



Introduction
Chronic aging associated disease (AAD) remains one of the defining medical challenges of our time, representing 95% of direct health costs for seniors and driving expected Medicare spending to over $1.2 trillion by 2024 [1, 2]. Further, patient care is complicated by the convolution of systemic factors, multiple diseases, and conflicting treatment plans. Indeed, patients co-presenting two or more AADs are common and costly, with patients managing 2 or more chronic conditions representing over 70% of healthcare spending [3]. This complexity is reflected at the molecular level, with numerous mechanisms implicated in the aging process. These mechanisms prominently include inflammation, oxidation, metabolic and mitochondrial dysfunction, telomere shortening, and cellular senescence; we direct readers to other reviews on the molecular drivers of aging [4, 5]. Despite strong research efforts, connecting the host of molecular changes to development of effective treatments for AAD remains challenging. Identifying and intervening in early stages of chronic disease remains difficult with the slow degeneration distributed over years, evaluation of molecular markers occurring long after pathogenesis, and convolution of many subtle pathway dysregulations. A major contributor to these challenges is the limitations of commonly used in vivo and in vitro models.
Animal models of aging broadly follow the phenotypes of human aging and can be used to model specific AAD [6]. However, specific mechanisms (e.g., immune function or telomere regulation) differ in important ways [7]. Further, many human AAD lack analogs in naturally occurring animal disease, especially in more cost-effective rodent models. Prime examples of this are cardiovascular disease [8], primary open angle glaucoma [9–11], and neurodegeneration [12]. While animal studies will remain an essential component of biomedical research for the foreseeable future, there is longstanding recognition of their limitations [13] and consideration of reduction strategies [14, 15].
Similarly, conventional two-dimensional in vitro culture has been indispensable in understanding the molecular mechanisms associated with aging [16]; advantages include cost-effectiveness, replicability, ease of chemical and genetic manipulation, and accessibility to analytical and imaging methods [17, 18]. Unfortunately, these advantages come with a number of known limitations including modified sensitivity to pharmacological agents, distorted expression profiles, abnormal morphology, and altered differentiation schema [7, 19, 20]. To address these limitations in both conventional in vitro and in vivo animal models, there has been increasing development of more physiologically representative in vitro models. Ideally, these models incorporate human cells and more accurately reflect the mechanical, physicochemical, biochemical, and cellular context of in vivo tissue. Models that mimic the heterogeneous cell composition and organization of native tissue are generally referred to as organotypic, a category that includes both ex vivo and in vitro models. Key examples of in vitro organotypic models include organoid, organ-on-a-chip, organotypic tissue slice, and tissue engineered organotypic models.
Organoid models are generated by a number of different source materials including tissue fragments and explants, reconstituted primary cells, and stem cells [7, 18]. While there is no single definition of organoid models, broadly speaking, they are constructed through the self-assembly of patient, primary, or stem cells; exhibit cellular and matrix organization mimetic of the in vivo environment; and a heterogeneous cell population mimetic of native tissue. Organ-on-a-chip models generally possess these same advantages, with additional potential features consisting of defined structural patterning of the cells, microfluidic or environmental control of the system, and incorporation of sensors or physiological readouts [21–23]. Organotypic tissue slice cultures use thinly sliced sections of tissue, preserving the cellular microenvironment and tissue organization; these have been used in a range of tissues, including heart, lung, liver, and most prominently, brain [24–30]. These model classes have enabled significant contributions to research and drug discovery, including in the aging field. A notable example is in brain, where organoids and organotypic slices have been used to research aging associated degeneration, Alzheimer’s, dementia, and Parkinson’s; the progress in brain organotypic models has been extensively reviewed by others [31, 32]. These model classes have enabled significant contributions to research and drug discovery, yet have notable limitations. For example, organoids and organ-on-a-chip models are typically small (sub-mm) due to the lack of vasculature and diffusion limits of oxygen and metabolites [33–35], although organ-on-a-chip models sometimes address this issue through microfluidic perfusion. Further, organoid and slice models often require patient or freshly isolated animal tissue that can be difficult to acquire; organ-on-a-chip models often rely on specialized microfabrication techniques that not all aging research labs can easily implement. Another culture category and topic of this review, tissue engineered organotypic culture, leverages the progress in tissue engineering to create tissue-scale and physiologically relevant in vitro models.
Tissue engineering, a term first coined over three decades ago, has long held promise for the in vitro creation of fully functional tissue grafts [36, 37], however, numerous challenges have limited development. In vitro development of skin grafts, one of the initial targets of the field [37], is only just now entering medical use as an adjunct to traditional therapy [38], with fully functional engineered skin still unavailable [39]. This is broadly representative of the current state of the field, which, despite significant research progress, have demonstrated limited clinical application of grafts. However, for the past two decades, researchers have repurposed engineered tissues towards research questions [14, 40–42]. Similar to organoid and organ-on-a-chip cultures, these models are constructed from organotypic cell populations, but typically offer a greater degree of control over the tissue architecture and included cell populations. Cells and structures can be patterned or allowed to self-assemble depending on the needs of the research [43, 44]; similarly, cell populations and sub-populations can be easily controlled or replaced to reflect tissue health and disease. Leaders in tissue engineering have urged the simplicity and cost-effectiveness of design [34, 45], and this is reflected into the increasing number of methods papers and decreasing costs of biomaterials [14, 40]. These models represent a powerful and accessible set of tools for aging research; and are likely to become increasingly relevant as the field moves towards bridging cellular and tissue-scale hallmarks of aging.
In this review, we summarize research efforts and potential for utilizing organotypic and tissue engineered models for aging and AAD. To streamline the review, it is broken into independent sections for skin, intestine, and skeletal muscle; which represent well-developed fields and are important tissues in physiological aging and AAD. Each section briefly covers important facets of the aging physiology in the tissue system, before describing current and emerging organotypic techniques and their application to aging. In each tissue section, we describe the advantages (and limitations) of organotypic models in elucidating aging mechanisms at the cellular and tissue scales, as well as highlighting the key methodological and accessibility factors.
Demonstrative organotypic models relevant to aging tissue
Skin
Native skin aging
Skin is one of the largest organs of the body and has functional roles in immune response, physical protection, and thermal regulation [46]. A simplification of skin anatomy is shown in Figure 1A. As aging occurs, skin function and healing capacity is reduced, with key aging changes summarized in Table 1. Skin aging is frequently divided into two related processes: intrinsic and extrinsic aging [47–50]. Intrinsic aging, also referred to as chronological aging, includes genetic and hormonal changes and the progression from cell maturity to cellular senescence [47, 50]. Extrinsic aging, also referred to as environmental aging, represents the impact of the environment, including: photoaging associated with sun exposure [47, 51, 52], cigarette smoking, pollution, chemical exposure, and trauma [50]. Due to the different underlying mechanisms, characteristics of each type of aged skin are different. Chronologically (intrinsically) aged skin presents as unblemished, smooth, pale, dry, lower elasticity, and has fine wrinkles while environmentally (extrinsically) aged skin has coarse wrinkling, rough textures, pigmentation changes, and lower elasticity [50, 53].
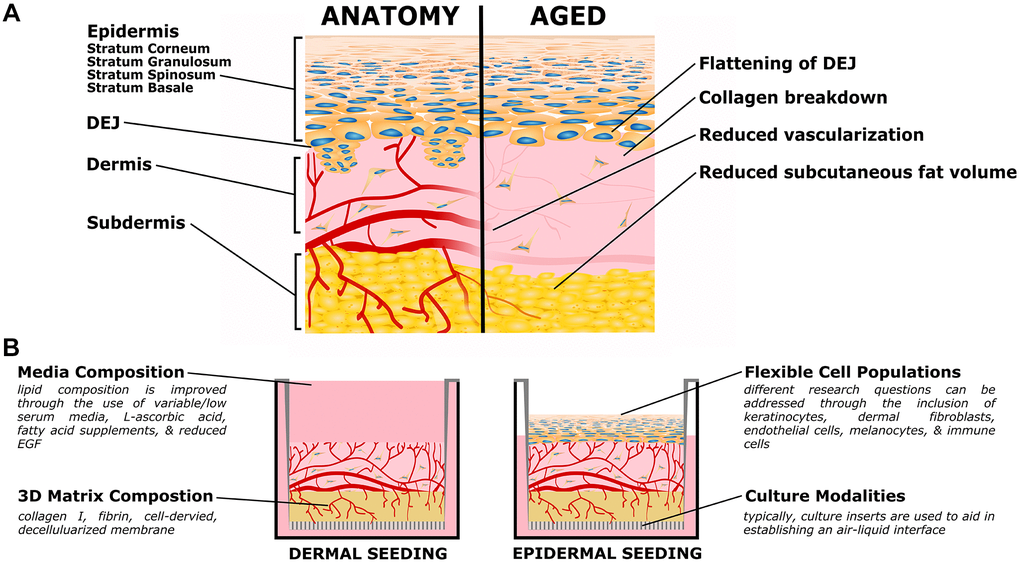
Figure 1. Organotypic models of skin aging. (A) Simplified skin anatomy and aging phenotypes. Skin can be separated into epidermal, dermal, and hypodermal layers. The epidermis is composed of Stratum Basale, Spinosum, Granulosum, and Corneum, composed of increasingly differentiated epidermal cells. The dermal-epidermal junction (DEJ) connects the basement membrane of the Stratum Basale to the upper (papillary) dermis, and is characterized by small dermal extensions (or papilla) into the epidermis. The DEJ flattens with age. The dermis is a collagen rich tissue supported by dermal fibroblasts. The subdermis (or hypodermis) is an important adipose compartment that contributes to overall metabolic function; this tends to thin with age. Both the dermis and subdermis are highly vascularized, important for thermal regulation; in age vascularization is reduced. The above schematic is simplified to focus on the level of current organotypic models, nerves, melanocytes, immune cells, and other components of in vivo skin are not pictured. (B) Organotypic skin models, also referred to as Human Skin Equivalents (HSE), typically consist of a dermal/subdermal culture grown on a permeable culture support (left), followed by seeding and differentiation of epidermis at the air-liquid interface (ALI). Benefits of this style is the accessibility of the culture format, ready customization of the specific cell populations (both immortalized or primary, patient specific, or transgenic disease models), and customization of the matrix and media formulations.
Table 1. Prominent phenotypes of aging skin.
| Prominent Aging Phenotypes | References |
| Lower elasticity, increased fragility, and wrinkle formation | [47, 50, 53, 54] |
| Increased collagen disorganization, accumulation of advanced glycation end products, and changes in (GAG) and (PG) concentrations/organization | [49, 53, 55–61] |
| Flattening of the dermal epidermal junction | [50, 52] |
| Decreased dermal vasculature | [62] |
| Reduced subcutaneous fat volume | [50] |
| Increased cellular senescence | [49, 63] |
| Decreased cell population and turnover, including melanocytes, epidermal cells, dermal fibroblasts, and immune cells | [50, 63, 64] |
| Reduced barrier function coupled with changes in the stratum corneum, lipid composition, and filaggrin expression | [65–69] |
Structural changes in intrinsically aged skin include decreased dermal vasculature [62]; changes in dermal elasticity and increased collagen disorganization [70, 71]; build-up of advanced glycation end products (AGEs) and changes in glycosaminoglycan (GAG) and proteoglycan (PG) concentrations/organization contributing to stiffening of dermal structure and frailty, and decreased hydration [49, 53, 55–61]; imbalance of tissue inhibitors and matrix metalloproteinases (MMPs) resulting in imbalance between collagen deposition and breakdown [50, 72]; and flattening of the dermal epidermal junction/loss of rete ridges [50, 52, 63, 64, 73]. Aging also contributes to variations in epidermal and dermal thickness [63, 64, 74, 75] and reduced subcutaneous fat volume [50]. There are also many changes related to cell population in all three main skin compartments (epidermal, dermal, hypodermal) including reduced epidermal cell turnover [50, 73], drop in number of active melanocytes [50]; decreases in dermal fibroblast concentrations [64], decreases in immune cells [63, 64] and immune function. Abnormalities of skin barrier (a major function of the epidermis) occur during aging and often present as dryness or skin irritation. In aged skin, barrier function has been studied in the context of decreases of filaggrin [65], increases in pH (5 to ~5.6), altered lipid presence [66, 67], and changes in cornified envelope arrangement [63, 68, 69, 76]. These changes add to fragility of older skin and increase chances of infection [54], it remains unclear exactly how these changes take place and what mechanisms are controlling them.
On the molecular scale, expression levels of soluble factors, proteins, and vitamins are both effects and contributors to aging phenotypes. Examples include upregulation of stress regulatory proteins (hypoxia-inducible factors, nuclear factor kappa-light chain-enhancer) [63], increases in AP-1 (leading to increased collagen breakdown via MMP activity) [52, 72], and declines in vitamin D production by the epidermis [63]. These changes are largely attributed to increases in reactive oxygen species (ROS) [52, 63], DNA mutations (including mitochondrial DNA), telomere shortening [63], increased cell senescence, and hormonal changes [49, 63]. Changes in skin aging have been associated with fluctuations in expression patterns of integrins including α6 and ß1 integrins [57, 59, 71, 77, 78]. In healthy human skin, α6 and ß1 (and other α/ß subunits) integrin expression are localized on the basal side of basal keratinocytes [57, 78]. Defects in integrin expression are present in human blistering skin diseases with supporting evidence in knockout mice [78] and also in aged human skin [57, 59], although further work is necessary to understand how integrin expression changes in aging.
Aging in the skin has sex-related differences as well, specifically, sex is linked to faster thinning of the dermis and collagen density decline in males as opposed to females [50, 79]. Males undergo a decline in androgen levels while estradiol levels are constant, these changes result in a linear decline of skin thickness and collagen content in men [70]. Women experience both androgen and estrogen decline linearly and an additional post-menopausal estrogen decline which is linked to lower collagen content, lower skin moisture and capacity to hold water, lessened wound healing response, thinner skin, and lower skin elasticity [50, 53, 70, 80]. Detailed summary and discussion of sex-related changes in skin aging have been previously reviewed [70].
These intrinsic mechanisms are compounded by environmental skin aging (extrinsic aging) [49, 52, 63]. A key example is the effects of ultraviolet (UV) irradiation (an extrinsic aging mechanism), which accelerates telomere shortening and DNA damage present with intrinsic aging [50, 81]. Other extrinsic aging and examples of compounding UV effects are discussed in previous literature [49, 71, 82–88]. Overall, skin aging at the molecular, cellular, and tissue levels continues to be a field of active research. While in vivo and traditional cell culture models remain important tools, there is increasing interest in more physiologically relevant culture models, and there is a growth in recent studies employing organotypic skin models (OSCs).
Tissue engineered skin models
Researchers have used organotypic models to study skin biology since the 1980s [89, 90], and the methodology are increasingly accessible. OSCs are also commonly referred to as human skin equivalents (HSEs) or full-thickness skin models; they typically have dermal and properly stratified epidermal layers (Figure 1B). These models have proven useful for studying skin development, evaluating cytotoxicity, studying wound healing, and more recently as disease and aging models. OSCs are highly customizable and allow for control of organotypic cell populations, genotypes, and culture conditions to enable carefully controlled studies on tissue-level biology. OSCs have the capacity to be used for in depth aging studies without the dangers of human trials or expensive animal models; with long-term culture stability for chronic studies (typical culture lengths of 8–12 weeks) [91–93]. Most commonly, OSCs contain dermal fibroblasts and keratinocytes and are cultured at an air-liquid interface for epidermal differentiation and stratification. However, with the growth of interest in heterogeneous cell-cell communication, an increasing number of models have been demonstrated with additional cell populations [71, 94, 95]. These include vascular endothelial cells [92, 93, 96–101], immune cells [102–105], adipose derived stem cells and adipocytes from adipose derived stem cells [106–108], embryonic stem cells [71], melanocytes [109–111] and melanocytes derived from induced pluripotent stem cells [112]. With this customizability and a growing number of accessible protocols, OSCs represent a useful tool for studying skin aging; exemplar applications are discussed below, first for disease generally and then with aging specifically.
OSCs have been used in a number of disease studies, both directly and as “hybrid” studies where a humanized OSC is grafted onto immunodeficient mice. Additionally, models have been shown useful for testing potential therapeutic techniques for debilitating skin disorders or injuries [113]. OSC skin disorder models include: psoriasis [114–116], recessive dystrophic epidermolysis bullosa [117, 118], lamellar ichthyosis [119], Netherton syndrome [120], congenital pachyonychia [121], Junctional epidermolysis bullosa [71, 122], and fibrosis [123–125]. Of these disease models, the fibrosis model by Varkey et al. is especially interesting for its potential to be adapted to use as an aging model. In this study, OSCs were generated using either deep dermal fibroblasts or superficial dermal fibroblasts in combination with normal human keratinocytes [123]. They found that the antifibrotic properties of deep dermal fibroblasts and the fibrotic properties of superficial fibroblasts influence OSC characteristics. Authors found that when compared to constructs with superficial or mixed fibroblast populations, OSCs with deep fibroblasts had higher levels of IL-6, reduced TGF-β1 production, higher PDGF expression, and epidermal formation was less defined and less continuous [123]. This model is potentially interesting as a platform for aging research, as TGF-β is implicated in skin aging through regulation of matrix metalloprotease activity [126, 127]. The work of Varkey et al. highlights the usefulness of OSCs to study signaling between specific cellular subpopulations in a controlled way; this approach could be readily adapted to aging studies. Given this potential, it is unsurprising that several research groups have used OSCs in aging research, which we highlight in the next section.
Tissue engineered skin models to study aging
As OSCs are stable for long culture periods (>17 weeks), using the extended culture time to study intrinsic aging is perhaps one of the most straightforward techniques and can be combined with other aging models and/or cell types [73]. With this model, authors demonstrated that extended culture (using a non-traditional matrix of collagen-glycosaminoglycan-chitosan porous polymer) exhibited several age-related aspects similar to those that occur with in vivo aging, including decreases in epidermal thickness, decreases in hyaluronan expression, increases of the aging biomarker p16Ink4a, decreases in keratinocyte proliferation over time, loss of expression of healthy epidermal markers, and basement membrane alterations. Another straightforward application of OSCs in aging is studying the impact of senescent cells. A number of studies have incorporated senescent fibroblasts into OSCs to generate models that recapitulate many of the features of in vivo aged skin. [74, 128, 129]. Diekmann and colleagues induced senescence in human dermal fibroblasts and keratinocytes using Mitomycin-C (MMC) treatment and incorporated the cells into OSCs [129]. When compared to mitotic OSCs, the senescent models demonstrated changes similar to aged in vivo skin, including a more compact stratum corneum (outer layer of the differentiated epidermis), reduced dermal fibroblast population, decreased collagen type I and III content, decreased elastin expression and looser elastin structures, increases in MMP1, and disordered epidermal differentiation. A similar study involving senescent fibroblasts used healthy fibroblasts that were exposed to H2O2 to induce senescence and then cultured the senescent fibroblasts in skin equivalents with healthy keratinocytes [128]. Aging phenotypes were characterized by changes in proliferation, differentiation of suprabasal epidermal layers, impairments of skin barrier function, and surface property modification. Further, authors found that fibroblasts exhibited senescence-associated secretory phenotype (SASP) markers including IL-6, GmCSF, and IL-1α. Interestingly, Weinmüllner et al. observed more Ki67 positive epidermal cells when senescent fibroblasts were present. More research is required to understand senescence in the dermis and how it may effect keratinocyte homeostasis [128]. Serial passaging of fibroblasts has also been employed to simulate aging in OSCs, showing that constructs generated with late passage fibroblasts were similar to in vivo aged skin [74]. OSCs were generated with 15-20% SA-β-gal positive fibroblasts cells in 2D culture prior to 3D seeding. Authors observed few changes in the epidermal compartment while the dermal component of OSCs presented a thinner dermis and increased MMP1, similar to in vivo aged skin [74]. Defects in epidermal-dermal junction in these OSCs were not observed and keratinocytes exhibited a healthy phenotype. Although not shown, authors noted that when greater than 30% SA-β-gal positive fibroblast cells in 2D were used to generate OSCs, the fibroblasts did not produce sufficient extracellular matrix (ECM) and constructs were not viable [74]. As Janson et al. found, generating an OSC using senescent cells is technically challenging since the percentage of senescent cells used to generate an OSC can alter skin structure and long-term culture health [74].
Other studies focused on the aging of the keratinocyte population. In OSCs generated from primary cells isolated from donors, cell donor age is an option for simulating intrinsic aging in vitro [71]. OSCs generated with either keratinocytes isolated from aged individuals or serially passaged keratinocyte cells have been used to examine the effects of replicative senescence [130]. Constructs generated with older keratinocytes (61 or 35-year-old donors) exhibited thinner epidermis compared to OSCs generated from 1-year old donor cells. Additionally, there were differences in epidermal organization, where constructs generated with young keratinocytes exhibiting more consistent organization and stratification than OSCs with older cells. This study also investigated the expression of epidermal stem cell markers. They found that when keratinocytes were passaged over six times (modeling in vitro cellular senescence), there was a decrease of stemness, indicated by high expression of α6 integrin and low expression of CD71 (a proliferation-associated cell surface marker) [130]. Likewise, in constructs generated with young (infant) keratinocytes, α6 integrin expression was observed in basal cells of epidermis while in constructs generated with adult and elderly cells there was faint and absent α6 integrin expression (respectively). These OSC findings demonstrated in both intrinsic aging (simulated from aged donor cells) and in vitro senescence induced by serial passaging results in depletion of epidermal stemness markers [130].
Epidermal changes associated with aging have also been shown in models generated through genetically altering expression of key components, for example p16Ink4a [131]. In vivo chronological human aging markers, p16Ink4a and its repressor BM1, are established markers of in vitro aging tissue [71, 73, 131, 132]. p16Ink4a is an inhibitor of cyclin-dependent kinases that blocks the progression from G1 phase to S phase of the cell cycle and promotes senescence onset. In vitro aged skin models can be generated from young donor keratinocytes cells by p16Ink4a overexpression [131]. Conversely, aging phenotypes observed in old donor keratinocytes can be rescued through silencing p16Ink4a. Aged models (both from older donors or p16Ink4a overexpression) resulted in thinner epidermis, loss of stratum corneum (the terminal epidermal layer), and atrophy [131].
OSCs also allow for studies of matrix and cell-matrix interactions in aging skin. Expression patterns of glycosaminoglycans (GAGs) and proteoglycans (PGs) are important in skin tissue mechanical integrity, and aging-related changes contribute to frailty in both intrinsically and extrinsically aged skin [53, 55, 133–137]. Glycation and the presence of advanced glycation end products (AGEs) increase in aging skin, and this has been leveraged in OSCs to create an aged skin model [57, 59]. In this model, collagen was glycated in vitro prior to construction of the OSC. This simulated intrinsic aging of the construct, resulting in modified integrin patterns in the suprabasal epidermal layers, activation of the dermal fibroblasts to increase the production of metalloproteinase, type III procollagen, and type IV collagen [57, 79]. Authors found that these morphological and molecular changes in the epidermis and dermis could be partially rescued by antiglycation agents such as aminoguanidine [57]. More investigation is necessary to understand exactly how GAGs and PGs are affected during skin aging. Open questions include how sex specific hormones may affect concentrations [53] and what downstream effects GAGs and PGs have on the expression of cytokines and growth factors [138]. As an accessible platform that can be customized with specific cell lines, biomolecules, and materials, OSCs are uniquely suited to elucidate aging mechanisms including detailed molecular studies regarding GAGs and PGs in skin.
In addition to researching aging biology, OSCs can also be employed as a testing platform for aging therapeutics [135, 138]. C-Xyloside is a xyloside derivative that has been investigated as therapeutic to improve dermal-epidermal junction (DEJ) morphology in aging skin [139, 140]. Sok et al. exposed OSCs to C-Xyloside and investigated the resulting DEJ morphology. C-Xyloside exposure resulted in higher basement membrane protein concentrations, specifically collagen IV, laminin 5, and collagen VII, and organization more similar to the microanatomy of healthy human skin. Further, C-Xyloside increased concentrations of dermal proteins such as pro-collagen I and fibrillin, which are key ECM proteins for the maintenance of skin elasticity. Since defects in the basement membrane, DEJ, or elasticity contribute to skin fragility in aging, this model has potential as a test bed for other aging therapeutics [135].
In the context of skin, tissue engineering has provided accessible and customizable models both for the direct research of aging phenotypes as well as models that can be readily adapted to aging questions. Further, there is demonstrated potential for therapeutic testing. Importantly, the cited models (or variants thereof) rely on commonly available cells, reagents, and techniques adaptable to many lab environments. Increasing use of these models in aging research holds promise to accelerate discovery and therapeutic goals. Despite this promise, there remain challenges to the use of OSCs in aging research, discussed below. Most notably, the power of OSCs comes from their intermediate status between simple in vitro models and in vivo models; there is an explicit tradeoff between increasing the complexity of the culture system and its cost or ease of use. While OSCs do allow customization by the researcher to focus on factors most important to their question, the tradeoff can be difficult to make for aging research. Some examples of OSC limitations relevant to aging research are provided below.
Limitations
The most predominant limitation of using tissue engineered organotypic models is that they typically do not match all cellular populations found in vivo. Nerves, sweat glands, stem cell niches, immune cells, subcutaneous adipose, and vasculature are important aspects of aging skin biology that are frequently missing in OSCs. While in many cases there is no strict technical reason for the absence of a specific component, any increase in complexity provides more challenge and cost. For example, inclusion of nerves requires a source of nerve cells, they must be maintained in culture while not losing their phenotype, and simply including cells in the OSC does not capture the complexity of the nervous system. However, progress is being made through iteration, providing researchers with increasingly powerful models that capture more of the relevant physiology. For example, wound healing is slowed in aged skin, and immune cells are vital in both physiological and pathological wounds. While fibrosis has been studied using OSCs, this is typically limited to observing fibroblast and keratinocyte responses; there is a recognized need for OSC models that include immune populations [102]. While not prevalent, some models do incorporate the immune system [104, 116, 141–143], demonstrating the trajectory of the field toward increased capability and flexibility. Similarly, changes in vasculature are prevalent in aged skin, but OSCs often lack vascular cells. While progress has been made in vascularizing OSCs and related models [92, 93, 96, 97, 99, 100, 141, 144–146], there is still a great deal of work to be done in applying this to aging questions.
