



Epigraph
“Growth stimulation leads to cellular senescence when the cell cycle is blocked” [1].
Arrest is not yet senescence
Not anything that causes arrest causes senescence. For example, serum withdrawal, contact inhibition, nutrient starvation and rapamycin cause reversible arrest (quiescence) instead of senescence. What these conditions have in common is that they inhibit cellular mass or volume growth and specifically inhibit the mTOR pathway. (Of note: in the cell culture, quiescent cells will eventually succumb to senescence, because even rapamycin does not suppress geroconversion completely).
To induce senescence, DNA-damaging agents p21 and p16 cause cell-cycle arrest. Freshly arrested cells do not have senescent phenotype. During several days, the arrested cells acquire a large, flat morphology, beta-Gal positivity and Senescence-Associated Secretory Phenotype (SASP) [2–4]. The acquisition of senescent phenotype in arrested cells is known as gerogenic conversion or geroconversion [4–8].
Geroconversion is a continuation of cellular growth, when the cell cycle is blocked [1]. It may also partially occur in proliferating cells and is overstimulated in cell culture conditions. Cellular mass (volume) growth is driven in part by growth-promoting pathways such as mTOR [6]. And this is how the anti-aging activity of rapamycin was predicted, before life-extension was shown in animals [9].
Despite the obvious (acquisition of senescent phenotype takes time via active process), the existence of geroconversion is largely ignored by scientific community. One of the reasons is that in cell culture, geroconversion occurs automatically, unless actively prevented by rapamycin, serum and nutrient withdrawal, contact inhibition, severe hypoxia and some other factors (discussed later). In 2011, it was pointed out that “In cell culture, cell cycle arrest typically leads to senescence, because the cell is overstimulated by serum, nutrients, oncogenes and so on. Therefore, cell cycle arrest is sufficient to cause senescence, especially in cancer cells. This is why arrest of cell cycle is confused with senescence” [10].
Growth stimulation drives senescence during cell cycle arrest
Nutrients, mitogens or growth factors (GF), hormones (e.g., insulin and testosterone), cytoplasmic oncoproteins, oxygen and other factors stimulate growth-promoting pathways such as mTOR and MAPK, which stimulate both cellular mass growth, cyclin D induction and cell cycle progression. In the absence of growth stimulation (e.g., GF or serum withdrawal), MAPK and mTOR are deactivated. This slows down both cellular mass growth and cell cycle progression, and the cell becomes quiescent. Re-addition of growth factors allows quiescent cells to re-start proliferation [5, 6].
In proliferating cells, mTOR drives cellular mass growth, and this growth in cell size is balanced by cell division (Figure 1A). In quiescent cells, mTOR is deactivated, and the cell cycle is arrested. What would happen if the cell cycle were arrested, but mTOR is still active?
