



Introduction
Mutations of prion protein gene (PRNP) are associated with a group of inherited prion diseases that are characterized clinically by dementia, ataxia, and myoclonus and pathologically by spongiform de-generation, astrocytic gliosis, and neuronal loss [1]. Like sporadic and acquired forms of prion diseases, the molecular hallmark of inherited prion diseases is the deposition in the central nervous system (CNS) of an abnormal isoform of prion protein (PrPSc) that is derived from a host-encoded cellular prion protein (PrPC) via a structural transition from α-helices into β-sheet structures [2]. However, unlike the other two forms of the diseases, the conversion of PrPC into the pathological PrPSc in inherited prion diseases is believed to be spontaneously triggered by the mutated PrP allele (PrPM). The wild-type PrP allele (PrPWt) may or may not be recruited into PrPSc by the PrPM allele [3-10]. Regardless of distinct etiologies, the PrPSc molecules present in all human prion diseases share some common structural, physicochemical, and biological properties, including a β-sheet-rich structure, resistance to proteinase K (PK) digestion, insolubility in non-denaturing detergents, and infectivity [2]. It has been well-documented that the co-existence of PrPC and PK-resistant PrPSc (rPrPSc) is a prerequisite for the pathogenesis of various prion diseases; however, what type of PrPSc conformers are directly responsible for the PrP deposition in the brain and the neuropathological changes in the prion diseases remains poorly understood [11].
Phenotypes of inherited prion diseases are mainly determined by specific mutations and a polymorphism at codon 129 (methionine (M) and valine (V)) of PRNP [12]. Most point mutations of PRNP are associated with inherited conditions exhibiting phenotypes similar to the well-characterized sporadic Creutzfeldt-Jakob disease (sCJD) with a fairly rapid course, deposition of a typical rPrPSc (designated PrP27-30 including PrPSc type 1 or type 2) in CNS, and widespread of spongiform degeneration. However, a few point mutations and a non-sense mutation are linked to the phenotype of Gerstmann-Sträussler-Scheinker disease (GSS), characterized by a relatively chronic clinical course and the presence of intense amyloid plaques composed of unique rPrPSc fragments (designated PrP7-8) in the affected brains. A third set of inherited prion diseases are associated with insertions of one to nine octapeptide repeats, except for three that has never been reported [13-18]. These mutations are located in a nonapeptide (R1) and four octapeptides (R2 to R4) of the form P(H/Q)GGG(-/G)WGQ between residues 51 and 91 of PrPWt [19-20]. The phenotypic heterogeneity and allelic origin of PrPSc linked to insertion of 6 octapeptide repeats of PrP have been extensively characterized [21-25, 5, 26-29]. Although deposition of PrP in the cerebellum of affected brains is strikingly consistent, phenotypes, neuropathological changes, levels of rPrPSc and transmissibility between individuals are variable, at least the electrophoretic pattern of PrP. PrP species in the insertion cases with undetectable rPrPSc by the conventional assay [24] have not been further characterized. Addressing these important issues may shed light on the correlation between highly variable neuropathological changes and the chameleon-like conformations of PrPSc.
Here we examined brain PrP and neuropathological changes in six cases carrying the 144-bp PrP insertion mutation. Neuropathologically, these cases exhibited spongiform degeneration, astrocytosis and multicore plaques with or without neuronal loss although the severity of these changes differed between cases or between areas of the same brain. However, all but one consistently had the deposits of PrP patches orientated perpendicular to the pial surface in the molecular layer of the cerebellum. Surprisingly, one of the cases displaying the cerebellar PrP patches revealed virtually no brain rPrPSc that was well represented in the other five cases. In contrast, this variant case was associated with a large amount of PrP species that was PK-sensitive but was captured by reagents including gene 5 protein (g5p) and sodium phosphotungstate (NaPTA), proven to specifically bind to insoluble and aggregated PrP regardless of its PK resistance [30-33].
Results
Clinical information of the six subjects examined
Sixcases with 144-bp insertion mutation (fCJDIns) were collected between 2000 and 2006 at the NPDPSC (Table 1). All six cases were female with average age at onset of 38.5 ± 9.8 years and highly variable disease durations ranging between 3 and 180 months. Two cases were methionine/methionine (M/M) homozygous at residue 129 of PrP, three valine/valine (V/V) homozygous and one M/V heterozygous. The insertion mutant allele was coupled with the 129M in the M/V heterozygous subject. The octarepeat region in the six fCJDIns cases examined included two types of sequences (Table 1).
Table 1. Clinical features of fCJDIns
| Case # | Codon 129-mut. Allele | Insertion typea | PrPSc type | Age at onset (yrs) | Duration (months) | Sex | Symptoms at onset | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | VV-129V | 1 | 1 | 51 | 6 | F | Ataxia associated with cognitive decline. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | VV-129V | 2 | 1 | 33 | 180 | F | Cognitive decline (mostly dyscalculia and visuospatial deficit). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | MM-129M | 2 | 2 | 26 | 144 | F | Jerks and progressive cognitive decline characterized by apraxia and dyscalculia. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | MV-129M | 1 | 1 | 39 | 9 | F | N/A | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | VV-129V | 2 | 2 | 49 | 3 | F | Headache and fatigue; weeks later, sleep disturbance and ataxia. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | MM-129M | 1 | Noneb | 33 | 60 | F | Cognitive decline and headache. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Case # | Symptoms during the disease course | EEGc | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Parkinsonism | Ataxia | Myoclonus | Focal weakness | Psychosis | Seizure | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | −d | +e | + | − | − | − | No typical, diffuse slowing | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Not reported | not reported | not reported | − | + | − | No typical, diffuse slowing | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | − | − | + | − | − | − | N/Af | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | + | + | + | − | − | − | No typical, diffuse slowing | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | + | + | + | − | + | − | No typical, diffuse slowing | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Case # | MRI | Family history of dementia | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | No typical signs of prion disease; brain atrophy particularly in the cerebellum and brainstem. | Mother: died of Alzheimer's disease at age 72. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | No typical signs of prion disease; severe atrophy in the parietal occipital and cerebellar cortex. | No family history of neurodegenerative diseases. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | N/A | High penetrance of autosomal dominant presenile dementia in the father's side. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | N/A | N/A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | No typical signs of prion disease. | Mother: died of corticobasal degeneration; mother's brother was diagnosed with Parkinson's disease. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | No typical signs of prion disease; severe brain atrophy 4 years after disease onset. | Similar dementia in her father who became demented at age 42 and died at age 52. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Two types of insertion mutations: 1: R1.R2.R2.R3g.R2.R2.R2.R2.R2.R3.R4; 2: R1.R2.R2.R3.R2.R2.R3g.R2.R2.R3.R4. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No detectable rPrPSc. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EEG: Electroencephalogram. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Absent. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Present. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Not available. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Detection of rPrPSc by conventional Western blot analysis
In the samples without PK-treatment, an extra band was observed migrating at ~38-40 kDa in the five cases with fCJDIns but not in PrPSc type 1 and type 2 controls from sporadic CJD (sCJD) (Fig. 1A, indicated by the arrow head). This extra high band represents the diglycosylated PrPIns with six extra octapeptide repeats and the monoglycosylated and non-glycosylated PrP species carrying the insertion were mixed with the three wild-type PrP bands, as indicated by the presence of multiple bands between 28 and 38 kDa compared to the relatively pure three bands in the samples from PrPSc type 1 and type 2 of sCJD (Fig. 1A). The bands below 29-30 kDa were endogenously N-terminally truncated PrP fragments such as in cases 2 and 3 (Fig. 1A). The amounts of samples loaded were monitored by the detection of β-actin (Fig. 1B). PK-resistant rPrPSc was detected in the brain homogenates from five out of six cases with fCJDIns by conventional Western blotting with 3F4, although in case 5 rPrPSc bands became visible only in the over-exposed film (Fig. 1C and 1D). The lower PrP band of rPrPSc had the gel mobility of 21 kDa (identical to that of PrPSc type 1) for cases 1, 2, and 4, or ~19 kDa (identical to that of PrPSc type 2) for cases 3 and 5 (Fig. 1B and 1C; Table 1). Case 6 exhibited no typical rPrPSc by the conventional Western blot analysis (Table 1), which was further characterized extensively by enrichment using g5p and NaPTA, ultracentrifugation-based sedimentation, and two-dimensional gel electrophoresis as described in detail below.
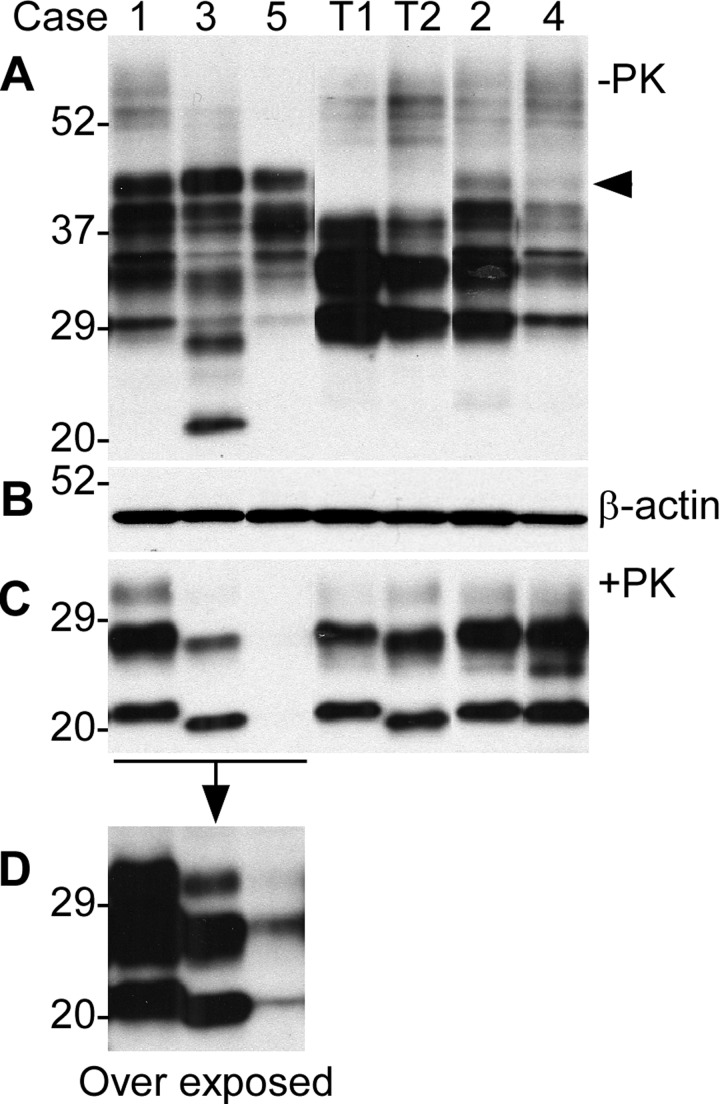
Figure 1. Detection of PrP in brains from five cases with six extra octapeptide repeats using Western blotting with 3F4. (A) Brain samples were from five cases (1 through 5) and were not treated with PK. T1: PrPSc type 1 control; T2: PrPSc type 2 control. (B) Western blot of β-actin, which was used to monitor the amounts of samples from each case. (C) The samples were treated with PK prior to SDS-PAGE and immunoblotting. The gel mobility of the PK-resistant PrP from the cases 1, 2, and 4 was similar to that of PrPSc type 1 control migrating at ~21 kDa while the case 3 was similar to PrPSc type 2 migrating at ~19 kDa. No PK-resistant PrP was visible in the case 5. (D) An over exposed smaller blot from the left part of the blot shown in C. The PK-resistant PrP bands from case 5 became detectable, the gel mobility of which was similar to that of case 2 migrating at ~19 kDa.
