



Introduction
As we all know, colorectal cancer (CRC) has a high incidence rate and mortality. According to statistics, in 2020, there will be about 1, 880, 000 new CRC cases and nearly 920, 000 CRC deaths worldwide [1]. Although great efforts have been made in early screening, which can effectively reduce the incidence of CRC [2], the morbidity and mortality of CRC have not been significantly improved, and the prognosis of patients with advanced CRC is still very poor [3]. It is estimated that there will be about 3, 200, 000 new CRC cases and 1, 600, 000 CRC deaths by 2040 [4]. At present, the mechanism of occurrence and development of CRC is still unclear. Therefore, it is particularly important to find new diagnostic and prognostic biomarkers for CRC patients. Metabolic reprogramming is generally considered to be one of the hallmarks of cancer [5].
In addition to classic glycolysis (Warburg effect) [6], researchers have focused on glutamine metabolism in recent years [7]. Studies have shown that glutamine is a non-essential amino acid with the highest content in blood [8]. In addition to providing a source of nitrogen, glutamine is a major source of carbon that supports cancer cell growth, especially when mitochondria are dysfunctional [9, 10]. Therefore, glutamine is one of the essential metabolites for cancer cells to maintain a malignant phenotype. Previous research has implicated that glutamine metabolism is associated with proliferation, invasion, autophagy, and immune evasion of cancer cells [11–15], and is a promising new target for cancer therapy [16, 17]. Therefore, a comprehensive analysis of GMRGs is imperative to find new promising biomarkers for the diagnosis and prognosis of CRC.
Glutamic pyruvic transaminase (GPT), also known as glutamic pyruvic transaminase 1, can reversibly catalyse alanine and α-ketoglutarate into pyruvate and glutamic acid and plays a pivotal role in the intermediate metabolism of amino acids and glucose [18]. Unlike mitochondrial GPT2, which has been extensively studied in cancer [19–21], GPT1 has been rarely reported in cancer. Current studies have shown that inhibiting GPT1 expression can reduce ATP production, thereby attenuating the malignant phenotype of HCC, which is a new therapeutic target for HCC [22]. In addition, GPT1 can be negatively regulated by SIRT4 to affect cervical cancer cell apoptosis by affecting glutamine metabolism. However, GPT1 has not been studied in other cancers, including CRC. Therefore, it is essential for the systematic study of GPT in CRC.
This study screened characteristic genes in the glutamine metabolism related family through multiple machine learning algorithms, and jointly analyzed their diagnostic and prognostic value, ultimately determining GPT as the key GMRGs in colorectal cancer. Then, we utilized bioinformatics to analyze the biological role of GPT in CRC and explore the relationship between its expression and tumor microenvironment (TME) and drug sensitivity. Finally, we also explored the differential expression, diagnostic performance, and prognostic value of GPT in pan cancer.
Materials and Methods
Data download and processing
The RNA-seq expression data and survival information from the CRC cohort (TCGA-COAD/READ) were obtained from the TCGA database (https://portal.gdc.cancer.gov/). Expression data and prognostic information of 31 cancers for pancancer analysis were downloaded from UCSC database (https://xena.ucsc.edu/). Single-cell RNA sequencing (scRNA-seq) data of 12 CRC patients were downloaded from GSE166555. The scRNA-seq data in GSE166555 were downloaded from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database. The glutamine metabolism-related genes involved in this study were obtained from previous studies published by He. et al. [23]. Analysis of the differential expression of GMRGs in the TCGA-CRC cohort using the R package “Limma”, and the screening condition was set to “|Foldchange > 2, P < 0.05|”.
Machine learning screening for differentially expressed GMRGs
For the differentially expressed GMRGs, we further used SVM-REF and RandomForest algorithm for separation to screen out the characteristic genes. SVM-REF is a powerful classification algorithm [24], that can be applied to characterize cancer, allowing the identification of biomarkers for diagnosis. Similarly, RandomForest is also a powerful classification algorithm [25] that ranks gene importance, and in this study, we picked genes with an importance score greater than 2. Finally, we use the common genes identified by these two machine learning algorithms for further research and consider them as characteristic genes of CRC.
scRNA-seq analysis of characteristic genes
The scRNA-seq data were preprocessed using the “Seurat” package. “PercentageFeatureSet” is used to calculate the percentage of mitochondrial genes. Cells with a gene count below 50 and a mitochondrial gene percentage above 5 were eliminated. The 1500 genes with the largest variation between cells were analyzed as cell clusters, the principal component was set to 20, and the resolution was 0.5.
Evaluation of diagnostic performance of characteristic genes
We further used the receiver operating characteristic curve (ROC) based on specificity and sensitivity to evaluate the diagnostic performance of the characteristic gene. The AUC indicated the diagnostic performance of the characteristic gene. Different AUC values mean different diagnostic values [26]. AUC = 1.0 means perfect diagnostic value; AUC = 0.9-1.0 means high diagnostic value; AUC = 0.7-0.9 means relative diagnostic value; AUC = 0.5-0.7 means low diagnostic value; AUC = 0.5 means no diagnostic value.
Evaluation of the prognostic value of characteristic genes
We performed a further analysis on the prognostic value of the characteristic genes. First, we observed the survival curves of different expression levels of the characteristic genes using the Kaplan-Meier method. Second, we further evaluated the ability of characteristic genes to affect the prognosis of CRC using univariate Cox regression analysis and displayed it using a forest plot. In the prognostic analysis, we excluded the samples with missing survival data and survival time ≤30.
External validation and experimental validation of core gene GPT expression
The GEPIA2 database analyzed the differential expression of the key gene GPT at the mRNA level of colon cancer and rectal cancer online (http://gepia2.cancer-pku.cn/#index) [27]. The UCLCAN database analyzed the expression difference of GPT at the protein level in the CPTAC database online (http://ualcan.path.uab.edu/index.html) [28]. The HPA database (https://www.proteinatlas.org/) obtained the immunohistochemical images of GPT in normal colorectal and CRC tissues [29], and obtained the basic information of the corresponding tissue samples.
CRC specimen collection, RNA extraction and quantitative real-time PCR (qRT-PCR) reaction
The 20 pairs of paracancerous normal tissues and matched CRC tissues were all from Shanghai Shidong Hospital. All patients provided written informed consent to participate in this study.
The qRT-PCR experiment was performed according to the previous procedure [30]. Briefly, total RNA from normal and CRC tissues was extracted using RNA-easy isolation reagent (Vazyme, China), and then the RNA was reversed to cDNA using PrimeScript™ RT Master Mix (Takara Bio, Japan). Finally, we performed qRT-PCR reactions using SYBR-Green qPCR Master Mix (Vazyme, China) and normalised using GAPDH. The experimental results were quantified relative to the 2-ΔΔCt method. Primer sequences were designed in this study as follows, The GAPDH primer sequences, forward: GTCTCCTCTGACTTCAACAGCG, reverse: ACCACCCTGTTGCTGTAGCCA. The GPT primer sequences, forward: CAAGAAGGTGCTCATGGAGATGG, reverse: TGTTCACCACCTCCACATAGCC.
Co-expression analysis and enrichment analysis
The co-expression network of GPT was analyzed using the LinkedOmics database (http://www.linkedomics.org/). GO analysis and KEGG analysis explored the biological processes involved in the co-expression network.
Assessment of the level of immune cell infiltration
Based on the gene expression profile of TCGA-CRC, we used CIBERSORT algorithm to evaluate the proportion of twenty-two types of immune cell [31], and samples with p < 0.05 were used for further analysis. Then, this research evaluated differences in the infiltration levels of 22 types of immune cells in CRC patients with different GPT expression.
Somatic mutation spectrum analysis
We used the R package “maftools” [32] to analyze the somatic gene mutation profile of TCGA-CRC patient samples. First, we analyzed the mutation status of characteristic genes in TCGA-CRC samples. Second, we further analyzed the 10 most mutated genes in samples with different levels of GPT expression.
Drug sensitivity analysis
We used R package “oncoPredict” [33] to analyze the half maximal inhibitory concentration (IC50) of TCGA-CRC samples based on gene expression data. The lower the IC50 value, the higher the drug sensitivity, and the larger the IC50 value, the lower the drug sensitivity.
Pancancer analysis of GPT
As previously described, we used the R package “Limma” to explore the expression differences of GPT in pancancer. Likewise, the diagnostic performance of GPT in pancancer was evaluated using ROC curves. The prognostic value of GPT in pancancer was evaluated using univariate Cox analysis and Kaplan-Meier method.
Statistical analysis
All bioinformatics research in this study was done by R language (version 4.1.2). Differences between groups were analyzed using the Wilcoxon test. The Kaplan-Meier method was used to draw survival curves. qRTPCR results were completed using Graphpad prism 9 software. p < 0.05 means that the results are statistically significantly different.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Results
Characteristic gene screening of GMRGs
First, we analyzed the differential expression of 118 glutamine metabolism family genes in the TCGA-CRC cohort, and the results showed that 31 GMRGs were differentially expressed genes (DEG), of which 4 GMRGs were down-regulated and 27 GMRGs were up-regulated. Figure 1A shows the volcano map of GMRG as well as the heat map of DEG (Figure 1B). Then, we performed two machine learning algorithms on DEG to screen out characteristic gene: RandomForest (Figure 1C, 1D) and SVM-RFE (Figure 1E). Detailed results are detailed in Supplementary Table 1. According to the results of the above two machine learning algorithms, we used the Venn diagram to obtain 9 overlapping genes and defined them as characteristic gene (Figure 1F). Finally, we explored the mutation status of CRC signature genes and found that CAD (6%), SLC3A2 (5%) and MET (4%) had the highest mutation rates (Figure 1G).