Further, OSCs tend to be structurally simplified. As mentioned, they typically lack nerves, glands, and other structures typical of skin. Building on the example of vasculature, even with appropriate vascular cells, OSCs often have a random or simplified organization; native cutaneous vasculature is organized into two horizontal plexus planes with connecting vessels between them along the apicobasal axis [147, 148]. In OSCs, this organization could be recapitulated through the inclusion of patterned or semi-patterned vasculature, although this is typically not done [149]. Additionally, decline of collagen density is an important aspect of skin aging, yet many OSCs are fabricated with collagen densities much lower than those found in vivo [79, 150]. While not common yet, OSCs can be fabricated from higher collagen densities through techniques such as dense collagen extractions [151], and compression of collagen cultures [152], to more closely represent the in vivo dermal matrix.
Another key limitation of current OSCs is loss of systemic factors present in vivo. For example, age-associated changes in sex hormone profiles impact skin physiology; e.g., post-menopausal decreases in collagen content, reduced elasticity, and lowered skin moisture in women. While changes in systemic factors can be addressed, they will invariably lack the full complexity of an in vivo model. For example, a recent study addressed the impact of exogenous estradiol on elastin synthesis using male and female dermal organotypic cultures [153]. Studies such as this highlight the tradeoffs in organotypic models, as reductionist culture models allow specific questions to be interrogated, they obviously lack the complexity inherent in aging at the organismal scale.
Intestine/gut
Native intestinal aging
In this section we focus on the gastrointestinal system and review relevant three-dimensional organotypic culture models. The small intestine is the primary organ for nutrient absorption from food, while the colon (or large intestine) is the primary organ for reabsorption of water [154]. Here, we focus on the small intestine, due to the larger number of in vitro three-dimensional models, but large intestine models are briefly discussed as well. The small intestine has a complex tissue structure involving crypts (valley points) and villi (mountain points); with the crypts providing a stem cell niche (Figure 2A). Stem cells located within crypts asymmetrically divide and the resultant epithelial cells migrate up toward villi and eventually slough off into the gut lumen. Multiple distinct epithelial populations arise from these stem cells, including microfold cells, enteroendocrine cells, enterocytes, goblet cells, Paneth cells, and tuft cells; this process of continual epithelial renewal and differentiation is integral to a healthy gut barrier. On the epithelial surface there is a brush boarder and single or bi-layered mucus layer depending on location within the gut [155]. Interacting with this surface is the microbiome which is made up of commensal bacteria and pathobionts (resident microbes with pathogenic potential) that constantly interact with the mucin layer of the gut [155]. Diversity of the gut microbiome has been established as an important factor in gut health and host health [156–165]. The diversity of the microbiota presents in different regions of the gastrointestinal tract depend on many factors including pH, host health, mucin composition, bacterial cooperation, nutrient availability, location within the gut, and age of the host [157]. Further, within the subepithelial and stromal tissue there are additional cells, including fibroblasts, smooth muscle cells, microvascular cells, and both circulating and resident immune cells (e.g., monocyte derived macrophages, neutrophils, dendritic cells, T cells). The immune cells are known to interact with and traverse the epithelial surface [166–168]. Given the complexity of the intestinal tissue and the number of host and bacterial cell types, it is unsurprising that many of the cellular interactions are poorly understood, especially in aging tissue where both the host tissue and microbiome can change [169].
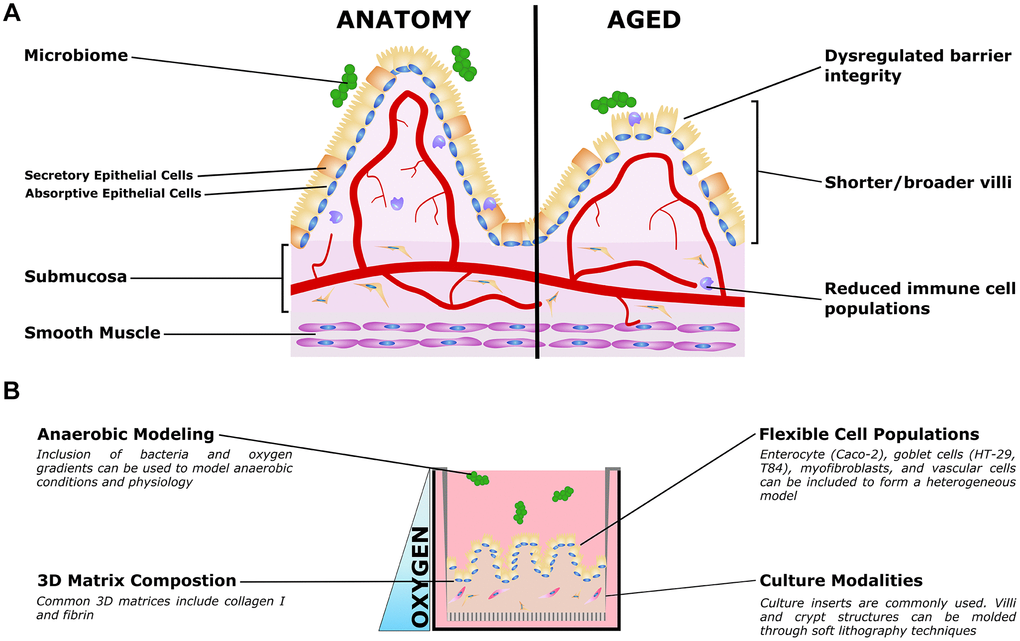
Figure 2. Organotypic models of gut aging. (A) Simplified gut anatomy and aging, focusing on the most commonly modeled components. A mixed epithelial population, described in the text, forms a simple cuboidal epithelial layer with both secretory and absorptive epithelium. A layer of mucus inside the gut lumen supports the host/microbiome interaction. The stroma underneath the epithelium, the submucosa, is host to nerves (not shown) blood vessels, fibroblasts, and immune cells important for gut function. Smooth muscle is required for gut peristalsis. In aging, the macrostructure of villi degrades, with villi becoming shorter and broader. Immune cell populations are disrupted, and reduced epithelial barrier integrity can lead to increased microbial infiltration into the submucosa and vasculature. (B) Organotypic models of the gut typically only model a small subset of these features, and are typically adapted to aspects that are relevant to specific questions. For example, epithelial and immune populations may be co-cultured to study intercellular interactions in a simple format. To study the influence of villous structures, soft lithography can be used to recreate the villi/crypt geometry. Microbiome co-cultures can be included, and microfluidic organ-on-a-chip models have been used to mimic the oxygen gradient from the vascularized submucosa to the anaerobic lumen.
Aging in the gut presents as reductions to nutrient ingestion, the tolerance of resident microbiota, and the response to infection (key aging phenotypes are summarized in Table 2). Often these co-present with dehydration and malnutrition [166]. Generally, there is a lower intake of macronutrients and micronutrients in aged individuals, although this lower intake could be attributed to lower physical activity, problems with teeth, impaired sense of taste and smell, psychological factors, income levels, and drug side effects [170–172]. Together, lessened nutrient intake, dehydration, and malnutrition contribute to overall healthy decline and morbidity in aged individuals [172]. Additionally, there is evidence showing that absorption of glucose and vitamins increases with age while some nutrients such as cholesterol and fatty acid decrease or slow; changes in absorption have been well reviewed in animals [170, 172] but continues to require more investigation in the human gut [172, 173]. It has been suggested that changes in nutrient absorption could also be tied to the changes in morphology found in aged animals and in humans [174].
Table 2. Prominent phenotypes of aging intestine.
| Prominent aging phenotypes | References |
| Increased microbial infiltration into submucosa and vasculature | [180–182] |
| Reductions to nutrient ingestion, tolerance of resident microbiota, and the response to infection. | [166] |
| Villi morphology changes, decreased cells per villus, decreased mucosal surface area, decreased crypt numbers | [174, 177, 178, 183–185] |
| Increased cell apoptosis, reduced cell proliferation and survival, decreased regenerative potential of stem cells | [166, 177, 184–187] |
| Disruption of Wnt Signaling | [177, 188–190] |
Morphologically, as the small intestine ages, numerous structural changes have been observed in several models. These structural changes are coupled to cellular changes, for example, the dynamics of cell life cycle from the crypt to extrusion at the villi [170, 175–177]. In one year old rabbits compared to young rabbits, there are morphological changes in the jejunum and ileum; villi shorten, number of cells/villus drops, and mucosal surface area declines in the jejunum while villus cell size remained constant in both areas [178]. Changes in villous height are associated with mucosal surface area at all ages [178] and these declines in surface area have been related to differences in nutrient absorption of aged individuals [174]. In healthy mice it takes around 4-5 days for a stem cell derived progenitor to move from the crypt, differentiating along the way, to the tip of the villus, where it ultimately undergoes apoptosis and extrusion. Morphological changes such as villi length increase and crypt number decrease lead to larger crypts with more cells and are coupled with less travel of progenitor cells to the tip of the villus as well as increased apoptotic events, decreased cell proliferation, and lower cell survival in aged mice [177]. Aging and how it effects wound healing in the small intestine has also been investigated in mouse models. Martin and colleagues studied the regenerative capacity of small intestinal epithelium after injury in young and old mice using full or partial body irradiation [179]. Authors found that after injury induced by full body irradiation, crypts of old mice were smaller than controls while young mice had larger crypts. After partial body irradiation, the crypts of young animals were found to be smaller, while the number of surviving crypts in old mice was lower than in young mice.
In rats, morphological changes such as increased numbers of crypts and villi are observed with aging, although size and cell production rate changes were not observed [183]. Atrophy of intestinal mucosa also occurs in aged rats and this contributes to decreased number of enterocytes [184, 185]. These changes can be localized to specific tissues; for example, mucosal atrophy in rats has been found in proximal regions of the small intestine, but not in the distal small intestine; similarly, the decline in villi height has been found in the ileum but not the duodenum [184]. Changes in morphology are thought to be closely tied to transport function across the gut barrier and may be tied to malabsorption of nutrients, but more evidence is needed to support this [169, 170, 174, 178]. Further, the association between aging and morphological changes is poorly understood in human intestine. Currently, there are few studies that have examined human intestinal morphology; Webster and colleagues found that elderly people have shorter villi and possibly broader villi when comparing shape and dimensions of proximal jejunal villi in young versus aged humans [174]. The villous changes in humans were not definitively linked to changes in intestinal function, but changes in surface area are thought to contribute to the nutrient absorption decline that aged individuals often experience [174].
Changes in enzyme distribution and brush border membrane makeup have been observed in mice [170], rats [185], and rabbits [178], but the conclusions differ by species and it is unclear whether these changes are associated with aging [170]. Briefly, in adult and aged mice there are similar activities and distribution of enzymes in the brush boarder membrane [170]; while in aged rats lower alkaline phosphatase activities have been found; conversely, higher sucrase/alkaline phosphatase in the brush boarder membrane have been found in adult vs. young rabbits. Differences in mucus structure and chemical composition have been tied to age changes [166, 170, 191]; specifically glycoproteins in the mucus change with age in rats [170, 191]. There is some evidence suggesting that the process of bacterial adhesion to mucus also changes with age, shown with bifidobacterial strains [166, 192–194]. However, gastric and duodenal mucus thickness does not change with age in healthy individuals [166, 195]; mechanical properties of mucus have been found to remain stable as well [166].
On a cellular level, differences have been observed with aging. Most prominently, stem cell changes have been observed in aged animal studies and in organoid cultures [177, 188]. In small intestinal tissue from mice, the intestinal stem cell markers Lgr5 and Olfm4 were examined but found to be similar in young and old samples, while the quiescent intestinal stem cell markers Lrig1 and Tert were reduced [177]. However, when examining numbers of stem cells in young versus old cultures, no difference was found [177]. Wnt signaling, an important aspect of self-renewal and proliferation in intestinal stem cells, is altered in aging gut [188–190]. Elevated Wnt activation can lead to intestinal tumorigenesis [196] and malformed crypts (less lobes and buds per crypt) in small intestine mouse organoid cultures [189]. However, there is conflicting literature on how elevated or lowered Wnt signaling effects stem cells in aged mice. Nalapareddy and colleagues found that during aging, intestinal stem cells, Paneth cells, and mesenchyme secrete less Wnt ligands which leads to overall reduced Wnt signaling and lower regenerative potential of stem cells [177]. Using organoid models derived from duodenal (proximal) crypts in mice, the decreased stem cell function can be rescued by endogenous Wnt in vitro [177]. There is evidence that the stem cells may lose fitness in maintaining differentiated cell populations; specifically Paneth cells, responsible for generating anti-microbial peptides [166]. The amount of Paneth cells and their secretory functions have been found to decline with age [166, 187], and this may be due to the age-related stem cell decline and reduced ability to generate Paneth cells [166, 179, 197].
The mucus is the site of antibody production (specifically, secretory immunoglobulin A; IgA) and is the first defense against harmful microorganisms [166]. Goblet cells, the primary contributor to the mucus layer, have a stable population in aging mice [166, 198]. As previously reviewed, the literature remains unclear on the effect of aging on IgA response, migration, and production [166]. Aging has been found to decrease secretory IgA amounts in animals (mice, rat, non-human primates) when exposed to cholera toxin [166, 199–202] and increase somatic hypermutation in mice [166, 203]. In contrast, other studies have shown no changes in serum or intestinal amounts of IgA in aged rats and mice; some results suggest that the lower levels of IgA are due to an overall homing decline rather than changes in amounts of IgA [166, 201, 204–207]. Dendritic cells present antigens to B and T cells in the intestinal immune system, and evidence points to decreasing cell numbers and function in aged mice [186]. Further, this plays a role in decline of regulatory immune functioning [166, 208, 209] and may play a role in low grade inflammation observed in the aging gut [166, 169, 210, 211].
The microbiome plays an important role in digestion, absorption, and nutrient processing [212], but it remains incompletely understood how the intestinal barrier and immune system interact with microbiota and how this system is affected by aging. In the study of microbiota, it remains unclear how gut diversity affects the aging process and how gut diversity changes with age. There is not enough evidence or investigation on age-related associations and gut health to determine causes/effects of gut on old age [164, 165], although there are many health practices that correlate with perturbations of the gut microbiome including drug/antibiotic usage and diet [164, 213]. There is evidence that the gut microbiome is affected by sex differences [212, 214–217], and this may be implicated in sex differences in aging-associated disease. Sex differences in the microbiome affect gut health but also risk of disease development including atherosclerosis, diabetes, hypertension, dyslipidemia, and obesity [212]. In general, aging and its relation to sex and hormonal differences requires more investigation, but there are indications that changes in the aging gut are sex-linked due to hormonal differences during early life, adulthood, and aging [214, 215]. In aging males, testosterone levels drop slightly from levels during adulthood while in aging females, there is a dramatic drop in estrogen from the oscillation range of adulthood [215]. The general effects of hormonal supply decline to the gut microbiome are unknown, but are likely sex-specific [215] and may be associated with the immune component of the gut [216].
Tissue engineered gut models
There are a few limitations to traditional intestinal models that can be addressed with 3D organotypic gut models (Figure 2B). 2D cultures on culture inserts are often used to model gut, but these cultures are unstable after 4 weeks due to cellular overgrowth and formation of multicellular layers [155]. To study enteric bacterial pathogens, researchers have often used human tissue explants; animal models [218]; and 2D cultures with cell lines such as T84 and HT-29 which mimic goblet cells, and Caco-2 which serve as enterocytes [219]. Although helpful in understanding microbiome-host responses, these models are typically inconsistent with the human anatomy and physiology in the gut [218, 220]. Similarly, mouse transgenic models are often used to study inflammatory gut diseases but mice do not develop some prevalent human diseases, such as ulcerative colitis or Barrett’s esophagus [221]. To address gaps in more traditional models, several 3D models have been established based on organoid, explant cultures, micro-fluidic chips, and organotypic gut models (OGMs) generated through self-assembly and partial villous molding. Intestinal tissue derived organoids are a popular model that has been used to study aging; these are called enteroids for small intestine, or colonoids for large intestine models. Enteroids consist of only epithelial cells and model crypt like populations or are often differentiated to model surface/villous epithelium [218]; these have been studied using monolayers on tissue culture inserts and embedded in extracellular matrix [218, 221]. Human induced pluripotent stem cell (iPSC) derived intestinal organoids, contain both epithelial and mesenchymal lineages and model both crypt and surface villus [218]. Models of differentiated intestinal organoids, although limit appropriate human scale, can include even the rare cells of intestine models including enteroendocrine, tuft, M cells, and Paneth cells [222].
3D cultures have been generated with both primary human cells and commercially available lines. OGMs have been generated with adult human intestinal stem cells [222], iPSC [222], Caco-2 [155, 222, 223], T84 [222], HT-29 [155, 222, 223], and myofibroblasts [155]. OGMs are only recently developed, but they have advantages over 2D models, micro-fluidic chips, explant cultures, and organoid structures because of their ability to mimic appropriate tissue length scales for oxygen diffusion and customizable cell and material properties [218]. Additionally, human based models that include human cells and relevant 3D microenvironments can be used to study diseases such as gastroesophageal reflux disease, Barrett’s esophagus, IBD, and ulcerative colitis; for therapeutic screening; and other aging associated research [221].
Incorporation of 3D villi in OGMs have been demonstrated to model the human system more closely [220] and help to understand the changes in crypt/villi that have been observed in aged animals [177, 178, 183, 184]. Several groups have generated 3D gut models with villous platforms though pre-culture molding of hydrogels and custom plate inserts [219, 220, 224]. These systems have been found to mimic mammalian intestines more closely than 2D cultures facilitating cell differentiation, absorption/metabolism, and have been used to evaluate drug permeability [220]. Yi and colleagues compared absorption and metabolism of enterocyte (Caco-2) 2D monolayer cultures and 3D villous collagen scaffolds covered with enterocytes. They found that in the 3D cultures, cell growth was higher (likely due to more surface area), there were more in vivo phenotypes such as lower expression of P-gp (efflux transporter protein, p-glycoprotein) which is overexpressed in 2D monolayers, and increased alkaline phosphatase expression (a metabolic enzyme and intestinal epithelial differentiation marker) [219]. To generate 3D collagen villi structures, multiple groups have used relatively stiff collagen and an alginate reverse molding method to create villous structures from collagen hydrogel [219, 220]. Yu and colleagues promoted a basement membrane like surface by coating the collagen with laminin. Villous structures were fabricated to match the density and depth of human villi and models were cultured for 14 days; a 21-day duration led to breakdown of villi [220]. Similar pre-culture molding of villous structures has been used in microfluidic-chips [225–227]; and as reviewed by others [225]. These models capture appropriate microanatomy of the intestinal surface and have the potential to elucidate the respective roles of structural and cellular changes in aging.
Organoid models have been used to study several diseases [189, 190, 222, 228, 229]; illustrating how 3D cultures provide a physiologically relevant model without the complexity of fully in vivo studies. Woo and colleagues demonstrate how a 3D model (specifically an intestinal organoid spheroid model) can be used to study the human disease dyskeratosis congenita. Dyskeratosis congenita causes intestinal defects (including stem cell failure) and is characterized by decreases in telomerase, telomere length, telomere capping, and Wnt activity [190]; it is particularly relevant to aging since some of these disease characteristics are similar to what happens in aged intestinal cells [188]. In organoids generated with the dyskeratosis congenita model cell line, there was incomplete and thin epithelia, overgrowth of mesenchymal cells, and inferior E-cadherin and beta-catenin expression; the organoids did not have proper budding crypts or cavitation [190]. Through CRISPR/CAS9-mediated repair and administration of Wnt agonists the authors were able to rescue the disease phenotype and demonstrate normal organoid formation in vitro. In other disease specific models, organoids made with cells derived from inflammatory bowel disease patients maintain characteristics of disease in vitro such as gene expression profiles that regulate absorption and secretion [222, 228]. Disease focused organoid studies [190] and other organoid models generated with aged mice cells [189] demonstrate the potential of more physiologically relevant in vitro models to address aging questions. By building off of these methods and incorporating human cell types, anatomies, and physiology it is possible to develop a human derived organotypic gut model [155] and avoid costly procedures involved in animal colonies [213].
Tissue engineered gut models to study aging
A recent study by Arnold and colleagues demonstrates the physiological relevance of 3D in vitro models for aging [230]. In vivo, older animals have higher ratios of non-saccharolytic v. saccharolytic bacteria and lower amounts of β-galactosidase when compared to younger animals. Pre-biotic galacto-oligosaccharides (GOS) have previously been found to have a positive impact on intestinal health and can be administered through diet. To study the effects of dietary GOS on aging in the gut, using young and old mice models of Clostridiodes difficile were used. In the aged mouse models, dietary GOS promoted changes in microbiome composition and transcriptomic analysis also revealed differences in gene expression. Aged mice that were fed a GOS diet had decreased intestinal permeability and increased mucus abundance and thickness when compared to aged mice not fed the GOS diet. These changes in permeability supported previous findings attributing the leaky gut to increased non-saccharolytic bacteria and lower amounts of key enzymes. Further, these results were additional tested in colonic organoids injected with stool samples from young and old mice. Using the colonic organoids generated from one young mouse and stool sample injection from experimental mouse models, authors showed that they were able to reproduce differences of age, minor differences of the GOS diet, and bifidogenic responses observed in the in vivo mouse models [230]. As the authors already showed a reproduction of aged phenotypes in organoid models, reproducing these characteristics in scalable and humanized organotypic models may be beneficial in research questions of how diet and microbiome affect aged humans.
The ability to culture anaerobic bacteria is an important step in modeling the microbiome of the gut in healthy tissue and to improving the understanding of how aging changes the host-microbiome interaction [158, 163, 164, 231–233]. Most in vitro models, including OGMs, only study a few relevant features of the complex physiology at a time; models that include microbiota are no exception. One study showed their ability to culture 5 different microbe types in vitro on a custom scaffold and evaluated for proliferation and biofilm formation [234]. It is important to recognize, that although this is a human microbiota gut model, it does not incorporate human gut cells or microanatomy. Combining microbiota and human 3D OGMs is an important step in modeling the human gut; some work on the combinations of microbiota and human gut cells has been carried out in microfluidic chips [225], but these tend to lack relevant villous anatomy and appropriate oxygen diffusion scales. These factors have been partially addressed in an innovative upright cylindrical culture system [155]. Authors generated the vertical lumen with an un-patterned surface and a threaded surface to mimic crypt and villi of the intestine. Their model includes epithelial cells (Caco-2 and mucus producing HT-29 cells) and myofibroblasts seeded on and into silk-based scaffolds, respectively. With this design, they achieved proximal-to-distal oxygen gradients and reached anaerobic conditions in patterned lumens. As a proof of concept, they cultured anaerobic bacteria using this model. Importantly, the patterned lumen model was stable for long-term culture (at least 8 weeks); they further showed continuous mucus production and accumulation (~10 μm average thickness of the mucus layer). Although this model does not incorporate aging phenotypes, aged cells, or differences due to aging in the microbiome, it highlights the recent progress in developing organotypic constructs that could be adapted to aging studies.
In vitro organoids are common in the gut/microbiome field of study [188, 218, 222, 235, 236] and have been used to assess intestinal stem cell function during chronological aging [177, 188–190, 237, 238]. Although there is conflicting literature on Wnt signaling in the intestine and how it effects intestinal stem cells, several recent studies have used organoid models to investigate aging and how it changes crypt/villi formation and stem cell function in the gut. Each study also presented a rescue method to restore normal Wnt signaling and gut formations [177, 189]. Cui et al. cultured organoids from aged mice and showed reduced differentiation and increased expression of Wnt target genes (Axin2 and Ascl2). The organoids generated from aged mice presented rounded cysts without typical differentiated cell types, in contrast to organoids generated from young mice, which demonstrated differentiation and formation of villus structures. These phenotypes matched organoid cultures of cells that exhibit overactivation of Wnt signaling (through seeding with adenomatous polyposis coli deficient cells). The decreased differentiation of intestinal stem cells and impaired structure could be rescued by reducing exposure to the Wnt agonist R-spondin-1 and thus reducing Wnt activity. Rescued organoids matched those generated with cells isolated from young mice. Nalpareddy and colleagues generated organoids from duodenal proximal crypts of aged and young mice as well as humans [177]. In humans, organoids were generated from people 12–16 and 62–77 years old. The authors found decreased formation of organoids in the aged group, which was improved by adding Wnt 3a (a Wnt pathway agonist). This data supported their findings in mice organoids where aged mice organoids had lower organoid formation rates after 3 passages and decreased stem cell function (determined by lower lobes and buds per crypt). Adding Wnt 3a increased organoid formation and expression of Wnt target genes (Axin1 and Ascl2) in the aged cultures [177]. While interpreting the apparently contradictory results of these studies is difficult, they do highlight the use of organotypic models in performing detailed signaling studies that would be challenging and expensive in animal models.
In vitro intestinal models have a particularly relevant potential impact on personalized medicine due to the person-to-person variability in gut health. Aside from genetics, variation in local community and world regions as well as day-to-day activities result in microbiome and inflammatory differences that are not yet understood [239]. Personalized medicine and patient derived organotypic models may help to address these parameters. One organotypic microfluidic chip model named iHuMiX has paved the way for personalized gut models [240]. The iHuMiX platform utilizes compartments including microbial, epithelial, and flow chambers and allows for study of specific bacteria on host specific physiology. While microfluidic systems often present technical barriers for non-specialist labs, these results highlight the customizability of organotypic models, including adaption to personalized medicine. As with OSCs described in the prior section, the tradeoff between complexity and capability for organotypic gut models results in several limitations.