Detection of PrPSc in the case with no typical rPrPSc
Case 6 was different from other five cases with the 144-bp insertion mutation (Fig. 1C) as no rPrPSc was detected by conventional Western blot in the brain homogenates from frontal cortex and cerebellum (Fig. 2A). Instead, there were multiple PrP bands prior to PK-treatment although the upper PrP band was also higher than that from non-CJD and sCJD controls (Fig. 2A). These observations were in agreement with the genetic finding that fCJDIns contains a PrPIns (Table 1). The PrPIns molecule may form additional set of three glycoforms including diglycosylated, mono-glycosylated, and unglycosylated PrPIns. Hence, there are at least 6 PrP bands that are assumed to be detected by 3F4 antibody in the brain homogenate from fCJDIns including differently glycosylated PrPWt and PrPIns. Nevertheless, it was surprising that no PK-resistant PrP fragments were detectable by the conventional analysis with 3F4 antibody (Fig. 2A). In addition, anti-C antibody also failed to detect PK-resistant PrP fragments (data not shown).
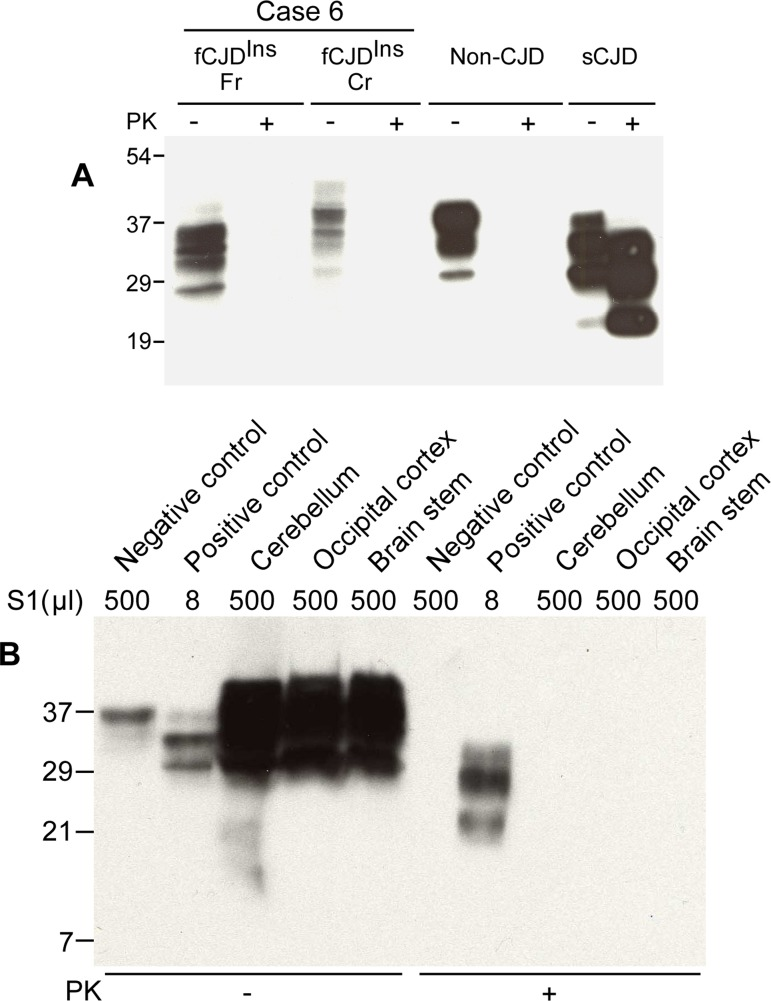
Figure 2. Detection of PK-sensitive PrPSc. (A) Conventional Western blotting of PrP treated with or without PK in case 6. Fr: frontal cortex; Cr: cerebellum. No PrP was observed after PK treatment in the samples from both fCJDIns (case 6) and non-CJD. The PK-resistant PrP27-30 was indicated in the sample from sCJD. The migration of PrP from the cerebellum of case 6 was slightly slower than that of PrP from both non-CJD and sCJD controls. (B) Precipitation of abnormal PrP by NaPTA. S1 from non-CJD (500 μl), sCJD (8 μl), and case 6 (three brain regions: 500 μl each) was incubated with NaPTA and then was subjected to SDS-PAGE and immunoblotting with 3F4. Although a small amount of PrP was precipitated from non-CJD brain sample (500 μl of S1), no PK-resistant PrP fragments were detected. NaPTA was able to precipitate PrP from 8 μl of sCJD S1 (62.5-fold less than non-CJD S1) and the precipitated PrP was resistant to PK-digestion. Compared to non-CJD sample, NaPTA precipitated large amounts of PrP from three different brain regions of case 6 including the cerebellum (Cr), occipital cortex (Oc) and brain stem (BS). After PK-treatment of the NaPTA-precipitated PrP from case 6, no PrP bands were observed.
NaPTA is a reagent that has been demonstrated to specifically precipitate both sPrPSc and rPrPSc [31, 32, 34]. In order to increase sensitivity of detection by Western blotting and ELISA, NaPTA has been used to enrich small amounts of PrPSc in prion-infected peripheral organs where no PK-resistant PrP can be detected by the conventional assays [32, 35, 36]. To detect sPrPSc species and determine if there is a small amount of rPrPSc in this fCJD subject, a relatively large amount of brain homogenate was used to incubate with NaPTA. Compared to the normal control, a larger amount of PrP was precipitated by NaPTA (Fig. 2B). Surprisingly, the precipitated PrP was virtually completely digested by PK (Fig. 2B). No typical rPrPSc was detectable even in the over-exposed film (data not shown).
Using the anti-PrP monoclonal antibody 1E4 that has an epitope N-terminally adjacent to the 3F4 epitope [37, 38], our recent study identified a novel PK-resistant PrPSc characterized by the presence of dominant PK-sensitive PrPSc in an atypical human prion disease termed variably protease-sensitive prionopathy (VPSPr) [34, 39]. Upon PK-treatment, virtually no rPrPSc was detected by conventional Western blotting with 3F4, whereas PrP with a five-step ladder-like gel profile was detected preferentially by 1E4 in VPSPr [34, 39]. On the 3F4 blots, similar to non-CJD and VPSPr, no rPrPSc was detectable in brain homogenates from this fCJDIns, whereas PK-rPrPSc was detected in sCJD type 1 and type 2 cases as well as in case 3 of fCJDIns (Fig. 3A). In contrast, on the 1E4 blots, except for non-CJD, PK-resistant PrPSc was detectable in cases 6 and 3 of fCJDIns, VPSPr, sCJD type 1 and type 2. Nevertheless, rPrPSc in case 6 was detectable in the samples treated at PK concentration equal to or less than 10 μg/ml and decreased significantly at PK equal to or greater than 25 μg/ml (Fig. 3B). The amount and gel profile of rPrPSc in case 3 was very similar to those of rPrPSc in sCJD type 2. As demonstrated in our previous study [34, 39], rPrPSc from VPSPr exhibited a five-step ladder-like gel profile (Fig. 3B). The rPrPSc in case 6 detected by 1E4 was more similar to that of sCJD type 1 at low PK concentrations. However, at high PK concentration, no rPrPSc was detected from case 6 while the profile of typical PrPSc type 1 appeared in the sCJD type 1 case (Fig. 3B). Therefore, although no typical rPrPSc was detected in both fCJDIns of case 6 and VPSPr cases, the gel profile of rPrPSc detected by 1E4 is clearly different from that detected in VPSPr, classic sCJD, and fCJDIns of case 3. Importantly, the amounts and PK-sensitivity of rPrPSc detected by 1E4 in this fCJDIns (case 6) were also significantly less than those in VPSPr, sCJD and typical fCJDIns (case 3).
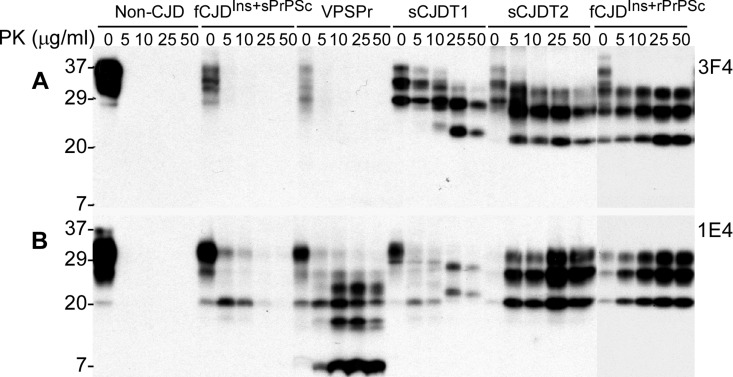
Figure 3. Determination of PK-resistant PrPSc with the 1E4 antibody. A: Brain homogenates from non-CJD, fCJDIns+sPrPSc (case 6), VPSPr with 129VV, sCJD type 1, sCJD type 2, and fCJDIns+rPrPSc were treated with a variety of concentrations of PK prior to SDS-PAGE and Western blotting with 3F4. PK-resistant PrP was only detected in sCJD type 1, type 2, or fCJDIns+rPrP but not in other cases. B: The PK-treated PrP from these cases were detected with 1E4. In contrast, PK-resistant PrP was also detected in both fCJDIns+sPrPSc and VPSPr, in addition to sCJD type 1, type 2, and fCJDIns+rPrPSc. However, the PK-resistant PrP in fCJDIns+sPrPSc was only detected when treated with lower amounts of PK less than 25 μg/ml.
Comparison of PrP oligomeric state between the two types of fCJD cases with the 144-bp insertion mutations
To determine whether the PrP molecule in case 6 has a different oligomeric state compared to other fCJDIns cases, we further conducted the sedimentation of PrPSc in the sucrose step gradients. PrP in three cases with readily detectable rPrPSc was mostly recovered in bottom fractions 9-12, but not in top fractions (Fig. 4A and 4C), similar to classic sCJD [33]. In contrast, PrP in case 6 was mostly recovered in top fractions 1 and 2, while no significant increases in the amounts of PrP were detected in bottom fractions (Fig. 4B and 4C).
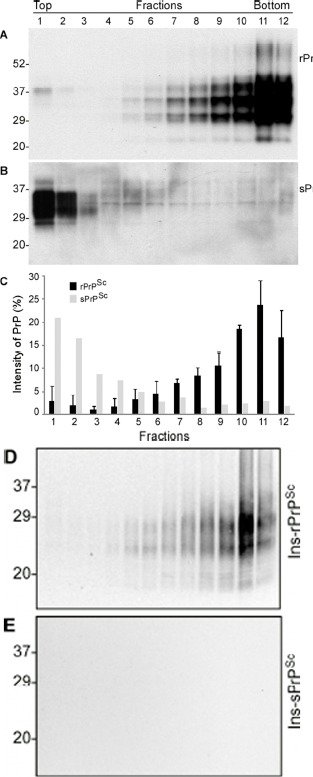
Figure 4. Comparison of oligomeric state of PrP in case 6 and other cases with rPrPSc by sucrose step gradient sedimentation. (A) Western blotting of PrP in individual fractions of sucrose gradient analysis of brain homogenate from case 3 with readily detectable rPrPSc. (B) Western blotting of PrP in individual fractions of sucrose gradient analysis of brain homogenate from case 6 with sPrPSc. C: Bar graph of PrP in individual fractions from three 144-bp insertion mutation cases with rPrPSc (average of PrP percentages from the three fCJDIns+rPrPSc cases) and case 6 with no rPrPSc. Blots were probed with 3F4 antibodies. D and E: PrP in individual fractions from cases 3 (D) and 6 (E) was detected by Western blotting after treatment with PK at 0.5 μg/ml. PrP was only detected in fCJDIns+rPrPSc but not in fCJDIns+sPrPSc.
To investigate whether there are any rPrPSc species in different fractions in case 6, we treated PrP in the fractions with an extremely low concentration of PK. After treatment with PK at 0.5 μg/ml, no PrP was detected in all fractions from case 6 while PrP was detected in bottom fractions from case 3 (Fig. 4D and 4E). Our result suggested that PrPSc from fCJDIns case 3 was sensitive to PK even at 0.5 μg/ml.