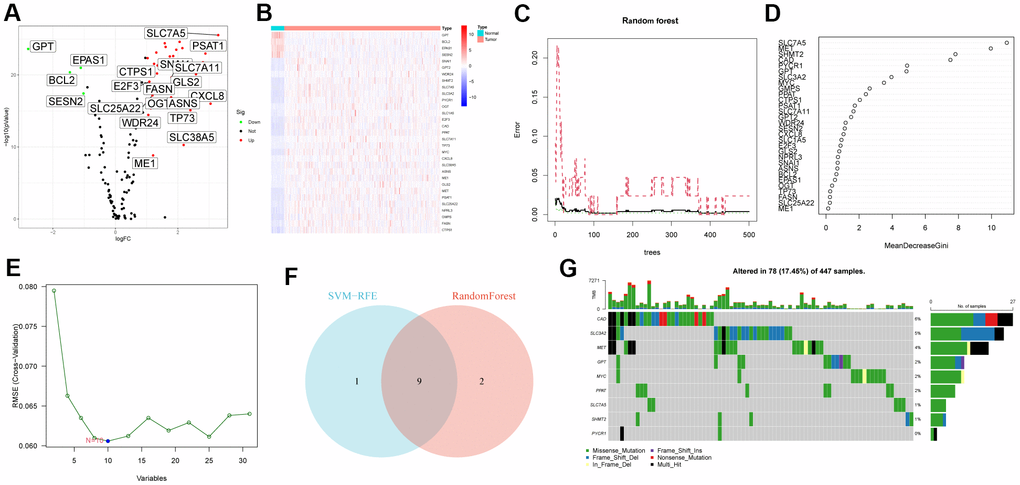
Figure 1. Characteristic gene screening. (A) Volcano map of differential expression of GMRGs. (B) Heatmap of DEG. (C, D) RandomForest evaluates the relative importance of DEG. (E) SVM–RFE algorithm. (F) Venn diagram of two machine learning algorithms. (G) Somatic mutation profiles of characteristic genes.
scRNA-seq analysis of characteristic genes
First, we processed single-cell sequencing data of CRC cancer tissues to classify cells into 18 clusters (Figure 2A). We then displayed the 10 genes that were significantly overexpressed in each cluster in Figure 2B. In addition, we automatically annotated cell types through the “SingleR” package, and finally determined that there are 9 main types of cells in CRC tissues, including T cells, B cells, Epithelial cells, Monocyte, Tissue stem cells, Smooth muscle cells, Endothelial cells, NK cells, Fibroblasts (Figure 2C).
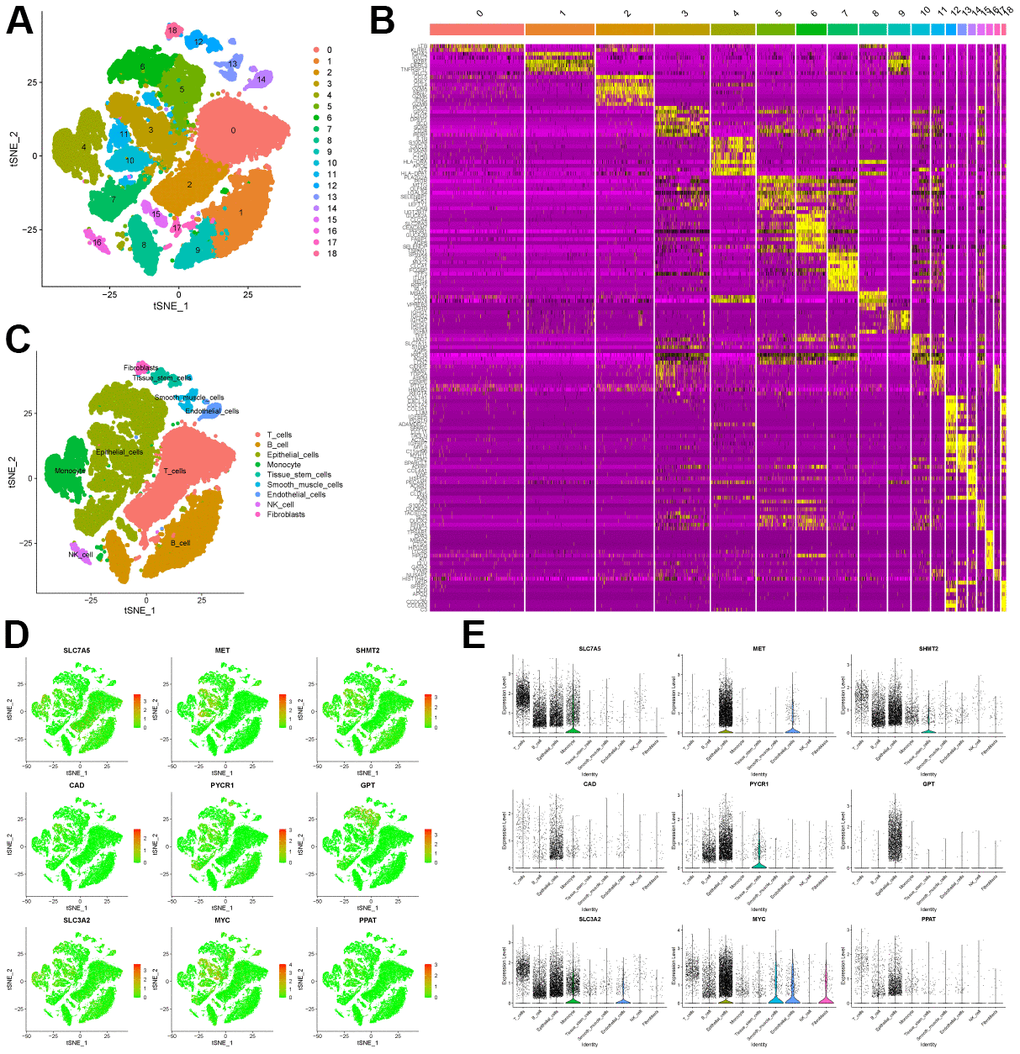
Figure 2. scRNA-seq analysis of CRC tissue. (A) tSNE analysis of CRC tissue scRNA-seq data to classify cell clusters. (B) Heatmap showing the top 10 genes highly expressed in each cell cluster. (C) The “Sin-gleR” package annotates the cell clusters into 9 cell types. (D) The distribution of the characteristic genes in the 9 cell types. (E) The bubble plot shows the expression levels of the characteristic genes in the 9 cell types.
Second, we explored the connections between cells using cell communication analysis. We visualized the number (Figure 3A) and weight (Figure 3B) of interactions between the 9 cell types. And centering on each cell type, the communication relationship between it and 9 cell types is shown separately (Figure 3C–3K). Furthermore, we use heatmaps to visualize the ligand-receptor molecules that mediate interactions between cells (Figure 3L).
