-mediated N6-methyladenosine modification of NLRP3 mRNA in kidney injury of diabetic nephropathy. Cell Mol Biol Lett. 2022; 27:51. https://doi.org/10.1186/s11658-022-00350-8 [PubMed]
- 43. Li C, Su F, Liang Z, Zhang L, Liu F, Fan W, Li Z. Macrophage M1 regulatory diabetic nephropathy is mediated by m6A methylation modification of lncRNA expression. Mol Immunol. 2022; 144:16–25. https://doi.org/10.1016/j.molimm.2022.02.008 [PubMed]
- 44. Li M, Deng L, Xu G. METTL14 promotes glomerular endothelial cell injury and diabetic nephropathy via m6A modification of α-klotho. Mol Med. 2021; 27:106. https://doi.org/10.1186/s10020-021-00365-5 [PubMed]
- 45. Liu S, Zhuo L, Wang J, Zhang Q, Li Q, Li G, Yan L, Jin T, Pan T, Sui X, Lv Q, Xie T. METTL3 plays multiple functions in biological processes. Am J Cancer Res. 2020; 10:1631–46. [PubMed]
- 46. Zeng C, Huang W, Li Y, Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020; 13:117. https://doi.org/10.1186/s13045-020-00951-w [PubMed]