Detection of allelic compositions in PrPC and PrPSc from fCJDIns with or without rPrPSc by sedimentation in detergents
To dissect the differences in the composition of PrPWt and PrPIns between the two types of fCJDIns with rPrPSc or sPrPSc (fCJDIns+rPrPSc or fCJDIns+sPrPSc), we analyzed the composition of PrPWt and PrPIns. We took advantage of the most effective method by which the soluble PrPC and insoluble PrPSc can be separated after ultracentrifugation in detergent buffer. After ultracentrifugation, PrP in the soluble and insoluble fractions was deglycosylated with PNGase F prior to detection by Western blotting with an antibody against N-terminal region of PrP called anti-N. In the samples from non-CJD and sCJD, only one PrP band was detected in supernatants (Fig. 5A, S2) and pellets (Fig. 5B, P2), migrating at ~28-29 kDa, which corresponds to the full-length deglycosylated PrPWt. In contrast, in the samples from fCJDIns, besides the ~28-29 kDa PrPWt band seen in non-CJD and sCJD samples, an additional PrP band migrating at ~33-34 kDa was detected in supernatants (Fig. 5A, S2) and pellets (Fig. 5B, P2), which corresponds to the full-length PrPIns with six octapeptide repeats containing 48 extra residues.
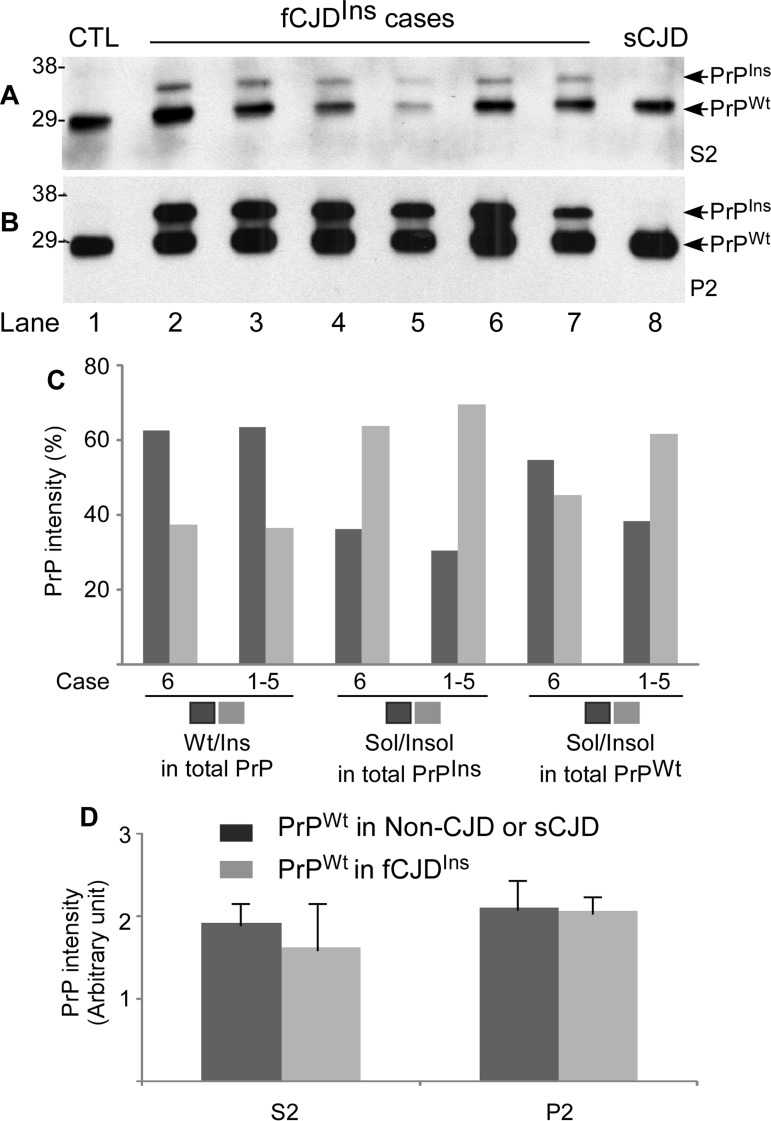
Figure 5. Determination of both PrPWt and PrPIns in detergent-soluble and -insoluble fractions by Western blotting. A and B: After ultracentrifugation in detergents, PrP in the detergent-soluble fraction (S2) (A) and -insoluble fraction (P2) (B) was detected by Western blotting with the anti-PrP antibody anti-N detecting the full-length PrP. Lane1: non-CJD control; Lane 2 fCJDIns+sPrPSc (case 6); Lanes 3 to 7: fCJDIns+rPrPSc (cases 1-5); Lane 8: sCJD control. C: Comparing compositions of total PrP, PrPIns, and PrPWt in fCJDIns+sPrPSc (case 6) and fCJDIns+rPrPSc (cases 1-5, mean) by densitometric analysis of PrP intensity detected with Western blotting as shown in A and B. The left four bars exhibit the percentage of PrPWt or PrPIns in total PrP including soluble and insoluble forms. The central four bars exhibit the percentage of soluble or insoluble form in total PrPIns. The right four bars exhibit the percentage of soluble or insoluble form in total PrPWt. There were no significant differences in the percentage of PrPWt or PrPIns in total PrP between fCJDIns+sPrPSc and fCJDIns+rPrPSc (62.5/37.5 vs 63.4/36.6). Also there were no significant differences in the percentage of the soluble or insoluble form in PrPIns between the two conditions (36.2/63.8 vs 30.5/69.5). In contrast, the percentage of the soluble form of PrPWt was remarkably greater in fCJDIns+sPrPSc than in fCJDIns+rPrP (54.7% vs 38.4%), while the percentage of the insoluble form of PrPWt was significantly smaller in fCJDIns+sPrPSc than in fCJDIns+rPrPSc (45.3% vs 61.6%). D: Comparison of PrPWt intensity in S2 and P2 fractions between fCJDIns and non-CJD or sCJD. No differences in the levels of PrPWt from soluble or insoluble fraction were detected between fCJDIns and non-CJD or sCJD.
By densitometric analysis, we quantified distributions of PrPWt and PrPIns in S2 and P2 fractions in the two types fCJDIns+rPrPSc and fCJDIns+sPrPSc. In both types of fCJDIns, the ratio of PrPWt or PrPIns to the total PrP was the same: PrPWt accounted for 63%, whereas PrPIns accounted for 37% (Fig. 5C). Moreover, there were no differences in ratios of soluble and insoluble PrPIns to the total PrPIns between the two types of fCJDIns (Fig. 5C). In contrast, the ratio of soluble PrPWt to total PrPWt was significantly greater in fCJDIns+sPrPSc than in fCJDIns+rPrPSc, whereas the ratio of insoluble PrPWt to total PrPWt was significantly smaller in fCJDIns+sPrPSc than in fCJDIns+rPrP (55% vs. 38% or 45% vs. 62%) (Fig. 5C).
Since fCJDIns contains only a single PrPWt allele whereas non-CJD or sCJD contains two PrPWt alleles, we assumed that the amount of PrPWt in fCJDIns should account for approximately half of total PrPWt detected in non-CJD or sCJD. To test for this possibility, we also quantified the intensity of the single PrPWt allele from fCJDIns and of total two PrPWt alleles from non-CJD and sCJD in both S2 and P2. Surprisingly, although fCJDIns contains only a single PrPWt allele, whereas non-CJD or sCJD contains two PrPWt alleles, the intensity of PrPWt detected in both S2 and P2 were similar between fCJDIns and non-CJD or sCJD (p > 0.05) (Fig. 5A, 5B and 5D).
Detection of allelic compositions of PrPC and PrPSc from the fCJDIns containing no rPrPSc by conformation-specific binding reagents
Taking advantage of anti-PrP antibody 6H4 and g5p that specifically recognizes either native PrPC or misfolded PrP [40, 41, 30, 33], we dissected allelic composition of sPrPSc and ratio of sPrPSc to total PrP in the detergent-insoluble fraction (P2) rich in PrPSc from fCJDIns. After specific capture of either PrPC by 6H4 or PrPSc by g5p, the samples were treated with or without PNGase F that significantly decreases the heterogeneity of the protein by removal of the glycans (Fig. 6A).
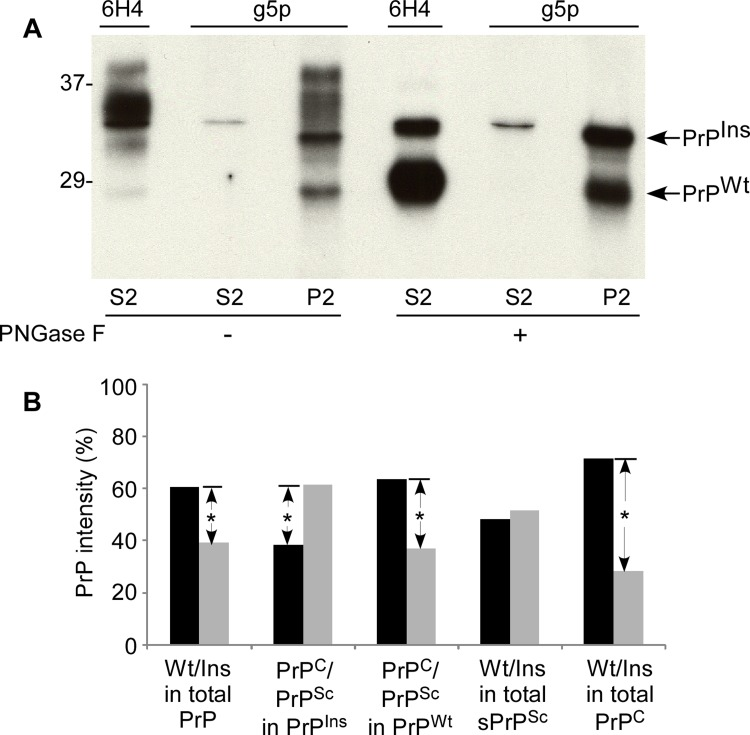
Figure 6. Allelic composition of PrPSc in fCJDIns+sPrPSc. (A) Western blot analysis of PrP precipitated by either 6H4 or g5p. PrP from S2 or P2 was precipitated with 6H4 or g5p, respectively. The precipitated PrP was subjected to deglycosylation with PNGase F and followed by SDS-PAGE and immunoblotting with anti-N antibody. Although there are different profiles of PrP precipitated by 6H4 in S2 and by g5p in P2, they exhibit two bands in both preparations containing an upper band corresponding to full-length PrPIns and a lower band corresponding to full-length PrPWt. (B) Quantitative analysis of allelic composition of PrPSc and PrPC from either PrPIns or PrPWt. The intensities of PrP treated with PNGase F on the blot shown in A were analyzed by densitometry. The PrP species precipitated from S2 by 6H4 was considered as PrPC while PrP precipitated from S2 and P2 by g5p was considered as PrPSc. Total PrP in fCJDIns+sPrPSc was composed of ~60% PrPWt and ~40% PrPIns. Approximately 62% PrPIns was converted into PrPSc while ~38% remained as PrPC. In contrast, approximately 64% PrPWt remained as PrPC while 36% was converted into PrPSc. PrPSc was composed of 52% PrPIns and 48% PrPWt while PrPC was composed of 28% PrPIns and 72% PrPWt. These data represent averages from three independent experiments. *p < 0.05.
Moreover, probing the captured proteins with anti-N antibody would simplify the recognition of the full-length PrPIns and PrPWt. Without PNGase F treatment, several bands migrating between 28-39 kDa were detected in both 6H4-captured preparation from S2 preparation rich in soluble normal PrP and g5p-captured preparation from P2 preparation rich in insoluble abnormal PrP (Fig. 6A). These bands represent glycosylated and unglycosylated full-length PrPWt and PrPIns. Notably, although g5p captured a large amount of misfolded PrP from P2 as expected, it also captured a small amount of PrP (a thin band) migrating at ~33-34 kDa that represents the unglycosylated full-length PrPIns.