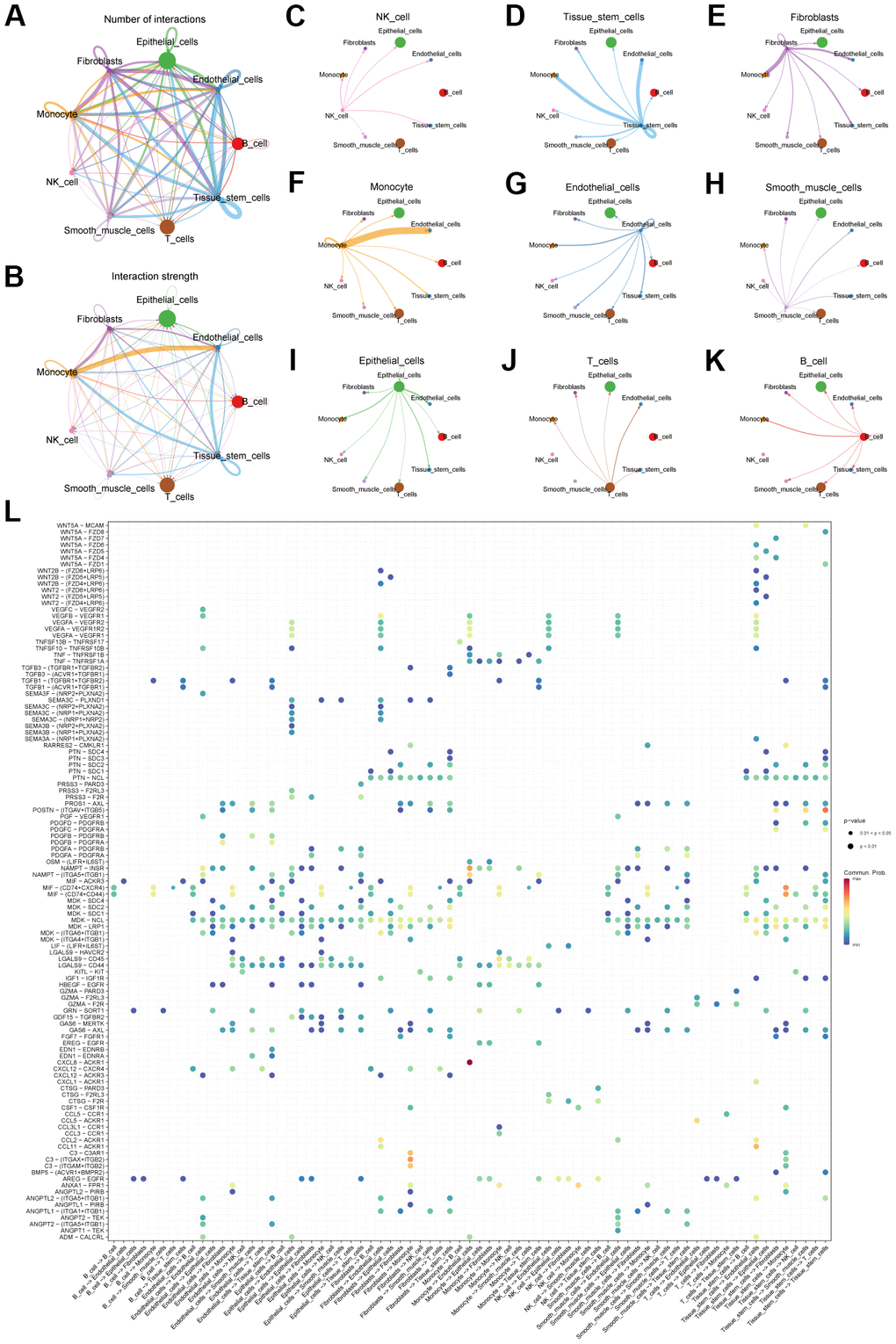
Figure 3. Cell communication analysis of scRNA-seq data. (A) Network diagram showing the number of connections between different cell types. (B) Network diagram showing the weight of connections between different cell types. (C–K) Diagram of the communication network between different cells and other cells. (L) Bubble plot showing genes involved in cell communication.
Finally, we explored the distribution of characteristic genes in the TME (Figure 2D). Bubble plots show the expression levels of signature genes in each cell type (Figure 2E). Combining the above results, we found that SLC7A5 was highly expressed in monocyte, B cells and T cells. MET is mainly expressed in endothelial cells and epithelial cells. SHMT2 has similar expression level in other cell types except T cells. CAD and PPAT were expressed at low levels in all cell types. PYCR1 is highly expressed in tissue stem cells, epithelial cells and fibroblasts. GPT is almost exclusively expressed in epithelial cells. SLC3A2 was highly expressed in monocytes, but very low in fibroblasts. MYC is mainly expressed in endothelial cells, smooth muscle cells and epithelial cells.
Diagnostic and prognostic value of characteristic genes
First, we analyzed the diagnostic performance of the characteristic genes using ROC curves. We found that the 9 characteristic genes all had high AUC values (Figure 2A–2I), specifically: CAD (AUC = 0.976), GPT (AUC = 0.973), MET (AUC = 0.983), MYC (AUC = 0.974), PPAT (AUC = 0.962), PYCR (AUC = 0.972), SHMT2 (AUC = 0.981), SLC3A2 (AUC = 0.968) and SLC7A5 (AUC = 0.992). These results imply that all characteristic genes are a potentially powerful class of diagnostic markers.
At the same time, we analyzed the survival curves of the 9 characteristic genes with different expression levels using the Kaplan-Meier method (Figure 2J–2U). We found that high expression of CAD and SLC7A5 had a worse prognosis, while high expression of GPT, MET, MYC, and PPAT had a better prognosis. The results showed that high expression of CAD and SLC7A5 had worse prognosis, while high expression of GPT, MET, MYC and PPAT had better prognosis. However, the prognosis of patients with different expression levels of PYCR, SHMT2, and SLC3A2 was not significantly different.
Finally, we used univariate Cox analysis and found only GPT as a protective factor affecting the prognosis, and none of the remaining 8 characteristic genes were factors affecting the prognosis in CRC (Figure 4A). Combining the above results, we regard GPT as the most important GMRGs in CRC and conduct further analysis.
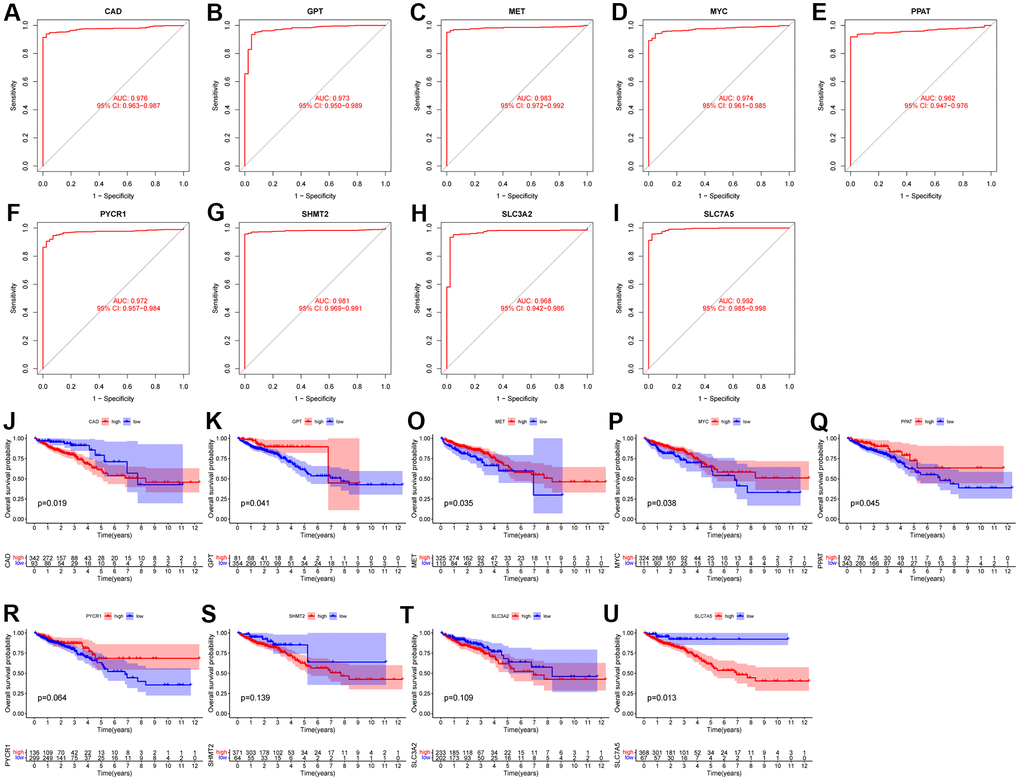
Figure 4. Evaluation of diagnostic performance and prognostic value of characteristic genes. (A–I) ROC curves of 9 characteristic genes. (J–U) KM survival curves of 9 characteristic genes.
Co-expression network and enrichment analysis of GPT in CRC
In order to fully understand the biological function of GPT in CRC, we analyzed the co-expression network of GPT in the LinkedOmics database online. The results showed that GPT expression was positively correlated with 3462 genes and negatively correlated with 5207 genes (Figure 5B). The heatmap shows the top 50 most significantly positively and negatively correlated genes with GPT expression, respectively (Figure 5C, 5D). We further performed GO analysis and KEGG analysis on the top 200 significantly positively correlated genes. The bubble charts show the top 15 GO analysis terms (including 5 BP, CC and MF terms respectively) and the 10 KEGG analysis terms (Figure 5E, 5F). GO analysis results showed that, in terms of BP, mainly enriched in hormone metabolic process; one−carbon metabolic process; cellular modified amino acid metabolic process; primary alcohol metabolic process; steroid metabolic process. in terms of CC, mainly enriched in brush border; brush border membrane; cluster of actin−based cell projections; apical part of cell; microvillus. in terms of MF, mainly enriched in carbonate dehydratase activity; oxidoreductase activity, acting on the CH−OH group of donors, NAD or NADP as acceptor; oxidoreductase activity, acting on CH−OH group of donors; steroid dehydrogenase activity, acting on the CH−OH group of donors, NAD or NADP as acceptor; carboxylic ester hydrolase activity. KEGG analysis mainly enriched in Pancreatic secretion; Bile secretion; Steroid hormone biosynthesis; Retinol metabolism; Glycerolipid metabolism. In short, the results suggest that GPT co-expressed genes are mainly involved in various metabolic pathways.