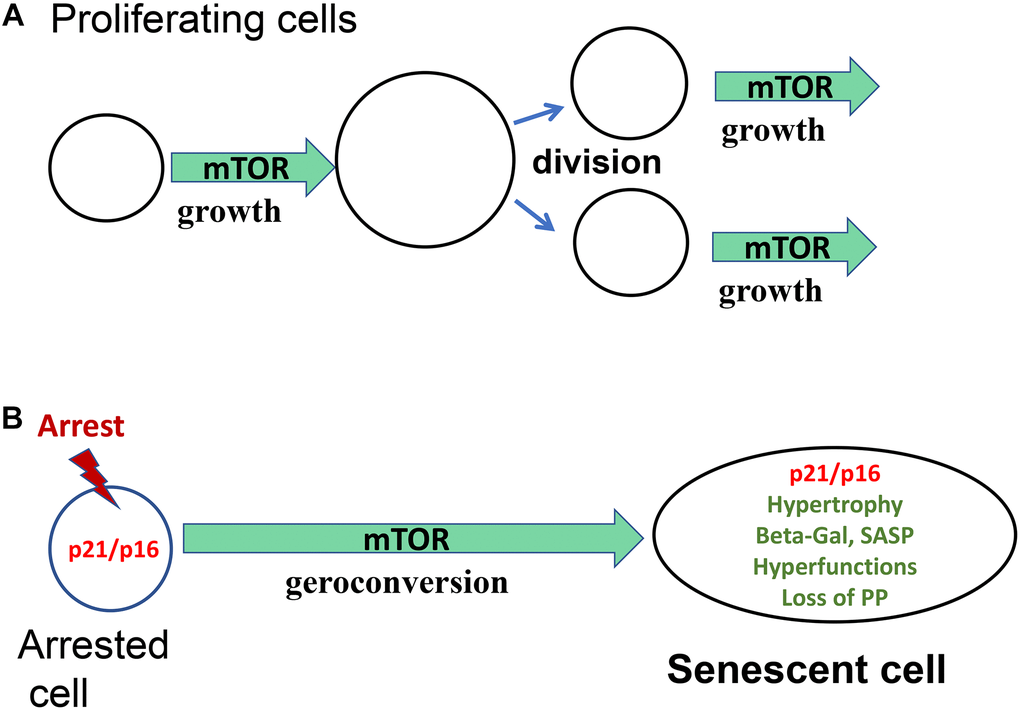
Figure 1. Geroconversion as a form of growth. (A) Proliferating cells. Cellular enlargement (growth) is followed by cell division. mTOR is shown as one of the drivers of growth. (B) Arrested cells. In the arrest cell (p21 and p16) cellular enlargement is followed by cell division. mTOR is shown as one of the drivers of geroconversion.
This condition can be caused by induction of CDK inhibitors (p21 and p16), which block the cell cycle, without affecting growth-promoting pathways such as mTOR and MAPK [6].
When the cell cycle is arrested by p21/p16, then mTOR drives growth in the absence of cell division, causing cellular hypertrophy (a large, flat cell morphology), lysosomal hyperfunction (beta-Gal-staining) and other hyperfunctions such as SASP (Figure 1B). It also increases tissue-specific hyperfunctions [6, 11].
Overactivated mTOR causes compensatory resistance to growth factors and insulin, via the pS6K1/IRS feedback loop [12, 13].
In a typical cell culture, cells are overstimulated by nutrients, serum and oxygen and grow in low cell density, making mTOR maximally active. For example, DMEM contains 5-fold higher than normal blood levels of glucose, higher than even in diabetic patients [13]. This is why it is sufficient to induce cell cycle arrest to induce senescence, unless mTOR-driven geroconversion is actively suppressed by serum withdrawal and contact inhibition, which deactivate mTOR [5, 14].
Pseudo-DNA-damage response in senescent cells
During geroconversion, overactivated kinases such as ATM phosphorylate H2AX, even in the absence of DNA damage [15]. As suggested by Rybak et al. [16] although DNA double-strand breaks always induce γH2AX, the reverse is not true: γH2AX is not an unequivocal marker of these breaks [16–18].
So, detection of γH2AX indicates that the cell may be senescent but does not indicate that it is necessarily caused by DNA damage. Unfortunately, it is not known to most scientists.
Acute DNA damage can cause arrest, but it’s not yet senescence
Acute DNA damage by radiation and DNA-damaging drugs activates DNA damage response (DDR). While DNA damage response (DDR) causes cell-cycle arrest, it is growth-promoting pathways such as mTOR that convert this arrest to senescent phenotype. (Figure 1B).
[Note: Life-long, gradual accumulation of DNA damage (accumulation of mutations) does not lead to cell-cycle arrest, but, in contrast, contributes to unlimited proliferation, robustness and immortality in cancer cells].
Once again, acute DNA damage or DDR in proliferating cells can lead to cellular senescence, because proliferating is associated with high activity of growth-promoting pathways necessary for geroconversion. When DDR causes arrest, these growth-promoting pathways drive geroconversion [19]. In serum-starved quiescent cells, mTOR is inactive and DNA damage cannot cause senescence. Growth stimulation with serum then drives geroconversion [19].