Limitations
As with OSCs and other organotypic models, the most prominent limitation is the lack of cell populations and structural features of the in vivo gut. While a great deal of the work described above has extensively modeled epithelial cells and their stem cell niches, the gut is much more complex; immune cells, vasculature, smooth muscle, and neuronal populations all contribute to the gut, and its physiology when aged. Further, the organization of the gut, most notably the crypts and villi, is well understood to influence function and disease; these features are only incompletely reflected in organotypic models [219, 220, 241]. More unique to the gut is the anaerobic microbiome, which is critical to understanding gut and organismal health [158, 163, 164, 231–233]. While there has been demonstrated inclusion of anaerobic microbiome in a gut model [225–227], the complexity of the system makes it challenging to broadly replicate in other labs. Indeed, the general challenges of creating and maintaining hypoxic and anoxic cultures significantly limits the ability of organotypic models to correctly match the lumen environment. Further, there is significant evidence that the microbiome is not restricted to the gut lumen, and translocation of commensal bacteria to surrounding tissues, including lymph nodes, is a driver of disease [242, 243]. While organotypic gut models may be suited to address some questions of bacterial translocation, none have reached the scale or complexity required to include lymphatics. While this is a single example, it does highlight the more general limitations on most organotypic models.
As with other organotypic models, sex differences are understudied. This is despite clear sex differences in aging associated gastrointestinal diseases [244, 245] and cancers [246, 247]. While sex differences local to the cell populations used could, and should, be studied using organotypic models, systemic factors including hormones remain a challenge. As a pertinent example in the gut, sex hormone levels are known to regulate the mucosal surface and barrier integrity [248]. While organotypic models to lend themselves to studying the impact of specific hormone levels, they clearly lack the complexity of overall systemic changes that come with aging and sex differences.
Skeletal muscle
Native skeletal muscle aging
Skeletal muscle is an abundant tissue, making up ~30–40% of body mass [249]. Healthy muscle regulates major physiological processes such as locomotion [250, 251], venous return [252–254] and metabolism [255–258]. From the 3rd to 8th decade of life fat-free mass declines by ~15%, even for healthy individuals, contributing to loss of independence and higher risk of injury and mortality. The age-associated loss of muscle mass, known as sarcopenia, is a major hallmark of human aging [259–261] with a complex etiology, resulting in muscular, vascular, and metabolic impairment [262–264]. Chronic inflammation [265–268], nutrient deficiencies [269–271], and decreased physical activity [272–274] are all contributing factors of sarcopenia, however, much remains unknown at the molecular, cellular, and tissue levels. Improved models of sarcopenia and other aging phenotypes are imperative for improving clinical outcomes and prophylaxis for the expanding geriatric populations.
In a healthy individual, skeletal muscle is composed of densely packed and aligned cylindrical myofibers individually sheathed in a specialized matrix called endomysium [275] (Figure 3A). Bundles of myofibers are encapsulated in a connective tissue layer known as the perimysium, while the whole muscle is surrounded in a thicker connective tissue layer called the epimysium. Myofibers are organized into fiber types (fast twitch and slow twitch) based on their metabolic, contractile, and morphological properties. Due to the unique signature of each fiber type, maintaining homeostatic fiber compositions is vital to muscle function [276]. Multiple muscle fibers and the corresponding motor neuron form a motor unit, with the overall force of muscle contraction controlled by activating more motor units. A dense vascular network that delivers nutrients and removes waste supports the high metabolic demands of muscle tissue.
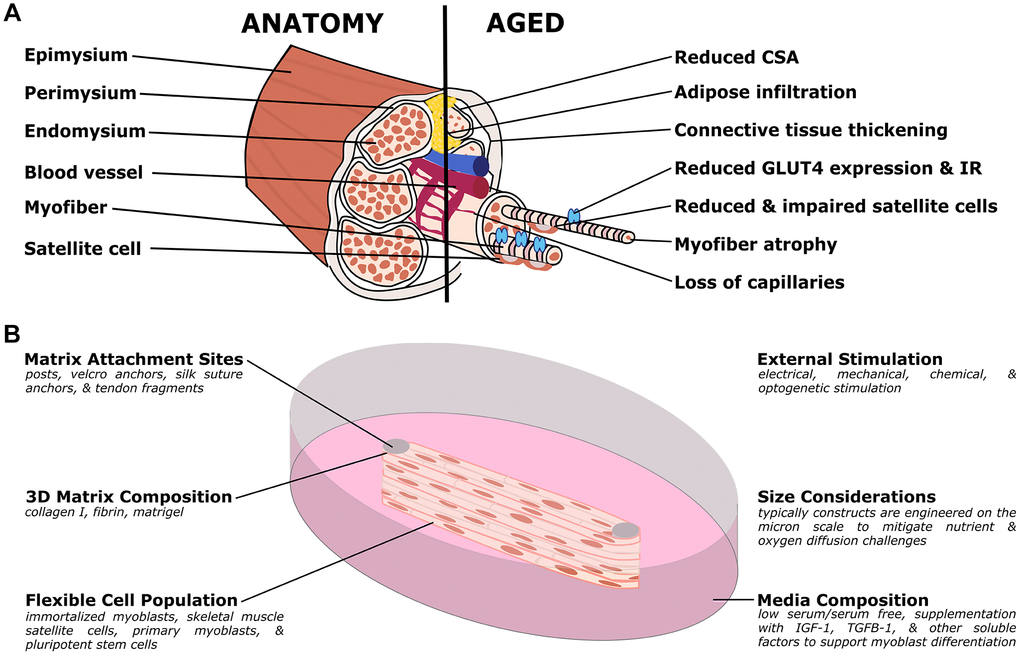
Figure 3. Organotypic models of skeletal muscle aging. (A) Simplified muscle anatomy and aging, focusing on the most commonly modeled components. The primary unit of muscle is the myofiber, a multinucleated cell responsible for contraction. Specialized matrix (endomysium, perimysium, and epimysium) support and organize the tissue. Satellite cells are an important stem cell population for the muscle, and the muscle is supported by a host of other cell types including nerves, fibroblasts, adipose, and vascular cells. In aged muscle, cross-sectional area (CSA) is reduced, in part due to myofiber atrophy, and decreasing capillary and satellite cell density. Conversely, there is increased infiltration of adipose and thickening of the connective tissues. At the molecular level, there is decreased expression of GLUT4, an important glucose transporter, and insulin resistance (IR) frequently develops. (B) Organotypic models of muscle have several unique challenges but have distinct advantages over other traditional models. Muscle cultures are contractile, and require anchoring to prevent collapse. Typical approaches include posts (although other methods are used) to provide points of resistance for the muscle to pull against. In order to study active contraction, researchers have used various stimulation methods, including electrical and optogenetic methods. Due to the high metabolic demand, the cultures are typically quite small, to allow nutrients and waste to diffuse more readily. As with other organotypic models, the matrix, cell population, and media can be customized for the research question.
Structural and cellular changes are prominent in aged muscle (summarized in Table 3). Structural changes include reduced muscle cross sectional area [277–280], thickening of the epimysium and endomysium connective tissue layers [281–284], increases in tissue fibrosis [285, 286], and decreased capillarization [278, 287, 288]. Further, reduction and atrophy of specific fiber types (particularly fast twitch/Type II fibers) has been observed, leading to altered fiber composition and increased percentages of slow twitch (Type I) fibers [289–292]. More specifically, Type II (fast) fiber atrophy is associated with reduced muscle mass and strength [289, 293]. Cellular changes include increased adipose infiltration into the muscle [294–296], and loss of motor units [297–299]; all result in decreased skeletal muscle force generation. Further, age associated changes in skeletal muscle satellite cell populations include a reduced progenitor pool [300–302], limited myogenic colony formation [303], loss of amplification and myofiber differentiation potential [285, 304–308], and an increased susceptibility to senescence and apoptosis [301]. Further, aged satellite cells have been shown to favor fibroblastic and adipogenic differentiation programs [285, 309–311], potentially explaining the observed increase in fibro-adipogenic progenitors in aged skeletal muscle [312–314]. Of course, aging muscle includes non-muscle cells, other skeletal muscle aging phenotypes include increased M2 macrophage presence [315–317] and endothelial apoptosis [318]. Together these cellular and microstructural changes contribute to loss of muscular and systemic function in the elderly population, motivating research into the molecular mechanisms underpinning these changes.
Table 3. Prominent phenotypes of aging skeletal muscle.
| Prominent Aging Phenotypes | References |
| Myofiber atrophy, reduced cross-sectional area, reduced mass, loss of motor units, and decreased strength | [277–280, 289, 293, 297–299] |
| Change in the ratio of fiber types (increased percentages of slow twitch/Type I fibers) | [289–292] |
| Decreased vascularization and increased endothelial cell apoptosis | [278, 287, 288, 318] |
| Increased fibrosis and thickening of connective tissue layers | [281–286] |
| Increased adipose infiltration and differentiation | [309–314] |
| Decreased progenitor pool and loss of regenerative capacity | [285, 300–302, 304–308] |
| Increased insulin resistance and metabolic dysfunction | [319–325] |
The above structural and cellular changes are coupled with molecular changes in the aged tissue. A loss of overall regenerative potential is likely largely influenced by a reduced satellite cell population and differentiation potential [306, 308]. Satellite cell activation is regulated by myogenic regulatory factors (MRFs). Primary examples of MRFs include: myogenin, myogenic determination factor (MyoD), myogenic factor 5 (Myf-5), and myogenic regulatory factor 4 (MRF4) [326]. In rats, MyoD and myogenin have been found to increase with age, indicating a potential compensatory role to attenuate loss of satellite cell activation [327]. Yet, human studies have observed a decrease in myogenin, Myf-5, and MyoD [328, 329]. Differential responses between organisms such as this emphasize the need for robust models of human muscle tissue. Myostatin, a member of the TGF-β superfamily, inhibits satellite cell proliferation (via upregulation of p21) and activation (via reduced MRF expression). Further, the elevation of myostatin contributes to muscle atrophy through glucocorticoid signaling [330–332]. Upregulation of myostatin is seen in aged individuals and is thought to contribute to age-associated loss of muscle mass [333–335]. Further, mitochondrial dysfunction and increased oxidative stress are hallmarks of aged muscle [336–339]. Mitochondria manage the cell’s energy supply, ROS generation, and apoptosis. Changes in mitochondrial bioenergetics lead to ROS accumulation, impaired quality control mechanisms, and apoptotic cell death [340–342]. ROS accumulation in aged muscle mitochondria contributes to protein and DNA damage [343–346]. This subsequent loss of mitochondria quality control mechanisms establishes a feedforward cycle of mitochondrial damage and muscle degeneration [347].
Of course, muscle is not separate from the systemic context, both being influenced by and influencing changes in the entire aged organism. Systemic changes contributing to skeletal muscle aging include altered cytokine and hormone signaling. Insulin-like growth factor (IGF) is both a circulating hormone and localized growth factor. IGF is predominantly produced by the liver and delivered systemically, although other tissues produce specific IGF splice variants; mechano growth factor (MGF) and IGF-1Ea are produced by skeletal muscle [348–350]. In skeletal muscle, IGF regulates muscle hypertrophy and growth, and concentrations are known to decline in elderly populations [327, 351, 352]. IGF and MGF are responsible for activating anabolic and anti-catabolic pathways via PI3K/Akt, ERK/MAPK, and PKC signaling, leading to increased protein synthesis and anabolic activity [351–353]. Examples of aging-associated dysregulation of IGF signaling includes evidence that mechanical loading of skeletal muscle results in MGF stimulation in young individuals, but not the elderly [354]. Inflammatory cytokines are also implicated in muscle aging. Elevated TNFα concentrations are found in aged muscle and cause increased apoptosis [355]. IL-6 is a pleotropic cytokine known to influence skeletal muscle function in a number of ways [356]. Elevated levels of IL-6 are strongly associated with diseased muscle, proinflammatory signaling, and a catabolic shift. In rats, with positive stress stimuli such as physical activity, IL-6 levels increase and may have anti-inflammatory effects [357]. In the context of aging there is evidence that in aged human muscle, chronically IL-6 elevated can initiate muscle wasting [358]. In contrast, local IL-6 expression appears in both young and aged individuals after exercise with beneficial effects, indicating a complex role for IL-6 in muscle homeostasis [359, 360].
Hormonally, testosterone and its precursor, dehydroepiandrosterone (DHEA), are key regulators of muscle mass. Androgens (including testosterone and DHEA) are important for maintaining muscle mass through hypertrophy via increases in myonuclear number and fiber cross-sectional area [361–363]. The mechanisms driving androgen mediated muscle growth are poorly understood, but there is evidence of impact on satellite cell commitment level and trophic signaling, discussed in more detail in other reviews [361, 362]. Relevant to the present work, androgen levels decrease in the elderly and contribute to reduced muscle mass [362, 364–367]. Thyroid hormones (TH), T3 and T4, are important regulators of metabolism, contractile function, and muscle differentiation [368, 369]. Expression of TH decreases with age [370], and this may be involved in the development of sarcopenia [371, 372].
Skeletal muscle also regulates systemic AAD. Skeletal muscle insulin resistance is a primary characteristic of Type II Diabetes (T2D) that presents years before the disease’s onset [323–325]. Yet, the mechanism connecting the pathogenesis of T2D and skeletal muscle insulin resistance is incompletely understood. Increases in mitochondrial dysregulation, oxidative stress, and inflammation are all known to contribute to diminished insulin sensitivity in skeletal muscle. Indeed, it has been demonstrated that elderly individuals have impaired glucose metabolism, and decreased expression of the insulin-mediated glucose transporter, GLUT4 [320–322]. Additionally, aged skeletal muscle exhibits reduced rates of mitochondrial oxidative phosphorylation and an inability to switch from lipid to glucose oxidation when stimulated with insulin [319]. Reduced insulin sensitivity of aged muscle contributes to the development of diabetes and other metabolic disorders. Importantly, the above molecular changes are not broadly conserved across species and gender, emphasizing the need to ensure research models match the morphological, functional, and biochemical characteristics observed in vivo. Overall, understanding human skeletal muscle aging remains a challenge, especially considering the diverse and interacting factors at the molecular, cellular, and tissue scales. Developing models that mimic the native tissue, while remaining accessible to experimental techniques, are needed to further push the field forward.
Tissue engineered muscle models
Tissue engineered skeletal muscle models, pioneered by Vandenburgh and colleagues [373], have been in use for over two decades. The earliest engineered constructs, termed bioartificial muscle (BAM), consist of skeletal myoblasts encapsulated in an ECM. The ECM is molded around artificial “tendons”, or posts, responsible for maintaining passive tension within the tissue (Figure 3B). As the myoblasts differentiate into highly contractile myotubes the cells align along the axis of tension and lift off the culture substrate. Myoblasts from a range of developmental stages are commonly sourced from muscle biopsies of organisms such as avian [374], mouse [375, 376], rat [377–379], and human [380–383]. Due to limited availability of primary cells, immortal myogenic lines, including C2C12 (mouse) and L6 (rat) cells, are commonly used due to ease of culture and availability [384–387]. Yet, immortal cell lines exhibit low excitability [388] and poor physiological relevance compared to primary cells [389–391]. Induced pluripotent stem cells (iPSCs) are a promising alternative to traditional primary and immortal cultures due to their high expansion capability and potential sourcing from specific genetic backgrounds [382, 392–397]. BAM models have been used to examine physiological events such as hypertrophy and atrophy in response to drugs and exercise [398–401], skeletal muscle wounding and regeneration [400, 402, 403], force production [404–407], cell signaling [408–410], and drug response [411–414]. Importantly, as different muscle cell sources have distinct costs and benefits, different cell populations can be readily interchanged in BAM models to suit specific research needs.
Further advances have been made in the field of skeletal muscle tissue engineering through other approaches, such as scaffold free assemblies, bioprinting, and chip based systems. Scaffold free assemblies use the contractile nature of myotubes to form 3D tissues. In these systems, differentiated skeletal muscle/fibroblast monolayers delaminate from the culture substrate are rolled in on itself and pinned down to form “myoids” or “myooids” [378, 380, 415, 416]. Myoid models recapitulate many structural and functional features of native muscle, such as production of ECM, microvessels, and spontaneous contractions [417]. Although myoid constructs have been reported to be stable for up to 40 days, drawbacks include long maturation times (3–4 weeks), inability to scale cultures [418], and low force generation [401]. Recent advances in bioprinting technology have led to the printing of biomimetic muscle tissues and have been reviewed extensively [419, 420]. Bioprinting skeletal muscle is an appealing technique due to its high precision in cell positioning and alignment; however, progress in this area is limited by broad challenges in the field such as cell viability, printing speed, and resolution [419–422]. Additionally, printing the soft materials necessary to recapitulate the skeletal muscle microenvironment remains a challenge [423]. Recent “muscle-on-a-chip” devices have shown several advantages, including avoiding perfusion required to feed thicker tissues. Using microfabricated cultures, researchers have demonstrated muscle viability and enhanced maturation in response to microtopographical and morphological cues [424–426]. Skeletal muscle-on-a-chip systems are a promising tool for drug toxicity studies, especially due to their low media consumption and extensibility to high throughput screenings. Recently, a 3D skeletal muscle microdevice has been coupled with a biosensing platform to monitor myokine secretion. The authors validated this system by measuring IL-6 and TNF-α levels in response to electrical and biological stimulation [427]. However, muscle microdevices are limited by the need for specialized training and equipment to fabricate and use these devices.
It is important to emphasize that most of the models described above largely consist of homogeneous cell populations that lack the organization of native tissue. Recent progress has been made in incorporating heterogeneous cell population in BAMs, including the addition of endothelial cells and demonstration of vascular network formation [410, 417, 428–433]. In a mixed muscle/vascular mouse myoid model, researchers found high levels of vascular endothelial markers such as VEGF, CD31, and VE-cadherin, indicating the survival and signaling of vascular cells. Yet, the extent of the network formation was not examined in this study [417]. Endothelial vessel formation has been demonstrated on engineered skeletal muscle scaffold systems; however, muscle cells do not align along one axis, limiting contractility and tissue function [431]. Applying uniaxial strain to a vascularized mouse BAM model has been shown to induce vascular tube formation, likely through increased VEGF secretion by the differentiating muscle [433]. In a human vascularized BAM model researchers identified optimal cell seeding ratios (50–70% muscle cells) and media blends (endothelial growth media) for generating endothelial tubes along with aligned myofibers [429, 430]. Despite these advances, further work should be done to characterize vessel structure, and nutrient and oxygen delivery in vascularized BAMs. As a model of muscle regeneration, macrophages have been added into rat BAMs to study the regenerative potential of satellite cells within the engineered tissue. The incorporation of bone marrow derived macrophages showed recovered Ca2+ transients after injury compared to muscle only controls. Muscle-macrophage constructs also had improved cell organization and regeneration of myofibers post injury. Further, the authors demonstrate impaired regeneration in adult derived engineered muscle compared to neonatal constructs. In the future, this model can be used to identify pro-regenerative treatments in adult muscle [434]. Continued development of heterogeneous muscle models is of interest to the field of aging research given the prevalence of dysregulated adipose, fibroblast, and macrophage signaling with age.
BAMs have been used to study physiological muscle function, pharmaceutical response, and human disease [378, 412, 414, 435, 436]. While few systems have been developed in the context of aging (discussed below), other BAM models of disease demonstrate the power of the technique. Disease models of skeletal muscle include Miyoshi myopathy, Duchenne, limb-girdle, congenital muscular dystrophy, Pompe disease, and amyotrophic lateral sclerosis [437–445]. One strategy that is readily applicable is incorporating cells isolated from diseased patients into tissue constructs. As an example, Bersini and colleagues engineered myobundles co-cultured with endothelial cells and muscle-derived fibroblasts isolated from patients with Duchenne muscular dystrophy (DMD) [446]. Tissues with DMD fibroblasts exhibited an increased fibrotic phenotype characterized by higher collagen I and fibronectin deposition compared to healthy and TGF-β (inducer of fibrotic response) treated controls. Further, samples with DMD fibroblasts exhibited increases of α-smooth muscle actin compared to controls, indicating a shift towards a myofibroblast phenotype, consistent with the in vivo disease. The ability to capture and assay fibrosis, as demonstrated in the above models, has clear applicability to many aging studies.
In another study, human iPSCs from patients with DMD and limb-girdle muscular dystrophy were used to engineer 3D disease models with muscle, vascular, and neuronal cells [444]. These engineered muscles recapitulated disease phenotypes seen in vivo including the nuclear elongation typical in laminopathies. As another key example, BAMs generated from primary muscle cells isolated from both healthy individuals and patients with Pompe disease were used to test potential therapies [442]. Pompe disease myobundles exhibited traits consistent with that of clinical data such as elevated glycogen content and low acid alpha-glucosidase (GAA) gene activity. Researchers compared tissue functionality between healthy and Pompe disease models, observing reduced fatigue resistance, tetanic force production, and glycogen mobilization. While the observed functional defects were not alleviated by treatment with recombinant human GAA (current standard of care) or AAV-mediated GAA expression, the use of similar platforms for screening therapies is promising. Disease models such as the above can be readily adapted to study aging phenotypes by incorporating cell populations derived from aged individuals. The ability to compare functional and mechanical properties of aged and young muscle is of special interest to aging research, as elderly people have reduced muscle functionality. Further, being able to screen pharmaceutical interventions in muscle specific AAD models represents a significant advancement in the field of aging biology.
Tissue engineered muscle models to study aging
In recent years, engineered muscle has been used to study specific aging and aging associated diseases. A key example is the role muscle plays in insulin sensitivity and the age-related disease, type 2 diabetes (T2D). As aged muscle displays reduced insulin sensitivity [322, 447], it is especially relevant to quantify insulin sensitivity in engineered muscle. To test this, Kondash and colleagues created human myobundle constructs using primary myoblasts, differentiated in a 3D matrix for 2 weeks [436]. The authors found that 3D engineered constructs displayed a significantly higher glucose uptake in response to insulin than similarly cultured 2D cells. Further, the usefulness of this model for elucidating therapeutic mechanisms was also tested. Metformin, a common pharmaceutical for hyperglycemia and T2D, led to similar increases in glucose uptake in the presence or absence of insulin; indicating that metformin does not impact insulin responsiveness in peripheral muscle tissue. Further, metformin was found to impair both twitch and tetanus force production as well as decrease fatigue resistance. Although the magnitude of insulin response observed in this study is lower than that of native muscle tissue, the authors demonstrate the importance of the 3D microenvironment for improving physiological relevance in T2D studies. Additional work performed by Acosta and colleagues used engineered muscle to test the effect of systemic metabolic changes on muscle health [448]. Using muscle precursor cells isolated from lean, obese, and diabetic rats, engineered constructs were maintained in either myogenic media or adipogenic media. The authors showed that constructs with diabetic muscle precursor cells had decreased creatine kinase activity, tissue compaction, myotube alignment, and reduced tensile strength when compared to lean control samples. Overall, these data indicate diabetic myogenic precursor cells reduce overall muscle integrity. Further, the authors showed increased adipogenic differentiation in diabetic samples. Increased adipose presence between muscle fibers is common in vivo with aging, where muscle precursor cells are a potential source of adipose tissue [448]. These examples demonstrate tissue engineered skeletal muscle can be readily applied to the study of aging phenotypes such as increased insulin resistance and adipose infiltration.
In addition to the genetic and systemic factors discussed above, models of aged muscle have also been generated similar to the BAM method described above [449–451]. Sharples and colleagues utilized late passage C2C12 myoblasts to replicate aging phenotypes, including reduced myofiber diameter, length, and peak force development [449]. The reduced force generation observed coincides with a decrease in construct differentiation and hypertrophy potential. The authors quantified transcript expression of muscle differentiation and hypertrophy markers throughout culture. In aged constructs, they observed an increase in myostatin and TNFα, genes associated with impaired differentiation potential and sarcopenia [449]. A study performed by Rajabian and colleagues takes this work a step further by measuring calcium handling and metabolic function in aged human engineered muscle tissue [450]. Human myoblasts were obtained from young and aged donors and seeded into engineered constructs. Tissues formed from aged myoblasts exerted lower contraction force compared to younger control samples, fail to respond to electrical stimulation and, consistent with a lack of muscle contraction, have lower Ca2+ and ATP concentrations. Further, to study regeneration in aged tissue, the authors induced muscle injury using cobra cardiotoxin (CTX). Samples made with young myoblasts regenerated myofibers within 5 d post CTX injury, while aged constructs did not regenerate, resulting in reduced myotube diameter. Indeed, the number of multipotent satellite cells (identified with positive staining for PAX7) did not change after CTX injury in pre-senescent tissues, indicating increased regenerative potential [450]. Overall, these studies demonstrate that engineered skeletal muscle replicates many of the basic phenotypes seen with aging in vivo.
An additional application of engineered muscle is to elucidate the molecular mechanisms of aging. Shahini and colleagues leveraged engineered skeletal muscle to test the role of NANOG expression in mitigating senescence-associated dysfunction [451]. These studies were built off prior work showing NANOG expression reversed senescent phenotypes in MSC populations [452, 453]. In the skeletal muscle study, late passage C2C12 myoblasts were engineered to express NANOG under the control of tetracycline and embedded in a 3D collagen/Matrigel matrix. The authors observed NANOG expression partially rescued myotube population levels, diameter, and length to that of early passage controls when compared to late passage constructs without NANOG. They further observed a restoration of differentiation markers MYHC and Actinin. A key advantage of engineered muscle models, demonstrated by the above studies, is the accessibility for targeted genetic and pharmacological manipulation. As with other models, the advantages of engineered muscle cultures are coupled to limitations, discussed below.