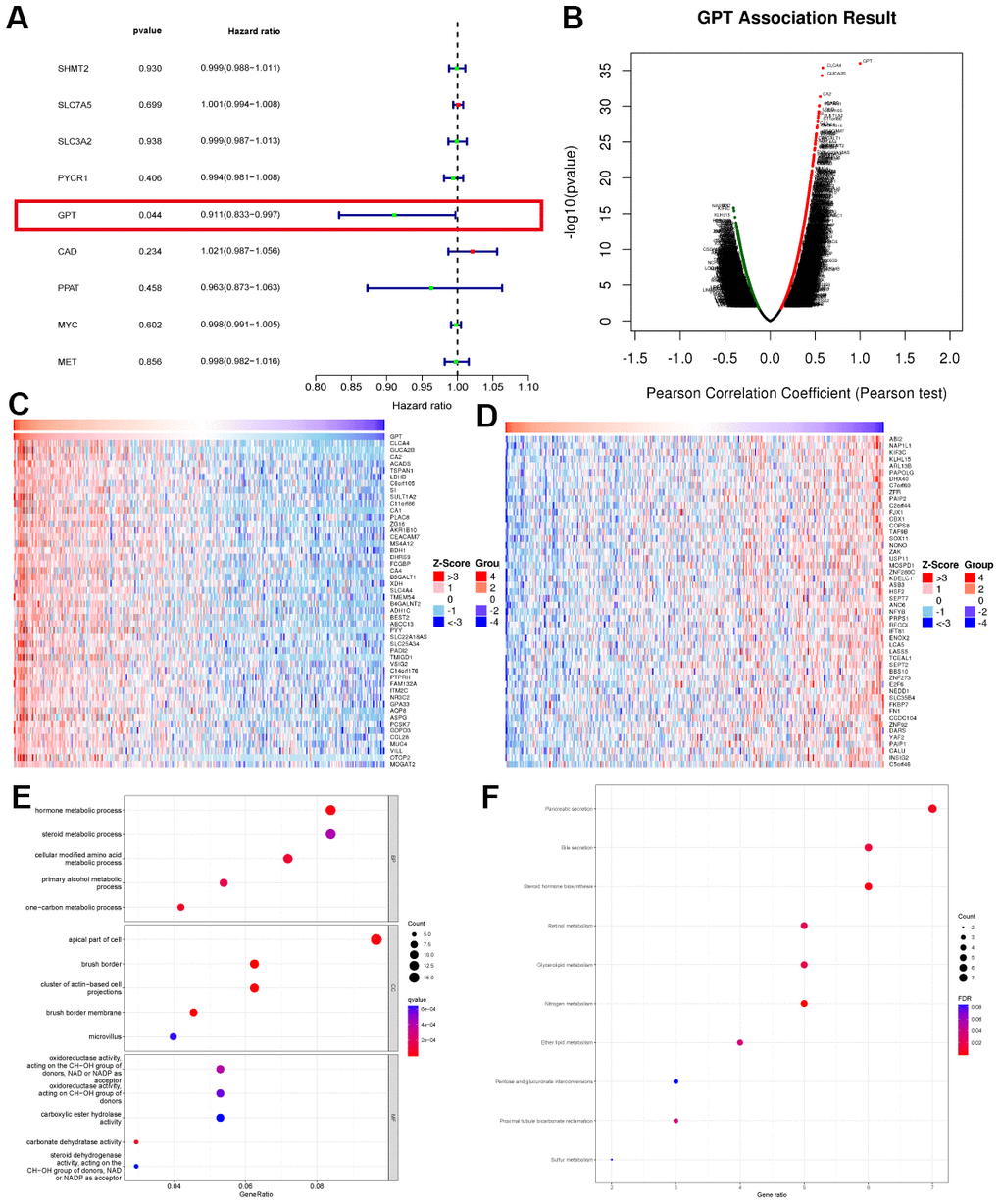
Figure 5. Co-expression analysis and enrichment analysis. (A) Univariate Cox regression analysis of characteristic genes. (B) Correlation of GPT ex pression with other genes. (C) Heatmap showing the 50 genes with the strongest positive correlation with GPT expression. (D) Heatmap showing the 50 genes most negatively correlated with GPT expression. (E) GO analysis. (F) KEGG analysis.
External validation and experimental validation of GPT expression
We first explored the differential expression of GPT in COAD and READ using the GEPIA2 database. We found that GPT mRNA expression levels were low in COAD and READ (Figure 6A). In addition, we detected GPT mRNA levels of GPT using qRT-PCR. The results also showed that compared with CRC tissues, GPT was highly expressed in normal tissues (Figure 6B). In addition, we also evaluated the expression difference of GPT at the protein level. First, we used the CAPTC database on UALCAN to analyze the differences in GPT protein expression levels, and found that GPT protein levels were significantly decreased in COAD tissues compared with normal tissues (Figure 6C). In addition, we obtained immunohistochemical images of normal colon and rectal tissues and CRC tissues from the HPA database. We found that the intensity of GPT in CRC tissues was significantly reduced compared to normal tissues (Figure 6D–6G).
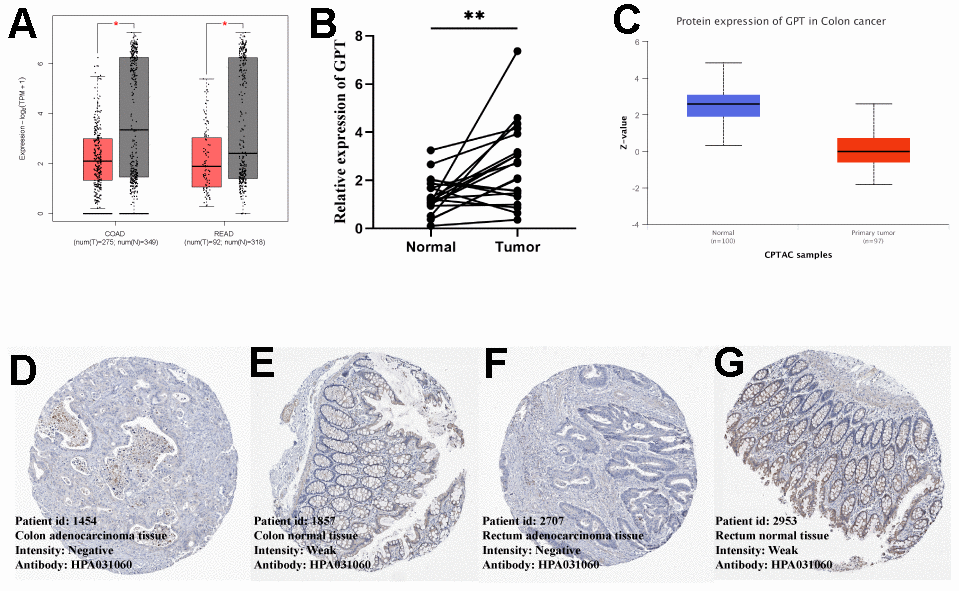
Figure 6. External data and experimental data verify GPT expression of GPT in CRC. (A) GEPIA2 database showing differential expression of GPT in COAD and READ (mRNA level). (B) qRT-PCR showed that GPT was significantly lower in CRC tissues than in adjacent normal tissues. (C) The UALCAN database showed that the low expression of GPT in COAD was correlated with cancer tissues (protein level). (D, E) HPA database showing IHC images of GPT in COAD and colon normal tissues. (F, G) HPA database showing IHC images of GPT in READ and rectal normal tissues.
Association analysis between GPT expression and TME
Our research assessed the infiltration levels of 22 immune cells in TCGA-CRC tissue samples using the CIBERSORT algorithm. We found that compared to the GPT low expression group, the infiltration levels increased significantly increased in the GPT high expression group, while the infiltration levels of T cells CD4, gamma delta of T cells gamma delta, Macrophages M0 and Macrophages M2 decreased significantly decreased (Figure 7A).