In the detergent-soluble fraction (S2) treated with PNGase F, two PrP bands were visualized by anti-N antibody in the preparation immunoprecipitated by 6H4: one with weaker intensity migrating at ~33-34 kDa corresponding to unglycosylated full-length PrPIns and another with stronger intensity migrating at ~28-29 kDa corresponding to unglycosylated full-length PrPWt, while only one thin PrP band corresponding to unglycosylated full-length PrPIns was captured by g5p (Fig. 6A). In the P2 fraction rich in detergent-insoluble PrP, there were also two bands visualized by anti-N antibody in the sample precipitated by g5p. The gel mobility of the captured two bands was similar to that of the bands immunoprecipitated by 6H4 from S2 (Fig. 6A). Therefore, not only PrPIns but also PrPWt molecule participated in the formation of sPrPSc in the fCJD with the PrP insertion mutation.
Quantitative analysis by densitometry from three independent experiments revealed that PrPIns precipitated by 6H4 in S2 and by g5p in S2 and P2 accounted for ~40 % of total PrP while PrPWt precipitated by the two reagents accounted for ~60% of total PrP (Fig 6B), which was in agreement with those observed by the direct loading of soluble and insoluble PrP in Fig. 5A through C, suggesting that most PrPIns and PrPWt molecules in either normal or pathological isoforms were recovered by 6H4 and g5p. A detailed quantitative analysis was conducted in order to dissect the PrP composition of various PrP species (Fig. 6B). We observed that the majority of PrPIns (~62%) was converted into PrPSc while the minority (~38%) remained as PrPC (Fig. 6B). In contrast, the majority of PrPWt (~64%) remained as PrPC while ~36% of it was converted into PrPSc. Also ~half of PrPSc was derived from PrPIns and another half from PrPWt (52% vs. 48%). However, PrPC was composed of ~72% of PrPWt and ~28% of PrPIns. Thus, these data were consistent with the results obtained in Fig. 5 and confirmed that the majority of PrPWt indeed was not converted into PrPSc.
Two-dimensional gel electrophoresis of PrP
To have a high resolution profile of PrP from fCJDIns containing both PrPWt and PrPIns, the PrP molecule treated with or without PNGase F was subjected to the two-dimensional (2D) gel electrophoresis that is capable of separating proteins based on both molecular weight and charge.
On 2D blots, PrP from non-CJD brain sample mainly was composed of two sets of PrP spots (Fig. 7B). The first set consisted of 13-14 PrP spots migrating between 33 and 42 kDa with pI 5.5-9.7, corresponding to diglycosylated, full-length PrP species (designated PrP 2D spots I). The second set comprised 9-10 PrP spots and was distributed between 29 and 32 kDa, pI 4.5-7.0, corresponding to diglycosylated, truncated PrP species (designated PrP 2D spots IV). This group of PrP species constituted the middle band of PrP on 1D blot.
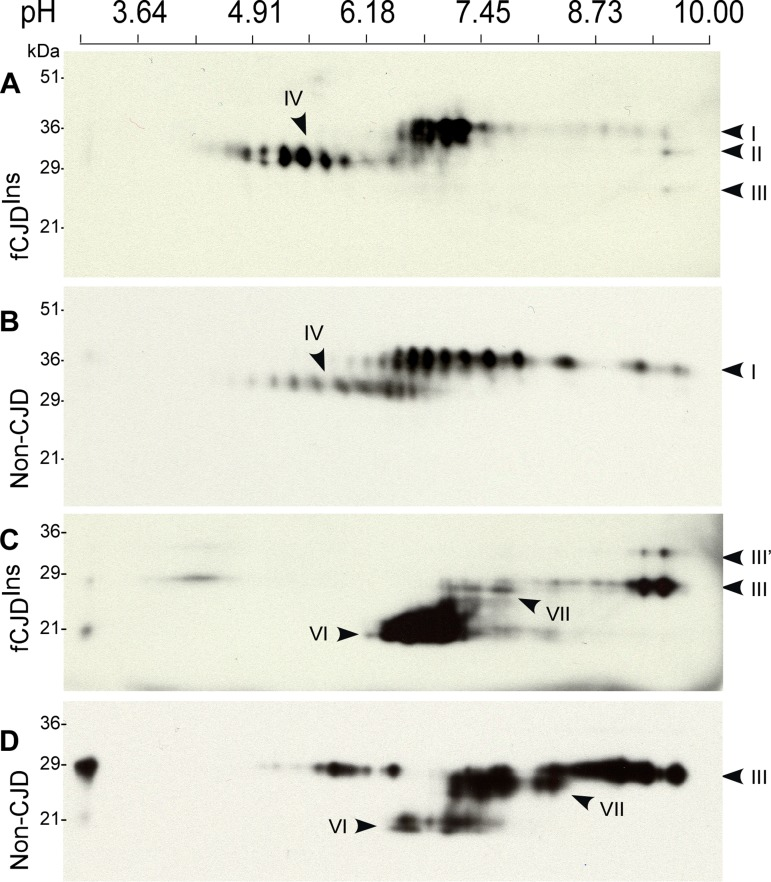
Figure 7. Two-dimensional Western blotting of PrP. (A) Comparison of untreated PrP from case 6 with fCJDIns+sPrPSc and non-CJD and sCJD. PrP from fCJDIns includes 2D spots I-IV and PrP from non-CJD mainly consists of 2D spots I and IV. (B) Comparison of PNGase F-treated PrP from fCJDIns and non-CJD. PrP from the two conditions comprises both full-length PrP (2D spots III) and N-terminally truncated PrP (2D spots VI); however, PrP from fCJDIns contains additional set of PrP spots (2D spots III’). The blots were probed with 3F4.
The 2D profile of PrP from case 6 with fCJDIns was basically similar to that of PrP from non-CJD (Fig. 7A). However, several differences between the two were still detectable. For instance, pIs of the predominant PrP species from fCJDIns were different from those of non-CJD sample. Most PrP from 2D spots I of fCJDIns were mainly localized between pI 6.5 and 7.3 while intense PrP spots from that of non-CJD spread from pI 6.3-9.1. Compared to PrP from non-CJD, slight increases in the intensity of PrP 2D spots II, III, and IV were also observed (Fig. 7A). Moreover, the PrP 2D spots IV were more intense in fCJDIns than in non-CJD. These differences may result from the intrinsic nature of PrP in fCJDIns, which is a mixture of PrPWt and PrPIns.
Various PrP species from fCJDIns and non-CJD were also compared on 2D blot after deglycosylation that often profoundly decreases PrP heterogeneity. As expected, by probing with 3F4 antibody, two major sets of PrP spots were observed in the deglycosylated PrP from non-CJD: PrP 2D spots III migrating at 27-29 kDa with pI 7.0-9.6 corresponding to full-length PrP, and PrP 2D spots VI migrating at ~19-22 kDa with pI 6.1-8.1 corresponding to the N-terminally truncated PrP (Fig. 7D). However, in contrast to PrP from non-CJD, PrP from fCJDIns had an additional set migrating at 31-33 kDa with pI 9.0-9.5 designated PrP 2D spots III’ (Fig. 7C), which fitted well with the full-length PrPIns molecule with 6 extra repeats in terms of their molecular weight. In addition, we also observed that there was a new set of PrP spots designated PrP 2D spots VII present in the two conditions including fCJDIns and non-CJD, migrating at ~26-27 kDa with pI 5.0-8.1 (Fig. 7C and 7D). This group of PrP spots was N-terminally truncated PrP species missing ~10 residues from the farthest N-terminal portion. While PrP 2D spots III from the non-CJD brain sample were predominant, PrP 2D spots VI from fCJDIns were predominant. It is also intriguing to note that each of the PrP 2D spots III from the two conditions had different pI of the most intense PrP spot. For example, pI of the most intense PrP spot in fCJDIns was about 9.6, and ~9.0 in non-CJD. In view of their identical full-length PrP sequence, whether this difference is associated with distinct anchors remains to be further determined.
Histopathology and immunohistochemistry
The five fCJDIns cases associated with rPrPSc showed various degree of spongiform degeneration (SD) and variable astrogliosis and neuronal loss in the cerebral cortex and basal ganglia (Table 2 and Figure 8). Plaques with single or multiple cores were detected only in one case of the four cases with adequate number of slides in the molecular layer of the cerebellum. Typical kuru plaques were observed in the cerebellar granule cell layer and white matter.
Table 2. Neuropathological features of fCJDIns
| Case # | Histopathology (H&E) | PrP immunostaining (IHC) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cerebrum | Cerebellum | Cerebrum | Cerebellum | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SDa | SD | Plaques | Synaptic with other pattern | Plaque/plaque-like | Plaque/plaque-like | Stripes | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | + | ++ | − | + | + | − | − | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | +++ | NA | NA | + | + | NA | NA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | + | ++ | − | + | + | − | ++b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | + | − | − | + | + | − | ++b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | + | − | + | + | + | +++/+c | +++ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | − | − | + | − | − | −/+++c | +++ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spongiform degeneration; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Not completely formed stripes; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Granular layer + white matter/molecular layer. The severity of the SD and deposition of PrP IHC was scored as follow: absent (−), mild (+), moderate (++), and severe (+++). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
PrP immunostaining showed the “synaptic” pattern in the cerebral cortex of all the cases of fCJDIns associated with rPrPSc (Table 2). However, the three cases VV-129 also showed the presence of prominent granules, micro plaques or plaque-like formations. Perineuronal staining was seen in the MV-129M and two VV-129V. Real plaques, often multicore, were seen in the 129-MM (case 6) and 129-VV (case 5) but not in the other cases.
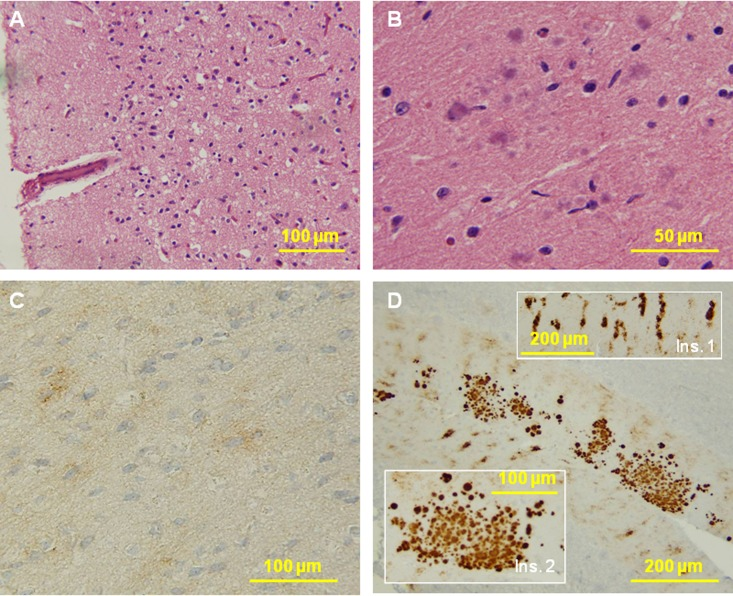
Figure 8. Histological and immunohistochemical examinations of brain tissues. (A) The cortex reveals no typical spongiform degeneration. (B) Several multicore plaques are present in the molecular layer of the cerebellum with a focal distribution. No pathology was seen in the pons and medulla. C: PrP immunostaining demonstrates weak staining with a synaptic pattern and occasional loose fine granular aggregates in the cerebral cortex. D: The molecular layer of the cerebellum shows a remarkable combination of strip-like staining (Ins. 1) and multicore plaques (Ins. 2). No immunostaining was seen in the pons and medulla.
Four of the five cases in which the cerebellum was available showed the distinctive stripe pattern in the molecular layer but the stripes of cases 3 and 4 of Table 2 were shorter or not completely formed. Stripes in the molecular layer could be either alone or associated with the synaptic or plaque-like formations in the granule cell layer and superficial white matter (VV-129, case 5). In the case lacking the stripe staining pattern, the staining of the molecular layer was synaptic (VV-129, case 1).
In fCJDIns case 6 lacking rPrPSc, the histology of the brain areas examined revealed no SD (Fig. 8A). Several multicore plaques were present in the molecular layer of the cerebellum with focal distribution (Fig. 8B).