Limitations
As with other organotypic models, exclusion of cell types present in vivo is a challenge for skeletal muscle as well. For example, common aging phenotypes of inflammation, reduced peripheral vascularization, and adipose infiltration require inclusion of immune cells, endothelial cells, and adipocytes. In addition to sourcing and maintaining these cells, co-culture with muscle cells presents additional challenges due to their high metabolic demand and contractility. Progress is being made, for example with inclusion of increasingly complex vascular components [410, 417, 428–433], but there are many areas needing improvement.
Further, skeletal muscle poses unique challenges for cell sourcing. Most in vitro models of aging skeletal muscle are established from primary cells that are derived from animal models and patients [450, 454–456]. Although primary cells offer increased physiological relevance relative to immortalized lines, the culture methods needed to isolate and expand these cells to populations suitable for organotypic studies rely on specialized techniques and restricted supplies, especially for human cells. Established cell lines are a more accessible sourced of aged myoblasts, and replicative senescence models have been established and used in 3D culture [449]. While the tradeoffs between primary cells and established cell lines are well documented for any in vitro culture system, the large number of cells needed for organotypic skeletal muscle models can make sourcing sufficient primary tissue difficult.
It is important to note that skeletal muscle is typically composed of multiple fiber types, with different physiology and function. In aging, fast twitch fibers preferentially atrophy, leading to changes in fiber composition. While an important phenotype, especially in aging, fiber type is typically not assessed or controlled in organotypic models, leading to an important capability gap [457]. Further, engineered skeletal muscle generates force several orders of magnitude lower than that of adult human muscle, with reduced myofiber diameters [458]. Methods to improve contractile properties in these models focus on co-culture with motor neurons, electrical and mechanical stimulation, and improved nutrient and gas delivery. Ultimately, better control of muscle differentiation and maturation will improve modeling of both healthy and aged tissues.
Finally, although both males and females exhibit loss of muscle mass with age, the pattern of decline is sex dependent. Similar to other tissues, organotypic constructs could be ideal platforms to isolate the impact of sex specific cells and specific hormone levels on muscle function [459, 460]; however, fully capturing the systemic sex differences in vitro is beyond the current capabilities of these models.
Discussion and outlook
Progress in tissue engineering has resulted in the development of three-dimensional organotypic models, and these have demonstrated potential to overcome several limitations of current aging models. Organotypic models, while not replacing animal models, have multiple advantages, including lower cost, increased accessibility, and human-specific biology. This allows for re-capitulation of human disease and aging phenotypes that animals may not experience naturally or may experience differently [7, 102]. Further, tissue engineered organotypic models have advantages over classic two-dimensional in vitro models as they incorporate physiologically important structural-cell and cell-cell interactions [71]. Additionally, tissue engineered cultures offer flexible scalability when compared to organoid and microchip culture formats. Appropriately scaled models are especially important when investigating aging; in many cases, aging contributes to breakdown of disruption and alterations of the overall tissue, and may include altered nutrient diffusion, organization, and cell-cell communication. In addition, tissue engineered models offer high customizability compared to conventional in vivo models, where specific cell populations or biomaterials can be easily selected or replaced to match research needs. In the three tissues that were addressed here, we highlighted studies that have specifically adapted these models to studying aging; where possible we have also highlighted the accessibility of these models to research groups that may not have prior experience. Importantly, organotypic models are straightforward to customize and, with some optimization, can be a reliable and powerful tool for any aging researcher to adapt to their needs and questions.
Author Contributions
MMS wrote sections on skin and gut and revised the manuscript. IAB wrote the section on skeletal muscle and revised the manuscript. WD wrote introduction and revised the manuscript. JTM conceived and revised the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
JTM is supported in part by National Science Foundation CAREER Award (2046093) and in part by the American Heart Association (19IPLOI34760636). WD is supported in part by a National Science Foundation Graduate Research Fellowship Program under Grant No. 2021307418. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. IAB is supported in part by a TRANSCEND fellowship from the California Institute of Regenerative Medicine (Award # EDUC4-12752). The content is solely the responsibility of the authors and does not necessarily represent the official views of CIRM.
References
- 1. Keehan SP, Cuckler GA, Sisko AM, Madison AJ, Smith SD, Stone DA, Poisal JA, Wolfe CJ, Lizonitz JM. National health expenditure projections, 2014-24: spending growth faster than recent trends. Health Aff (Millwood). 2015; 34:1407–17. https://doi.org/10.1377/hlthaff.2015.0600 [PubMed]
- 2. Centers for Disease Control and Prevention. The State of Aging & Health in America 2013. Atlanta, GA: Centers for Disease Control and Prevention, US Dept of Health and Human Services. 2013. https://www.cdc.gov/aging/pdf/state-aging-health-in-america-2013.pdf.
- 3. Gerteis J, Izrael D, Deitz D, LeRoy L, Ricciardi R, Miller T, Basu J. Multiple Chronic Conditions Chartbook. Agency for Healthcare Research and Quality. 2014.
- 4. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 5. Partridge L, Deelen J, Slagboom PE. Facing up to the global challenges of ageing. Nature. 2018; 561:45–56. https://doi.org/10.1038/s41586-018-0457-8 [PubMed]
- 6. Mitchell SJ, Scheibye-Knudsen M, Longo DL, de Cabo R. Animal models of aging research: implications for human aging and age-related diseases. Annu Rev Anim Biosci. 2015; 3:283–303. https://doi.org/10.1146/annurev-animal-022114-110829 [PubMed]
- 7. Hu JL, Todhunter ME, LaBarge MA, Gartner ZJ. Opportunities for organoids as new models of aging. J Cell Biol. 2018; 217:39–50. https://doi.org/10.1083/jcb.201709054 [PubMed]
- 8. Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012; 32:1104–15. https://doi.org/10.1161/ATVBAHA.111.237693 [PubMed]
- 9. Almasieh M, Levin LA. Neuroprotection in Glaucoma: Animal Models and Clinical Trials. Annu Rev Vis Sci. 2017; 3:91–120. https://doi.org/10.1146/annurev-vision-102016-061422 [PubMed]
- 10. Biswas S, Wan KH. Review of rodent hypertensive glaucoma models. Acta Ophthalmol. 2019; 97:e331–40. https://doi.org/10.1111/aos.13983 [PubMed]
- 11. Pang IH, Clark AF. Inducible rodent models of glaucoma. Prog Retin Eye Res. 2020; 75:100799. https://doi.org/10.1016/j.preteyeres.2019.100799 [PubMed]
- 12. Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci. 2018; 21:1370–9. https://doi.org/10.1038/s41593-018-0236-8 [PubMed]
- 13. Knight A. Systematic reviews of animal experiments demonstrate poor human clinical and toxicological utility. Altern Lab Anim. 2007; 35:641–59. https://doi.org/10.1177/026119290703500610 [PubMed]
- 14. Bédard P, Gauvin S, Ferland K, Caneparo C, Pellerin È, Chabaud S, Bolduc S. Innovative Human Three-Dimensional Tissue-Engineered Models as an Alternative to Animal Testing. Bioengineering (Basel). 2020; 7:115. https://doi.org/10.3390/bioengineering7030115 [PubMed]
- 15. de Boo J, Hendriksen C. Reduction strategies in animal research: a review of scientific approaches at the intra-experimental, supra-experimental and extra-experimental levels. Altern Lab Anim. 2005; 33:369–77. https://doi.org/10.1177/026119290503300404 [PubMed]
- 16. Cristofalo VJ. Ten years later: what have we learned about human aging from studies of cell cultures? Gerontologist. 1996; 36:737–41. https://doi.org/10.1093/geront/36.6.737 [PubMed]
- 17. Jensen C, Teng Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front Mol Biosci. 2020; 7:33. https://doi.org/10.3389/fmolb.2020.00033 [PubMed]
- 18. Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. 2020; 21:571–84. https://doi.org/10.1038/s41580-020-0259-3 [PubMed]
- 19. Costa EC, Moreira AF, de Melo-Diogo D, Gaspar VM, Carvalho MP, Correia IJ. 3D tumor spheroids: an overview on the tools and techniques used for their analysis. Biotechnol Adv. 2016; 34:1427–41. https://doi.org/10.1016/j.biotechadv.2016.11.002 [PubMed]
- 20. Langhans SA. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol. 2018; 9:6. https://doi.org/10.3389/fphar.2018.00006 [PubMed]
- 21. Ahadian S, Civitarese R, Bannerman D, Mohammadi MH, Lu R, Wang E, Davenport-Huyer L, Lai B, Zhang B, Zhao Y, Mandla S, Korolj A, Radisic M. Organ-On-A-Chip Platforms: A Convergence of Advanced Materials, Cells, and Microscale Technologies. Adv Healthc Mater. 2018; 7. https://doi.org/10.1002/adhm.201800734 [PubMed]
- 22. Low LA, Mummery C, Berridge BR, Austin CP, Tagle DA. Organs-on-chips: into the next decade. Nat Rev Drug Discov. 2021; 20:345–61. https://doi.org/10.1038/s41573-020-0079-3 [PubMed]
- 23. Zhang B, Korolj A, Lai BFL, Radisic M. Advances in organ-on-a-chip engineering. Nat Rev Mater. 2018; 3:257–78. https://doi.org/10.1038/s41578-018-0034-7
- 24. Sundstrom L, Morrison B
3rd , Bradley M, Pringle A. Organotypic cultures as tools for functional screening in the CNS. Drug Discov Today. 2005; 10:993–1000. https://doi.org/10.1016/S1359-6446(05)03502-6 [PubMed] - 25. Viana F, O'Kane CM, Schroeder GN. Precision-cut lung slices: A powerful ex vivo model to investigate respiratory infectious diseases. Mol Microbiol. 2022; 117:578–88. https://doi.org/10.1111/mmi.14817 [PubMed]
- 26. Soldatow VY, Lecluyse EL, Griffith LG, Rusyn I. In vitro models for liver toxicity testing. Toxicol Res (Camb). 2013; 2:23–39. https://doi.org/10.1039/C2TX20051A [PubMed]
- 27. Weitz JR, Tiriac H, Hurtado de Mendoza T, Wascher A, Lowy AM. Using Organotypic Tissue Slices to Investigate the Microenvironment of Pancreatic Cancer: Pharmacotyping and Beyond. Cancers (Basel). 2021; 13:4991. https://doi.org/10.3390/cancers13194991 [PubMed]
- 28. Hughes DL, Hughes A, Soonawalla Z, Mukherjee S, O'Neill E. Dynamic Physiological Culture of Ex Vivo Human Tissue: A Systematic Review. Cancers (Basel). 2021; 13:2870. https://doi.org/10.3390/cancers13122870 [PubMed]
- 29. de Boer TP, Camelliti P, Ravens U, Kohl P. Myocardial tissue slices: organotypic pseudo-2D models for cardiac research & development. Future Cardiol. 2009; 5:425–30. https://doi.org/10.2217/fca.09.32 [PubMed]
- 30. Watson SA, Terracciano CM, Perbellini F. Myocardial Slices: an Intermediate Complexity Platform for Translational Cardiovascular Research. Cardiovasc Drugs Ther. 2019; 33:239–44. https://doi.org/10.1007/s10557-019-06853-5 [PubMed]
- 31. Grenier K, Kao J, Diamandis P. Three-dimensional modeling of human neurodegeneration: brain organoids coming of age. Mol Psychiatry. 2020; 25:254–74. https://doi.org/10.1038/s41380-019-0500-7 [PubMed]
- 32. Humpel C. Organotypic brain slice cultures: A review. Neuroscience. 2015; 305:86–98. https://doi.org/10.1016/j.neuroscience.2015.07.086 [PubMed]
- 33. Auger FA, Gibot L, Lacroix D. The pivotal role of vascularization in tissue engineering. Annu Rev Biomed Eng. 2013; 15:177–200. https://doi.org/10.1146/annurev-bioeng-071812-152428 [PubMed]
- 34. Hirt MN, Hansen A, Eschenhagen T. Cardiac tissue engineering: state of the art. Circ Res. 2014; 114:354–67. https://doi.org/10.1161/CIRCRESAHA.114.300522 [PubMed]
- 35. Montgomery M, Zhang B, Radisic M. Cardiac Tissue Vascularization: From Angiogenesis to Microfluidic Blood Vessels. J Cardiovasc Pharmacol Ther. 2014; 19:382–93. https://doi.org/10.1177/1074248414528576 [PubMed]
- 36. Langer R, Vacanti JP. Tissue engineering. Science. 1993; 260:920–6. https://doi.org/10.1126/science.8493529 [PubMed]
- 37. Nerem RM. Cellular engineering. Ann Biomed Eng. 1991; 19:529–45. https://doi.org/10.1007/BF02367396 [PubMed]
- 38. Chua AW, Khoo YC, Tan BK, Tan KC, Foo CL, Chong SJ. Skin tissue engineering advances in severe burns: review and therapeutic applications. Burns Trauma. 2016; 4:3. https://doi.org/10.1186/s41038-016-0027-y [PubMed]
- 39. Vig K, Chaudhari A, Tripathi S, Dixit S, Sahu R, Pillai S, Dennis VA, Singh SR. Advances in Skin Regeneration Using Tissue Engineering. Int J Mol Sci. 2017; 18:789. https://doi.org/10.3390/ijms18040789 [PubMed]
- 40. Gibbons MC, Foley MA, Cardinal KO. Thinking inside the box: keeping tissue-engineered constructs in vitro for use as preclinical models. Tissue Eng Part B Rev. 2013; 19:14–30. https://doi.org/10.1089/ten.TEB.2012.0305 [PubMed]
- 41. Tissue-engineered disease models. Nat Biomed Eng. 2018; 2:879–80. https://doi.org/10.1038/s41551-018-0339-2 [PubMed]
- 42. Verbridge SS, Chandler EM, Fischbach C. Tissue-engineered three-dimensional tumor models to study tumor angiogenesis. Tissue Eng Part A. 2010; 16:2147–52. https://doi.org/10.1089/ten.tea.2009.0668 [PubMed]
- 43. Athanasiou KA, Eswaramoorthy R, Hadidi P, Hu JC. Self-organization and the self-assembling process in tissue engineering. Annu Rev Biomed Eng. 2013; 15:115–36. https://doi.org/10.1146/annurev-bioeng-071812-152423 [PubMed]
- 44. Varner VD, Nelson CM. Toward the directed self-assembly of engineered tissues. Annu Rev Chem Biomol Eng. 2014; 5:507–26. https://doi.org/10.1146/annurev-chembioeng-060713-040016 [PubMed]
- 45. Johnson PC, Mikos AG, Fisher JP, Jansen JA. Strategic directions in tissue engineering. Tissue Eng. 2007; 13:2827–37. https://doi.org/10.1089/ten.2007.0335 [PubMed]
- 46. Mathes SH, Ruffner H, Graf-Hausner U. The use of skin models in drug development. Adv Drug Deliv Rev. 2014; 69-70:81–102. https://doi.org/10.1016/j.addr.2013.12.006 [PubMed]
- 47. Fenske NA, Lober CW. Structural and functional changes of normal aging skin. J Am Acad Dermatol. 1986; 15:571–85. https://doi.org/10.1016/s0190-9622(86)70208-9 [PubMed]
- 48. Makrantonaki E, Zouboulis CC. Molecular mechanisms of skin aging: state of the art. Ann N Y Acad Sci. 2007; 1119:40–50. https://doi.org/10.1196/annals.1404.027 [PubMed]
- 49. Naylor EC, Watson RE, Sherratt MJ. Molecular aspects of skin ageing. Maturitas. 2011; 69:249–56. https://doi.org/10.1016/j.maturitas.2011.04.011 [PubMed]
- 50. Tobin DJ. Introduction to skin aging. J Tissue Viability. 2017; 26:37–46. https://doi.org/10.1016/j.jtv.2016.03.002 [PubMed]
- 51. Han A, Chien AL, Kang S. Photoaging. Dermatol Clin. 2014; 32:291–9. https://doi.org/10.1016/j.det.2014.03.015 [PubMed]
- 52. Helfrich YR, Sachs DL, Voorhees JJ. Overview of skin aging and photoaging. Dermatol Nurs. 2008; 20:177–83. [PubMed]
- 53. Lee DH, Oh JH, Chung JH. Glycosaminoglycan and proteoglycan in skin aging. J Dermatol Sci. 2016; 83:174–81. https://doi.org/10.1016/j.jdermsci.2016.05.016 [PubMed]
- 54. Blume-Peytavi U, Kottner J, Sterry W, Hodin MW, Griffiths TW, Watson RE, Hay RJ, Griffiths CE. Age-Associated Skin Conditions and Diseases: Current Perspectives and Future Options. Gerontologist. 2016 (Suppl 2); 56:S230–42. https://doi.org/10.1093/geront/gnw003 [PubMed]
- 55. Li Y, Liu Y, Xia W, Lei D, Voorhees JJ, Fisher GJ. Age-dependent alterations of decorin glycosaminoglycans in human skin. Sci Rep. 2013; 3:2422. https://doi.org/10.1038/srep02422 [PubMed]
- 56. Pageon H, Zucchi H, Rousset F, Monnier VM, Asselineau D. Skin aging by glycation: lessons from the reconstructed skin model. Clin Chem Lab Med. 2014; 52:169–74. https://doi.org/10.1515/cclm-2013-0091 [PubMed]
- 57. Pageon H, Bakala H, Monnier VM, Asselineau D. Collagen glycation triggers the formation of aged skin in vitro. Eur J Dermatol. 2007; 17:12–20. https://doi.org/10.1684/ejd.2007.0102 [PubMed]
- 58. Pageon H. Reaction of glycation and human skin: the effects on the skin and its components, reconstructed skin as a model. Pathol Biol (Paris). 2010; 58:226–31. https://doi.org/10.1016/j.patbio.2009.09.009 [PubMed]
- 59. Pageon H, Técher MP, Asselineau D. Reconstructed skin modified by glycation of the dermal equivalent as a model for skin aging and its potential use to evaluate anti-glycation molecules. Exp Gerontol. 2008; 43:584–8. https://doi.org/10.1016/j.exger.2008.04.004 [PubMed]
- 60. Gkogkolou P, Böhm M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinol. 2012; 4:259–70. https://doi.org/10.4161/derm.22028 [PubMed]
- 61. Bucala R, Cerami A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol. 1992; 23:1–34. https://doi.org/10.1016/s1054-3589(08)60961-8 [PubMed]
- 62. Chung JH, Eun HC. Angiogenesis in skin aging and photoaging. J Dermatol. 2007; 34:593–600. https://doi.org/10.1111/j.1346-8138.2007.00341.x [PubMed]
- 63. Mansouri P, Chalangari R, Chalangari KM, Saffarian Z. Skin Aging and Immune System. In: Massoud A, Rezaei N, editors. Immunology of Aging. Berlin, Heidelberg: Springer. 2014; 339–68. https://doi.org/10.1007/978-3-642-39495-9_25
- 64. Bennett MF, Robinson MK, Baron ED, Cooper KD. Skin immune systems and inflammation: protector of the skin or promoter of aging? J Investig Dermatol Symp Proc. 2008; 13:15–9. https://doi.org/10.1038/jidsymp.2008.3 [PubMed]
- 65. Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009; 122:1285–94. https://doi.org/10.1242/jcs.033969 [PubMed]
- 66. Ponec M, Weerheim A, Kempenaar J, Mulder A, Gooris GS, Bouwstra J, Mommaas AM. The formation of competent barrier lipids in reconstructed human epidermis requires the presence of vitamin C. J Invest Dermatol. 1997; 109:348–55. https://doi.org/10.1111/1523-1747.ep12336024 [PubMed]
- 67. Boyce ST, Williams ML. Lipid supplemented medium induces lamellar bodies and precursors of barrier lipids in cultured analogues of human skin. J Invest Dermatol. 1993; 101:180–4. https://doi.org/10.1111/1523-1747.ep12363678 [PubMed]
- 68. Jungersted JM. Stratum Corneum Lipids and Filaggrin. In: Thyssen JP, Maibach HI, editors. Filaggrin: Basic Science, Epidemiology, Clinical Aspects and Management. Berlin, Heidelberg: Springer. 2014; 23–6. https://doi.org/10.1007/978-3-642-54379-1_3
- 69. Wang Z, Man MQ, Li T, Elias PM, Mauro TM. Aging-associated alterations in epidermal function and their clinical significance. Aging (Albany NY). 2020; 12:5551–65. https://doi.org/10.18632/aging.102946 [PubMed]
- 70. Farage MA, Miller KW, Zouboulis CC, Piérard GE, Maibach HI. Gender differences in skin aging and the changing profile of the sex hormones with age. J Steroids Horm Sci. 2012; 3:109. https://doi.org/10.4172/2157-7536.1000109
- 71. Ali N, Hosseini M, Vainio S, Taïeb A, Cario-André M, Rezvani HR. Skin equivalents: skin from reconstructions as models to study skin development and diseases. Br J Dermatol. 2015; 173:391–403. https://doi.org/10.1111/bjd.13886 [PubMed]
- 72. Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001; 20:2413–23. https://doi.org/10.1038/sj.onc.1204380 [PubMed]
- 73. Dos Santos M, Metral E, Boher A, Rousselle P, Thepot A, Damour O. In vitro 3-D model based on extending time of culture for studying chronological epidermis aging. Matrix Biol. 2015; 47:85–97. https://doi.org/10.1016/j.matbio.2015.03.009 [PubMed]
- 74. Janson D, Rietveld M, Willemze R, El Ghalbzouri A. Effects of serially passaged fibroblasts on dermal and epidermal morphogenesis in human skin equivalents. Biogerontology. 2013; 14:131–40. https://doi.org/10.1007/s10522-013-9416-9 [PubMed]
- 75. Varani J, Dame MK, Rittie L, Fligiel SE, Kang S, Fisher GJ, Voorhees JJ. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006; 168:1861–8. https://doi.org/10.2353/ajpath.2006.051302 [PubMed]
- 76. Rinnerthaler M, Richter K. The Influence of Calcium on the Skin pH and Epidermal Barrier During Aging. Curr Probl Dermatol. 2018; 54:79–86. https://doi.org/10.1159/000489521 [PubMed]
- 77. Sonnenberg A, Calafat J, Janssen H, Daams H, van der Raaij-Helmer LM, Falcioni R, Kennel SJ, Aplin JD, Baker J, Loizidou M. Integrin alpha 6/beta 4 complex is located in hemidesmosomes, suggesting a major role in epidermal cell-basement membrane adhesion. J Cell Biol. 1991; 113:907–17. https://doi.org/10.1083/jcb.113.4.907 [PubMed]
- 78. Kaur P, Li A. Adhesive properties of human basal epidermal cells: an analysis of keratinocyte stem cells, transit amplifying cells, and postmitotic differentiating cells. J Invest Dermatol. 2000; 114:413–20. https://doi.org/10.1046/j.1523-1747.2000.00884.x [PubMed]
- 79. Shuster S, Black MM, McVitie E. The influence of age and sex on skin thickness, skin collagen and density. Br J Dermatol. 1975; 93:639–43. https://doi.org/10.1111/j.1365-2133.1975.tb05113.x [PubMed]
- 80. Saville CR, Hardman MJ. The Role of Estrogen Deficiency in Skin Aging and Wound Healing. In: Farage MA, Miller KW, Fugate Woods N, Maibach HI, editors. Skin, Mucosa and Menopause: Management of Clinical Issues. Berlin, Heidelberg: Springer. 2015; 71–88. https://doi.org/10.1007/978-3-662-44080-3_6
- 81. Kosmadaki MG, Gilchrest BA. The role of telomeres in skin aging/photoaging. Micron. 2004; 35:155–9. https://doi.org/10.1016/j.micron.2003.11.002 [PubMed]
- 82. Duval C, Schmidt R, Regnier M, Facy V, Asselineau D, Bernerd F. The use of reconstructed human skin to evaluate UV-induced modifications and sunscreen efficacy. Exp Dermatol. 2003 (Suppl 2); 12:64–70. https://doi.org/10.1034/j.1600-0625.12.s2.10.x [PubMed]
- 83. Lee KE, Nho YH, Yun SK, Park SM, Kang S, Yeo H. Caviar Extract and Its Constituent DHA Inhibits UVB-Irradiated Skin Aging by Inducing Adiponectin Production. Int J Mol Sci. 2020; 21:3383. https://doi.org/10.3390/ijms21093383 [PubMed]