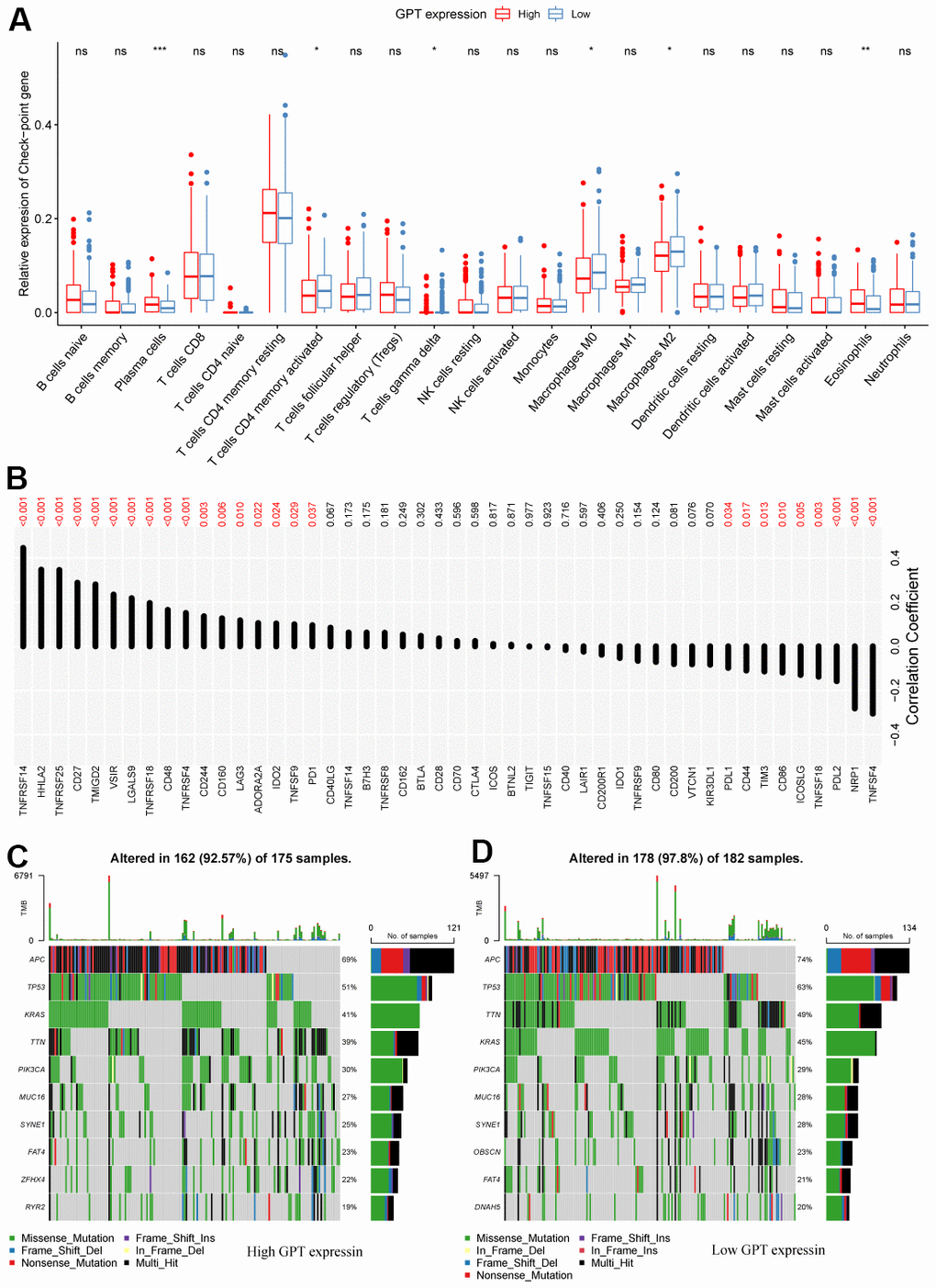
Figure 7. Correlation between GPT expression and the tumor microenvironment. (A) Differential analysis of immune cell infiltration levels with different levels of GPT expression. (B) Correlation between GPT expression and immune checkpoint molecules. (C, D) Somatic mutation profiles at different GPT expression levels.
We also analyzed the correlation between GPT expression and 48 immune checkpoint molecules (ICM), in which GPT expression was correlated with 26 ICM, including positive correlations with 17 ICM and negative correlations with 9 ICM (Figure 7B).
Furthermore, we explored the somatic mutation profile in patients with CRC and identified the 10 most mutated genes in different GPT expression level groups. The results showed that APC, TP53, KRAS, TTN, PIK3CA, MUC16, and SYNE1 were the 8 most mutated genes in different groups of expression level groups, and APC had the highest mutation rate (Figure 7C, 7D).
Prediction of potentially sensitive drugs
In order to explore the sensitivity of patients with different GPT expression groups to common chemotherapy drugs, we used the R package “oncoPredict” to analyze the IC50 values of CRC patients in the TCGA-CRC cohort to 5−Fluorouracil, Oxaliplatin, Gefitinib, Tamoxifen, Sorafenib and Dabrafenib. The results showed that the IC50 values of 5−Fluorouracil (Figure 8A), Oxaliplatin (Figure 8B), Gefitinib (Figure 8C), Tamoxifen (Figure 8D) and Sorafenib (Figure 8E) were higher in the GPT low expression group, whereas those of Dabrafenib (Figure 8F) were lower.
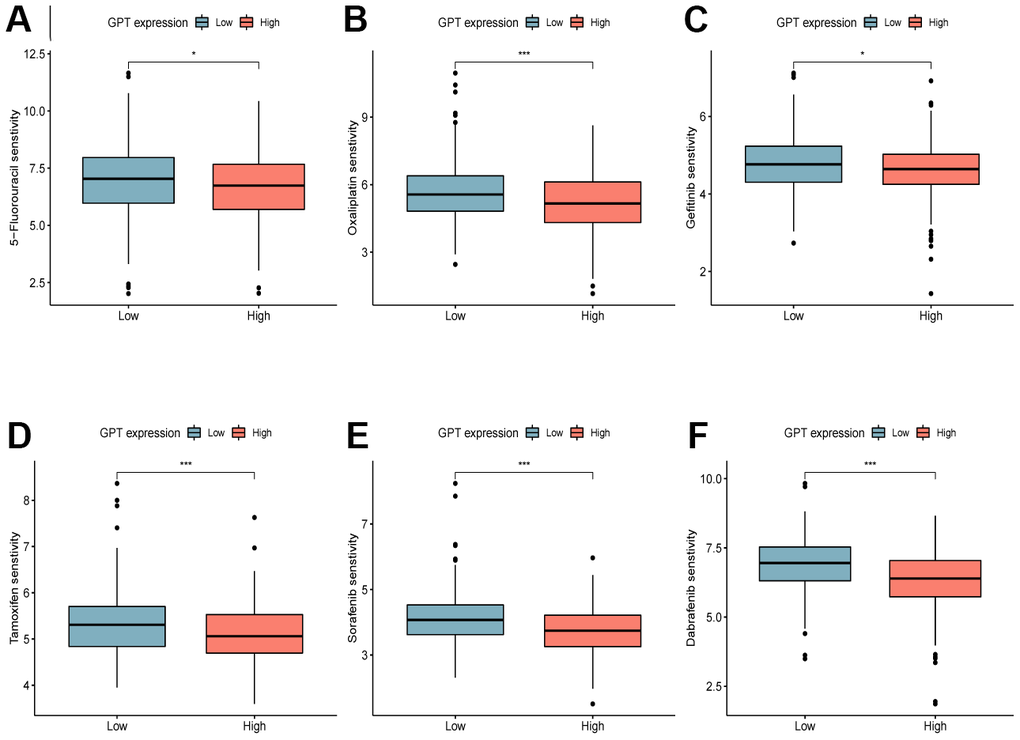
Figure 8. Drug sensitivity analysis. Sensitivity analysis of 5-Fluorouracil (A), Oxaliplatin (B), Gefitinib (C), Tamoxifen (D), Sorafenib (E), Dabrafenib (F) in different GPT expression levels.
Diagnostic performance of GPT in pancancer
As mentioned above, the diagnostic performance of GPT in pancancer is evaluated by the ROC curve. We found that GPT had excellent diagnostic performance in CHOL (AUC: 0.985). In BRCA (AUC: 0.711), CESC (AUC: 0.790), ESCA (AUC: 0.785), GBM (AUC: 0.780), HNSC (AUC: 0.894), KICH (AUC: 0.775), KIRP (AUC: 0.827), LUAD (AUC: 0.767), LUSC (AUC: 0.748), PCPG (AUC: 0.887), THCA (AUC: 0.716) and THYM (AUC: 0.786) had higher diagnostic performance (Figure 9).
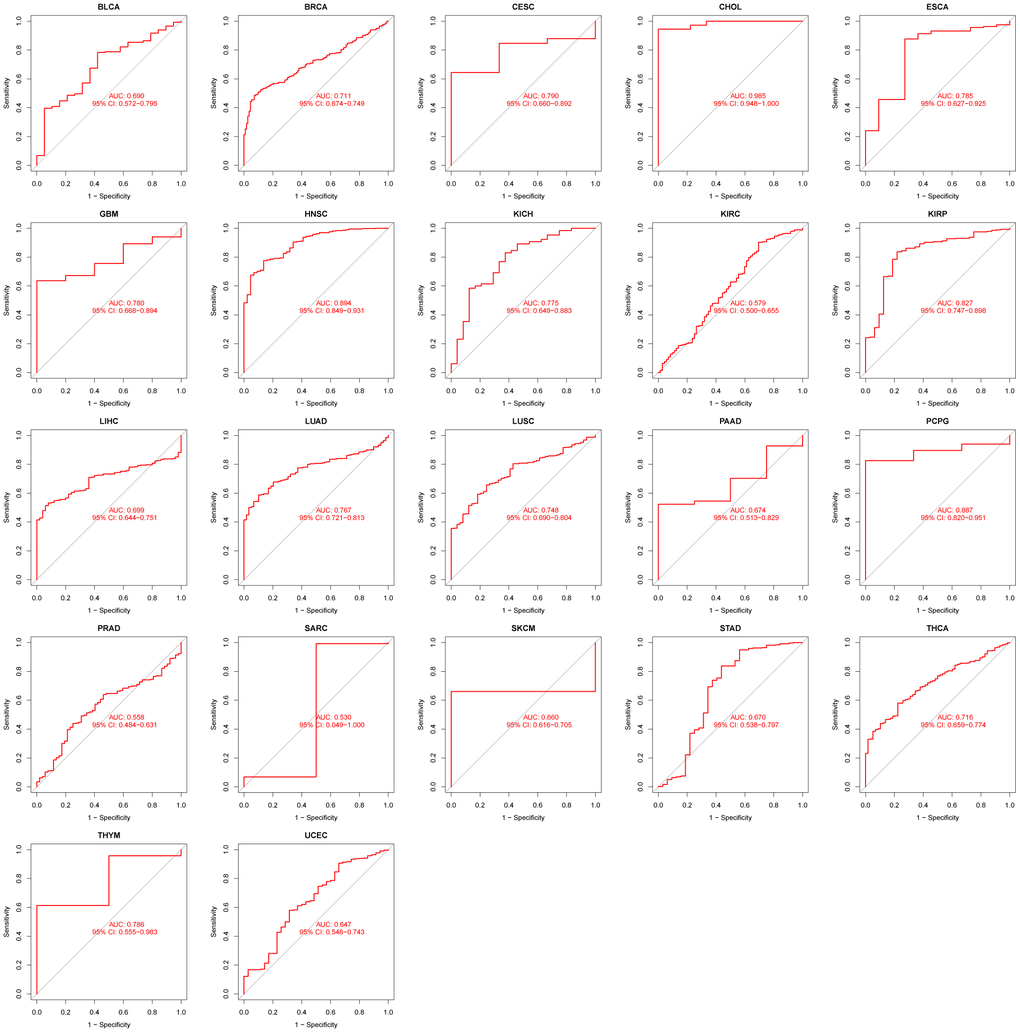
Figure 9. RUC curve to evaluate the diagnostic performance of GPT expression in 22 cancer types.