PrP immunostaining demonstrated weak staining with a focal synaptic pattern and occasional loose fine granular aggregates in the cerebral cortex (Fig. 8C). The molecular layer of the cerebellum had a remarkable combination of stripe-like staining so called PrP patches that are pathognostic for insertion mutation (Fig. 8D, ins. 1) and multicore plaques (Fig. 8D, ins. 2).
In conclusion, the case with sPrPSc (case 6) differed from those associated with rPrPSc by the lack of typical SD, and the presence of multicore plaques in limited regions of the cerebellar molecular layer. However had similar PrP stripes in the cerebellar molecular layer as the fCJD cases associated with rPrPSc.
Histoblotting
We investigated the PK-resistance of the PrPSc forming the stripes with two histoblot procedures: one based on IHC principles, the other similar to WB. Without PK-treatment, PrP was detected in the blots from cases with or without rPrPSc by both methods (Fig. 9, A-D). After PK-treatment with 10 or more μg/ml, PrP was detected only in cases with rPrPSc (Fig. 9B and 9D) but not in case 6 with sPrPSc (Fig. 9A and 9C). In contrast, after treatment with thermolysin, PrP was detected in all cases with fCJDIns regardless of the presence or absence of rPrPSc but not in normal controls (Fig. 9 E through G). Therefore, the histoblot results were consistent with the data from WB and with the conclusion that in case 6 the cerebellar stripes were made of sPrPSc.
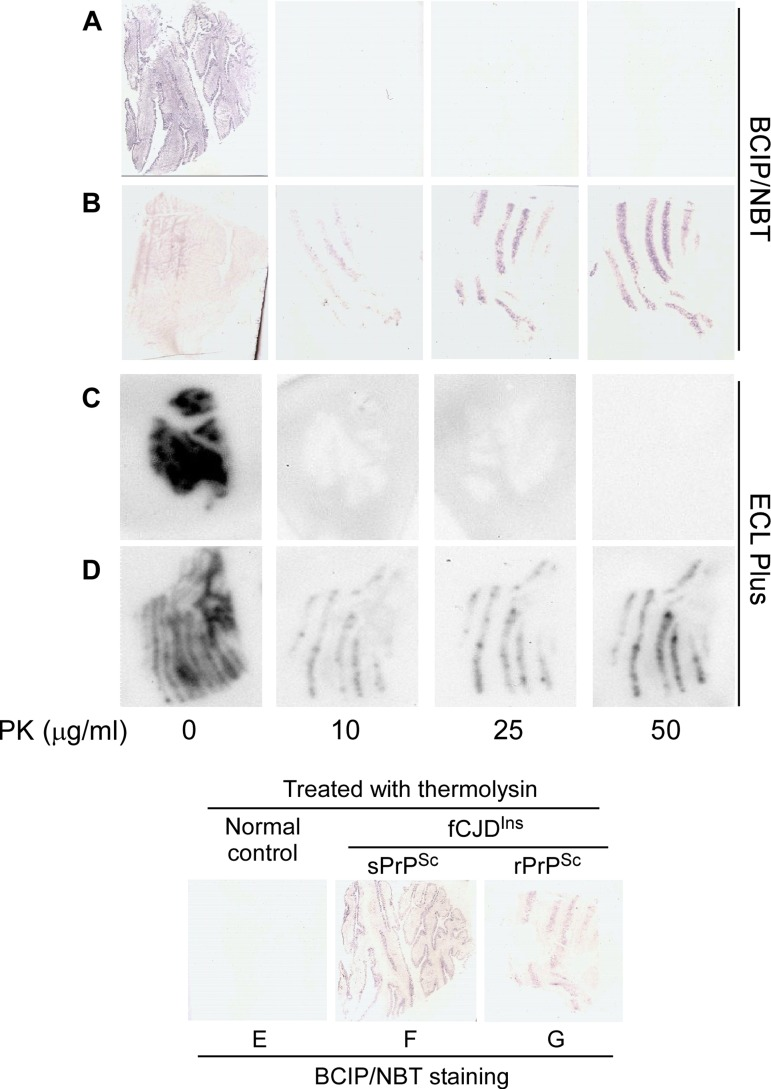
Figure 9. Histoblotting of fCJDIns+sPrPSc and fCJDIns+rPrPSc. The nitrocellulose membranes blotted with tissue sections from the cerebellar cortex of fCJDIns+sPrPSc (A, C, and F) or fCJDIns+rPrPSc (B, D, and G) were developed with BCIP/NBT similar to classic IHC in A, B, E, F, and G while the membranes were developed with ECL Plus similar to Western blotting in C and D. PrP staining was detected in all blots without PK-treatment. After PK-treatment at either 10, 25, or 50 μg/ml, no PrP staining was detected in the samples from fCJDIns+sPrPSc (A and C), whereas PrP was detected in the samples from fCJDIns+rPrP (B and D). In contrast, PrP staining was detected in fCJDIns+sPrPSc (F) in addition to fCJDIns+rPrP (G) but not in normal controls after thermolysin that was reported to digest PrPC only (E).
Discussion
Neurodegenerative disorders are all associated with misfolding of various cellular proteins [42-44]. Human prion diseases including sporadic, inherited and infectious forms are highly heterogeneous in terms of their broad range of clinical and pathological phenotypes [12]. In addition to the transmissible spongiform encephalopathy (TSE), non-transmissible prion diseases have also been reported [45-47], which has brought about a proposal that the spectrum of prion diseases should be beyond the classic definition of TSE [48]. The high heterogeneity of prion diseases may be associated with the chameleon-like conformations of the PrPSc molecule [11], the only component identified in the infectious prion pathogen to date [2]. Presence of a variety of rPrPSc including PrPSc type 1 and type 2 in sCJD and PrP7-8 in GSS detected by Western blotting and distinct brain PrP deposits detected by immunohistochemistry might be attributable to the variable PrPSc conformation [11, 12]. Recently, novel PK-resistant PrP species with a distinctive ladder-like gel profile have also been identified in a new prion disease termed VPSPr [34, 39]. These newly-identified PK-resistant PrP fragments are preferentially detected by 1E4 but much less immunoreactivity with the widely used 3F4, which is similar to those PK-resistant PrP species detected in the normal brain and uninfected cultured cells [33, 37, 49, 50]. Studies on the correlation between the phenotypic heterogeneity of the diseases and the chameleon-like conformation of PrPSc molecule are often complicated by the diversity in the etiologies of the diseases. Therefore, an investigation on cases with a single etiology will be critical for understanding the molecular mechanism responsible for this high heterogeneity in disease phenotypes.
Inherited CJD linked to the 144 bp insertion mutation provides us with an excellent opportunity to limit the influence of variable etiologies on phenotype. Since the disease is significantly and tightly linked to this single mutation [21, 22], the phenotypes of the disease should be mainly determined by the de novo generated-pathogen itself, mutation-containing PrPSc. Compared to the observation by Mead et al. [24], our six cases had a similar mean age of death (44.0 ± 5.7 vs. 45.1 ± 7.3) while an elder mean age of onset (39.7 ± 8.8 vs. 34.9 ± 6.93). In our fCJDIns cases, five cases showed rPrPSc either similar to PrPSc type 1 or type 2 of sCJD. Strikingly, one case had no typical rPrPSc albeit the presence of clinical and phenotypic characteristics similar to other five cases. Instead, this case was demonstrated to be rich in sPrPSc. The identification of the fCJDIns case rich in sPrPSc but lacking in typical rPrPSc raises several issues and implications as to molecular bases underlying the phenotypic heterogeneity and neuropathological changes of prion diseases.
Effect of the levels and types of rPrPSc and sPrPSc on deposits of PrP patches and neuropathological changes
So far more than 100 individuals from at least eight families affected by the 144-bp insertion mutation have been identified around the world [21, 22, 51, 16, 24-29]. However, the reported examination of the brain PrP with Western blotting has been no more than nine cases in total to date, only six of which exhibited a detectable rPrPSc [24-26, 28]. The rPrPSc species examined with Western blotting were three heterozygotes with 129-M/V polymorphism and 144-bp insertion, in which two were similar to the Gambetti et al. sCJD PrPSc type 1 in both size and ratio of the three major PrP glycoforms [25] and one similar to PrP7-8 detected in GSS [28]. Although the two fCJD cases had an identical PRNP, one with a 4-year course exhibited only minimal focal spongiform degeneration and another with a 10-year course showed significant astrocytosis, neuronal loss and pronounced spongiform degeneration [25]. The levels of rPrPSc were five-fold less in the former than in the latter. Nevertheless, the deposits of PrP patches were equally detected in the molecular layer of the cerebellum of the two cases. In another study involving the largest known kindred so far with 86 affected individuals, PrP was examined by Western blotting in total five cases, of which a heterozygote with 129-M/V had the Collinge et al. PrPSc type 2 (corresponding to Gambetti et al. type 1 in terms of its gel mobility) and a homozygote with 129-M/M contained the Collinge et al. type 3 (corresponding to Gambetti et al. type 2) [24]. In addition, three out of five cases examined revealed no detectable rPrPSc by conventional Western blotting [24]. Interestingly, the heterozygote with Collinge et al. type 2 and a 19-year course (VI. 23 or their case 10) and the homozygote (129-M/M) with no rPrPSc and a 9-year course (VII.25 or their case 4) shared the same intense PrP patches in the cerebellum while the former exhibited slightly severer spongiosis and astrocytosis [23, 24]. Another case with144-bp insertion examined by Western blotting was reported by Gelpi et al. [26]. This case showed rPrPSc in a pattern resembling Gambetti et al. PrPSc type 1 although PrPSc type 2 might also be present. Neurohistologically, this case exhibited numerous eosinophilic globular structures in the molecular layer and the parahippocampal gyrus in addition to the spongiform changes, slight neuronal loss and gliosis as well as PrP diffuse synaptic staining [26]. The latest case examined by Vital et al was a heterozygote with 129-M/V polymorphism and was of GSS phenotype with PrP7-8 [28].
Our current study confirmed the marked variability in severity of neuropathological changes and levels of rPrPSc and the predictability in PrP patches staining, similar to previous observations [23-26, 28]. Nevertheless, our cases did not seem to reveal the close correlation either between the amount of rPrPSc and the length of disease duration or between the amount of rPrPSc and the severity of neuropathological changes. For instance, although the cases 2 and 3 had the longest and second longest disease durations (15- and 11-year, respectively) among the six cases we examined, the amounts of rPrPSc in the two cases were not the largest. The current characterization on six fCJDIns cases including two PrPSc type 2 and three PrPSc type 1 and one with no typical rPrPSc clearly demonstrated that the similar PrP patches are always equally detectable in fCJDIns regardless of the levels and types of rPrPSc except a case. It is conceivable that rPrPSc conformer should not be the main component of this type of PrP deposits and the abnormal sPrPSc conformer containing PrPIns participates in the formation of the PrP patches instead.
It is known that Western blot analysis is more sensitive than immunohistochemistry in terms of detection of a protein. However, for detection of the abnormal PrP, it has been reported in at least two conditions that immunohistochemistry readily detected the abnormal PrP staining in the samples in which the conventional Western blot analysis showed no detectable rPrPSc [34, 36]. For instance, by immunohistochemistry PrP deposition was readily detected in the olfactory mucosa of sCJD, where the amount of rPrPSc only accounted for as small as 8% of brain rPrPSc [36]. In these tissues, no rPrPSc was detected by conventional Western blotting. It is possible that the majority of PrPSc was composed of sPrPSc in the olfactory mucosa. Indeed, this was the case in VPSPr [34, 39]. Although there was strong PrP immunostaining with 3F4 in brains from the subjects with VPSPr, no rPrPSc was detected by the conventional Western blotting probed with the same antibody. The quantitative analysis revealed that the amount of rPrPSc was very small (as small as 10% of brain rPrPSc in sCJD) [34]. Therefore, it is most likely that PrP staining in the tissue section detected by immunohistochemistry is the signature of sPrPSc instead of rPrPSc alone. The pathognostic PrP patches in the fCJDIns may comprise sPrPSc deriving from both PrPIns and PrPWt. Indeed, using histoblotting after PK- or thermolysin-treatment, we confirmed that staining of sPrPSc can be eliminated by PK but not by thermolysin. The latter is the enzyme that has been reported to specifically degrade PrPC but not PrPSc including sPrPSc [52, 53]. Thus, our results suggest that sPrPSc may preferentially attack the cerebellum compared to the cerebrum.