In the organism, acute DNA damage, or DDR, can lead to cell senescence by arresting proliferating cells. This is an age-independent cellular senescence that may occur at any age. This is also called non-adaptive cell senescence [20].
In contrast, age-dependent cellular senescence may be driven by life-long hyperfunction of growth-promoting pathways, especially in arrested (post-mitotic) cells.
Proliferative potential
At first, the freshly arrested cells retain proliferative potential (PP) and can re-start proliferation, if cell-cycle arrest is lifted. Following geroconversion, senescent cells cannot proliferate, even when cell-cycle arrest is lifted. The senescent cell may re-enter the cell cycle but cannot progress further or die in mitosis [2–4]. Loss of PP is a marker of the senescent phenotype, and rapamycin partially prevents loss of PP, as it partially prevents other markers of senescent phenotype such as cell hypertrophy, beta-Gal and SASP. Proliferative potential should not be confused with proliferation. For example, rapamycin inhibits proliferation but preserves PP. When p16 and p21 were induced for one day and then switched off, the cells resumed proliferation. If p16 was switched off after six days, cells remained phenotypically senescent and could not restart proliferation [2, 3]. Serum starvation [1, 19, 21] and mTOR inhibitors [1, 4, 22], prevent loss of PP during arrest, caused by switchable p21/p16 and the synthetic CDK4/6 inhibitor Palbociclib (PD0332991).
The irreversibility of cell cycle arrest should not be confused with Loss of PP. For example, Doxorubicin, a DNA-damaging drug, can render cell-cycle arrest irreversible, because doxorubicin cannot be easily washed out from the cell. If arrest is irreversible, it is impossible to know whether the cell retained (or not) the proliferative potential.
Cell hypertrophy (enlargement) as a marker of senescence
The large senescent morphology is the most noticeable feature of senescence in cell culture [23] and in the organism [24]. And it is not coincidental. Geroconversion is a continuation (quasi-program) of cellular growth [25]. At the beginning of geroconversion in p21-arrested cells, cellular mass (protein per well) is increased exponentially, and then growth becomes linear in p21-arrested cells [26]. In agreement, Neurohr et al. showed that within 9 days after doxorubicin-induced arrest, cell size increased linearly 8-fold [21]. Similarly, linear increase in cell volume was observed during arrest caused by the CDKi Palbociclib, and this increase was completely prevented by serum starvation [21]. Rapamycin partially decreases hypertrophy during cell-cycle arrest caused by either p21 or synthetic CDK inhibitors [4, 26]. Pan-mTOR inhibitors more potently suppressed hypertrophy than rapamycin [27, 28].
Thus, hypertrophy is only partially rapamycin-sensitive [26, 27].
Excessive cell growth as a marker of geroconversion
Geroconversion can occur not only in arrested but also in proliferating cells, if growth stimulation is excessive. For example, stem cells are small, and their size is increased with aging [29], and excessive growth stimulation drives stem cell geroconversion [7, 8].
It was even suggested that an increase in cell size by itself can cause senescence [21, 29, 30]. According to the geroconversion concept, excessive activation of growth-promoting pathways (MAPK, mTOR, etc.,) drives both excessive growth and other hyperfunctions (SASP, lysosomal hyperfunction (beta-Gal), hyper-differentiation). Furthermore, overactivated MAPK and mTOR pathways may induce p53/p21 and cycle arrest [31]. Following cell-cycle arrest, growth becomes even more excessive. Excessive growth and other manifestations of geroconversion are difficult to dissociate, because the manipulations that decrease growth (serum/nutrient starvation, rapamycin) also block MAPK/mTOR network that drives ALL manifestations together. This may suggest that cell size drives senescence rather than hyperfunctional growth-signaling drives senescence-associated hyprertrophy. As suggested, excessive mitogen/growth-stimulation may lead to hypermitogenic arrest [32] and then full-blown cell senescence [9, 31].