Expression of GPT in pancancer
GPT was differentially expressed in 16 cancers compared to normal tissues. Specifically, GPT was low expressed in BRCA, CHOL, ESCA, GBM, HNSC, KICH, KIRC, KIRP, LIHC, LUAD, LUSC, PCPG, STAD, UCS, and highly expressed in BLCA, LUAD, LUSC, THCA (Figure 10A). Furthermore, we sorted the expression of the tumor tissues, we found that GPT had the lowest expression level in TGCT and the highest expression level in LIHC (Figure 10B).
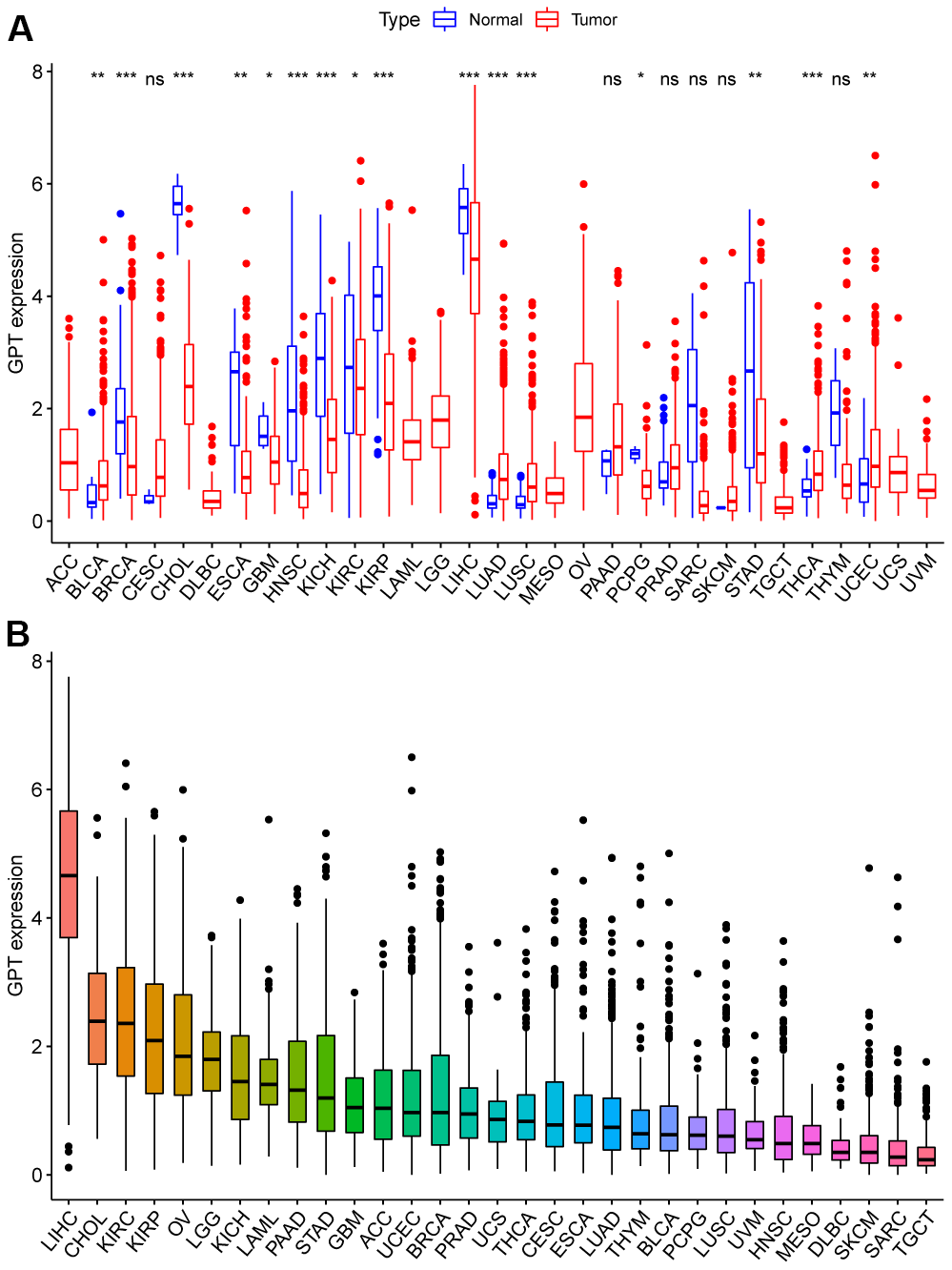
Figure 10. GPT expression patterns in pancancer. (A) Different expression of GPT in pancancer. (B) Ranking map of GPT expression in tumor tissues (from high to low).
Prognostic value of GPT in pancancer
As previously mentioned, we explored the role of GPT expression in the prognosis of pancancer using univariate Cox analysis and drew a forest plot. We found that GPT expression can affect the prognosis of ACC, BRCA, KIRC, KIRP, LGG, LIHC, and THYM. Specifically, GPT acts as a protective factor affecting the prognosis of ACC (HR: 0.373), BRCA (HR: 0.832), KIRC (HR: 0.869), KIRP (HR: 0.720), LGG (HR: 0.739), LIHC (HR: 0.840) and acts as a risk factor affecting the prognosis of THYM (HR: 2.495) (Figure 11A). Finally, for these 7 cancer types, we used the Kaplan-Meier method to analyze the survival curves of different expression levels of GPT, and the results showed that the GPT high expression group had a better prognosis in ACC (Figure 11B), BRCA (Figure 11C), KIRC (Figure 11D), KIRP (Figure 11E), LGG (Figure 11F), LIHC (Figure 11G), and in THYM (Figure 11H) The prognosis is poor.
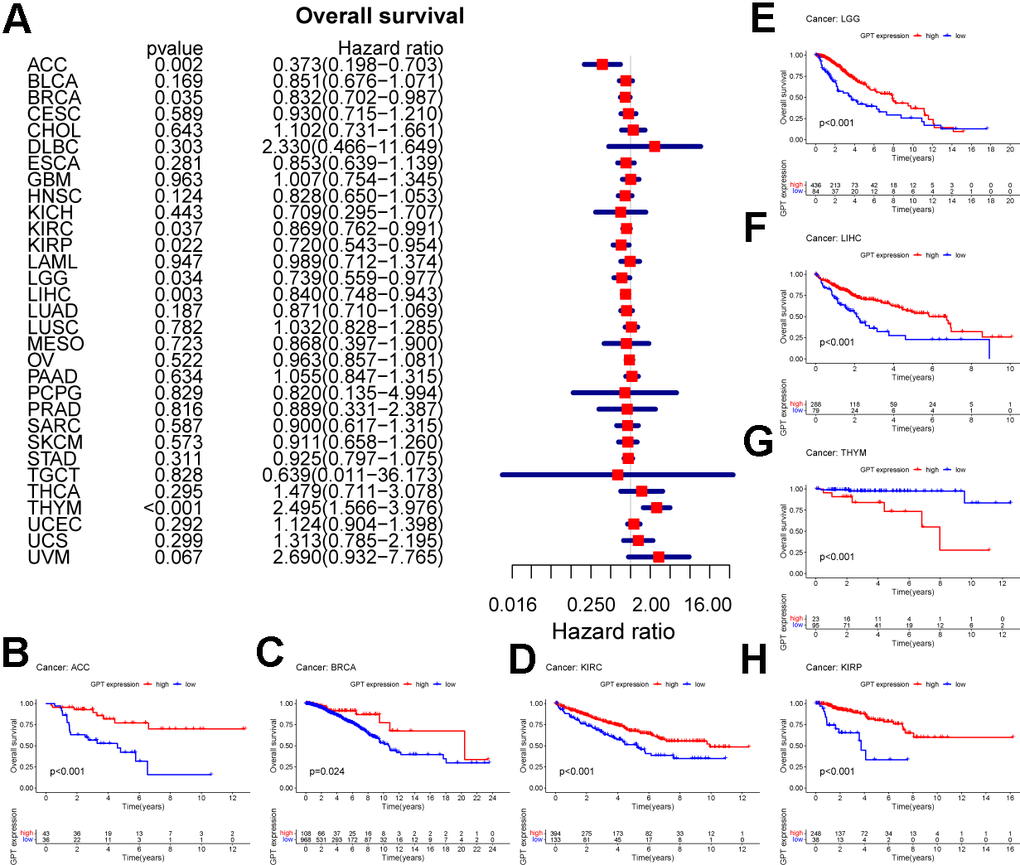
Figure 11. Prognostic value of GPT in pancancer. (A) Univariate Cox analysis to evaluate the correlation between GPT expression and OS in the patient. (B–H) Survival curves of GPT expression in 7 cancers.