- 84. Tanaka K, Asamitsu K, Uranishi H, Iddamalgoda A, Ito K, Kojima H, Okamoto T. Protecting skin photoaging by NF-kappaB inhibitor. Curr Drug Metab. 2010; 11:431–5. https://doi.org/10.2174/138920010791526051 [PubMed]
- 85. Bernerd F, Asselineau D. UVA exposure of human skin reconstructed in vitro induces apoptosis of dermal fibroblasts: subsequent connective tissue repair and implications in photoaging. Cell Death Differ. 1998; 5:792–802. https://doi.org/10.1038/sj.cdd.4400413 [PubMed]
- 86. Bernerd F, Asselineau D. Successive alteration and recovery of epidermal differentiation and morphogenesis after specific UVB-damages in skin reconstructed in vitro. Dev Biol. 1997; 183:123–38. https://doi.org/10.1006/dbio.1996.8465 [PubMed]
- 87. Bernerd F, Vioux C, Asselineau D. Evaluation of the protective effect of sunscreens on in vitro reconstructed human skin exposed to UVB or UVA irradiation. Photochem Photobiol. 2000; 71:314–20. https://doi.org/10.1562/0031-8655(2000)071<0314:EOTPEO>2.0.CO;2 [PubMed]
- 88. Amano S, Ogura Y, Akutsu N, Matsunaga Y, Kadoya K, Adachi E, Nishiyama T. Protective effect of matrix metalloproteinase inhibitors against epidermal basement membrane damage: skin equivalents partially mimic photoageing process. Br J Dermatol. 2005 (Suppl 2); 153:37–46. https://doi.org/10.1111/j.1365-2133.2005.06968.x [PubMed]
- 89. Bell E, Ehrlich HP, Sher S, Merrill C, Sarber R, Hull B, Nakatsuji T, Church D, Buttle DJ. Development and use of a living skin equivalent. Plast Reconstr Surg. 1981; 67:386–92. https://doi.org/10.1097/00006534-198103000-00024 [PubMed]
- 90. Bell E, Sher S, Hull B, Merrill C, Rosen S, Chamson A, Asselineau D, Dubertret L, Coulomb B, Lapiere C, Nusgens B, Neveux Y. The reconstitution of living skin. J Invest Dermatol. 1983; 81:2s–10s. https://doi.org/10.1111/1523-1747.ep12539993 [PubMed]
- 91. El Ghalbzouri A, Commandeur S, Rietveld MH, Mulder AA, Willemze R. Replacement of animal-derived collagen matrix by human fibroblast-derived dermal matrix for human skin equivalent products. Biomaterials. 2009; 30:71–8. https://doi.org/10.1016/j.biomaterials.2008.09.002 [PubMed]
- 92. Sanchez MM, Orneles DN, Park BH, Morgan JT. Automated epidermal thickness quantification of in vitro human skin equivalents using optical coherence tomography. Biotechniques. 2022; 72:194–200. https://doi.org/10.2144/btn-2021-0123 [PubMed]
- 93. Sanchez MM, Morgan JT. Generation of Self-assembled Vascularized Human Skin Equivalents. J Vis Exp. 2021; e62125. https://doi.org/10.3791/62125 [PubMed]
- 94. Shevchenko RV, James SL, James SE. A review of tissue-engineered skin bioconstructs available for skin reconstruction. J R Soc Interface. 2010; 7:229–58. https://doi.org/10.1098/rsif.2009.0403 [PubMed]
- 95. Goodarzi P, Falahzadeh K, Nematizadeh M, Farazandeh P, Payab M, Larijani B, Beik AT, Arjmand B. Tissue Engineered Skin Substitutes. In: Turksen K, editor. Cell Biology and Translational Medicine, Volume 3. Advances in Experimental Medicine and Biology. Cham: Springer International Publishing. 2018; 143–88. https://doi.org/10.1007/5584_2018_226
- 96. Baltazar T, Merola J, Catarino C, Xie CB, Kirkiles-Smith NC, Lee V, Hotta S, Dai G, Xu X, Ferreira FC, Saltzman WM, Pober JS, Karande P. Three Dimensional Bioprinting of a Vascularized and Perfusable Skin Graft Using Human Keratinocytes, Fibroblasts, Pericytes, and Endothelial Cells. Tissue Engineering Part A. 2019; 26:227–38. https://doi.org/10.1089/ten.TEA.2019.0201
- 97. Black AF, Berthod F, L'heureux N, Germain L, Auger FA. In vitro reconstruction of a human capillary-like network in a tissue-engineered skin equivalent. FASEB J. 1998; 12:1331–40. https://doi.org/10.1096/fasebj.12.13.1331 [PubMed]
- 98. Hudon V, Berthod F, Black AF, Damour O, Germain L, Auger FA. A tissue-engineered endothelialized dermis to study the modulation of angiogenic and angiostatic molecules on capillary-like tube formation in vitro. Br J Dermatol. 2003; 148:1094–104. https://doi.org/10.1046/j.1365-2133.2003.05298.x [PubMed]
- 99. Klar AS, Güven S, Biedermann T, Luginbühl J, Böttcher-Haberzeth S, Meuli-Simmen C, Meuli M, Martin I, Scherberich A, Reichmann E. Tissue-engineered dermo-epidermal skin grafts prevascularized with adipose-derived cells. Biomaterials. 2014; 35:5065–78. https://doi.org/10.1016/j.biomaterials.2014.02.049 [PubMed]
- 100. Martins-Green M, Li QJ, Yao M. A new generation organ culture arising from cross-talk between multiple primary human cell types. FASEB J. 2005; 19:222–4. https://doi.org/10.1096/fj.04-1725fje [PubMed]
- 101. Tremblay PL, Hudon V, Berthod F, Germain L, Auger FA. Inosculation of tissue-engineered capillaries with the host's vasculature in a reconstructed skin transplanted on mice. Am J Transplant. 2005; 5:1002–10. https://doi.org/10.1111/j.1600-6143.2005.00790.x [PubMed]
- 102. Bergers LIJ, Reijnders CMA, van den Broek LJ, Spiekstra SW, de Gruijl TD, Weijers EM, Gibbs S. Immune-competent human skin disease models. Drug Discov Today. 2016; 21:1479–88. https://doi.org/10.1016/j.drudis.2016.05.008 [PubMed]
- 103. Bechetoille N, Dezutter-Dambuyant C, Damour O, André V, Orly I, Perrier E. Effects of solar ultraviolet radiation on engineered human skin equivalent containing both Langerhans cells and dermal dendritic cells. Tissue Eng. 2007; 13:2667–79. https://doi.org/10.1089/ten.2006.0405 [PubMed]
- 104. Linde N, Gutschalk CM, Hoffmann C, Yilmaz D, Mueller MM. Integrating macrophages into organotypic co-cultures: a 3D in vitro model to study tumor-associated macrophages. PLoS One. 2012; 7:e40058. https://doi.org/10.1371/journal.pone.0040058 [PubMed]
- 105. Dezutter-Dambuyant C, Black A, Bechetoille N, Bouez C, Maréchal S, Auxenfans C, Cenizo V, Pascal P, Perrier E, Damour O. Evolutive skin reconstructions: from the dermal collagen-glycosaminoglycan-chitosane substrate to an immunocompetent reconstructed skin. Biomed Mater Eng. 2006; 16:S85–94. [PubMed]
- 106. Vermette M, Trottier V, Ménard V, Saint-Pierre L, Roy A, Fradette J. Production of a new tissue-engineered adipose substitute from human adipose-derived stromal cells. Biomaterials. 2007; 28:2850–60. https://doi.org/10.1016/j.biomaterials.2007.02.030 [PubMed]
- 107. Son WC, Yun JW, Kim BH. Adipose-derived mesenchymal stem cells reduce MMP-1 expression in UV-irradiated human dermal fibroblasts: therapeutic potential in skin wrinkling. Biosci Biotechnol Biochem. 2015; 79:919–25. https://doi.org/10.1080/09168451.2015.1008972 [PubMed]
- 108. Trottier V, Marceau-Fortier G, Germain L, Vincent C, Fradette J. IFATS collection: Using human adipose-derived stem/stromal cells for the production of new skin substitutes. Stem Cells. 2008; 26:2713–23. https://doi.org/10.1634/stemcells.2008-0031 [PubMed]
- 109. Topol BM, Haimes HB, Dubertret L, Bell E. Transfer of melanosomes in a skin equivalent model in vitro. J Invest Dermatol. 1986; 87:642–7. https://doi.org/10.1111/1523-1747.ep12456314 [PubMed]
- 110. Okazaki M, Suzuki Y, Yoshimura K, Harii K. Construction of pigmented skin equivalent and its application to the study of congenital disorders of pigmentation. Scand J Plast Reconstr Surg Hand Surg. 2005; 39:339–43. https://doi.org/10.1080/02844310500300362 [PubMed]
- 111. Boyce ST, Medrano EE, Abdel-Malek Z, Supp AP, Dodick JM, Nordlund JJ, Warden GD. Pigmentation and inhibition of wound contraction by cultured skin substitutes with adult melanocytes after transplantation to athymic mice. J Invest Dermatol. 1993; 100:360–5. https://doi.org/10.1111/1523-1747.ep12471822 [PubMed]
- 112. Kim Y, Park N, Rim YA, Nam Y, Jung H, Lee K, Ju JH. Establishment of a complex skin structure via layered co-culture of keratinocytes and fibroblasts derived from induced pluripotent stem cells. Stem Cell Res Ther. 2018; 9:217. https://doi.org/10.1186/s13287-018-0958-2 [PubMed]
- 113. Gingras M, Paradis I, Berthod F. Nerve regeneration in a collagen-chitosan tissue-engineered skin transplanted on nude mice. Biomaterials. 2003; 24:1653–61. https://doi.org/10.1016/s0142-9612(02)00572-0 [PubMed]
- 114. Barker CL, McHale MT, Gillies AK, Waller J, Pearce DM, Osborne J, Hutchinson PE, Smith GM, Pringle JH. The development and characterization of an in vitro model of psoriasis. J Invest Dermatol. 2004; 123:892–901. https://doi.org/10.1111/j.0022-202X.2004.23435.x [PubMed]
- 115. Guerrero-Aspizua S, García M, Murillas R, Retamosa L, Illera N, Duarte B, Holguín A, Puig S, Hernández MI, Meana A, Jorcano JL, Larcher F, Carretero M, Del Río M. Development of a bioengineered skin-humanized mouse model for psoriasis: dissecting epidermal-lymphocyte interacting pathways. Am J Pathol. 2010; 177:3112–24. https://doi.org/10.2353/ajpath.2010.100078 [PubMed]
- 116. van den Bogaard EH, Tjabringa GS, Joosten I, Vonk-Bergers M, van Rijssen E, Tijssen HJ, Erkens M, Schalkwijk J, Koenen HJP. Crosstalk between keratinocytes and T cells in a 3D microenvironment: a model to study inflammatory skin diseases. J Invest Dermatol. 2014; 134:719–27. https://doi.org/10.1038/jid.2013.417 [PubMed]
- 117. Gache Y, Baldeschi C, Del Rio M, Gagnoux-Palacios L, Larcher F, Lacour JP, Meneguzzi G. Construction of skin equivalents for gene therapy of recessive dystrophic epidermolysis bullosa. Hum Gene Ther. 2004; 15:921–33. https://doi.org/10.1089/hum.2004.15.921 [PubMed]
- 118. Di Nunzio F, Maruggi G, Ferrari S, Di Iorio E, Poletti V, Garcia M, Del Rio M, De Luca M, Larcher F, Pellegrini G, Mavilio F. Correction of laminin-5 deficiency in human epidermal stem cells by transcriptionally targeted lentiviral vectors. Mol Ther. 2008; 16:1977–85. https://doi.org/10.1038/mt.2008.204 [PubMed]
- 119. Aufenvenne K, Rice RH, Hausser I, Oji V, Hennies HC, Rio MD, Traupe H, Larcher F. Long-term faithful recapitulation of transglutaminase 1-deficient lamellar ichthyosis in a skin-humanized mouse model, and insights from proteomic studies. J Invest Dermatol. 2012; 132:1918–21. https://doi.org/10.1038/jid.2012.65 [PubMed]
- 120. Di WL, Larcher F, Semenova E, Talbot GE, Harper JI, Del Rio M, Thrasher AJ, Qasim W. Ex-vivo gene therapy restores LEKTI activity and corrects the architecture of Netherton syndrome-derived skin grafts. Mol Ther. 2011; 19:408–16. https://doi.org/10.1038/mt.2010.201 [PubMed]
- 121. García M, Larcher F, Hickerson RP, Baselga E, Leachman SA, Kaspar RL, Del Rio M. Development of skin-humanized mouse models of pachyonychia congenita. J Invest Dermatol. 2011; 131:1053–60. https://doi.org/10.1038/jid.2010.353 [PubMed]
- 122. Larcher F, Espada J, Díaz-Ley B, Jaén P, Juarranz A, Quintanilla M. New experimental models of skin homeostasis and diseases. Actas Dermosifiliogr. 2015; 106:17–28. https://doi.org/10.1016/j.ad.2014.03.008 [PubMed]
- 123. Varkey M, Ding J, Tredget EE. Fibrotic remodeling of tissue-engineered skin with deep dermal fibroblasts is reduced by keratinocytes. Tissue Eng Part A. 2014; 20:716–27. https://doi.org/10.1089/ten.TEA.2013.0434 [PubMed]
- 124. Moulin VJ. Reconstitution of skin fibrosis development using a tissue engineering approach. Methods Mol Biol. 2013; 961:287–303. https://doi.org/10.1007/978-1-62703-227-8_19 [PubMed]
- 125. Berthod F, Germain L, Li H, Xu W, Damour O, Auger FA. Collagen fibril network and elastic system remodeling in a reconstructed skin transplanted on nude mice. Matrix Biol. 2001; 20:463–73. https://doi.org/10.1016/s0945-053x(01)00162-7 [PubMed]
- 126. Quan T, He T, Shao Y, Lin L, Kang S, Voorhees JJ, Fisher GJ. Elevated cysteine-rich 61 mediates aberrant collagen homeostasis in chronologically aged and photoaged human skin. Am J Pathol. 2006; 169:482–90. https://doi.org/10.2353/ajpath.2006.060128 [PubMed]
- 127. Borg M, Brincat S, Camilleri G, Schembri-Wismayer P, Brincat M, Calleja-Agius J. The role of cytokines in skin aging. Climacteric. 2013; 16:514–21. https://doi.org/10.3109/13697137.2013.802303 [PubMed]
- 128. Weinmüllner R, Zbiral B, Becirovic A, Stelzer EM, Nagelreiter F, Schosserer M, Lämmermann I, Liendl L, Lang M, Terlecki-Zaniewicz L, Andriotis O, Mildner M, Golabi B, et al. Organotypic human skin culture models constructed with senescent fibroblasts show hallmarks of skin aging. NPJ Aging Mech Dis. 2020; 6:4. https://doi.org/10.1038/s41514-020-0042-x [PubMed]
- 129. Diekmann J, Alili L, Scholz O, Giesen M, Holtkötter O, Brenneisen P. A three-dimensional skin equivalent reflecting some aspects of in vivo aged skin. Exp Dermatol. 2016; 25:56–61. https://doi.org/10.1111/exd.12866 [PubMed]
- 130. Youn SW, Kim DS, Cho HJ, Jeon SE, Bae IH, Yoon HJ, Park KC. Cellular senescence induced loss of stem cell proportion in the skin in vitro. J Dermatol Sci. 2004; 35:113–23. https://doi.org/10.1016/j.jdermsci.2004.04.002 [PubMed]
- 131. Adamus J, Aho S, Meldrum H, Bosko C, Lee JM. p16INK4A influences the aging phenotype in the living skin equivalent. J Invest Dermatol. 2014; 134:1131–3. https://doi.org/10.1038/jid.2013.468 [PubMed]
- 132. Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Dürr P, Wlaschek M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006; 5:379–89. https://doi.org/10.1111/j.1474-9726.2006.00231.x [PubMed]
- 133. Oh JH, Kim YK, Jung JY, Shin JE, Kim KH, Cho KH, Eun HC, Chung JH. Intrinsic aging- and photoaging-dependent level changes of glycosaminoglycans and their correlation with water content in human skin. J Dermatol Sci. 2011; 62:192–201. https://doi.org/10.1016/j.jdermsci.2011.02.007 [PubMed]
- 134. Maquart FX, Brézillon S, Wegrowski Y. Proteoglycans in Skin Aging. In: Farage MA, Miller KW, Maibach HI (eds), Textbook of Aging Skin. Berlin, Heidelberg: Springer. 2010. https://doi.org/10.1007/978-3-540-89656-2_11
- 135. Sok J, Pineau N, Dalko-Csiba M, Breton L, Bernerd F. Improvement of the dermal epidermal junction in human reconstructed skin by a new c-xylopyranoside derivative. Eur J Dermatol. 2008; 18:297–302. [PubMed]
- 136. Nomura Y. Structural change in decorin with skin aging. Connect Tissue Res. 2006; 47:249–55. https://doi.org/10.1080/03008200600846606 [PubMed]
- 137. Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997; 136:729–43. https://doi.org/10.1083/jcb.136.3.729 [PubMed]
- 138. Henriet E, Jäger S, Tran C, Bastien P, Michelet JF, Minondo AM, Formanek F, Dalko-Csiba M, Lortat-Jacob H, Breton L, Vivès RR. A jasmonic acid derivative improves skin healing and induces changes in proteoglycan expression and glycosaminoglycan structure. Biochim Biophys Acta Gen Subj. 2017; 1861:2250–60. https://doi.org/10.1016/j.bbagen.2017.06.006 [PubMed]
- 139. Deloche C, Minondo AM, Bernard BA, Bernerd F, Salas F, Garnier J, Tancrède E. Effect of C-xyloside on morphogenesis of the dermal epidermal junction in aged female skin. An ultrastuctural pilot study. Eur J Dermatol. 2011; 21:191–6. https://doi.org/10.1684/ejd.2010.1225 [PubMed]
- 140. Vassal-Stermann E, Duranton A, Black AF, Azadiguian G, Demaude J, Lortat-Jacob H, Breton L, Vivès RR. A New C-Xyloside induces modifications of GAG expression, structure and functional properties. PLoS One. 2012; 7:e47933. https://doi.org/10.1371/journal.pone.0047933 [PubMed]
- 141. Kreimendahl F, Marquardt Y, Apel C, Bartneck M, Zwadlo-Klarwasser G, Hepp J, Jockenhoevel S, Baron JM. Macrophages significantly enhance wound healing in a vascularized skin model. J Biomed Mater Res A. 2019; 107:1340–50. https://doi.org/10.1002/jbm.a.36648 [PubMed]
- 142. Chau DY, Johnson C, MacNeil S, Haycock JW, Ghaemmaghami AM. The development of a 3D immunocompetent model of human skin. Biofabrication. 2013; 5:035011. https://doi.org/10.1088/1758-5082/5/3/035011 [PubMed]
- 143. Ouwehand K, Spiekstra SW, Waaijman T, Scheper RJ, de Gruijl TD, Gibbs S. Technical advance: Langerhans cells derived from a human cell line in a full-thickness skin equivalent undergo allergen-induced maturation and migration. J Leukoc Biol. 2011; 90:1027–33. https://doi.org/10.1189/jlb.0610374 [PubMed]
- 144. Marino D, Luginbühl J, Scola S, Meuli M, Reichmann E. Bioengineering dermo-epidermal skin grafts with blood and lymphatic capillaries. Sci Transl Med. 2014; 6:221ra14. https://doi.org/10.1126/scitranslmed.3006894 [PubMed]
- 145. Kim BS, Gao G, Kim JY, Cho DW. 3D Cell Printing of Perfusable Vascularized Human Skin Equivalent Composed of Epidermis, Dermis, and Hypodermis for Better Structural Recapitulation of Native Skin. Adv Healthc Mater. 2019; 8:e1801019. https://doi.org/10.1002/adhm.201801019 [PubMed]
- 146. Grebenyuk S, Ranga A. Engineering Organoid Vascularization. Front Bioeng Biotechnol. 2019; 7:39. https://doi.org/10.3389/fbioe.2019.00039
- 147. Braverman IM, Yen A. Ultrastructure of the capillary loops in the dermal papillae of psoriasis. J Invest Dermatol. 1977; 68:53–60. https://doi.org/10.1111/1523-1747.ep12485169 [PubMed]
- 148. Braverman IM. The cutaneous microcirculation. J Investig Dermatol Symp Proc. 2000; 5:3–9. https://doi.org/10.1046/j.1087-0024.2000.00010.x [PubMed]
- 149. Lovett M, Lee K, Edwards A, Kaplan DL. Vascularization strategies for tissue engineering. Tissue Eng Part B Rev. 2009; 15:353–70. https://doi.org/10.1089/ten.TEB.2009.0085 [PubMed]
- 150. Grinnell F, Lamke CR. Reorganization of hydrated collagen lattices by human skin fibroblasts. J Cell Sci. 1984; 66:51–63. https://doi.org/10.1242/jcs.66.1.51 [PubMed]
- 151. Cross VL, Zheng Y, Won Choi N, Verbridge SS, Sutermaster BA, Bonassar LJ, Fischbach C, Stroock AD. Dense type I collagen matrices that support cellular remodeling and microfabrication for studies of tumor angiogenesis and vasculogenesis in vitro. Biomaterials. 2010; 31:8596–607. https://doi.org/10.1016/j.biomaterials.2010.07.072 [PubMed]
- 152. Braziulis E, Diezi M, Biedermann T, Pontiggia L, Schmucki M, Hartmann-Fritsch F, Luginbühl J, Schiestl C, Meuli M, Reichmann E. Modified plastic compression of collagen hydrogels provides an ideal matrix for clinically applicable skin substitutes. Tissue Eng Part C Methods. 2012; 18:464–74. https://doi.org/10.1089/ten.TEC.2011.0561 [PubMed]
- 153. Moset Zupan A, Nietupski C, Schutte SC. Cyclic Adenosine Monophosphate Eliminates Sex Differences in Estradiol-Induced Elastin Production from Engineered Dermal Substitutes. Int J Mol Sci. 2021; 22:6358. https://doi.org/10.3390/ijms22126358 [PubMed]
- 154. Kim Y, Pritts TA. The Gastrointestinal Tract. In: Luchette F, Yelon J, editors. Geriatric Trauma and Critical Care. Cham: Springer. 2017; 35–43. https://doi.org/10.1007/978-3-319-48687-1_5
- 155. Chen Y, Lin Y, Davis KM, Wang Q, Rnjak-Kovacina J, Li C, Isberg RR, Kumamoto CA, Mecsas J, Kaplan DL. Robust bioengineered 3D functional human intestinal epithelium. Sci Rep. 2015; 5:13708. https://doi.org/10.1038/srep13708 [PubMed]
- 156. Kong F, Deng F, Li Y, Zhao J. Identification of gut microbiome signatures associated with longevity provides a promising modulation target for healthy aging. Gut Microbes. 2019; 10:210–15. https://doi.org/10.1080/19490976.2018.1494102 [PubMed]
- 157. Wallace TC, Guarner F, Madsen K, Cabana MD, Gibson G, Hentges E, Sanders ME. Human gut microbiota and its relationship to health and disease. Nutr Rev. 2011; 69:392–403. https://doi.org/10.1111/j.1753-4887.2011.00402.x [PubMed]
- 158. Galkin F, Mamoshina P, Aliper A, Putin E, Moskalev V, Gladyshev VN, Zhavoronkov A. Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. iScience. 2020; 23:101199. https://doi.org/10.1016/j.isci.2020.101199 [PubMed]
- 159. Brillanti S, Alterini G. Chapter 6 - The role of gut microbiota in chronic liver diseases. In: Stasi C, editor. The Complex Interplay Between Gut-Brain, Gut-Liver, and Liver-Brain Axes. Academic Press. 2021; 123–8. https://www.sciencedirect.com/science/article/pii/B9780128219270000036.
- 160. Tran SM, Mohajeri MH. The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease. Nutrients. 2021; 13:732. https://doi.org/10.3390/nu13030732 [PubMed]
- 161. DeJong EN, Surette MG, Bowdish DME. The Gut Microbiota and Unhealthy Aging: Disentangling Cause from Consequence. Cell Host Microbe. 2020; 28:180–89. https://doi.org/10.1016/j.chom.2020.07.013 [PubMed]
- 162. Vaiserman AM, Koliada AK, Marotta F. Gut microbiota: A player in aging and a target for anti-aging intervention. Ageing Res Rev. 2017; 35:36–45. https://doi.org/10.1016/j.arr.2017.01.001 [PubMed]
- 163. Kim S, Jazwinski SM. The Gut Microbiota and Healthy Aging: A Mini-Review. Gerontology. 2018; 64:513–20. https://doi.org/10.1159/000490615 [PubMed]
- 164. Nagpal R, Mainali R, Ahmadi S, Wang S, Singh R, Kavanagh K, Kitzman DW, Kushugulova A, Marotta F, Yadav H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr Healthy Aging. 2018; 4:267–85. https://doi.org/10.3233/NHA-170030 [PubMed]
- 165. O'Toole PW, Jeffery IB. Gut microbiota and aging. Science. 2015; 350:1214–5. https://doi.org/10.1126/science.aac8469 [PubMed]
- 166. Man AL, Gicheva N, Nicoletti C. The impact of ageing on the intestinal epithelial barrier and immune system. Cell Immunol. 2014; 289:112–8. https://doi.org/10.1016/j.cellimm.2014.04.001 [PubMed]
- 167. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011; 9:356–68. https://doi.org/10.1038/nrmicro2546 [PubMed]
- 168. Hatoum OA, Binion DG. The vasculature and inflammatory bowel disease: contribution to pathogenesis and clinical pathology. Inflamm Bowel Dis. 2005; 11:304–13. https://doi.org/10.1097/01.mib.0000160772.78951.61 [PubMed]
- 169. Mabbott NA. A breakdown in communication? Understanding the effects of aging on the human small intestine epithelium. Clin Sci (Lond). 2015; 129:529–31. https://doi.org/10.1042/CS20150364 [PubMed]
- 170. Thomson AB, Keelan M. The aging gut. Can J Physiol Pharmacol. 1986; 64:30–8. https://doi.org/10.1139/y86-004 [PubMed]
- 171. O'Hanlon P, Kohrs MB. Dietary studies of older Americans. Am J Clin Nutr. 1978; 31:1257–69. https://doi.org/10.1093/ajcn/31.7.1257 [PubMed]
- 172. Drozdowski L, Thomson AB. Aging and the intestine. World J Gastroenterol. 2006; 12:7578–84. https://doi.org/10.3748/wjg.v12.i47.7578 [PubMed]
- 173. Holt PR. Intestinal absorption and malabsorption. The Aging Gut. Masson Publ. 1983; 33–56.