Geroconversion as terminal differentiation
Geroconversion can also be viewed as hypertrophic differentiation. For example, chondrocytes, responsible for bone growth in length, become hypertrophic and undergo senescence [33–36]. Like geroconvesrsion, terminal differentiation is an active process associated with decrease of proliferative potential [37], possible beta-Gal-positivity [38] as well as hypertrophy [39, 40] and increase of cellular functions, mainly tissue-specific functions.
Developmentally programmed cell senescence
While cell senescence is a quasi-programmed in aging, it may be programed in development [20, 41–45]. During mammalian embryonic development, senescent cells are cleared by macrophages, resulting in tissue remodeling [41].
Oncogene-induced senescence
Hyper-mitogenic stimulation may trigger cell-cycle arrest and simultaneously promote size growth [32, 46–49].
How should we define cellular senescence?
Cellular senescence is neither functional decline nor caused by chronic accumulation of molecular damage. In contrast, cellular senescence is characterized by universal hyperfunctions such as SASP plus tissue-specific hyperfunctions (senescent beta-cells as an example). Second, whether accumulation of molecular damages (mutations) lead to cancer, cancer cells tend to be immortal. A common definition of cellular senescence as permanent loss of proliferative potential does not recapitulate the most important features of the senescent phenotype, such as hypertrophy and hyperfunctions (e.g., SASP).
Cell senescence is a proliferation-like state in non-proliferating cells. Growth-promoting pathways, including mTOR and MEK/MAPK, drive both growth and geroconversion. When actual growth is completed, growth-promoting pathways drive cellular senescence (Figure 1). Thus, a program of growth becomes a quasi-program of senescence. (Quasi- means pseudo- or “resembling but not real”). Senescent cells resemble proliferating cells but do not proliferate [5]. As “Growth stimulation leads to cellular senescence when the cell cycle is blocked” the molecular hallmark of senescent cells is presented: high levels of p21/p16, phospho-S6 and cyclin D1 [50]. Cell senescence is associated with constitutive, proliferative-like activity of nutrient-sensing and growth-promoting pathways such as mTOR in non-proliferative (arrested) cells.
David Gems and Carina Kern suggested replacing the term cellular senescence with remodeling activation, and SASP with RASP [20]. The key word is activation. According to hyperfunction theory, cellular senescence (or remodeling activation) can be viewed as hyperactivation, hyperfunction, hypertrophy, hyper-differentiation.
In 2003, I proposed “that simultaneous stimulation of mitogen-activated pathways and downstream inhibition of cyclin-dependent kinases leads, ultimately, to cell senescence” [32]. In other words, senescence occurs when growth stimulation meets cell cycle arrest. In agreement, Rapamycin and other rapalogs (Everolimus and Ridaforolimus), pan-mTOR inhibitors [27, 28] and, to a lesser extent, MEK, PI3K, mdm-2 and S6K inhibitors all slow down geroconversion in mammalian cells [1, 22, 26, 51–55].
Numerous studies further confirmed that mTOR is involved in the senescence phenotype [56–69].
Organismal aging as quasi-program of developmental growth
Like geroconversion is a continuation of cellular growth, the organismal aging is a continuation of developmental growth (see Figure 1 in reference [70]). Aging is not programmed, it is quasi-programmed. A quasi-program is a purposeless continuation of programs that were not turned off upon their completion. This has been discussed in detail [9, 50, 71–75].
Growth and aging are driven by overlapping signaling pathways. As suggested in 2007, “mTOR stands out because (a) it is a hub in the signaling network, (b) it is conserved from plants to animals (c) its inhibitors, rapamycin (Sirolimus) and everolimus, are clinically available drugs” [76]. To be clinically useful, the hyperfunction theory is mTOR-centric.
Conflicts of Interest
The author declares no conflicts of interest related to this study.