Allelic composition of total PrP and PrPSc in fCJDIns
By using a PK-sensitivity assay, the rPrPSc molecule in fCJD with 144-bp insertion has been demonstrated to comprise both PrPIns and PrPWt alleles in a case with detectable rPrPSc [5]. However, whether both PrPWt and PrPIns participate in the formation of sPrPSc or not remains unknown. The ratios of PrPWt and PrPIns to the total PrP in fCJDIns are also unclear. We demonstrated that like rPrPSc in the typical fCJDIns+rPrPSc, sPrPSc in fCJDIns+sPrPSc also derived from both PrPWt (~48%) and PrPIns (~52%). Although PrPWt and PrPIns accounted for 60% and 40% of total PrP, respectively, most of PrPIns (~62%) was converted into sPrPSc and 64% of PrPWt remained as PrPC. It is worth noting that less PrPWt became insoluble in the fCJDIns+sPrPSc case than in fCJDIns+rPrPSc (PrPWt: ~45% vs. ~62%). Whether this is the reason that the insoluble PrPWt and PrPIns in the fCJDIns+sPrPSc did not form the typical rPrPSc remains to be confirmed. Compared to the previously reported case [5], the current study showed a greater percentage of insoluble PrPWt (~62% vs. ~57%) and smaller PrPIns (~70% vs. ~94%) in fCJDIns+rPrP [5]. The difference between the current study and the previous one may result from that we examined five cases here while only one case was examined in the previous study. Our 2D study further confirmed that PrPWt is predominant in fCJDIns+sPrPSc. Whether PrPIns is less-expressed or it is readily degraded in fCJDIns remains to be determined. Surprisingly, both full-length PrPIns and PrPWt molecules share a similar pI at 9.0-9.5 and the difference between the two seems to be in the molecular weight only but not in the molecular charge. Another interesting finding is that although there are both PrPWt and PrPIns in fCJDIns, the amount of PrPWt in fCJDIns is almost similar to that of PrPWt in non-CJD and sCJD.
Pathophysiology of rPrPSc and sPrPSc species
The correlation between rPrPSc and the neuropathological changes is still controversial. The rPrPSc species detected in prion-infected brains are surprisingly not neurotoxic and PrP-knockout mice are resistant to prion infection [54, 55]. Moreover, subclinical forms of prion diseases have been observed in experimentally or naturally infected animals that harbor high levels of infectivity and PrPSc but are asymptomatic during a normal life-span [56, 57]. Conversely, wild-type mice inoculated with PrPSc of bovine spongiform encephalopathy showed no detectable rPrPSc in the brain despite the presence of neurological symptoms and neuronal death [58]. These conditions were observed not only in animals but also in humans. Fatal familial insomnia or GSS with substitution of valine for alanine at residue 117 (A117V) revealed striking clinical manifestations but little or undetectable PK-resistant PrP [59, 60]. Therefore, the molecular features of the neurotoxic forms of PrP remain to be determined. Several potentially toxic PrP isoforms have been studied in prion-infected transgenic mice, rodents and humans including transmembrane, cytosolic and PK-sensitive forms of abnormal PrP [61, 31, 62, 30]. Based on the “refolding” or “seeding” models, PrPC may unfold to an intermediate before it refolds under the influence of PrPSc or the conversion of PrPC into PrPSc requires a PrPSc-like form (PrP*) [63, 64]. The intermediates have been widely observed in cell-based and cell-free models [65, 66, 41, 67]. These intermediates generated in the process of conversion of PrPC to PrPSc could be the neurotoxic PrP species.
The PK-sensitive sPrPSc was initially proposed in experimentally infected animals using PTA-based ELISA by Safar et al. [31]. To our knowledge, our previous study was the first to demonstrate that human PrPSc is composed of both rPrPSc and sPrPSc and that the majority of PrPSc in GSS is sensitive to PK-digestion by using a PrPSc-specific antibody-based Western blot analysis [30]. Currently the physiochemical features and pathophysiology of sPrPSc are poorly understood. The possibility cannot be ruled out that sPrPSc is an intermediate in the formation of the terminal product rPrPSc and it is responsible for the prion-related neurotoxicity. Remarkably, VPSPr-129VV that we recently identified is rich in sPrPSc and virally lacks the typical rPrPSc type 1 and type 2 and PrP7-8 in the cerebral cortex by conventional Western blot analysis [34, 39]. In the scrapie-infected hamsters, it has been shown that sPrPSc forms smaller oligomers while the rPrPSc forms the larger aggregates [62]. Our current finding that an fCJDIns with typical clinical and neuropathological characteristics is lack of typical rPrPSc but full of sPrPSc favors the hypothesis that the sPrPSc comprising small oligomers is most likely responsible for the neuropathological changes. Indeed, in Alzheimer's disease, the toxicity was originally thought to be a property of the fibrillar form of Aβ, consistent with the widespread notion at the time that the amyloid fibril itself was pathogenic [68, 69]. However, subsequent studies revealed that Aβ fractions containing protofibrillar comprising soluble oligomers, but not fibrillar, material retained their toxicity [70-72].
Like in non-CJD, uninfected cultured cells, and VPSPr, we detected rPrP in fCJDIns+sPrPSc as well when the 1E4 antibody-based Western blotting was used [33, 37, 34, 39, 50]. Although the gel profile of rPrP detected in non-CJD with 1E4 is similar to that of fCJDIns+sPrPSc, the intensity of rPrP is much lower in non-CJD than in fCJDIns+sPrPSc. The rPrP species in non-CJD was only visible in over-exposed films (data not shown). The gel profile of rPrP in fCJDIns+sPrPSc is different from that of VPSPr. Interestingly, it is more similar to that of rPrPSc in sCJD type 1at the lower PK concentrations ranging from 5 to 10 μg/ml with an unglycosylated PrP band migrating at ~19 kDa similar to sCJD type 2 while the typical gel profile of sCJD type 1 with an unglycosylated PrP band migrating at ~21 kDa becomes dominant at higher PK-concentrations greater than 25 μg/ml. It would be interesting to further investigate whether all 1E4-preferentially detected rPrP species originate from a similar precursor.
Materials and Methods
Reagents and antibodies
Sodium phosphotungstic acid (NaPTA), proteinase K (PK), and phenylmethylsulfonyl fluoride (PMSF) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Thermolysin was purchased from Sigma. Peptide N-glycosidase F (PNGase F) was from New England Biolabs Inc. and used following manufacturer protocol. Urea, CHAPS, DL-dithiothreitol (DTT), Iodoacetamide (IAA), tributylphosphine (TBP), Ampholine pH 3-10, and immobilized pH gradient (IPG) strips (pH 3-10, 11 cm long) were from Bio-Rad (Richmond, CA, USA). Reagents for enhanced chemiluminescence (ECL Plus) were from Amersham Pharmacia Biotech, Inc. (Piscataway, NJ, USA). Magnetic beads (Dynabeads M-280, tosylactivated) were from Dynal Co. (Oslo, Norway). Anti-PrP antibodies, including rabbit anti-N-terminal antiserum against human PrP residues 23-40 (B. Ghetti, Indiana University, USA), anti-C-terminal antiserum immuno-reactive to human PrP residues 220-231 [59], and mouse monoclonal antibody 3F4 against human PrP residues 106-110 [73, 38], were used.
Human brain tissues
Consent to use autopsy material for research purposes had been obtained for all samples. Autopsy was performed within 20 hours from the death. Clinical data and relevant hospital records were examined. Cases of proved non-CJD, sporadic CJD, and GSS diagnosed at the National Prion Disease Pathology Surveillance Center (NPDPSC, Cleveland, USA), were used as controls.
Brains, sent to the NPDPSC for suspected prion disease diagnosis, were obtained at autopsy and one half was immediately frozen and stores at −80 °C. The remaining tissue was fixed in formalin for 10 days, kept in 98 % formic acid for 1 h and again in formalin until sampling for neuropathological examination and PrP immuno-histochemistry [39]. The presence of PrPSc from frozen tissues of frontal (FC) and occipital (OC) cortices, brain stem (BS), and cerebellum (CE) were determined by western blotting. In addition, paraffin blocks of tissues from FC, OC, BS, and CE were prepared for histology and immunohistochemistry.
Molecular genetics
The genomic DNA was extracted from frozen brain tissues. The open reading frame (ORF) of the PRNP was amplified by the polymerase chain reaction (PCR) using 20 ng of genomic DNA and primers PrPO-F (GTCATYATGGCGAACCTTGG, Y=C+T) and PrPO-R (CTCATCCCACKATCAGGAAG, K=T+G) (PCR cycles: 94°C for 3 min; 94°C for 1 min, 57°C for 1 min, 72°C for 1 min, 30 cycles; 72°C for 10 min). The PCR products were separated on a 1.0% agarose gel. Both the larger band (~0.9 kb) and the wild type size band (0.77 kb) were recovered separately from the gel using the QIAGEN gel extraction kit, and subjected to automated sequencing with primers PrPO-F, PrPO-R, and HP306R (CATGTTGGTTTTTGGCTTAC TC). For some samples where direct sequencing of PCR products did not give conclusive sequences, the PCR products were cloned then sequenced. The sequences were compared with that of the wild type human PrP using the LALIGN program (https://www.igh.cnrs.fr/fr/). The sequence of R3g is CCC CAT GGT GGT GGC TGG GGg CAG as defined by Goldfarb et al. [74].
Preparation of brain homogenate, S2, and P2 fractions
The 10% (w/v) brain homogenates were prepared in 9 volumes of lysis buffer (10mM Tris, 150 mM NaCl, 0.5% Nonidet P-40, 0.5% deoxycholate, 5mM EDTA, pH 7.4) with pestle on ice. When required, brain homogenates were centrifuged at 1,000 g for 10 min at 4°C. In order to prepare S2 and P2 fractions, the supernatants (S1) were further centrifuged at 35,000 rpm (100,000 g) for 1 h at 4°C. After the ultracentrifugation, the detergent-soluble fraction was recovered in the supernatants (S2) while the detergent-insoluble fraction (P2) was recovered in the pellets that were resuspended in lysis buffer as described [33].
Specific capture of PrPC and PrPSc by 6H4 and g5p
The anti-PrP antibody 6H4 or DNA binding protein g5p (100 μg each) were conjugated to 7×108 tosyl activated superparamagnetic beads (Dynabeads M-280, Dynal Co.) in 1 ml of phosphate-buffered saline (PBS) at 37 °C for 20 h, respectively. The conjugated beads were incubated with 0.1 % bovine serum albumin (BSA) in 0.2 M Tris-HCl at pH 8.5 to block non-specific binding. The prepared beads were stable for at least 3 months at 4°C. Brain homogenate (10%, w/v) was prepared in lysis buffer, followed by centrifugation at 3,000 g for 10 min at 4°C to remove debris. The specific capture of PrPC or PrPSc by 6H4 or g5p was performed as described [41, 33] using brain homogenate and conjugated beads (10 μg mAb or g5p/6×107 beads) in 1 ml of binding buffer (3% Tween-20, 3% Nonidet-40 in PBS, pH 7.4). After incubation with constant rotation for 3 h at room temperature, the beads were attracted to the sidewall of the plastic tubes by external magnetic force, allowing easy removal of all unbound materials in the solution. After three washes in wash buffer (2% Tween-20 and 2% Nonidet P-40 in PBS, pH 7.5), the beads were collected and were heated at 95°C for 5 min in SDS sample buffer (3% sodium dodecyl sulfate (SDS), 2 mM EDTA, 10% glycerol, 50 mM Tris-HCl, pH 6.8). The proteins eluted from the beads were subjected to SDS-PAGE and immunoblotting as described below.