Discussion
CRC is a malignant tumour of the digestive system with high incidence rate, and its morbidity and mortality are still on the rise. CRC is a highly heterogeneous malignant tumor, so it is necessary to find new biomarkers as the diagnosis, prognosis, and treatment target of CRC. Current research shows that glutamine metabolism plays a key role in the growth, metastasis, recurrence and drug resistance of CRC [34–36]. Therefore, in this study, we systematically analyzed the GMRGs in order to find powerful and promising biomarkers for diagnosis and prognosis. In this study, we first performed differential analysis on 113 GMRGs, and found that a total of 27 GMRGs were highly expressed in the TCGA-CRC cohort, and 4 GMRGs were lowly expressed. Then, we performed two machine learning algorithms, SVM-REF and RandomForest, on the 31 DEG to find more representative eigengenes. Based on the two algorithms, we found that 9 GMRGs can be regarded as characteristic genes of CRC. Furthermore, we further explored the cellular composition of the tumor microenvironment in CRC in order to understand the heterogeneity of CRC tissue. The results showed that CRC tissue was mainly composed of 9 types of cells, and epithelial cells, B cells, and T cells dominated, suggesting that immune cells may play an important role in the occurrence and development of CRC. We also explored the expression of 9 GMRGs in cells. Notably, GPT is expressed almost exclusively in epithelial cells. On this basis, we explored the prognostic value of signature genes by Kaplan-Meier method and univariate Cox analysis, and found that only GPT showed some prognostic value in the TCGA-CRC cohort. CRC patients with high GPT expression had a better prognosis than those with low GPT expression and were a protective factor for the prognosis of CRC. Based on this, we evaluated the expression of GPT in CRC again, and we double verified the low expression of GPT in CRC at the mRNA level and protein level by GEPIA2, UALCAN, HPA and qRT-PCR experiments. In addition, GO analysis and KEGG results based on GPT co-expressed genes indicated that GPT may affect multiple metabolic pathways.
Previous studies have shown that glutamine metabolism and the tumor immune microenvironment closely interact with each other [15, 37]. Interestingly, we also found that GPT expression was associated with the tumor microenvironment in CRC through bioinformatics. First, in our study, we found that GPT expression can affect the infiltration levels of various immune cells through the CIBERSORT algorithm. Studies have shown that Plasma cells and Eosinophils play an important role in anti-tumor, which means better clinical outcomes [38–41]. Whereas Macrophages M2 generally have tumor-promoting effects [42–44], implying a worse prognosis. These results may explain the better prognosis of low GPT expression in CRC. Immunotherapy is currently the mainstay of cancer treatment, and drug development based on immune checkpoint inhibitors is a promising strategy. Therefore, our research found that GPT expression was negatively correlated with 9 ICM and positively correlated with 17 ICM. This close correlation suggests that GPT may also be a promising immunotherapy marker. Finally, we explored the somatic mutations in different GPT expression levels, and found that the mutation rates of TP53 and TTN were decreased in the GPT high expression group compared with the GPT low expression group (TP53: 51% vs. 63%, TTN: 39% vs. 49%). Previous studies have shown that TP53, TTN mutations usually mean poorer prognosis in CRC [45, 46]. This may also be one of the high-expression group of reasons why the GPT had a better prognosis than the low-expression group of GPT.
We also predicted the correlation between GPT expression and the efficacy of common drugs. In this study, we predicted the sensitivity of 5-FU and Oxaliplatin, which are widely used in the first-line treatment of CRC [47, 48]. However, due to the frequent drug resistance events, we also analyzed the sensitivity of other common chemotherapeutic drugs. These results may provide potential therapeutic drugs for CRC patients with different levels of GPT expression.
Given that GPT is rarely reported in cancer, we also performed a pancancer analysis of GPT. First, we found that GPT was significantly differentially expressed in a variety of cancers (16/33). On the whole, GPT expression was reduced in most cancers. Through the ROC curve, we found that GPT showed relatively excellent diagnostic performance in 13 cancer types. Taken together, these results suggest that GPT may show promising promise in pancancer diagnostics. Finally, we also investigated the correlation between GPT expression and pancancer prognosis and found that GPT expression was a protective factor in 6 types of cancer and a risk factor in THYM. It shows that GPT plays different functions in different types of cancer.
This study, like other studies, has some limitations. First of all, this study did not verify the carcinogenesis of GPT in CRC through in vivo and in vitro experiments. Second, the specific mechanism by which GPT affects CRC has not been elucidated. Our future research will focus on the carcinogenesis and mechanism of GPT in CRC.
In summary, our systematic analysis of GMRGs screened for GPT as a potential diagnostic and prognostic marker for CRC, revealed the correlation between GPT and the tumor microenvironment, and explored the relationship between GPT expression and sensitivity to common drugs. These correlations between them suggest that GPT may be a potential therapeutic target for CRC. Finally, we also explored the diagnostic performance and prognostic value of GPT in pancancer, providing new insights for the function of GPT in pancancer.
Supplementary Materials
Author Contributions
XX was responsible for the design of the study. WD and WM were responsible for data download and analysis and wrote the manuscript. WX and DH were responsible for picture arrangement and typesetting. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical Statement and Consent
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Shanghai Shidong Hospital. All patients provided written informed consent to participate in this study.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82002539) and the General Project of Yangpu District, Shanghai (YPM202117).
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence
References
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71:209–49. https://doi.org/10.3322/caac.21660 [PubMed]
- 2. Levin TR, Corley DA, Jensen CD, Schottinger JE, Quinn VP, Zauber AG, Lee JK, Zhao WK, Udaltsova N, Ghai NR, Lee AT, Quesenberry CP, Fireman BH, Doubeni CA. Effects of Organized Colorectal Cancer Screening on Cancer Incidence and Mortality in a Large Community-Based Population. Gastroenterology. 2018; 155:1383–91.e5. https://doi.org/10.1053/j.gastro.2018.07.017 [PubMed]
- 3. Seymour MT, Thompson LC, Wasan HS, Middleton G, Brewster AE, Shepherd SF, O’Mahony MS, Maughan TS, Parmar M, Langley RE, FOCUS2 Investigators, and National Cancer Research Institute Colorectal Cancer Clinical Studies Group. Chemotherapy options in elderly and frail patients with metastatic colorectal cancer (MRC FOCUS2): an open-label, randomised factorial trial. Lancet. 2011; 377:1749–59. https://doi.org/10.1016/S0140-6736(11)60399-1 [PubMed]
- 4. Morgan E, Arnold M, Gini A, Lorenzoni V, Cabasag CJ, Laversanne M, Vignat J, Ferlay J, Murphy N, Bray F. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. 2023; 72:338–44. https://doi.org/10.1136/gutjnl-2022-327736 [PubMed]
- 5. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012; 21:297–308. https://doi.org/10.1016/j.ccr.2012.02.014 [PubMed]
- 6. Vaupel P, Schmidberger H, Mayer A. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol. 2019; 95:912–9. https://doi.org/10.1080/09553002.2019.1589653 [PubMed]
- 7. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016; 16:619–34. https://doi.org/10.1038/nrc.2016.71 [PubMed]
- 8. Yang L, Venneti S, Nagrath D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu Rev Biomed Eng. 2017; 19:163–94. https://doi.org/10.1146/annurev-bioeng-071516-044546 [PubMed]
- 9. Dai W, Xu L, Yu X, Zhang G, Guo H, Liu H, Song G, Weng S, Dong L, Zhu J, Liu T, Guo C, Shen X. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020; 72:909–23. https://doi.org/10.1016/j.jhep.2019.12.015 [PubMed]