References
- 1. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7:3355–61. https://doi.org/10.4161/cc.7.21.6919 [PubMed]
- 2. Dai CY, Enders GH. p16 INK4a can initiate an autonomous senescence program. Oncogene. 2000; 19:1613–22. https://doi.org/10.1038/sj.onc.1203438 [PubMed]
- 3. Chang BD, Broude EV, Fang J, Kalinichenko TV, Abdryashitov R, Poole JC, Roninson IB. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000; 19:2165–70. https://doi.org/10.1038/sj.onc.1203573 [PubMed]
- 4. Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013; 12:3063–9. https://doi.org/10.4161/cc.26130 [PubMed]
- 5. Blagosklonny MV. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle. 2018; 17:2655–65. https://doi.org/10.1080/15384101.2018.1554781 [PubMed]
- 6. Blagosklonny MV. Cell senescence, rapamycin and hyperfunction theory of aging. Cell Cycle. 2022; 21:1456–67. https://doi.org/10.1080/15384101.2022.2054636 [PubMed]
- 7. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, Perdiguero E, Muñoz-Cánoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014; 506:316–21. https://doi.org/10.1038/nature13013 [PubMed]
- 8. Sousa-Victor P, Perdiguero E, Muñoz-Cánoves P. Geroconversion of aged muscle stem cells under regenerative pressure. Cell Cycle. 2014; 13:3183–90. https://doi.org/10.4161/15384101.2014.965072 [PubMed]
- 9. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5:2087–102. https://doi.org/10.4161/cc.5.18.3288 [PubMed]
- 10. Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY). 2011; 3:94–101. https://doi.org/10.18632/aging.100281 [PubMed]
- 11. Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, Fixler Y, Shreibman D, Zamir A, et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016; 22:412–20. https://doi.org/10.1038/nm.4054 [PubMed]
- 12. Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004; 14:1650–6. https://doi.org/10.1016/j.cub.2004.08.026 [PubMed]
- 13. Leontieva OV, Demidenko ZN, Blagosklonny MV. Rapamycin reverses insulin resistance (IR) in high-glucose medium without causing IR in normoglycemic medium. Cell Death Dis. 2014; 5:e1214. https://doi.org/10.1038/cddis.2014.178 [PubMed]
- 14. Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A. 2014; 111:8832–7. https://doi.org/10.1073/pnas.1405723111 [PubMed]
- 15. Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009; 8:4112–8. https://doi.org/10.4161/cc.8.24.10215 [PubMed]
- 16. Rybak P, Hoang A, Bujnowicz L, Bernas T, Berniak K, Zarębski M, Darzynkiewicz Z, Dobrucki J. Low level phosphorylation of histone H2AX on serine 139 (γH2AX) is not associated with DNA double-strand breaks. Oncotarget. 2016; 7:49574–87. https://doi.org/10.18632/oncotarget.10411 [PubMed]
- 17. Oricchio E, Saladino C, Iacovelli S, Soddu S, Cundari E. ATM is activated by default in mitosis, localizes at centrosomes and monitors mitotic spindle integrity. Cell Cycle. 2006; 5:88–92. https://doi.org/10.4161/cc.5.1.2269 [PubMed]
- 18. Pankotai T, Hoffbeck AS, Boumendil C, Soutoglou E. DNA damage response in the absence of DNA lesions continued. Cell Cycle. 2009; 8:4025–6. [PubMed]
- 19. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2:924–35. https://doi.org/10.18632/aging.100265 [PubMed]
- 20. Gems D, Kern CC. Is "cellular senescence" a misnomer? Geroscience. 2022; 44:2461–9. https://doi.org/10.1007/s11357-022-00652-x [PubMed]
- 21. Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, Miettinen TP, Vaites LP, Soares LM, Paulo JA, Harper JW, Buratowski S, Manalis S, et al. Excessive Cell Growth Causes Cytoplasm Dilution And Contributes to Senescence. Cell. 2019; 176:1083–97.e18. https://doi.org/10.1016/j.cell.2019.01.018 [PubMed]