- 174. Webster SG, Leeming JT. The appearance of the small bowel mucosa in old age. Age Ageing. 1975; 4:168–74. https://doi.org/10.1093/ageing/4.3.168 [PubMed]
- 175. Lesher S, Sacher GA. Effects of age on cell proliferation in mouse duodenal crypts. Exp Gerontol. 1968; 3:211–7. https://doi.org/10.1016/0531-5565(68)90004-1 [PubMed]
- 176. Thrasher JD, Greulich RC. The duodenal progenitor population. I. Age related increase in the duration of the cryptal progenitor cycle. J Exp Zool. 1965; 159:39–46. https://doi.org/10.1002/jez.1401590105 [PubMed]
- 177. Nalapareddy K, Nattamai KJ, Kumar RS, Karns R, Wikenheiser-Brokamp KA, Sampson LL, Mahe MM, Sundaram N, Yacyshyn MB, Yacyshyn B, Helmrath MA, Zheng Y, Geiger H. Canonical Wnt Signaling Ameliorates Aging of Intestinal Stem Cells. Cell Rep. 2017; 18:2608–21. https://doi.org/10.1016/j.celrep.2017.02.056 [PubMed]
- 178. Keelan M, Walker K, Thomson AB. Intestinal morphology, marker enzymes and lipid content of brush border membranes from rabbit jejunum and ileum: effect of aging. Mech Ageing Dev. 1985; 31:49–68. https://doi.org/10.1016/0047-6374(85)90026-0 [PubMed]
- 179. Martin K, Potten CS, Roberts SA, Kirkwood TB. Altered stem cell regeneration in irradiated intestinal crypts of senescent mice. J Cell Sci. 1998; 111:2297–303. https://doi.org/10.1242/jcs.111.16.2297 [PubMed]
- 180. Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, Schertzer JD, Larché MJ, Davidson DJ, et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe. 2017; 21:455–66.e4. https://doi.org/10.1016/j.chom.2017.03.002 [PubMed]
- 181. Wilson QN, Wells M, Davis AT, Sherrill C, Tsilimigras MCB, Jones RB, Fodor AA, Kavanagh K. Greater Microbial Translocation and Vulnerability to Metabolic Disease in Healthy Aged Female Monkeys. Sci Rep. 2018; 8:11373. https://doi.org/10.1038/s41598-018-29473-9 [PubMed]
- 182. Mitchell EL, Davis AT, Brass K, Dendinger M, Barner R, Gharaibeh R, Fodor AA, Kavanagh K. Reduced Intestinal Motility, Mucosal Barrier Function, and Inflammation in Aged Monkeys. J Nutr Health Aging. 2017; 21:354–61. https://doi.org/10.1007/s12603-016-0725-y [PubMed]
- 183. Ecknauer R, Vadakel T, Wepler R. Intestinal morphology and cell production rate in aging rats. J Gerontol. 1982; 37:151–5. https://doi.org/10.1093/geronj/37.2.151 [PubMed]
- 184. Jakab L, Pénzes L. Relationship between glucose absorption and villus height in ageing. Experientia. 1981; 37:740–2. https://doi.org/10.1007/BF01967955 [PubMed]
- 185. Höhn P, Gabbert H, Wagner R. Differentiation and aging of the rat intestinal mucosa. II. Morphological, enzyme histochemical and disc electrophoretic aspects of the aging of the small intestinal mucosa. Mech Ageing Dev. 1978; 7:217–26. https://doi.org/10.1016/0047-6374(78)90068-4 [PubMed]
- 186. Hubbard RE, Goodwin VA, Llewellyn DJ, Warmoth K, Lang IA. Frailty, financial resources and subjective well-being in later life. Arch Gerontol Geriatr. 2014; 58:364–9. https://doi.org/10.1016/j.archger.2013.12.008 [PubMed]
- 187. Valenkevich IN, Zhukova NM. [The structure of the mucous membrane of the human duodenum with aging]. Arkh Patol. 1976; 38:58–61. [PubMed]
- 188. Larrick JW, Mendelsohn AR. Roads to the Fountain of Youth? Rejuvenating Intestinal Stem Cells. Rejuvenation Res. 2019; 22:342–7. https://doi.org/10.1089/rej.2019.2251 [PubMed]
- 189. Cui H, Tang D, Garside GB, Zeng T, Wang Y, Tao Z, Zhang L, Tao S. Wnt Signaling Mediates the Aging-Induced Differentiation Impairment of Intestinal Stem Cells. Stem Cell Rev Rep. 2019; 15:448–55. https://doi.org/10.1007/s12015-019-09880-9 [PubMed]
- 190. Woo DH, Chen Q, Yang TL, Glineburg MR, Hoge C, Leu NA, Johnson FB, Lengner CJ. Enhancing a Wnt-Telomere Feedback Loop Restores Intestinal Stem Cell Function in a Human Organotypic Model of Dyskeratosis Congenita. Cell Stem Cell. 2016; 19:397–405. https://doi.org/10.1016/j.stem.2016.05.024 [PubMed]
- 191. Shub MD, Pang KY, Swann DA, Walker WA. Age-related changes in chemical composition and physical properties of mucus glycoproteins from rat small intestine. Biochem J. 1983; 215:405–11. https://doi.org/10.1042/bj2150405 [PubMed]
- 192. He F, Ouwehand AC, Isolauri E, Hashimoto H, Benno Y, Salminen S. Comparison of mucosal adhesion and species identification of bifidobacteria isolated from healthy and allergic infants. FEMS Immunol Med Microbiol. 2001; 30:43–7. https://doi.org/10.1111/j.1574-695X.2001.tb01548.x [PubMed]
- 193. He F, Ouwehand AC, Isolauri E, Hosoda M, Benno Y, Salminen S. Differences in composition and mucosal adhesion of bifidobacteria isolated from healthy adults and healthy seniors. Curr Microbiol. 2001; 43:351–4. https://doi.org/10.1007/s002840010315 [PubMed]
- 194. Ouwehand AC, Isolauri E, Kirjavainen PV, Salminen SJ. Adhesion of four Bifidobacterium strains to human intestinal mucus from subjects in different age groups. FEMS Microbiol Lett. 1999; 172:61–4. https://doi.org/10.1111/j.1574-6968.1999.tb13450.x [PubMed]
- 195. Newton JL, Jordan N, Pearson J, Williams GV, Allen A, James OF. The adherent gastric antral and duodenal mucus gel layer thins with advancing age in subjects infected with Helicobacter pylori. Gerontology. 2000; 46:153–7. https://doi.org/10.1159/000022151 [PubMed]
- 196. Leedham SJ, Rodenas-Cuadrado P, Howarth K, Lewis A, Mallappa S, Segditsas S, Davis H, Jeffery R, Rodriguez-Justo M, Keshav S, Travis SP, Graham TA, East J, et al. A basal gradient of Wnt and stem-cell number influences regional tumour distribution in human and mouse intestinal tracts. Gut. 2013; 62:83–93. https://doi.org/10.1136/gutjnl-2011-301601 [PubMed]
- 197. Kirkwood TB. Intrinsic ageing of gut epithelial stem cells. Mech Ageing Dev. 2004; 125:911–5. https://doi.org/10.1016/j.mad.2004.09.004 [PubMed]
- 198. Kobayashi A, Donaldson DS, Erridge C, Kanaya T, Williams IR, Ohno H, Mahajan A, Mabbott NA. The functional maturation of M cells is dramatically reduced in the Peyer's patches of aged mice. Mucosal Immunol. 2013; 6:1027–37. https://doi.org/10.1038/mi.2012.141 [PubMed]
- 199. Fulton JR, Cuff CF. Mucosal and systemic immunity to intestinal reovirus infection in aged mice. Exp Gerontol. 2004; 39:1285–94. https://doi.org/10.1016/j.exger.2004.06.013 [PubMed]
- 200. Thoreux K, Owen RL, Schmucker DL. Intestinal lymphocyte number, migration and antibody secretion in young and old rats. Immunology. 2000; 101:161–7. https://doi.org/10.1046/j.1365-2567.2000.00095.x [PubMed]
- 201. Schmucker DL, Daniels CK, Wang RK, Smith K. Mucosal immune response to cholera toxin in ageing rats. I. Antibody and antibody-containing cell response. Immunology. 1988; 64:691–5. [PubMed]
- 202. Taylor LD, Daniels CK, Schmucker DL. Ageing compromises gastrointestinal mucosal immune response in the rhesus monkey. Immunology. 1992; 75:614–8. [PubMed]
- 203. Lindner C, Wahl B, Föhse L, Suerbaum S, Macpherson AJ, Prinz I, Pabst O. Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J Exp Med. 2012; 209:365–77. https://doi.org/10.1084/jem.20111980 [PubMed]
- 204. Santiago AF, Alves AC, Oliveira RP, Fernandes RM, Paula-Silva J, Assis FA, Carvalho CR, Weiner HL, Faria AM. Aging correlates with reduction in regulatory-type cytokines and T cells in the gut mucosa. Immunobiology. 2011; 216:1085–93. https://doi.org/10.1016/j.imbio.2011.05.007 [PubMed]
- 205. Arranz E, O'Mahony S, Barton JR, Ferguson A. Immunosenescence and mucosal immunity: significant effects of old age on secretory IgA concentrations and intraepithelial lymphocyte counts. Gut. 1992; 33:882–6. https://doi.org/10.1136/gut.33.7.882 [PubMed]
- 206. Haq JA, Szewczuk MR. Differential effect of aging on B-cell immune responses to cholera toxin in the inductive and effector sites of the mucosal immune system. Infect Immun. 1991; 59:3094–100. https://doi.org/10.1128/iai.59.9.3094-3100.1991 [PubMed]
- 207. Senda S, Cheng E, Kawanishi H. Aging-associated changes in murine intestinal immunoglobulin A and M secretions. Scand J Immunol. 1988; 27:157–64. https://doi.org/10.1111/j.1365-3083.1988.tb02334.x [PubMed]
- 208. Moretto MM, Lawlor EM, Khan IA. Aging mice exhibit a functional defect in mucosal dendritic cell response against an intracellular pathogen. J Immunol. 2008; 181:7977–84. https://doi.org/10.4049/jimmunol.181.11.7977 [PubMed]
- 209. Kato H, Fujihashi K, Kato R, Dohi T, Fujihashi K, Hagiwara Y, Kataoka K, Kobayashi R, McGhee JR. Lack of oral tolerance in aging is due to sequential loss of Peyer's patch cell interactions. Int Immunol. 2003; 15:145–58. https://doi.org/10.1093/intimm/dxg011 [PubMed]
- 210. Baylis D, Bartlett DB, Patel HP, Roberts HC. Understanding how we age: insights into inflammaging. Longev Healthspan. 2013; 2:8. https://doi.org/10.1186/2046-2395-2-8 [PubMed]
- 211. Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007; 128:92–105. https://doi.org/10.1016/j.mad.2006.11.016 [PubMed]
- 212. Ahmed S, Spence JD. Sex differences in the intestinal microbiome: interactions with risk factors for atherosclerosis and cardiovascular disease. Biol Sex Differ. 2021; 12:35. https://doi.org/10.1186/s13293-021-00378-z [PubMed]
- 213. Verhulsel M, Simon A, Bernheim-Dennery M, Gannavarapu VR, Gérémie L, Ferraro D, Krndija D, Talini L, Viovy JL, Vignjevic DM, Descroix S. Developing an advanced gut on chip model enabling the study of epithelial cell/fibroblast interactions. Lab on a chip. 2020; 21:365–77. https://doi.org/10.1039/d0lc00672f [PubMed]
- 214. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013; 339:1084–8. https://doi.org/10.1126/science.1233521 [PubMed]
- 215. Jašarević E, Morrison KE, Bale TL. Sex differences in the gut microbiome-brain axis across the lifespan. Philos Trans R Soc Lond B Biol Sci. 2016; 371:20150122. https://doi.org/10.1098/rstb.2015.0122 [PubMed]
- 216. Gomez A, Luckey D, Taneja V. The gut microbiome in autoimmunity: Sex matters. Clin Immunol. 2015; 159:154–62. https://doi.org/10.1016/j.clim.2015.04.016 [PubMed]
- 217. Shin JH, Park YH, Sim M, Kim SA, Joung H, Shin DM. Serum level of sex steroid hormone is associated with diversity and profiles of human gut microbiome. Res Microbiol. 2019; 170:192–201. https://doi.org/10.1016/j.resmic.2019.03.003 [PubMed]
- 218. Ranganathan S, Smith EM, Foulke-Abel JD, Barry EM. Research in a time of enteroids and organoids: how the human gut model has transformed the study of enteric bacterial pathogens. Gut Microbes. 2020; 12:1795492. https://doi.org/10.1080/19490976.2020.1795389 [PubMed]
- 219. Yi B, Shim KY, Ha SK, Han J, Hoang HH, Choi I, Park S, Sung JH. Three-dimensional in vitro gut model on a villi-shaped collagen scaffold. BioChip J. 2017; 11:219–31. https://doi.org/10.1007/s13206-017-1307-8
- 220. Yu J, Peng S, Luo D, March JC. In vitro 3D human small intestinal villous model for drug permeability determination. Biotechnol Bioeng. 2012; 109:2173–8. https://doi.org/10.1002/bit.24518 [PubMed]
- 221. Hartman KG, Bortner JD, Falk GW, Yu J, Martín MG, Rustgi AK, Lynch JP. Modeling inflammation and oxidative stress in gastrointestinal disease development using novel organotypic culture systems. Stem Cell Res Ther. 2013 (Suppl 1); 4:S5. https://doi.org/10.1186/scrt366 [PubMed]
- 222. Poletti M, Arnauts K, Ferrante M, Korcsmaros T. Organoid-based Models to Study the Role of Host-microbiota Interactions in IBD. J Crohns Colitis. 2021; 15:1222–35. https://doi.org/10.1093/ecco-jcc/jjaa257 [PubMed]
- 223. Dosh RH, Jordan-Mahy N, Sammon C, Le Maitre CL. Use of l-pNIPAM hydrogel as a 3D-scaffold for intestinal crypts and stem cell tissue engineering. Biomater Sci. 2019; 7:4310–24. https://doi.org/10.1039/c9bm00541b [PubMed]
- 224. Kim HJ, Ingber DE. Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr Biol (Camb). 2013; 5:1130–40. https://doi.org/10.1039/c3ib40126j [PubMed]
- 225. Bein A, Shin W, Jalili-Firoozinezhad S, Park MH, Sontheimer-Phelps A, Tovaglieri A, Chalkiadaki A, Kim HJ, Ingber DE. Microfluidic Organ-on-a-Chip Models of Human Intestine. Cell Mol Gastroenterol Hepatol. 2018; 5:659–68. https://doi.org/10.1016/j.jcmgh.2017.12.010 [PubMed]
- 226. Jalili-Firoozinezhad S, Gazzaniga FS, Calamari EL, Camacho DM, Fadel CW, Bein A, Swenor B, Nestor B, Cronce MJ, Tovaglieri A, Levy O, Gregory KE, Breault DT, et al. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat Biomed Eng. 2019; 3:520–31. https://doi.org/10.1038/s41551-019-0397-0 [PubMed]
- 227. Jalili-Firoozinezhad S, Bein A, Gazzaniga FS, Fadel CW, Novak R, Ingber DE. Establishment of a Modular Anaerobic Human Intestine Chip. Methods Mol Biol. 2022; 2373:69–85. https://doi.org/10.1007/978-1-0716-1693-2_5 [PubMed]
- 228. Dotti I, Mora-Buch R, Ferrer-Picón E, Planell N, Jung P, Masamunt MC, Leal RF, Martín de Carpi J, Llach J, Ordás I, Batlle E, Panés J, Salas A. Alterations in the epithelial stem cell compartment could contribute to permanent changes in the mucosa of patients with ulcerative colitis. Gut. 2017; 66:2069–79. https://doi.org/10.1136/gutjnl-2016-312609 [PubMed]
- 229. Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, Kou B, Opekun AR, Burrin D, et al. Replication of human noroviruses in stem cell-derived human enteroids. Science. 2016; 353:1387–93. https://doi.org/10.1126/science.aaf5211 [PubMed]
- 230. Arnold JW, Roach J, Fabela S, Moorfield E, Ding S, Blue E, Dagher S, Magness S, Tamayo R, Bruno-Barcena JM, Azcarate-Peril MA. The pleiotropic effects of prebiotic galacto-oligosaccharides on the aging gut. Microbiome. 2021; 9:31. https://doi.org/10.1186/s40168-020-00980-0 [PubMed]
- 231. Greenhalgh K, Meyer KM, Aagaard KM, Wilmes P. The human gut microbiome in health: establishment and resilience of microbiota over a lifetime. Environ Microbiol. 2016; 18:2103–16. https://doi.org/10.1111/1462-2920.13318 [PubMed]
- 232. Peniche AG, Spinler JK, Boonma P, Savidge TC, Dann SM. Aging impairs protective host defenses against Clostridioides (Clostridium) difficile infection in mice by suppressing neutrophil and IL-22 mediated immunity. Anaerobe. 2018; 54:83–91. https://doi.org/10.1016/j.anaerobe.2018.07.011 [PubMed]
- 233. Maffei VJ, Kim S, Blanchard E
4th , Luo M, Jazwinski SM, Taylor CM, Welsh DA. Biological Aging and the Human Gut Microbiota. J Gerontol A Biol Sci Med Sci. 2017; 72:1474–82. https://doi.org/10.1093/gerona/glx042 [PubMed] - 234. Biagini F, Calvigioni M, Lapomarda A, Vecchione A, Magliaro C, De Maria C, Montemurro F, Celandroni F, Mazzantini D, Mattioli-Belmonte M, Ghelardi E, Vozzi G. A novel 3D in vitro model of the human gut microbiota. Sci Rep. 2020; 10:21499. https://doi.org/10.1038/s41598-020-78591-w [PubMed]
- 235. Date S, Sato T. Mini-gut organoids: reconstitution of the stem cell niche. Annu Rev Cell Dev Biol. 2015; 31:269–89. https://doi.org/10.1146/annurev-cellbio-100814-125218 [PubMed]
- 236. Almeqdadi M, Mana MD, Roper J, Yilmaz ÖH. Gut organoids: mini-tissues in culture to study intestinal physiology and disease. Am J Physiol Cell Physiol. 2019; 317:C405–19. https://doi.org/10.1152/ajpcell.00300.2017 [PubMed]
- 237. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009; 459:262–5. https://doi.org/10.1038/nature07935 [PubMed]
- 238. Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, Freinkman E, Dickey AS, La Spada AR, et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell. 2018; 22:769–78.e4. https://doi.org/10.1016/j.stem.2018.04.001 [PubMed]
- 239. Paul W, Marta C, Tom VW. Resolving host-microbe interactions in the gut: the promise of in vitro models to complement in vivo research. Curr Opin Microbiol. 2018; 44:28–33. https://doi.org/10.1016/j.mib.2018.07.001 [PubMed]
- 240. Eain MMG, Baginska J, Greenhalgh K, Fritz JV, Zenhausern F, Wilmes P. Engineering Solutions for Representative Models of the Gastrointestinal Human-Microbe Interface. Engineering. 2017; 3:60–5. https://doi.org/10.1016/J.ENG.2017.01.011
- 241. Sung JH, Yu J, Luo D, Shuler ML, March JC. Microscale 3-D hydrogel scaffold for biomimetic gastrointestinal (GI) tract model. Lab Chip. 2011; 11:389–92. https://doi.org/10.1039/c0lc00273a [PubMed]
- 242. Fung TC, Artis D, Sonnenberg GF. Anatomical localization of commensal bacteria in immune cell homeostasis and disease. Immunol Rev. 2014; 260:35–49. https://doi.org/10.1111/imr.12186 [PubMed]
- 243. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S, Ouwehand A, Langella P, Rautonen N, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med. 2011; 3:559–72. https://doi.org/10.1002/emmm.201100159 [PubMed]
- 244. Regan JC, Khericha M, Dobson AJ, Bolukbasi E, Rattanavirotkul N, Partridge L. Sex difference in pathology of the ageing gut mediates the greater response of female lifespan to dietary restriction. Elife. 2016; 5:e10956. https://doi.org/10.7554/eLife.10956 [PubMed]
- 245. Chang L, Heitkemper MM. Gender differences in irritable bowel syndrome. Gastroenterology. 2002; 123:1686–701. https://doi.org/10.1053/gast.2002.36603 [PubMed]
- 246. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387 [PubMed]
- 247. Kim SE, Paik HY, Yoon H, Lee JE, Kim N, Sung MK. Sex- and gender-specific disparities in colorectal cancer risk. World J Gastroenterol. 2015; 21:5167–75. https://doi.org/10.3748/wjg.v21.i17.5167 [PubMed]
- 248. Grishina I, Fenton A, Sankaran-Walters S. Gender differences, aging and hormonal status in mucosal injury and repair. Aging Dis. 2014; 5:160–9. https://doi.org/10.14336/AD.2014.0500160 [PubMed]
- 249. Janssen I, Heymsfield SB, Wang ZM, Ross R. Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J Appl Physiol (1985). 2000; 89:81–8. https://doi.org/10.1152/jappl.2000.89.1.81 [PubMed]
- 250. Kawakami Y, Fukunaga T. New insights into in vivo human skeletal muscle function. Exerc Sport Sci Rev. 2006; 34:16–21. https://doi.org/10.1097/00003677-200601000-00005 [PubMed]
- 251. Pandy MG, Andriacchi TP. Muscle and joint function in human locomotion. Annu Rev Biomed Eng. 2010; 12:401–33. https://doi.org/10.1146/annurev-bioeng-070909-105259 [PubMed]
- 252. Miller JD, Pegelow DF, Jacques AJ, Dempsey JA. Skeletal muscle pump versus respiratory muscle pump: modulation of venous return from the locomotor limb in humans. J Physiol. 2005; 563:925–43. https://doi.org/10.1113/jphysiol.2004.076422 [PubMed]
- 253. Delp MD, Laughlin MH. Regulation of skeletal muscle perfusion during exercise. Acta Physiol Scand. 1998; 162:411–9. https://doi.org/10.1046/j.1365-201X.1998.0324e.x [PubMed]
- 254. Berlin DA, Bakker J. Understanding venous return. Intensive Care Med. 2014; 40:1564–6. https://doi.org/10.1007/s00134-014-3379-4 [PubMed]
- 255. Wolfe RR. The underappreciated role of muscle in health and disease. Am J Clin Nutr. 2006; 84:475–82. https://doi.org/10.1093/ajcn/84.3.475 [PubMed]
- 256. Periasamy M, Herrera JL, Reis FCG. Skeletal Muscle Thermogenesis and Its Role in Whole Body Energy Metabolism. Diabetes Metab J. 2017; 41:327–36. https://doi.org/10.4093/dmj.2017.41.5.327 [PubMed]
- 257. Zurlo F, Nemeth PM, Choksi RM, Sesodia S, Ravussin E. Whole-body energy metabolism and skeletal muscle biochemical characteristics. Metabolism. 1994; 43:481–6. https://doi.org/10.1016/0026-0495(94)90081-7 [PubMed]
- 258. Zurlo F, Larson K, Bogardus C, Ravussin E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest. 1990; 86:1423–7. https://doi.org/10.1172/JCI114857 [PubMed]
- 259. Miljkovic N, Lim JY, Miljkovic I, Frontera WR. Aging of skeletal muscle fibers. Ann Rehabil Med. 2015; 39:155–62. https://doi.org/10.5535/arm.2015.39.2.155 [PubMed]
- 260. Evans WJ. Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am J Clin Nutr. 2010; 91:1123S–7S. https://doi.org/10.3945/ajcn.2010.28608A [PubMed]
- 261. Evans WJ, Campbell WW. Sarcopenia and age-related changes in body composition and functional capacity. J Nutr. 1993; 123:465–8. https://doi.org/10.1093/jn/123.suppl_2.465 [PubMed]
- 262. Poehlman ET, Toth MJ, Fishman PS, Vaitkevicius P, Gottlieb SS, Fisher ML, Fonong T. Sarcopenia in aging humans: the impact of menopause and disease. J Gerontol A Biol Sci Med Sci. 1995; 50:73–7. https://doi.org/10.1093/gerona/50a.special_issue.73 [PubMed]
- 263. Kenney WL, Buskirk ER. Functional consequences of sarcopenia: effects on thermoregulation. J Gerontol A Biol Sci Med Sci. 1995; 50:78–85. https://doi.org/10.1093/gerona/50a.special_issue.78 [PubMed]
- 264. Kim SH, Jeong JB, Kang J, Ahn DW, Kim JW, Kim BG, Lee KL, Oh S, Yoon SH, Park SJ, Lee DH. Association between sarcopenia level and metabolic syndrome. PLoS One. 2021; 16:e0248856. https://doi.org/10.1371/journal.pone.0248856 [PubMed]
- 265. Cesari M, Kritchevsky SB, Baumgartner RN, Atkinson HH, Penninx BW, Lenchik L, Palla SL, Ambrosius WT, Tracy RP, Pahor M. Sarcopenia, obesity, and inflammation--results from the Trial of Angiotensin Converting Enzyme Inhibition and Novel Cardiovascular Risk Factors study. Am J Clin Nutr. 2005; 82:428–34. https://doi.org/10.1093/ajcn.82.2.428 [PubMed]
- 266. Cesari M, Penninx BW, Pahor M, Lauretani F, Corsi AM, Rhys Williams G, Guralnik JM, Ferrucci L. Inflammatory markers and physical performance in older persons: the InCHIANTI study. J Gerontol A Biol Sci Med Sci. 2004; 59:242–8. https://doi.org/10.1093/gerona/59.3.m242 [PubMed]
- 267. Beyer I, Mets T, Bautmans I. Chronic low-grade inflammation and age-related sarcopenia. Curr Opin Clin Nutr Metab Care. 2012; 15:12–22. https://doi.org/10.1097/MCO.0b013e32834dd297 [PubMed]
- 268. Jensen GL. Inflammation: roles in aging and sarcopenia. JPEN J Parenter Enteral Nutr. 2008; 32:656–9. https://doi.org/10.1177/0148607108324585 [PubMed]
- 269. Remelli F, Vitali A, Zurlo A, Volpato S. Vitamin D Deficiency and Sarcopenia in Older Persons. Nutrients. 2019; 11:2861. https://doi.org/10.3390/nu11122861 [PubMed]