- 10. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017; 36:1302–15. https://doi.org/10.15252/embj.201696151 [PubMed]
- 11. Mestre-Farrera A, Bruch-Oms M, Peña R, Rodríguez-Morató J, Alba-Castellón L, Comerma L, Quintela-Fandino M, Duñach M, Baulida J, Pozo ÓJ, García de Herreros A. Glutamine-Directed Migration of Cancer-Activated Fibroblasts Facilitates Epithelial Tumor Invasion. Cancer Res. 2021; 81:438–51. https://doi.org/10.1158/0008-5472.CAN-20-0622 [PubMed]
- 12. Brunner JS, Finley LWS. SnapShot: Cancer metabolism. Mol Cell. 2021; 81:3878–8.e1. https://doi.org/10.1016/j.molcel.2021.06.021 [PubMed]
- 13. van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG, Deng N, Harvey K, Beith JM, Selinger CI, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2016; 35:3201–8. https://doi.org/10.1038/onc.2015.381 [PubMed]
- 14. Bodineau C, Tomé M, Murdoch PDS, Durán RV. Glutamine, MTOR and autophagy: a multiconnection relationship. Autophagy. 2022; 18:2749–50. https://doi.org/10.1080/15548627.2022.2062875 [PubMed]
- 15. Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, Li D, Wang L, Chen Y, Liang Y, Niu H. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal. 2022; 20:114. https://doi.org/10.1186/s12964-022-00909-0 [PubMed]
- 16. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010; 35:427–33. https://doi.org/10.1016/j.tibs.2010.05.003 [PubMed]
- 17. Yang WH, Qiu Y, Stamatatos O, Janowitz T, Lukey MJ. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer. 2021; 7:790–804. https://doi.org/10.1016/j.trecan.2021.04.003 [PubMed]
- 18. Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, Sirota-Madi A, Olender T, Golan Y, et al. GeneCards Version 3: the human gene integrator. Database (Oxford). 2010; 2010:baq020. https://doi.org/10.1093/database/baq020 [PubMed]
- 19. Mitra D, Vega-Rubin-de-Celis S, Royla N, Bernhardt S, Wilhelm H, Tarade N, Poschet G, Buettner M, Binenbaum I, Borgoni S, Vetter M, Kantelhardt EJ, Thomssen C, et al. Abrogating GPT2 in triple-negative breast cancer inhibits tumor growth and promotes autophagy. Int J Cancer. 2021; 148:1993–2009. https://doi.org/10.1002/ijc.33456 [PubMed]
- 20. Wu J, Miao C, Wang Y, Wang S, Wang Z, Liu Y, Wang X, Wang Z. SPTBN1 abrogates renal clear cell carcinoma progression via glycolysis reprogramming in a GPT2-dependent manner. J Transl Med. 2022; 20:603. https://doi.org/10.1186/s12967-022-03805-w [PubMed]
- 21. Kim M, Gwak J, Hwang S, Yang S, Jeong SM. Mitochondrial GPT2 plays a pivotal role in metabolic adaptation to the perturbation of mitochondrial glutamine metabolism. Oncogene. 2019; 38:4729–38. https://doi.org/10.1038/s41388-019-0751-4 [PubMed]
- 22. Guo W, Tan HY, Li S, Wang N, Feng Y. Glutamic-Pyruvic Transaminase 1 Facilitates Alternative Fuels for Hepatocellular Carcinoma Growth-A Small Molecule Inhibitor, Berberine. Cancers (Basel). 2020; 12:1854. https://doi.org/10.3390/cancers12071854 [PubMed]
- 23. He S, Zhang S, Yao Y, Xu B, Niu Z, Liao F, Wu J, Song Q, Li M, Liu Z. Turbulence of glutamine metabolism in pan-cancer prognosis and immune microenvironment. Front Oncol. 2022; 12:1064127. https://doi.org/10.3389/fonc.2022.1064127 [PubMed]
- 24. Huang S, Cai N, Pacheco PP, Narrandes S, Wang Y, Xu W. Applications of Support Vector Machine (SVM) Learning in Cancer Genomics. Cancer Genomics Proteomics. 2018; 15:41–51. https://doi.org/10.21873/cgp.20063 [PubMed]
- 25. Ishwaran H, Kogalur UB. Consistency of Random Survival Forests. Stat Probab Lett. 2010; 80:1056–64. https://doi.org/10.1016/j.spl.2010.02.020 [PubMed]
- 26. Wang M, Zhu J, Ye Y, Li P, Sun W, Zhang M. N6AMT1 is a novel potential diagnostic, prognostic and immunotherapy response biomarker in pan-cancer. Aging (Albany NY). 2023; 15:6526–44. https://doi.org/10.18632/aging.204868 [PubMed]
- 27. Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019; 47:W556–60. https://doi.org/10.1093/nar/gkz430 [PubMed]
- 28. Chandrashekar DS, Karthikeyan SK, Korla PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne U, Creighton CJ, Varambally S. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia. 2022; 25:18–27. https://doi.org/10.1016/j.neo.2022.01.001 [PubMed]
- 29. Colwill K, Gräslund S, and Renewable Protein Binder Working Group. A roadmap to generate renewable protein binders to the human proteome. Nat Methods. 2011; 8:551–8. https://doi.org/10.1038/nmeth.1607 [PubMed]
- 30. Dai W, Li Y, Sun W, Ji M, Bao R, Chen J, Xu S, Dai Y, Chen Y, Liu W, Ge C, Sun W, Mo W, et al. Silencing of OGDHL promotes liver cancer metastasis by enhancing hypoxia inducible factor 1 α protein stability. Cancer Sci. 2023; 114:1309–23. https://doi.org/10.1111/cas.15540 [PubMed]
- 31. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015; 12:453–7. https://doi.org/10.1038/nmeth.3337 [PubMed]
- 32. Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018; 28:1747–56. https://doi.org/10.1101/gr.239244.118 [PubMed]
- 33. Maeser D, Gruener RF, Huang RS. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021; 22:bbab260. https://doi.org/10.1093/bib/bbab260 [PubMed]
- 34. Song Z, Wei B, Lu C, Li P, Chen L. Glutaminase sustains cell survival via the regulation of glycolysis and glutaminolysis in colorectal cancer. Oncol Lett. 2017; 14:3117–23. https://doi.org/10.3892/ol.2017.6538 [PubMed]
- 35. Sun H, Zhang C, Zheng Y, Liu C, Wang X, Cong X. Glutamine deficiency promotes recurrence and metastasis in colorectal cancer through enhancing epithelial-mesenchymal transition. J Transl Med. 2022; 20:330. https://doi.org/10.1186/s12967-022-03523-3 [PubMed]
- 36. Chen P, Liu XQ, Lin X, Gao LY, Zhang S, Huang X. Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism. Mol Ther Oncolytics. 2021; 20:228–39. https://doi.org/10.1016/j.omto.2021.01.001 [PubMed]
- 37. Zhu L, Zhu X, Wu Y. Effects of Glucose Metabolism, Lipid Metabolism, and Glutamine Metabolism on Tumor Microenvironment and Clinical Implications. Biomolecules. 2022; 12:580. https://doi.org/10.3390/biom12040580 [PubMed]
- 38. Wouters MCA, Nelson BH. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin Cancer Res. 2018; 24:6125–35. https://doi.org/10.1158/1078-0432.CCR-18-1481 [PubMed]
- 39. Berntsson J, Nodin B, Eberhard J, Micke P, Jirström K. Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int J Cancer. 2016; 139:1129–39. https://doi.org/10.1002/ijc.30138 [PubMed]
- 40. Davis BP, Rothenberg ME. Eosinophils and cancer. Cancer Immunol Res. 2014; 2:1–8. https://doi.org/10.1158/2326-6066.CIR-13-0196 [PubMed]
- 41. Loktionov A. Eosinophils in the gastrointestinal tract and their role in the pathogenesis of major colorectal disorders. World J Gastroenterol. 2019; 25:3503–26. https://doi.org/10.3748/wjg.v25.i27.3503 [PubMed]
- 42. Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. Int J Mol Sci. 2021; 22:6995. https://doi.org/10.3390/ijms22136995 [PubMed]
- 43. Zhao S, Mi Y, Guan B, Zheng B, Wei P, Gu Y, Zhang Z, Cai S, Xu Y, Li X, He X, Zhong X, Li G, et al. Tumor-derived exosomal miR-934 induces macrophage M2 polarization to promote liver metastasis of colorectal cancer. J Hematol Oncol. 2020; 13:156. https://doi.org/10.1186/s13045-020-00991-2 [PubMed]
- 44. Lan J, Sun L, Xu F, Liu L, Hu F, Song D, Hou Z, Wu W, Luo X, Wang J, Yuan X, Hu J, Wang G. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019; 79:146–58. https://doi.org/10.1158/0008-5472.CAN-18-0014 [PubMed]
- 45. Schulz-Heddergott R, Stark N, Edmunds SJ, Li J, Conradi LC, Bohnenberger H, Ceteci F, Greten FR, Dobbelstein M, Moll UM. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell. 2018; 34:298–314.e7. https://doi.org/10.1016/j.ccell.2018.07.004 [PubMed]
- 46. Zhou Y, Cheng X, Zhang F, Chen Q, Chen X, Shen Y, Lai C, Kota VG, Sun W, Huang Q, Yuan Y, Wang J, Lai M, Zhang D. Integrated multi-omics data analyses for exploring the co-occurring and mutually exclusive gene alteration events in colorectal cancer. Hum Mutat. 2020; 41:1588–99. https://doi.org/10.1002/humu.24059 [PubMed]
- 47. Li Y, Gan Y, Liu J, Li J, Zhou Z, Tian R, Sun R, Liu J, Xiao Q, Li Y, Lu P, Peng Y, Peng Y, et al. Downregulation of MEIS1 mediated by ELFN1-AS1/EZH2/DNMT3a axis promotes tumorigenesis and oxaliplatin resistance in colorectal cancer. Signal Transduct Target Ther. 2022; 7:87. https://doi.org/10.1038/s41392-022-00902-6 [PubMed]
- 48. Kishore C, Bhadra P. Current advancements and future perspectives of immunotherapy in colorectal cancer research. Eur J Pharmacol. 2021; 893:173819. https://doi.org/10.1016/j.ejphar.2020.173819 [PubMed]