- 22. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8:1888–95. https://doi.org/10.4161/cc.8.12.8606 [PubMed]
- 23. Polymenis M, Kennedy BK. Unbalanced Growth, Senescence and Aging. Adv Exp Med Biol. 2017; 1002:189–208. https://doi.org/10.1007/978-3-319-57127-0_8 [PubMed]
- 24. Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z, Krizhanovsky V. Quantitative identification of senescent cells in aging and disease. Aging Cell. 2017; 16:661–71. https://doi.org/10.1111/acel.12592 [PubMed]
- 25. Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY). 2009; 1:357–62. https://doi.org/10.18632/aging.100040 [PubMed]
- 26. Demidenko ZN, Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging (Albany NY). 2009; 1:1008–16. https://doi.org/10.18632/aging.100115 [PubMed]
- 27. Leontieva OV, Blagosklonny MV. Gerosuppression by pan-mTOR inhibitors. Aging (Albany NY). 2016; 8:3535–51. https://doi.org/10.18632/aging.101155 [PubMed]
- 28. Walters HE, Deneka-Hannemann S, Cox LS. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY). 2016; 8:231–44. https://doi.org/10.18632/aging.100872 [PubMed]
- 29. Lengefeld J, Cheng CW, Maretich P, Blair M, Hagen H, McReynolds MR, Sullivan E, Majors K, Roberts C, Kang JH, Steiner JD, Miettinen TP, Manalis SR, et al. Cell size is a determinant of stem cell potential during aging. Sci Adv. 2021; 7:eabk0271. https://doi.org/10.1126/sciadv.abk0271 [PubMed]
- 30. Lanz MC, Zatulovskiy E, Swaffer MP, Zhang L, Ilerten I, Zhang S, You DS, Marinov G, McAlpine P, Elias JE, Skotheim JM. Increasing cell size remodels the proteome and promotes senescence. Mol Cell. 2022; 82:3255–69.e8. https://doi.org/10.1016/j.molcel.2022.07.017 [PubMed]
- 31. Blagosklonny MV. Cell senescence: hypertrophic arrest beyond the restriction point. J Cell Physiol. 2006; 209:592–7. https://doi.org/10.1002/jcp.20750 [PubMed]
- 32. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4:358–62. https://doi.org/10.1038/sj.embor.embor806 [PubMed]
- 33. Nilsson O, Baron J. Fundamental limits on longitudinal bone growth: growth plate senescence and epiphyseal fusion. Trends Endocrinol Metab. 2004; 15:370–4. https://doi.org/10.1016/j.tem.2004.08.004 [PubMed]
- 34. Forcinito P, Andrade AC, Finkielstain GP, Baron J, Nilsson O, Lui JC. Growth-inhibiting conditions slow growth plate senescence. J Endocrinol. 2011; 208:59–67. https://doi.org/10.1677/JOE-10-0302 [PubMed]
- 35. Dreier R. Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res Ther. 2010; 12:216. https://doi.org/10.1186/ar3117 [PubMed]
- 36. Hallett SA, Ono W, Ono N. The hypertrophic chondrocyte: To be or not to be. Histol Histopathol. 2021; 36:1021–36. https://doi.org/10.14670/HH-18-355 [PubMed]
- 37. Minoo P, Sullivan W, Solomon LR, Martin TE, Toft DO, Scott RE. Loss of proliferative potential during terminal differentiation coincides with the decreased abundance of a subset of heterogeneous ribonuclear proteins. J Cell Biol. 1989; 109:1937–46. https://doi.org/10.1083/jcb.109.5.1937 [PubMed]
- 38. Untergasser G, Gander R, Rumpold H, Heinrich E, Plas E, Berger P. TGF-beta cytokines increase senescence-associated beta-galactosidase activity in human prostate basal cells by supporting differentiation processes, but not cellular senescence. Exp Gerontol. 2003; 38:1179–88. https://doi.org/10.1016/j.exger.2003.08.008 [PubMed]