- 270. Robinson S, Cooper C, Aihie Sayer A. Nutrition and sarcopenia: a review of the evidence and implications for preventive strategies. J Aging Res. 2012; 2012:510801. https://doi.org/10.1155/2012/510801 [PubMed]
- 271. Visser M, Deeg DJ, Lips P, and Longitudinal Aging Study Amsterdam. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): the Longitudinal Aging Study Amsterdam. J Clin Endocrinol Metab. 2003; 88:5766–72. https://doi.org/10.1210/jc.2003-030604 [PubMed]
- 272. Kortebein P, Ferrando A, Lombeida J, Wolfe R, Evans WJ. Effect of 10 days of bed rest on skeletal muscle in healthy older adults. JAMA. 2007; 297:1772–4. https://doi.org/10.1001/jama.297.16.1772-b [PubMed]
- 273. Montero-Fernández N, Serra-Rexach JA. Role of exercise on sarcopenia in the elderly. Eur J Phys Rehabil Med. 2013; 49:131–43. [PubMed]
- 274. Doherty TJ. Invited review: Aging and sarcopenia. J Appl Physiol (1985). 2003; 95:1717–27. https://doi.org/10.1152/japplphysiol.00347.2003 [PubMed]
- 275. Light N, Champion AE. Characterization of muscle epimysium, perimysium and endomysium collagens. Biochem J. 1984; 219:1017–26. https://doi.org/10.1042/bj2191017 [PubMed]
- 276. Zierath JR, Hawley JA. Skeletal muscle fiber type: influence on contractile and metabolic properties. PLoS Biol. 2004; 2:e348. https://doi.org/10.1371/journal.pbio.0020348 [PubMed]
- 277. Kent-Braun JA, Ng AV, Young K. Skeletal muscle contractile and noncontractile components in young and older women and men. J Appl Physiol (1985). 2000; 88:662–8. https://doi.org/10.1152/jappl.2000.88.2.662 [PubMed]
- 278. Frontera WR, Hughes VA, Fielding RA, Fiatarone MA, Evans WJ, Roubenoff R. Aging of skeletal muscle: a 12-yr longitudinal study. J Appl Physiol (1985). 2000; 88:1321–6. https://doi.org/10.1152/jappl.2000.88.4.1321 [PubMed]
- 279. Williams GN, Higgins MJ, Lewek MD. Aging skeletal muscle: physiologic changes and the effects of training. Phys Ther. 2002; 82:62–8. https://doi.org/10.1093/ptj/82.1.62 [PubMed]
- 280. Lang T, Cauley JA, Tylavsky F, Bauer D, Cummings S, Harris TB, and Health ABC Study. Computed tomographic measurements of thigh muscle cross-sectional area and attenuation coefficient predict hip fracture: the health, aging, and body composition study. J Bone Miner Res. 2010; 25:513–9. https://doi.org/10.1359/jbmr.090807 [PubMed]
- 281. Ballak SB, Degens H, de Haan A, Jaspers RT. Aging related changes in determinants of muscle force generating capacity: a comparison of muscle aging in men and male rodents. Ageing Res Rev. 2014; 14:43–55. https://doi.org/10.1016/j.arr.2014.01.005 [PubMed]
- 282. Gao Y, Kostrominova TY, Faulkner JA, Wineman AS. Age-related changes in the mechanical properties of the epimysium in skeletal muscles of rats. J Biomech. 2008; 41:465–9. https://doi.org/10.1016/j.jbiomech.2007.09.021 [PubMed]
- 283. Zhang C, Gao Y. Effects of aging on the lateral transmission of force in rat skeletal muscle. J Biomech. 2014; 47:944–8. https://doi.org/10.1016/j.jbiomech.2014.01.026 [PubMed]
- 284. Zhang Y, Chen JS, He Q, He X, Basava RR, Hodgson J, Sinha U, Sinha S. Microstructural analysis of skeletal muscle force generation during aging. Int J Numer Method Biomed Eng. 2020; 36:e3295. https://doi.org/10.1002/cnm.3295 [PubMed]
- 285. Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007; 317:807–10. https://doi.org/10.1126/science.1144090 [PubMed]
- 286. Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Muñoz-Cánoves P. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011; 1:21. https://doi.org/10.1186/2044-5040-1-21 [PubMed]
- 287. Groen BB, Hamer HM, Snijders T, van Kranenburg J, Frijns D, Vink H, van Loon LJ. Skeletal muscle capillary density and microvascular function are compromised with aging and type 2 diabetes. J Appl Physiol (1985). 2014; 116:998–1005. https://doi.org/10.1152/japplphysiol.00919.2013 [PubMed]
- 288. Prior SJ, Ryan AS, Blumenthal JB, Watson JM, Katzel LI, Goldberg AP. Sarcopenia Is Associated With Lower Skeletal Muscle Capillarization and Exercise Capacity in Older Adults. J Gerontol A Biol Sci Med Sci. 2016; 71:1096–101. https://doi.org/10.1093/gerona/glw017 [PubMed]
- 289. Kirkendall DT, Garrett WE
Jr . The effects of aging and training on skeletal muscle. Am J Sports Med. 1998; 26:598–602. https://doi.org/10.1177/03635465980260042401 [PubMed] - 290. Lexell J, Downham D. What is the effect of ageing on type 2 muscle fibres? J Neurol Sci. 1992; 107:250–1. https://doi.org/10.1016/0022-510x(92)90297-x [PubMed]
- 291. Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab. 2007; 292:E151–7. https://doi.org/10.1152/ajpendo.00278.2006 [PubMed]
- 292. Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci. 1995; 50:11–6. https://doi.org/10.1093/gerona/50a.special_issue.11 [PubMed]
- 293. Brunner F, Schmid A, Sheikhzadeh A, Nordin M, Yoon J, Frankel V. Effects of aging on Type II muscle fibers: a systematic review of the literature. J Aging Phys Act. 2007; 15:336–48. https://doi.org/10.1123/japa.15.3.336 [PubMed]
- 294. Delmonico MJ, Harris TB, Visser M, Park SW, Conroy MB, Velasquez-Mieyer P, Boudreau R, Manini TM, Nevitt M, Newman AB, Goodpaster BH, and Health, Aging, and Body. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am J Clin Nutr. 2009; 90:1579–85. https://doi.org/10.3945/ajcn.2009.28047 [PubMed]
- 295. Goodpaster BH, Kelley DE, Thaete FL, He J, Ross R. Skeletal muscle attenuation determined by computed tomography is associated with skeletal muscle lipid content. J Appl Physiol (1985). 2000; 89:104–10. https://doi.org/10.1152/jappl.2000.89.1.104 [PubMed]
- 296. Rahemi H, Nigam N, Wakeling JM. The effect of intramuscular fat on skeletal muscle mechanics: implications for the elderly and obese. J R Soc Interface. 2015; 12:20150365. https://doi.org/10.1098/rsif.2015.0365 [PubMed]
- 297. Kaya RD, Nakazawa M, Hoffman RL, Clark BC. Interrelationship between muscle strength, motor units, and aging. Exp Gerontol. 2013; 48:920–5. https://doi.org/10.1016/j.exger.2013.06.008 [PubMed]
- 298. McNeil CJ, Doherty TJ, Stashuk DW, Rice CL. Motor unit number estimates in the tibialis anterior muscle of young, old, and very old men. Muscle Nerve. 2005; 31:461–7. https://doi.org/10.1002/mus.20276 [PubMed]
- 299. Dalton BH, McNeil CJ, Doherty TJ, Rice CL. Age-related reductions in the estimated numbers of motor units are minimal in the human soleus. Muscle Nerve. 2008; 38:1108–15. https://doi.org/10.1002/mus.20984 [PubMed]
- 300. Zwetsloot KA, Childs TE, Gilpin LT, Booth FW. Non-passaged muscle precursor cells from 32-month old rat skeletal muscle have delayed proliferation and differentiation. Cell Prolif. 2013; 46:45–57. https://doi.org/10.1111/cpr.12007 [PubMed]
- 301. García-Prat L, Sousa-Victor P, Muñoz-Cánoves P. Functional dysregulation of stem cells during aging: a focus on skeletal muscle stem cells. FEBS J. 2013; 280:4051–62. https://doi.org/10.1111/febs.12221 [PubMed]
- 302. Renault V, Thornell LE, Eriksson PO, Butler-Browne G, Mouly V. Regenerative potential of human skeletal muscle during aging. Aging Cell. 2002; 1:132–9. https://doi.org/10.1046/j.1474-9728.2002.00017.x [PubMed]
- 303. Day K, Shefer G, Shearer A, Yablonka-Reuveni Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev Biol. 2010; 340:330–43. https://doi.org/10.1016/j.ydbio.2010.01.006 [PubMed]
- 304. Shefer G, Van de Mark DP, Richardson JB, Yablonka-Reuveni Z. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Dev Biol. 2006; 294:50–66. https://doi.org/10.1016/j.ydbio.2006.02.022 [PubMed]
- 305. Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007; 25:885–94. https://doi.org/10.1634/stemcells.2006-0372 [PubMed]
- 306. Brack AS, Bildsoe H, Hughes SM. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J Cell Sci. 2005; 118:4813–21. https://doi.org/10.1242/jcs.02602 [PubMed]
- 307. Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003; 302:1575–7. https://doi.org/10.1126/science.1087573 [PubMed]
- 308. Chargé SB, Brack AS, Hughes SM. Aging-related satellite cell differentiation defect occurs prematurely after Ski-induced muscle hypertrophy. Am J Physiol Cell Physiol. 2002; 283:C1228–41. https://doi.org/10.1152/ajpcell.00206.2002 [PubMed]
- 309. Taylor-Jones JM, McGehee RE, Rando TA, Lecka-Czernik B, Lipschitz DA, Peterson CA. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev. 2002; 123:649–61. https://doi.org/10.1016/s0047-6374(01)00411-0 [PubMed]
- 310. Sanna M, Franzin C, Pozzobon M, Favaretto F, Rossi CA, Calcagno A, Scarda A, Dal Prà C, Pilon C, Milan G, Federspil G, De Coppi P, Vettor R. Adipogenic potential of skeletal muscle satellite cells. Clinical Lipidology. 2009; 4:245–65. https://doi.org/10.2217/clp.09.8
- 311. Shefer G, Wleklinski-Lee M, Yablonka-Reuveni Z. Skeletal muscle satellite cells can spontaneously enter an alternative mesenchymal pathway. J Cell Sci. 2004; 117:5393–404. https://doi.org/10.1242/jcs.01419 [PubMed]
- 312. Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010; 12:153–63. https://doi.org/10.1038/ncb2015 [PubMed]
- 313. Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010; 12:143–52. https://doi.org/10.1038/ncb2014 [PubMed]
- 314. Parker E, Hamrick MW. Role of fibro-adipogenic progenitor cells in muscle atrophy and musculoskeletal diseases. Curr Opin Pharmacol. 2021; 58:1–7. https://doi.org/10.1016/j.coph.2021.03.003 [PubMed]
- 315. Cui CY, Driscoll RK, Piao Y, Chia CW, Gorospe M, Ferrucci L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell. 2019; 18:e13032. https://doi.org/10.1111/acel.13032 [PubMed]
- 316. Wang Y, Wehling-Henricks M, Samengo G, Tidball JG. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell. 2015; 14:678–88. https://doi.org/10.1111/acel.12350 [PubMed]
- 317. Przybyla B, Gurley C, Harvey JF, Bearden E, Kortebein P, Evans WJ, Sullivan DH, Peterson CA, Dennis RA. Aging alters macrophage properties in human skeletal muscle both at rest and in response to acute resistance exercise. Exp Gerontol. 2006; 41:320–7. https://doi.org/10.1016/j.exger.2005.12.007 [PubMed]
- 318. Wang H, Listrat A, Meunier B, Gueugneau M, Coudy-Gandilhon C, Combaret L, Taillandier D, Polge C, Attaix D, Lethias C, Lee K, Goh KL, Béchet D. Apoptosis in capillary endothelial cells in ageing skeletal muscle. Aging Cell. 2014; 13:254–62. https://doi.org/10.1111/acel.12169 [PubMed]
- 319. Petersen KF, Morino K, Alves TC, Kibbey RG, Dufour S, Sono S, Yoo PS, Cline GW, Shulman GI. Effect of aging on muscle mitochondrial substrate utilization in humans. Proc Natl Acad Sci U S A. 2015; 112:11330–4. https://doi.org/10.1073/pnas.1514844112 [PubMed]
- 320. Meneilly GS, Elliott T, Tessier D, Hards L, Tildesley H. NIDDM in the elderly. Diabetes Care. 1996; 19:1320–5. https://doi.org/10.2337/diacare.19.12.1320 [PubMed]
- 321. Petersen KF, Shulman GI. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am J Cardiol. 2002; 90:11G–8G. https://doi.org/10.1016/s0002-9149(02)02554-7 [PubMed]
- 322. Shou J, Chen PJ, Xiao WH. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol Metab Syndr. 2020; 12:14. https://doi.org/10.1186/s13098-020-0523-x [PubMed]
- 323. Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Targher G, Alberiche M, Bonadonna RC, Muggeo M. Prevalence of insulin resistance in metabolic disorders: the Bruneck Study. Diabetes. 1998; 47:1643–9. https://doi.org/10.2337/diabetes.47.10.1643 [PubMed]
- 324. Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, Papademetris X, Rothman DL, Shulman GI. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007; 104:12587–94. https://doi.org/10.1073/pnas.0705408104 [PubMed]
- 325. Dela F, Kjaer M. Resistance training, insulin sensitivity and muscle function in the elderly. Essays Biochem. 2006; 42:75–88. https://doi.org/10.1042/bse0420075 [PubMed]
- 326. Buckingham M. Myogenic progenitor cells and skeletal myogenesis in vertebrates. Curr Opin Genet Dev. 2006; 16:525–32. https://doi.org/10.1016/j.gde.2006.08.008 [PubMed]
- 327. Ryall JG, Schertzer JD, Lynch GS. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology. 2008; 9:213–28. https://doi.org/10.1007/s10522-008-9131-0 [PubMed]
- 328. Bigot A, Jacquemin V, Debacq-Chainiaux F, Butler-Browne GS, Toussaint O, Furling D, Mouly V. Replicative aging down-regulates the myogenic regulatory factors in human myoblasts. Biol Cell. 2008; 100:189–99. https://doi.org/10.1042/BC20070085 [PubMed]
- 329. Pietrangelo T, Puglielli C, Mancinelli R, Beccafico S, Fanò G, Fulle S. Molecular basis of the myogenic profile of aged human skeletal muscle satellite cells during differentiation. Exp Gerontol. 2009; 44:523–31. https://doi.org/10.1016/j.exger.2009.05.002 [PubMed]
- 330. Dalbo VJ, Roberts MD, Sunderland KL, Poole CN, Stout JR, Beck TW, Bemben M, Kerksick CM. Acute loading and aging effects on myostatin pathway biomarkers in human skeletal muscle after three sequential bouts of resistance exercise. J Gerontol A Biol Sci Med Sci. 2011; 66:855–65. https://doi.org/10.1093/gerona/glr091 [PubMed]
- 331. Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, Ketelslegers JM, Thissen JP. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology. 2007; 148:452–60. https://doi.org/10.1210/en.2006-0539 [PubMed]
- 332. Ma K, Mallidis C, Bhasin S, Mahabadi V, Artaza J, Gonzalez-Cadavid N, Arias J, Salehian B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab. 2003; 285:E363–71. https://doi.org/10.1152/ajpendo.00487.2002 [PubMed]
- 333. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997; 387:83–90. https://doi.org/10.1038/387083a0 [PubMed]
- 334. Yarasheski KE, Bhasin S, Sinha-Hikim I, Pak-Loduca J, Gonzalez-Cadavid NF. Serum myostatin-immunoreactive protein is increased in 60-92 year old women and men with muscle wasting. J Nutr Health Aging. 2002; 6:343–8. [PubMed]
- 335. Morissette MR, Stricker JC, Rosenberg MA, Buranasombati C, Levitan EB, Mittleman MA, Rosenzweig A. Effects of myostatin deletion in aging mice. Aging Cell. 2009; 8:573–83. https://doi.org/10.1111/j.1474-9726.2009.00508.x [PubMed]
- 336. Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, Kandarian SC. Identification of a molecular signature of sarcopenia. Physiol Genomics. 2005; 21:253–63. https://doi.org/10.1152/physiolgenomics.00249.2004 [PubMed]
- 337. Brzeszczyńska J, Johns N, Schilb A, Degen S, Degen M, Langen R, Schols A, Glass DJ, Roubenoff R, Greig CA, Jacobi C, Fearon KCH, Ross JA. Loss of oxidative defense and potential blockade of satellite cell maturation in the skeletal muscle of patients with cancer but not in the healthy elderly. Aging (Albany NY). 2016; 8:1690–702. https://doi.org/10.18632/aging.101006 [PubMed]
- 338. Joseph AM, Adhihetty PJ, Leeuwenburgh C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J Physiol. 2016; 594:5105–23. https://doi.org/10.1113/JP270659 [PubMed]
- 339. Gomes MJ, Martinez PF, Pagan LU, Damatto RL, Cezar MDM, Lima ARR, Okoshi K, Okoshi MP. Skeletal muscle aging: influence of oxidative stress and physical exercise. Oncotarget. 2017; 8:20428–40. https://doi.org/10.18632/oncotarget.14670 [PubMed]
- 340. Calvani R, Joseph AM, Adhihetty PJ, Miccheli A, Bossola M, Leeuwenburgh C, Bernabei R, Marzetti E. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem. 2013; 394:393–414. https://doi.org/10.1515/hsz-2012-0247 [PubMed]
- 341. Gouspillou G, Bourdel-Marchasson I, Rouland R, Calmettes G, Biran M, Deschodt-Arsac V, Miraux S, Thiaudiere E, Pasdois P, Detaille D, Franconi JM, Babot M, Trézéguet V, et al. Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell. 2014; 13:39–48. https://doi.org/10.1111/acel.12147 [PubMed]
- 342. Carter HN, Chen CC, Hood DA. Mitochondria, muscle health, and exercise with advancing age. Physiology (Bethesda). 2015; 30:208–23. https://doi.org/10.1152/physiol.00039.2014 [PubMed]
- 343. Gianni P, Jan KJ, Douglas MJ, Stuart PM, Tarnopolsky MA. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp Gerontol. 2004; 39:1391–400. https://doi.org/10.1016/j.exger.2004.06.002 [PubMed]
- 344. Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005; 102:5618–23. https://doi.org/10.1073/pnas.0501559102 [PubMed]
- 345. Tanhauser SM, Laipis PJ. Multiple deletions are detectable in mitochondrial DNA of aging mice. J Biol Chem. 1995; 270:24769–75. https://doi.org/10.1074/jbc.270.42.24769 [PubMed]
- 346. Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006; 79:469–80. https://doi.org/10.1086/507132 [PubMed]
- 347. Ferri E, Marzetti E, Calvani R, Picca A, Cesari M, Arosio B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int J Mol Sci. 2020; 21:5236. https://doi.org/10.3390/ijms21155236 [PubMed]
- 348. Cohick WS, Clemmons DR. The insulin-like growth factors. Annu Rev Physiol. 1993; 55:131–53. https://doi.org/10.1146/annurev.ph.55.030193.001023 [PubMed]
- 349. Sjögren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Törnell J, Isaksson OG, Jansson JO, Ohlsson C. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U S A. 1999; 96:7088–92. https://doi.org/10.1073/pnas.96.12.7088 [PubMed]
- 350. Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999; 96:7324–9. https://doi.org/10.1073/pnas.96.13.7324 [PubMed]
- 351. Barclay RD, Burd NA, Tyler C, Tillin NA, Mackenzie RW. The Role of the IGF-1 Signaling Cascade in Muscle Protein Synthesis and Anabolic Resistance in Aging Skeletal Muscle. Front Nutr. 2019; 6:146. https://doi.org/10.3389/fnut.2019.00146
- 352. Huffman DM, Farias Quipildor G, Mao K, Zhang X, Wan J, Apontes P, Cohen P, Barzilai N. Central insulin-like growth factor-1 (IGF-1) restores whole-body insulin action in a model of age-related insulin resistance and IGF-1 decline. Aging Cell. 2016; 15:181–6. https://doi.org/10.1111/acel.12415 [PubMed]
- 353. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001; 3:1009–13. https://doi.org/10.1038/ncb1101-1009 [PubMed]
- 354. Owino V, Yang SY, Goldspink G. Age-related loss of skeletal muscle function and the inability to express the autocrine form of insulin-like growth factor-1 (MGF) in response to mechanical overload. FEBS Lett. 2001; 505:259–63. https://doi.org/10.1016/s0014-5793(01)02825-3 [PubMed]
- 355. Dirks AJ, Leeuwenburgh C. Tumor necrosis factor alpha signaling in skeletal muscle: effects of age and caloric restriction. J Nutr Biochem. 2006; 17:501–8. https://doi.org/10.1016/j.jnutbio.2005.11.002 [PubMed]
- 356. Roubenoff R. Catabolism of aging: is it an inflammatory process? Curr Opin Clin Nutr Metab Care. 2003; 6:295–9. https://doi.org/10.1097/01.mco.0000068965.34812.62 [PubMed]
- 357. Haddad F, Zaldivar F, Cooper DM, Adams GR. IL-6-induced skeletal muscle atrophy. J Appl Physiol (1985). 2005; 98:911–7. https://doi.org/10.1152/japplphysiol.01026.2004 [PubMed]
- 358. Visser M, Pahor M, Taaffe DR, Goodpaster BH, Simonsick EM, Newman AB, Nevitt M, Harris TB. Relationship of interleukin-6 and tumor necrosis factor-alpha with muscle mass and muscle strength in elderly men and women: the Health ABC Study. J Gerontol A Biol Sci Med Sci. 2002; 57:M326–32. https://doi.org/10.1093/gerona/57.5.m326 [PubMed]
- 359. Belizário JE, Fontes-Oliveira CC, Borges JP, Kashiabara JA, Vannier E. Skeletal muscle wasting and renewal: a pivotal role of myokine IL-6. Springerplus. 2016; 5:619. https://doi.org/10.1186/s40064-016-2197-2 [PubMed]
- 360. Pedersen M, Steensberg A, Keller C, Osada T, Zacho M, Saltin B, Febbraio MA, Pedersen BK. Does the aging skeletal muscle maintain its endocrine function? Exerc Immunol Rev. 2004; 10:42–55. [PubMed]
- 361. Dubois V, Laurent M, Boonen S, Vanderschueren D, Claessens F. Androgens and skeletal muscle: cellular and molecular action mechanisms underlying the anabolic actions. Cell Mol Life Sci. 2012; 69:1651–67. https://doi.org/10.1007/s00018-011-0883-3 [PubMed]
- 362. Herbst KL, Bhasin S. Testosterone action on skeletal muscle. Curr Opin Clin Nutr Metab Care. 2004; 7:271–7. https://doi.org/10.1097/00075197-200405000-00006 [PubMed]
- 363. Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. Am J Physiol Endocrinol Metab. 2002; 283:E154–64. https://doi.org/10.1152/ajpendo.00502.2001 [PubMed]
- 364. Vermeulen A, Goemaere S, Kaufman JM. Testosterone, body composition and aging. J Endocrinol Invest. 1999; 22:110–6. [PubMed]
- 365. Allan CA, Strauss BJ, Burger HG, Forbes EA, McLachlan RI. Testosterone therapy prevents gain in visceral adipose tissue and loss of skeletal muscle in nonobese aging men. J Clin Endocrinol Metab. 2008; 93:139–46. https://doi.org/10.1210/jc.2007-1291 [PubMed]
- 366. Morley JE, Kaiser F, Raum WJ, Perry HM
3rd , Flood JF, Jensen J, Silver AJ, Roberts E. Potentially predictive and manipulable blood serum correlates of aging in the healthy human male: progressive decreases in bioavailable testosterone, dehydroepiandrosterone sulfate, and the ratio of insulin-like growth factor 1 to growth hormone. Proc Natl Acad Sci U S A. 1997; 94:7537–42. https://doi.org/10.1073/pnas.94.14.7537 [PubMed] - 367. Khosla S, Melton LJ
3rd , Atkinson EJ, O'Fallon WM, Klee GG, Riggs BL. Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J Clin Endocrinol Metab. 1998; 83:2266–74. https://doi.org/10.1210/jcem.83.7.4924 [PubMed] - 368. Bloise FF, Oliveira TS, Cordeiro A, Ortiga-Carvalho TM. Thyroid Hormones Play Role in Sarcopenia and Myopathies. Front Physiol. 2018; 9:560. https://doi.org/10.3389/fphys.2018.00560
- 369. Bloise FF, Cordeiro A, Ortiga-Carvalho TM. Role of thyroid hormone in skeletal muscle physiology. J Endocrinol. 2018; 236:R57–68. https://doi.org/10.1530/JOE-16-0611 [PubMed]
- 370. da Costa VM, Moreira DG, Rosenthal D. Thyroid function and aging: gender-related differences. J Endocrinol. 2001; 171:193–8. https://doi.org/10.1677/joe.0.1710193 [PubMed]
- 371. Simonides WS, van Hardeveld C. Thyroid hormone as a determinant of metabolic and contractile phenotype of skeletal muscle. Thyroid. 2008; 18:205–16. https://doi.org/10.1089/thy.2007.0256 [PubMed]