Precipitation of PrPSc by sodium phosphotungstate
Precipitation of PrPSc by sodium phosphotungstate (NaPTA) was conducted as described [32, 33] with mild modification. Briefly, 10% (w/v) brain homogenates from brain tissues were prepared in Dulbecco's sterile phosphate buffered saline (PBS) lacking Ca2+ and Mg2+. The gross cellular debris was removed by centrifugation at 1,000 rpm (80 g) for 1 min. Supernatant (500 μl) was mixed with an equal volume of 4% (w/v) sarkosyl prepared in PBS pH 7.4 and incubated for 10 min at 37°C with constant agitation. Samples were adjusted to final concentrations of 50 units/ml Benzonase (Benzon nuclease, purity 1; Merck) and 1 mM MgCl2 and incubated for 30 min at 37°C with constant agitation. Subsequently, the samples were adjusted with 81.3 μl of a stock solution containing 4% (w/v) NaPTA and 170 mM MgCl2 (prepared in water and titrated to pH 7.4 with sodium hydroxide) to give a final concentration in the sample of 0.3% (w/v) NaPTA. This stock solution was pre-warmed to 37°C before use. Samples were incubated at 37°C for 30 min with constant agitation before centrifugation at 14 000 rpm for 30 min. After careful isolation of the supernatant, the pellet was resuspended to 60 μl final volume of 1 X lysis buffer. The samples were incubated with PK at a final concentration of 50 μg/ml at 37°C for 1 h. Digestion was terminated by the addition of PMSF (3 mM final concentration) and boiling for 10 minutes in an equal volume of electrophoresis sample buffer (3% SDS, 2mM EDTA, 10% glycerol, 2.5% β-mercaptoethanol in 62.5 mM Tris, pH 6.8). After cooling for 2 min, the samples were incubated with a five-fold volume of pre-chilled methanol at −20°C for 2 h and centrifuged at 14,000 rpm for 20 min at 4°C. The supernatant was discarded and the pellet was resuspended in 30 μl sample buffer. The latter was subjected to SDS-PAGE and immunoblotting.
Velocity sedimentation in sucrose step gradients
Brain homogenates (10%, w/v) in 1X Dulbecco's PBS pH 7.4 were mixed with an equal volume of 2X lysis buffer, then centrifuged for 10 min at 3,000 rpm at 4°C. Supernatants were collected and sarkosyl was added to 1% final concentration. Each sample was loaded atop of 10-60% step sucrose gradients and centrifuged at 200,000 × g in the SW55 rotor for 1 h at 4°C as described with minor modification [33]. After centrifugation, the content of the centrifuge tubes was sequentially removed from the top to the bottom to collect 12 fractions which were subjected to immunoblotting as described below.
One- and two-dimensional gel electrophoresis and immunoblotting
Brain homogenates treated with or without PK were resolved either on 15% Tris-HCl Criterion (Bio-Rad) for one-dimensional (1D) PAGE or on pH gradient (IPG) strips for two-dimensional (2D) PAGE. The latter was performed as described by the supplier with minor modifications using the PROTEIN IEF cell (Bio-Rad) [33]. Briefly, for the 1D PAGE, samples boiled in 2X electrophoresis sample buffer were precipitated by 5-fold volume of pre-chilled methanol at −20 °C for 2 h, followed by centrifugation at 14,000 rpm for 20 min at 4°C. Pellets were resuspended in reducing buffer (8 M urea, 2% CHAPS, 5 mM TBP, and 20 mM Tris, pH 8.0) for 1 h at room temperature and then incubated with 20 mM IAA for 1 h. The samples were incubated with 5-fold volume of pre-chilled methanol at −20°C for 2 h and centrifuged at 14,000 rpm for 20 min at 4°C. Pellets were resuspended in 200 μl of rehydration buffer (7M urea, 2 M thiourea, 1% DTT, 1% CHAPS, 1% Triton X-100, 1% Ampholine pH 3-10, and trace amounts of bromophenol blue). Samples dissolved in rehydration buffer were incubated with IPG strips for 14 h at room temperature with shaking. The rehydrated strips were focused for about 40 kVh. For the second dimension, the focused IPG strips were equilibrated for 15 min in equilibration buffer 1 containing 6M urea, 2% SDS, 20% glycerol, 130 mM DTT, and 375 mM Tris pH 8.8, and then in equilibration buffer 2 containing 6M urea, 2%SDS, 20% glycerol, 135 mM iodoacetamide and 375 mM Tris pH 8.8 for another 15 min. The equilibrated strips were loaded onto 8-16% Tris-glycine Criterion gel (Bio-Rad).
The proteins on the gels were transferred to either Immobilon-P (PVDF, Millipore) or Immobilon-FL membranes (PVDF, LI-COR) for 2 h at 70V. For probing the PrP molecule, the membranes were incubated for 2 h at room temperature with 3F4 (1:40,000) or anti-C-terminal antibody (1:4,000) as primary antibody. Incubation with a secondary antibody was performed either with the horseradish peroxidase-conjugated goat anti-mouse antibody (1: 3,000) or IRDye 800CW conjugated goat anti-mouse (LI-COR). The PrP bands or PrP spots were visualized on either Kodak film by the ECL Plus as described by the manufacturer or Odyssey infrared imaging system (LI-COR® Biosciences, NE, USA).
PrP Immunohistochemistry and histoblotting
Tissue was fixed in formalin for 3 weeks. The following procedures were performed as described [33]. For histological preparations, brain sections were embedded in paraffin and stained with hematoxylin-eosin. For immunohistochemistry sections were deparaffinized, rehydrated, and immersed in 98% formic acid for 1 h at room temperature. Endogenous peroxidase was blocked by immersion in 8% hydrogen peroxide in methanol for 10 min. Sections were completely immersed in 1.5 mM hydrochloric acid and microwaved for 10 min. After rinsing, they were incubated with the mouse monoclonal antibody 3F4 at 1:600, washed and incubated with bridge antibody (goat anti-mouse, Cappel, 1:50) followed by incubation with mouse PAP complex (Sternberger, Meyer Immunocytochemicals, 1:250). Diaminobenzidine tetrahydrochloride was used to visualize the immunoreactivity.
The histoblot was prepared as previously described [75]. Four 8-mm-thick cryosections from each block were transferred to nitrocellulose membranes that had been dampened with lysis buffer (0.5% NP-40, 0.5% sodium deoxycholate, 100 mM NaCl, 10 mM EDTA, 100 mM Tris HCl pH 8.0). The membranes were thoroughly air dried, rehydrated for 30 min in 0.5% Tween 20 with phosphate-buffered saline (PBS-T), dampened with Guanidine hydrochloride 2M (in Millipore H2O) 25 min and handled as follows: 1) no proteases-treatment, 2) digestion with proteases: PK at 10, 25 and 50 μg/ml in lysis buffer (37°C for 1 h), or thermolysin 150 μg/ml (70°C, 1 h). The membranes were then treated in PBS-T containing 5% nonfat dry milk for blocking (30 min at room temperature). After being washed with PBS-T, membranes were incubated with the 3F4 monoclonal antibody (1:10,000) for 2 h at 37°C or overnight at 4°C. Binding was detected by incubation with either peroxidase-conjugated goat anti-mouse IgG antibody for 1 h at 37°C, and the reaction was developed with a chemiluminescence detection kit (ECL Plus, Amersham Biosciences, USA) or by phosphatase-conjugate goat anti-mouse IgG antibody for 1 h at 37°C, and the reaction was developed with BCIP/NBT(5-Bromo-4-chloro-3-indolyl hosphate/Nitro blue tetrazolium).
Acknowledgments
The authors want to thank Diane Kofskey and Phyllis Scalzo for technical support in studies of histology and immunohistochemistry and thank Kay Edmonds, Yovonda Rease and Janis Blevins for coordinating brain tissues and clinical information. The authors also want to thank Drs. Geoff Kneale and John McGeehan from University of Portsmouth, UK, for kindly providing g5p. This study was supported by grants to W.Q.Z. from the National Institutes of Health (NIH) R01NS062787, the CJD Foundation, Alliance BioSecure, the University Center on Aging and Health with the support of the McGregor Foundation and the President's Discretionary Fund (Case Western Reserve University), grants to P.G. from NIH AG-14359, and Center for Disease Control and Prevention Contract UR8/CCU515004, as well as a grant to Q.K. from NIH NS-052319.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1. Prusiner SB. Biology and genetics of prion diseases. Annu Rev Microbiol. 1994; 48: 655 -686. [PubMed] .
- 2. Prusiner SB. Prions. Natl Acad Sci U S A. 1998; 95: 13363 -13383. .
- 3. Tagliavini F, Prelli F, Porro M, Rossi G, Giaccone G, Farlow MR, Dlouhy SR, Ghetti B, Bugiani O, Frangione B. Amyloid fibrils in Gerstmann-Sträussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell. 1994; 79: 695 -703. [PubMed] .
- 4. Gabizon R, Telling G, Meiner Z, Halimi M, Kahana I, Prusiner SB. Insoluble wild-type and protease-resistant mutant prion protein in brains of patients with inherited prion disease. Nat Med. 1996; 2: 59 -64. [PubMed] .
- 5. Chen SG, Parchi P, Brown P, Capellari S, Zou W, Cochran EJ, Vnencak-Jones CL, Julien J, Vital C, Mikol J, Lugaresi E, Autilio-Gambetti L, Gambetti P. Allelic origin of the abnormal prion protein isoform in inherited prion diseases. Nat Med. 1997; 3: 1009 -1015. [PubMed] .
- 6. Silvestrini MC, Cardone F, Maras B, Pucci P, Barra D, Brunori M, Pocchiari M. Identification of the prion protein allotypes which accumulate in the brain of sporadic and inherited Creutzfeldt-Jakob disease patients. Nat Med. 1997; 3: 521 -525. [PubMed] .
- 7. Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci U S A. 1998; 95: 8322 -8327. [PubMed] .
- 8. Rossi G, Giaccone G, Giampaolo L, Iussich S, Puoti G, Frigo M, Cavaletti G, Frattola L, Bugiani O, Tagliavini F. Creutzfeldt-Jakob disease with a novel four extra-repeat insertional mutation in the PrP gene. Neurology. 2000; 55: 405 -410. [PubMed] .
- 9. Tagliavini F, Lievens PM, Tranchant C, Warter JM, Mohr M, Giaccone G, Perini F, Rossi G, Salmona M, Piccardo P, Ghetti B, Beavis RC, Bugiani O, Frangione B, Prelli F. 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Sträussler-Scheinker disease A117V. J Biol Chem. 2001; 276: 6009 -6015. [PubMed] .
- 10. Pietrini V, Puoti G, Limido L, Rossi G, Di Fede G, Giaccone G, Mangieri M, Tedeschi F, Bondavalli A, Mancia D, Bugiani O, Tagliavini F. Creutzfeldt-Jakob disease with a novel extra-repeat insertional mutation in the PRNP gene. Neurology. 2003; 61: 1288 -1291. [PubMed] .
- 11. Zou WQ and Gambetti P. Prion: the chameleon protein. Cell Mol Life Sci. 2007; 64: 3266 -3270. [PubMed] .
- 12. Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and inherited CJD: classification and characterisation. Br Med Bull. 2003; 66: 213 -239. [PubMed] .
- 13. Goldfarb LG, Brown P, Little BW, Cervenáková L, Kenney K, Gibbs CJ Jr, Gajdusek DC. A new (two-repeat) octapeptide coding insert mutation in Creutzfeldt-Jakob disease. Neurology. 1993; 43: 2392 -2394. [PubMed] .
- 14. Laplanche JL, Delasnerie-Lauprêtre N, Brandel JP, Dussaucy M, Chatelain J, Launay JM. Two novel insertions in the prion protein gene in patients with late-onset dementia. Hum Mol Genet. 1995; 4: 1109 -1111. [PubMed] .