- 39. Ding L, Yin Y, Han L, Li Y, Zhao J, Zhang W. TSC1-mTOR signaling determines the differentiation of islet cells. J Endocrinol. 2017; 232:59–70. https://doi.org/10.1530/JOE-16-0276 [PubMed]
- 40. Facchini A, Borzì RM, Olivotto E, Platano D, Pagani S, Cetrullo S, Flamigni F. Role of polyamines in hypertrophy and terminal differentiation of osteoarthritic chondrocytes. Amino Acids. 2012; 42:667–78. https://doi.org/10.1007/s00726-011-1041-9 [PubMed]
- 41. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. Programmed cell senescence during mammalian embryonic development. Cell. 2013; 155:1104–18. https://doi.org/10.1016/j.cell.2013.10.019 [PubMed]
- 42. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013; 155:1119–30. https://doi.org/10.1016/j.cell.2013.10.041 [PubMed]
- 43. Davaapil H, Brockes JP, Yun MH. Conserved and novel functions of programmed cellular senescence during vertebrate development. Development. 2017; 144:106–14. https://doi.org/10.1242/dev.138222 [PubMed]
- 44. Da Silva-Álvarez S, Guerra-Varela J, Sobrido-Cameán D, Quelle A, Barreiro-Iglesias A, Sánchez L, Collado M. Developmentally-programmed cellular senescence is conserved and widespread in zebrafish. Aging (Albany NY). 2020; 12:17895–901. https://doi.org/10.18632/aging.103968 [PubMed]
- 45. Rhinn M, Ritschka B, Keyes WM. Cellular senescence in development, regeneration and disease. Development. 2019; 146:dev151837. https://doi.org/10.1242/dev.151837 [PubMed]
- 46. Zhu H, Blake S, Kusuma FK, Pearson RB, Kang J, Chan KT. Oncogene-induced senescence: From biology to therapy. Mech Ageing Dev. 2020; 187:111229. https://doi.org/10.1016/j.mad.2020.111229 [PubMed]
- 47. Anerillas C, Abdelmohsen K, Gorospe M. Regulation of senescence traits by MAPKs. Geroscience. 2020; 42:397–408. https://doi.org/10.1007/s11357-020-00183-3 [PubMed]
- 48. Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, Haupt Y, Hannan RD, Pearson RB. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. 2012; 31:1949–62. https://doi.org/10.1038/onc.2011.394 [PubMed]
- 49. Jung SH, Hwang HJ, Kang D, Park HA, Lee HC, Jeong D, Lee K, Park HJ, Ko YG, Lee JS. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene. 2019; 38:1639–50. https://doi.org/10.1038/s41388-018-0521-8 [PubMed]
- 50. Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle. 2010; 9:1859–62. https://doi.org/10.4161/cc.9.10.11872 [PubMed]
- 51. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107:9660–4. https://doi.org/10.1073/pnas.1002298107 [PubMed]
- 52. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012; 109:13314–8. https://doi.org/10.1073/pnas.1205690109 [PubMed]
- 53. Leontieva OV, Demidenko ZN, Blagosklonny MV. S6K in geroconversion. Cell Cycle. 2013; 12:3249–52. https://doi.org/10.4161/cc.26248 [PubMed]
- 54. Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013; 20:1241–9. https://doi.org/10.1038/cdd.2013.86 [PubMed]
- 55. Leontieva OV, Blagosklonny MV. Tumor promoter-induced cellular senescence: cell cycle arrest followed by geroconversion. Oncotarget. 2014; 5:12715–27. https://doi.org/10.18632/oncotarget.3011 [PubMed]
- 56. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015; 17:1049–61. https://doi.org/10.1038/ncb3195 [PubMed]
- 57. Wang R, Sunchu B, Perez VI. Rapamycin and the inhibition of the secretory phenotype. Exp Gerontol. 2017; 94:89–92. https://doi.org/10.1016/j.exger.2017.01.026 [PubMed]
- 58. Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, Caples K, Bradley L, Beaver LM, Ho E, Löhr CV, Perez VI. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017; 16:564–74. https://doi.org/10.1111/acel.12587 [PubMed]