- 372. Salvatore D, Simonides WS, Dentice M, Zavacki AM, Larsen PR. Thyroid hormones and skeletal muscle--new insights and potential implications. Nat Rev Endocrinol. 2014; 10:206–14. https://doi.org/10.1038/nrendo.2013.238 [PubMed]
- 373. Vandenburgh HH, Karlisch P, Farr L. Maintenance of highly contractile tissue-cultured avian skeletal myotubes in collagen gel. In Vitro Cell Dev Biol. 1988; 24:166–74. https://doi.org/10.1007/BF02623542 [PubMed]
- 374. Chromiak JA, Shansky J, Perrone C, Vandenburgh HH. Bioreactor perfusion system for the long-term maintenance of tissue-engineered skeletal muscle organoids. In Vitro Cell Dev Biol Anim. 1998; 34:694–703. https://doi.org/10.1007/s11626-998-0065-2 [PubMed]
- 375. Prüller J, Mannhardt I, Eschenhagen T, Zammit PS, Figeac N. Satellite cells delivered in their niche efficiently generate functional myotubes in three-dimensional cell culture. PLoS One. 2018; 13:e0202574. https://doi.org/10.1371/journal.pone.0202574 [PubMed]
- 376. van der Schaft DW, van Spreeuwel AC, Boonen KJ, Langelaan ML, Bouten CV, Baaijens FP. Engineering skeletal muscle tissues from murine myoblast progenitor cells and application of electrical stimulation. J Vis Exp. 2013; e4267. https://doi.org/10.3791/4267 [PubMed]
- 377. Kosnik PE, Faulkner JA, Dennis RG. Functional development of engineered skeletal muscle from adult and neonatal rats. Tissue Eng. 2001; 7:573–84. https://doi.org/10.1089/107632701753213192 [PubMed]
- 378. Dennis RG, Kosnik PE
2nd . Excitability and isometric contractile properties of mammalian skeletal muscle constructs engineered in vitro. In Vitro Cell Dev Biol Anim. 2000; 36:327–35. https://doi.org/10.1290/1071-2690(2000)036<0327:EAICPO>2.0.CO;2 [PubMed] - 379. Saxena AK, Marler J, Benvenuto M, Willital GH, Vacanti JP. Skeletal muscle tissue engineering using isolated myoblasts on synthetic biodegradable polymers: preliminary studies. Tissue Eng. 1999; 5:525–32. https://doi.org/10.1089/ten.1999.5.525 [PubMed]
- 380. Dennis RG, Kosnik PE
2nd , Gilbert ME, Faulkner JA. Excitability and contractility of skeletal muscle engineered from primary cultures and cell lines. Am J Physiol Cell Physiol. 2001; 280:C288–95. https://doi.org/10.1152/ajpcell.2001.280.2.C288 [PubMed] - 381. Koning M, Werker PM, van der Schaft DW, Bank RA, Harmsen MC. MicroRNA-1 and microRNA-206 improve differentiation potential of human satellite cells: a novel approach for tissue engineering of skeletal muscle. Tissue Eng Part A. 2012; 18:889–98. https://doi.org/10.1089/ten.TEA.2011.0191 [PubMed]
- 382. Pantelic MN, Larkin LM. Stem Cells for Skeletal Muscle Tissue Engineering. Tissue Eng Part B Rev. 2018; 24:373–91. https://doi.org/10.1089/ten.TEB.2017.0451 [PubMed]
- 383. Scime A, Caron AZ, Grenier G. Advances in myogenic cell transplantation and skeletal muscle tissue engineering. Front Biosci (Landmark Ed). 2009; 14:3012–23. https://doi.org/10.2741/3431 [PubMed]
- 384. Koffler J, Kaufman-Francis K, Shandalov Y, Egozi D, Pavlov DA, Landesberg A, Levenberg S. Improved vascular organization enhances functional integration of engineered skeletal muscle grafts. Proc Natl Acad Sci U S A. 2011; 108:14789–94. https://doi.org/10.1073/pnas.1017825108 [PubMed]
- 385. Okano T, Matsuda T. Tissue engineered skeletal muscle: preparation of highly dense, highly oriented hybrid muscular tissues. Cell Transplant. 1998; 7:71–82. https://doi.org/10.1177/096368979800700110 [PubMed]
- 386. Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977; 270:725–7. https://doi.org/10.1038/270725a0 [PubMed]
- 387. Zhang M, Guo B. Electroactive 3D Scaffolds Based on Silk Fibroin and Water-Borne Polyaniline for Skeletal Muscle Tissue Engineering. Macromol Biosci. 2017; 17:1700147. https://doi.org/10.1002/mabi.201700147 [PubMed]
- 388. Donnelly K, Khodabukus A, Philp A, Deldicque L, Dennis RG, Baar K. A novel bioreactor for stimulating skeletal muscle in vitro. Tissue Eng Part C Methods. 2010; 16:711–8. https://doi.org/10.1089/ten.TEC.2009.0125 [PubMed]
- 389. Dennis RG, Dow DE. Excitability of skeletal muscle during development, denervation, and tissue culture. Tissue Eng. 2007; 13:2395–404. https://doi.org/10.1089/ten.2006.0367 [PubMed]
- 390. Duffy RM, Sun Y, Feinberg AW. Understanding the Role of ECM Protein Composition and Geometric Micropatterning for Engineering Human Skeletal Muscle. Ann Biomed Eng. 2016; 44:2076–89. https://doi.org/10.1007/s10439-016-1592-8 [PubMed]
- 391. Hindi L, McMillan JD, Afroze D, Hindi SM, Kumar A. Isolation, Culturing, and Differentiation of Primary Myoblasts from Skeletal Muscle of Adult Mice. Bio Protoc. 2017; 7:e2248. https://doi.org/10.21769/BioProtoc.2248 [PubMed]
- 392. Darabi R, Pan W, Bosnakovski D, Baik J, Kyba M, Perlingeiro RC. Functional myogenic engraftment from mouse iPS cells. Stem Cell Rev Rep. 2011; 7:948–57. https://doi.org/10.1007/s12015-011-9258-2 [PubMed]
- 393. Langridge B, Griffin M, Butler PE. Regenerative medicine for skeletal muscle loss: a review of current tissue engineering approaches. J Mater Sci Mater Med. 2021; 32:15. https://doi.org/10.1007/s10856-020-06476-5 [PubMed]
- 394. McCullagh KJ, Perlingeiro RC. Coaxing stem cells for skeletal muscle repair. Adv Drug Deliv Rev. 2015; 84:198–207. https://doi.org/10.1016/j.addr.2014.07.007 [PubMed]
- 395. Rao L, Qian Y, Khodabukus A, Ribar T, Bursac N. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat Commun. 2018; 9:126. https://doi.org/10.1038/s41467-017-02636-4 [PubMed]
- 396. Wang J, Khodabukus A, Rao L, Vandusen K, Abutaleb N, Bursac N. Engineered skeletal muscles for disease modeling and drug discovery. Biomaterials. 2019; 221:119416. https://doi.org/10.1016/j.biomaterials.2019.119416 [PubMed]
- 397. van der Wal E, Herrero-Hernandez P, Wan R, Broeders M, In't Groen SLM, van Gestel TJM, van IJcken WFJ, Cheung TH, van der Ploeg AT, Schaaf GJ, Pijnappel WWM. Large-Scale Expansion of Human iPSC-Derived Skeletal Muscle Cells for Disease Modeling and Cell-Based Therapeutic Strategies. Stem Cell Reports. 2018; 10:1975–90. https://doi.org/10.1016/j.stemcr.2018.04.002 [PubMed]
- 398. Chen Z, Li B, Zhan RZ, Rao L, Bursac N. Exercise mimetics and JAK inhibition attenuate IFN-γ-induced wasting in engineered human skeletal muscle. Sci Adv. 2021; 7:eabd9502. https://doi.org/10.1126/sciadv.abd9502 [PubMed]
- 399. Khodabukus A, Madden L, Prabhu NK, Koves TR, Jackman CP, Muoio DM, Bursac N. Electrical stimulation increases hypertrophy and metabolic flux in tissue-engineered human skeletal muscle. Biomaterials. 2019; 198:259–69. https://doi.org/10.1016/j.biomaterials.2018.08.058 [PubMed]
- 400. Khodabukus A, Kaza A, Wang J, Prabhu N, Goldstein R, Vaidya VS, Bursac N. Tissue-Engineered Human Myobundle System as a Platform for Evaluation of Skeletal Muscle Injury Biomarkers. Toxicol Sci. 2020; 176:124–36. https://doi.org/10.1093/toxsci/kfaa049 [PubMed]
- 401. Lee PH, Vandenburgh HH. Skeletal muscle atrophy in bioengineered skeletal muscle: a new model system. Tissue Eng Part A. 2013; 19:2147–55. https://doi.org/10.1089/ten.TEA.2012.0597 [PubMed]
- 402. Juhas M, Engelmayr GC
Jr , Fontanella AN, Palmer GM, Bursac N. Biomimetic engineered muscle with capacity for vascular integration and functional maturation in vivo. Proc Natl Acad Sci U S A. 2014; 111:5508–13. https://doi.org/10.1073/pnas.1402723111 [PubMed] - 403. Tiburcy M, Markov A, Kraemer LK, Christalla P, Rave-Fraenk M, Fischer HJ, Reichardt HM, Zimmermann WH. Regeneration competent satellite cell niches in rat engineered skeletal muscle. FASEB Bioadv. 2019; 1:731–46. https://doi.org/10.1096/fba.2019-00013 [PubMed]
- 404. Bian W, Juhas M, Pfeiler TW, Bursac N. Local tissue geometry determines contractile force generation of engineered muscle networks. Tissue Eng Part A. 2012; 18:957–67. https://doi.org/10.1089/ten.TEA.2011.0313 [PubMed]
- 405. Cheng CS, Davis BN, Madden L, Bursac N, Truskey GA. Physiology and metabolism of tissue-engineered skeletal muscle. Exp Biol Med (Maywood). 2014; 239:1203–14. https://doi.org/10.1177/1535370214538589 [PubMed]
- 406. Moon du G, Christ G, Stitzel JD, Atala A, Yoo JJ. Cyclic mechanical preconditioning improves engineered muscle contraction. Tissue Eng Part A. 2008; 14:473–82. https://doi.org/10.1089/tea.2007.0104 [PubMed]
- 407. Sato M, Ito A, Kawabe Y, Nagamori E, Kamihira M. Enhanced contractile force generation by artificial skeletal muscle tissues using IGF-I gene-engineered myoblast cells. J Biosci Bioeng. 2011; 112:273–8. https://doi.org/10.1016/j.jbiosc.2011.05.007 [PubMed]
- 408. Martin NRW, Turner MC, Farrington R, Player DJ, Lewis MP. Leucine elicits myotube hypertrophy and enhances maximal contractile force in tissue engineered skeletal muscle in vitro. J Cell Physiol. 2017; 232:2788–97. https://doi.org/10.1002/jcp.25960 [PubMed]
- 409. Nakamura T, Takagi S, Okuzaki D, Matsui S, Fujisato T. Hypoxia transactivates cholecystokinin gene expression in 3D-engineered muscle. J Biosci Bioeng. 2021; 132:64–70. https://doi.org/10.1016/j.jbiosc.2021.03.006 [PubMed]
- 410. Osaki T, Sivathanu V, Kamm RD. Crosstalk between developing vasculature and optogenetically engineered skeletal muscle improves muscle contraction and angiogenesis. Biomaterials. 2018; 156:65–76. https://doi.org/10.1016/j.biomaterials.2017.11.041 [PubMed]
- 411. Davis BN, Santoso JW, Walker MJ, Cheng CS, Koves TR, Kraus WE, Truskey GA. Human, Tissue-Engineered, Skeletal Muscle Myobundles to Measure Oxygen Uptake and Assess Mitochondrial Toxicity. Tissue Eng Part C Methods. 2017; 23:189–99. https://doi.org/10.1089/ten.tec.2016.0264 [PubMed]
- 412. Madden L, Juhas M, Kraus WE, Truskey GA, Bursac N. Bioengineered human myobundles mimic clinical responses of skeletal muscle to drugs. Elife. 2015; 4:e04885. https://doi.org/10.7554/eLife.04885 [PubMed]
- 413. Vandenburgh H. High-content drug screening with engineered musculoskeletal tissues. Tissue Eng Part B Rev. 2010; 16:55–64. https://doi.org/10.1089/ten.TEB.2009.0445 [PubMed]
- 414. Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L, Valentini R, Crawford G. Drug-screening platform based on the contractility of tissue-engineered muscle. Muscle Nerve. 2008; 37:438–47. https://doi.org/10.1002/mus.20931 [PubMed]
- 415. Baker EL, Dennis RG, Larkin LM. Glucose transporter content and glucose uptake in skeletal muscle constructs engineered in vitro. In Vitro Cell Dev Biol Anim. 2003; 39:434–9. https://doi.org/10.1290/1543-706X(2003)039<0434:GTCAGU>2.0.CO;2 [PubMed]
- 416. Strohman RC, Bayne E, Spector D, Obinata T, Micou-Eastwood J, Maniotis A. Myogenesis and histogenesis of skeletal muscle on flexible membranes in vitro. In Vitro Cell Dev Biol. 1990; 26:201–8. https://doi.org/10.1007/BF02624113 [PubMed]
- 417. Carosio S, Barberi L, Rizzuto E, Nicoletti C, Del Prete Z, Musarò A. Generation of eX vivo-vascularized Muscle Engineered Tissue (X-MET). Sci Rep. 2013; 3:1420. https://doi.org/10.1038/srep01420 [PubMed]
- 418. Mertens JP, Sugg KB, Lee JD, Larkin LM. Engineering muscle constructs for the creation of functional engineered musculoskeletal tissue. Regen Med. 2014; 9:89–100. https://doi.org/10.2217/rme.13.81 [PubMed]
- 419. Ostrovidov S, Salehi S, Costantini M, Suthiwanich K, Ebrahimi M, Sadeghian RB, Fujie T, Shi X, Cannata S, Gargioli C, Tamayol A, Dokmeci MR, Orive G, et al. 3D Bioprinting in Skeletal Muscle Tissue Engineering. Small. 2019; 15:e1805530. https://doi.org/10.1002/smll.201805530 [PubMed]
- 420. Zhuang P, An J, Chua CK, Tan LP. Bioprinting of 3D in vitro skeletal muscle models: A review. Materials & Design. 2020; 193:108794. https://doi.org/10.1016/j.matdes.2020.108794
- 421. Kaushik G, Leijten J, Khademhosseini A. Concise Review: Organ Engineering: Design, Technology, and Integration. Stem Cells. 2017; 35:51–60. https://doi.org/10.1002/stem.2502 [PubMed]
- 422. Sears NA, Seshadri DR, Dhavalikar PS, Cosgriff-Hernandez E. A Review of Three-Dimensional Printing in Tissue Engineering. Tissue Eng Part B Rev. 2016; 22:298–310. https://doi.org/10.1089/ten.TEB.2015.0464 [PubMed]
- 423. Lee VK, Dai G. Printing of Three-Dimensional Tissue Analogs for Regenerative Medicine. Ann Biomed Eng. 2017; 45:115–31. https://doi.org/10.1007/s10439-016-1613-7 [PubMed]
- 424. Agrawal G, Aung A, Varghese S. Skeletal muscle-on-a-chip: an in vitro model to evaluate tissue formation and injury. Lab Chip. 2017; 17:3447–61. https://doi.org/10.1039/c7lc00512a [PubMed]
- 425. Anene-Nzelu CG, Peh KY, Fraiszudeen A, Kuan YH, Ng SH, Toh YC, Leo HL, Yu H. Scalable alignment of three-dimensional cellular constructs in a microfluidic chip. Lab Chip. 2013; 13:4124–33. https://doi.org/10.1039/c3lc50730k [PubMed]
- 426. Shimizu K, Araki H, Sakata K, Tonomura W, Hashida M, Konishi S. Microfluidic devices for construction of contractile skeletal muscle microtissues. J Biosci Bioeng. 2015; 119:212–6. https://doi.org/10.1016/j.jbiosc.2014.07.003 [PubMed]
- 427. Ortega MA, Fernández-Garibay X, Castaño AG, De Chiara F, Hernández-Albors A, Balaguer-Trias J, Ramón-Azcón J. Muscle-on-a-chip with an on-site multiplexed biosensing system for in situ monitoring of secreted IL-6 and TNF-α. Lab Chip. 2019; 19:2568–80. https://doi.org/10.1039/c9lc00285e [PubMed]
- 428. Chen S, Kawazoe N, Chen G. Biomimetic Assembly of Vascular Endothelial Cells and Muscle Cells in Microgrooved Collagen Porous Scaffolds. Tissue Eng Part C Methods. 2017; 23:367–76. https://doi.org/10.1089/ten.TEC.2017.0088 [PubMed]
- 429. Gholobova D, Decroix L, Van Muylder V, Desender L, Gerard M, Carpentier G, Vandenburgh H, Thorrez L. Endothelial Network Formation Within Human Tissue-Engineered Skeletal Muscle. Tissue Eng Part A. 2015; 21:2548–58. https://doi.org/10.1089/ten.TEA.2015.0093 [PubMed]
- 430. Gholobova D, Terrie L, Mackova K, Desender L, Carpentier G, Gerard M, Hympanova L, Deprest J, Thorrez L. Functional evaluation of prevascularization in one-stage versus two-stage tissue engineering approach of human bio-artificial muscle. Biofabrication. 2020; 12:035021. https://doi.org/10.1088/1758-5090/ab8f36 [PubMed]
- 431. Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, Marini R, van Blitterswijk CA, Mulligan RC, D'Amore PA, Langer R. Engineering vascularized skeletal muscle tissue. Nat Biotechnol. 2005; 23:879–84. https://doi.org/10.1038/nbt1109 [PubMed]
- 432. Perry L, Flugelman MY, Levenberg S. Elderly Patient-Derived Endothelial Cells for Vascularization of Engineered Muscle. Mol Ther. 2017; 25:935–48. https://doi.org/10.1016/j.ymthe.2017.02.011 [PubMed]
- 433. van der Schaft DW, van Spreeuwel AC, van Assen HC, Baaijens FP. Mechanoregulation of vascularization in aligned tissue-engineered muscle: a role for vascular endothelial growth factor. Tissue Eng Part A. 2011; 17:2857–65. https://doi.org/10.1089/ten.TEA.2011.0214 [PubMed]
- 434. Juhas M, Abutaleb N, Wang JT, Ye J, Shaikh Z, Sriworarat C, Qian Y, Bursac N. Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat Biomed Eng. 2018; 2:942–54. https://doi.org/10.1038/s41551-018-0290-2 [PubMed]
- 435. Hinds S, Bian W, Dennis RG, Bursac N. The role of extracellular matrix composition in structure and function of bioengineered skeletal muscle. Biomaterials. 2011; 32:3575–83. https://doi.org/10.1016/j.biomaterials.2011.01.062 [PubMed]
- 436. Kondash ME, Ananthakumar A, Khodabukus A, Bursac N, Truskey GA. Glucose Uptake and Insulin Response in Tissue-engineered Human Skeletal Muscle. Tissue Eng Regen Med. 2020; 17:801–13. https://doi.org/10.1007/s13770-020-00242-y [PubMed]
- 437. Tanaka A, Woltjen K, Miyake K, Hotta A, Ikeya M, Yamamoto T, Nishino T, Shoji E, Sehara-Fujisawa A, Manabe Y, Fujii N, Hanaoka K, Era T, et al. Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling Miyoshi Myopathy in vitro. PLoS One. 2013; 8:e61540. https://doi.org/10.1371/journal.pone.0061540 [PubMed]
- 438. Vandenburgh H, Shansky J, Benesch-Lee F, Skelly K, Spinazzola JM, Saponjian Y, Tseng BS. Automated drug screening with contractile muscle tissue engineered from dystrophic myoblasts. FASEB J. 2009; 23:3325–34. https://doi.org/10.1096/fj.09-134411 [PubMed]
- 439. Smith AST, Davis J, Lee G, Mack DL, Kim DH. Muscular dystrophy in a dish: engineered human skeletal muscle mimetics for disease modeling and drug discovery. Drug Discov Today. 2016; 21:1387–98. https://doi.org/10.1016/j.drudis.2016.04.013 [PubMed]
- 440. Osaki T, Uzel SGM, Kamm RD. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci Adv. 2018; 4:eaat5847. https://doi.org/10.1126/sciadv.aat5847 [PubMed]
- 441. Yoshida T, Awaya T, Jonouchi T, Kimura R, Kimura S, Era T, Heike T, Sakurai H. A Skeletal Muscle Model of Infantile-onset Pompe Disease with Patient-specific iPS Cells. Sci Rep. 2017; 7:13473. https://doi.org/10.1038/s41598-017-14063-y [PubMed]
- 442. Wang J, Zhou CJ, Khodabukus A, Tran S, Han SO, Carlson AL, Madden L, Kishnani PS, Koeberl DD, Bursac N. Three-dimensional tissue-engineered human skeletal muscle model of Pompe disease. Commun Biol. 2021; 4:524. https://doi.org/10.1038/s42003-021-02059-4 [PubMed]
- 443. Ebrahimi M, Lad H, Fusto A, Tiper Y, Datye A, Nguyen CT, Jacques E, Moyle LA, Nguyen T, Musgrave B, Chávez-Madero C, Bigot A, Chen C, et al. De novo revertant fiber formation and therapy testing in a 3D culture model of Duchenne muscular dystrophy skeletal muscle. Acta Biomater. 2021; 132:227–44. https://doi.org/10.1016/j.actbio.2021.05.020 [PubMed]
- 444. Maffioletti SM, Sarcar S, Henderson ABH, Mannhardt I, Pinton L, Moyle LA, Steele-Stallard H, Cappellari O, Wells KE, Ferrari G, Mitchell JS, Tyzack GE, Kotiadis VN, et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018; 23:899–08. https://doi.org/10.1016/j.celrep.2018.03.091 [PubMed]
- 445. Moyle LA, Jacques E, Gilbert PM. Engineering the next generation of human skeletal muscle models: From cellular complexity to disease modeling. Curr Opin Biomed Eng. 2020; 16:9–18. https://doi.org/10.1016/j.cobme.2020.05.006
- 446. Bersini S, Gilardi M, Ugolini GS, Sansoni V, Talò G, Perego S, Zanotti S, Ostano P, Mora M, Soncini M, Vanoni M, Lombardi G, Moretti M. Engineering an Environment for the Study of Fibrosis: A 3D Human Muscle Model with Endothelium Specificity and Endomysium. Cell Rep. 2018; 25:3858–68.e4. https://doi.org/10.1016/j.celrep.2018.11.092 [PubMed]
- 447. Iossa S, Mollica MP, Lionetti L, Crescenzo R, Tasso R, Liverini G. A possible link between skeletal muscle mitochondrial efficiency and age-induced insulin resistance. Diabetes. 2004; 53:2861–6. https://doi.org/10.2337/diabetes.53.11.2861 [PubMed]
- 448. Acosta FM, Jia UA, Stojkova K, Howland KK, Guda T, Pacelli S, Brey EM, Rathbone CR. Diabetic Conditions Confer Metabolic and Structural Modifications to Tissue-Engineered Skeletal Muscle. Tissue Eng Part A. 2021; 27:549–60. https://doi.org/10.1089/ten.TEA.2020.0138 [PubMed]
- 449. Sharples AP, Player DJ, Martin NR, Mudera V, Stewart CE, Lewis MP. Modelling in vivo skeletal muscle ageing in vitro using three-dimensional bioengineered constructs. Aging Cell. 2012; 11:986–95. https://doi.org/10.1111/j.1474-9726.2012.00869.x [PubMed]
- 450. Rajabian N, Shahini A, Asmani M, Vydiam K, Choudhury D, Nguyen T, Ikhapoh I, Zhao R, Lei P, Andreadis ST. Bioengineered Skeletal Muscle as a Model of Muscle Aging and Regeneration. Tissue Eng Part A. 2021; 27:74–86. https://doi.org/10.1089/ten.TEA.2020.0005 [PubMed]
- 451. Shahini A, Choudhury D, Asmani M, Zhao R, Lei P, Andreadis ST. NANOG restores the impaired myogenic differentiation potential of skeletal myoblasts after multiple population doublings. Stem Cell Res. 2018; 26:55–66. https://doi.org/10.1016/j.scr.2017.11.018 [PubMed]
- 452. Han J, Mistriotis P, Lei P, Wang D, Liu S, Andreadis ST. Nanog reverses the effects of organismal aging on mesenchymal stem cell proliferation and myogenic differentiation potential. Stem Cells. 2012; 30:2746–59. https://doi.org/10.1002/stem.1223 [PubMed]
- 453. Shahini A, Mistriotis P, Asmani M, Zhao R, Andreadis ST. NANOG Restores Contractility of Mesenchymal Stem Cell-Based Senescent Microtissues. Tissue Eng Part A. 2017; 23:535–45. https://doi.org/10.1089/ten.TEA.2016.0494 [PubMed]
- 454. Lee AS, Anderson JE, Joya JE, Head SI, Pather N, Kee AJ, Gunning PW, Hardeman EC. Aged skeletal muscle retains the ability to fully regenerate functional architecture. Bioarchitecture. 2013; 3:25–37. https://doi.org/10.4161/bioa.24966 [PubMed]
- 455. Lacraz G, Rouleau AJ, Couture V, Söllrald T, Drouin G, Veillette N, Grandbois M, Grenier G. Increased Stiffness in Aged Skeletal Muscle Impairs Muscle Progenitor Cell Proliferative Activity. PLoS One. 2015; 10:e0136217. https://doi.org/10.1371/journal.pone.0136217 [PubMed]
- 456. Snijders T, Nederveen JP, McKay BR, Joanisse S, Verdijk LB, van Loon LJC, Parise G. Satellite cells in human skeletal muscle plasticity. Front Physiol. 2015; 6:283. https://doi.org/10.3389/fphys.2015.00283 [PubMed]
- 457. Khodabukus A. Tissue-Engineered Skeletal Muscle Models to Study Muscle Function, Plasticity, and Disease. Front Physiol. 2021; 12:619710. https://doi.org/10.3389/fphys.2021.619710 [PubMed]
- 458. Khodabukus A, Prabhu N, Wang J, Bursac N. In Vitro Tissue-Engineered Skeletal Muscle Models for Studying Muscle Physiology and Disease. Adv Healthc Mater. 2018; 7:e1701498. https://doi.org/10.1002/adhm.201701498 [PubMed]
- 459. Anderson LJ, Liu H, Garcia JM. Sex Differences in Muscle Wasting. Adv Exp Med Biol. 2017; 1043:153–97. https://doi.org/10.1007/978-3-319-70178-3_9 [PubMed]
- 460. Brown M. Skeletal muscle and bone: effect of sex steroids and aging. Adv Physiol Educ. 2008; 32:120–6. https://doi.org/10.1152/advan.90111.2008 [PubMed]