- 59. Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, Rideau D, Parpaleix A, Huang J, Amsellem V, Vienney N, Validire P, Maitre B, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018; 3:93203. https://doi.org/10.1172/jci.insight.93203 [PubMed]
- 60. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, Gutkind JS. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012; 11:401–14. https://doi.org/10.1016/j.stem.2012.06.007 [PubMed]
- 61. Sodagam L, Lewinska A, Wnuk M, Rattan SIS. Chronic exposure to rapamycin and episodic serum starvation modulate ageing of human fibroblasts in vitro. Biogerontology. 2017; 18:841–54. https://doi.org/10.1007/s10522-017-9730-8 [PubMed]
- 62. Nie D, Zhang J, Zhou Y, Sun J, Wang W, Wang JH. Rapamycin Treatment of Tendon Stem/Progenitor Cells Reduces Cellular Senescence by Upregulating Autophagy. Stem Cells Int. 2021; 2021:6638249. https://doi.org/10.1155/2021/6638249 [PubMed]
- 63. Maskey RS, Wang F, Lehman E, Wang Y, Emmanuel N, Zhong W, Jin G, Abraham RT, Arndt KT, Myers JS, Mazurek A. Sustained mTORC1 activity during palbociclib-induced growth arrest triggers senescence in ER+ breast cancer cells. Cell Cycle. 2021; 20:65–80. https://doi.org/10.1080/15384101.2020.1859195 [PubMed]
- 64. Gao C, Ning B, Sang C, Zhang Y. Rapamycin prevents the intervertebral disc degeneration via inhibiting differentiation and senescence of annulus fibrosus cells. Aging (Albany NY). 2018; 10:131–43. https://doi.org/10.18632/aging.101364 [PubMed]
- 65. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z, Guo G, Xia Y, Zhu X, Shi G, Cheng C. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016; 8:1102–14. https://doi.org/10.18632/aging.100925 [PubMed]
- 66. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015; 17:1205–17. https://doi.org/10.1038/ncb3225 [PubMed]
- 67. Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, Liu L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014; 97:20–9. https://doi.org/10.1097/TP.0b013e3182a7fcf8 [PubMed]
- 68. Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012; 11:2391–401. https://doi.org/10.4161/cc.20683 [PubMed]
- 69. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R, Pestell RG, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012; 181:278–93. https://doi.org/10.1016/j.ajpath.2012.03.017 [PubMed]
- 70. Blagosklonny MV. Does rapamycin slow down time? Oncotarget. 2018; 9:30210–2. https://doi.org/10.18632/oncotarget.25788 [PubMed]
- 71. Gems D, de la Guardia Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2013; 19:321–9. https://doi.org/10.1089/ars.2012.4840 [PubMed]
- 72. Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013; 75:621–44. https://doi.org/10.1146/annurev-physiol-030212-183712 [PubMed]
- 73. Wang H, Zhang Z, Gems D. Monsters in the uterus: teratoma-like tumors in senescent C. elegans result from a parthenogenetic quasi-program. Aging (Albany NY). 2018; 10:1188–9. https://doi.org/10.18632/aging.101486 [PubMed]
- 74. Gems D. The hyperfunction theory: An emerging paradigm for the biology of aging. Ageing Res Rev. 2022; 74:101557. https://doi.org/10.1016/j.arr.2021.101557 [PubMed]
- 75. Blagosklonny MV. The hyperfunction theory of aging: three common misconceptions. Oncoscience. 2021; 8:103–7. https://doi.org/10.18632/oncoscience.545 [PubMed]
- 76. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Discov Today. 2007; 12:218–24. https://doi.org/10.1016/j.drudis.2007.01.004 [PubMed]