- 15. Croes EA, Theuns J, Houwing-Duistermaat JJ, Dermaut B, Sleegers K, Roks G, Van den Broeck M, van Harten B, van Swieten JC, Cruts M, Van Broeckhoven C, van Duijn CM. Octapeptide repeat insertions in the prion protein gene and early onset dementia. J Neurol Neurosurg Psychiatry. 2004; 75: 1166 -1170. [PubMed] .
- 16. Oda T, Kitamoto T, Tateishi J, Mitsuhashi T, Iwabuchi K, Haga C, Oguni E, Kato Y, Tominaga I, Yanai K, Kashima H, Kogure T, Hori K, Ogino K. Prion disease with 144 base pair insertion in a Japanese family line. Acta Neuropathol. 1995; 90: 80 -86. [PubMed] .
- 17. van Gool WA, Hensels GW, Hoogerwaard EM, Wiezer JH, Wesseling P, Bolhuis PA. Hypokinesia and presenile dementia in a Dutch family with a novel insertion in the prion protein gene. Brain. 1995; 118: 1565 -1571. [PubMed] .
- 18. Laplanche JL, Hachimi KH, Durieux I, Thuillet P, Defebvre L, Delasnerie-Lauprêtre N, Peoc'h K, Foncin JF, Destée A. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain. 1999; 122: 2375 -2386. [PubMed] .
- 19. Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986; 46: 417 -428. [PubMed] .
- 20. Bendheim PE and Bolton DC. A 54-kDa normal cellular protein may be the precursor of the scrapie agent protease-resistant protein. Proc Natl Acad Sci U S A. 1986; 83: 2214 -2218. [PubMed] .
- 21. Poulter M, Baker HF, Frith CD, Leach M, Lofthouse R, Ridley RM, Shah T, Owen F, Collinge J, Brown J, Hardy J, Mullan MJ, Harding AE, Bennett C, Doshi R, Crow TJ. Inherited prion disease with 144 base pair gene insertion. 1. Genealogical and molecular studies. Brain. 1992; 115: 675 -685. [PubMed] .
- 22. Collinge J, Brown J, Hardy J, Mullan M, Rossor MN, Baker H, Crow TJ, Lofthouse R, Poulter M, Ridley R, Owen F, Bennett C, Dunn G, Harding AE, Quinn N, Doshi B, Roberts GW, Honavar M, Janota I, Lantos PL. Inherited prion disease with 144 base pair gene insertion. 2. Clinical and pathological features. Brain. 1992; 115: 687 -710. [PubMed] .
- 23. King A, Doey L, Rossor M, Mead S, Collinge J, Lantos P. Phenotypic variability in the brains of a family with a prion disease characterized by a 144-base pair insertion in the prion protein gene. Neuropathol Appl Neurobiol. 2003; 29: 98 -105. [PubMed] .
- 24. Mead S, Poulter M, Beck J, Webb TE, Campbell TA, Linehan JM, Desbruslais M, Joiner S, Wadsworth JD, King A, Lantos P, Collinge J. Inherited prion disease with six octapeptide repeat insertional mutation--molecular analysis of phenotypic heterogeneity. Brain. 2006; 129: 2297 -2317. [PubMed] .
- 25. Capellari S, Vital C, Parchi P, Petersen RB, Ferrer X, Jarnier D, Pegoraro E, Gambetti P, Julien J. Inherited prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology. 1997; 49: 133 -141. [PubMed] .
- 26. Gelpi E, Kovacs GG, Ströbel T, Koperek O, Voigtländer T Liberski PP, Budka H. Prion disease with a 144 base pair insertion: unusual cerebellar prion protein immunoreactivity. Acta Neuropathol. 2005; 110: 513 -519. [PubMed] .
- 27. Kovács T, Beck JA, Papp MI, Lantos PL, Arányi Z, Szirmai IG, Farsang M, Stuke A, Csillik A, Collinge J. Familial prion disease in a Hungarian family with a novel 144-base pair insertion in the prion protein gene. J Neurol Neurosurg Psychiatry. 2007; 78: 321 -323. [PubMed] .
- 28. Vital A, Laplanche JL, Bastard JR, Xiao X, Zou WQ, Vital C. A case of Gerstmann-Sträussler-Scheinker disease with a novel six octapeptide repeat insertion. Neuropathol Appl Neurobiol. 2011; 37: 554 -559. [PubMed] .
- 29. Alner K, Hyare H, Mead S, Rudge P, Wroe S, Rohrer JD, Ridgway GR, Ourselin S, Clarkson M, Hunt H, Fox NC, Webb T, Collinge J, Cipolotti L. Distinct neuropsychological profiles correspond to distribution of cortical thinning in inherited prion disease caused by insertional mutation. J Neurol Neurosurg Psychiatry. 2012; 83: 109 -114. [PubMed] .
- 30. Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG. Antibody to DNA detects scrapie but not normal prion protein. Proc Natl Acad Sci U S A. 2004; 101: 1380 -1385. [PubMed] .
- 31. Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998; 4: 1157 -1165. [PubMed] .
- 32. Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet. 2001; 358: 171 -180. [PubMed] .
- 33. Yuan J, Xiao X, McGeehan J, Dong Z, Cali I, Fujioka H, Kong Q, Kneale G, Gambetti P, Zou WQ. Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J Biol Chem. 2006; 281: 34848 -34858. [PubMed] .
- 34. Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D, Belay ED, Schonberger LB, Marder K, Harris C, Burke JR, Montine T, Wisniewski T, Dickson DW, Soto C, Hulette CM, Mastrianni JA, Kong Q, Zou WQ. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008; 63: 697 -708. [PubMed] .
- 35. Glatzel M, Abela E, Maissen M, Aguzzi A. Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. N Engl J Med. 2003; 349: 1812 -1820. [PubMed] .
- 36. Zanusso G, Ferrari S, Cardone F, Zampieri P, Gelati M, Fiorini M, Farinazzo A, Gardiman M, Cavallaro T, Bentivoglio M, Righetti PG, Pocchiari M, Rizzuto N, Monaco S. Detection of pathologic prion protein in the olfactory epithelium in sporadic Creutzfeldt-Jakob disease. N Engl J Med. 2003; 348: 711 -719. [PubMed] .
- 37. Yuan J, Dong Z, Guo JP, McGeehan J, Xiao X, Wang J, Cali I, McGeer PL, Cashman NR, Bessen R, Surewicz WK, Kneale G, Petersen RB, Gambetti P, Zou WQ. Accessibility of a critical prion protein region involved in strain recognition and its implications for the early detection of prions. Cell Mol Life Sci. 2008; 65: 631 -643. [PubMed] .
- 38. Zou WQ, Langeveld J, Xiao X, Chen S, McGeer PL, Yuan J, Payne MC, Kang HE, McGeehan J, Sy MS, Greenspan NS, Kaplan D, Wang GX, Parchi P, Hoover E, Kneale G, Telling G, Surewicz WK, Kong Q, Guo JP. PrP conformational transitions alter species preference of a PrP-specific antibody. J Biol Chem. 2010; 285: 13874 -13884. [PubMed] .
- 39. Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, Shimoji M, Langeveld JP, Castellani R, Notari S, Crain B, Schmidt RE, Geschwind M, Dearmond SJ, Cairns NJ, Dickson D, Honig L, Torres JM, Mastrianni J, Capellari S, Giaccone G, Belay ED, Schonberger LB, Cohen M, Perry G, Kong Q, Parchi P, Tagliavini F, Gambetti P. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010; 68: 162 -172. [PubMed] .
- 40. Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, Schulz-Schaeffer W, Kretzschmar H, Raeber A, Braun U, Ehrensperger F, Hornemann S, Glockshuber R, Riek R, Billeter M, Wüthrich K, Oesch B. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature. 1997; 390: 74 -77. [PubMed] .
- 41. Zou WQ and Cashman NR. Acidic pH and detergents enhance in vitro conversion of human brain PrPC to a PrPSc-like form. J Biol Chem. 2002; 277: 43942 -43947. [PubMed] .
- 42. Holmes BB and Diamond MI. Cellular mechanisms of protein aggregate propagation. Curr Opin Neurol. 2012; 25: 721 -726. [PubMed] .
- 43. Kumar S and Walter J. Phosphorylation of amyloid beta (Aβ) peptides - a trigger for formation of toxic aggregates in Alzheimer's disease. Aging (Albany NY). 2011; 3: 803 -812. [PubMed] .
- 44. Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, Ross CA. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle. 2012; 11: 2006 -2021. [PubMed] .
- 45. Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994; 35: 513 -529. [PubMed] .
- 46. Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of -Jakob disease and related diseases to rodents. Neurology. 1996; 46: 532 -537. [PubMed] .
- 47. Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A. 2007; 104: 4712 -4717. [PubMed] .
- 48. Zou WQ. Transmissible spongiform encephalopathy and beyond (E-letter). Science. http://www.sciencemag.org/content/308/5727/1420.long/reply#sci_el_10316 2007; .
- 49. Zou RS, Fujioka H, Guo JP, Xiao X, Shimoji M, Kong C, Chen C, Tasnadi M, Voma C, Yuan J, Moudjou M, Laude H, Petersen RB, Zou WQ. Characterization of spontaneously generated prion-like conformers in cultured cells. (Albany NY). 2011; 3: 968 -984. .
- 50. Zou WQ, Zhou X, Yuan J, Xiao X. Insoluble cellular prion protein and its association with prion and Alzheimer diseases. Prion. 2011; 5: 172 -178. [PubMed] .
- 51. Nicholl D, Windl O, de Silva R, Sawcer S, Dempster M, Ironside JW, Estibeiro JP, Yuill GM, Lathe R, Will RG. Inherited Creutzfeldt-Jakob disease in a British family associated with a novel 144 base pair insertion of the prion protein gene. J Neurol Neurosurg Psychiatry. 1995; 58: 65 -69. [PubMed] .
- 52. Owen JP, Maddison BC, Whitelam GC, Gough KC. Use of thermolysin in the of prion diseases. Mol Biotechnol. 2007; 35: 161 -170. [PubMed] .
- 53. Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, Wadsworth JD. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J. 2008; 416: 297 -305. [PubMed] .
- 54. Brandner S, Raeber A, Sailer A, Blättler T, Fischer M, Weissmann C, Aguzzi A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci U S A. 1996; 93: 13148 -13151. [PubMed] .
- 55. Race R and Chesebro B. Scrapie infectivity found in resistant species. Nature. 1998; 392: 770 [PubMed] .
- 56. Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci U S A. 2000; 97: 10248 -10253. [PubMed] .
- 57. Lasmézas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science. 1997; 275: 402 -405. [PubMed] .
- 58. Collinge J, Owen F, Poulter M, Leach M, Crow TJ, Rossor MN, Hardy J, Mullan MJ, Janota I, Lantos PL. Prion dementia without characteristic pathology. Lancet. 1990; 336: 7 -9. [PubMed] .
- 59. Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, Gray F, Cortelli P, Montagna P, Ghetti B, Goldfarbt LG, Gajdusekt DC, Lugaresit E, Gambetti P, Autilio-Gambetti L. Fatal inherited insomnia and inherited Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A. 1994; 91: 2839 -2842. [PubMed] .
- 60. Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998; 279: 827 -834. [PubMed] .
- 61. Ma J and Lindquist S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 2002; 298: 1785 -1788. [PubMed] .
- 62. Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, Gabizon R, Taraboulos A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002; 41: 12868 -12875. [PubMed] .
- 63. Prusiner SB. Molecular biology of prion diseases. Science. 1991; 252: 1515 -1522. [PubMed] .
- 64. Jarrett JT and Lansbury PT Jr.. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993; 73: 1055 -1058. [PubMed] .
- 65. Daude N, Lehmann S, Harris DA. Identification of intermediate steps in the conversion of a mutant prion protein to a scrapie-like form in cultured cells. J Biol Chem. 1997; 272: 11604 -11612. [PubMed] .
- 66. Jansen K, Schäfer O, Birkmann E, Post K, Serban H, Prusiner SB, Riesner D. Structural intermediates in the putative pathway from the cellular prion protein to the pathogenic form. Biol Chem. 2001; 382: 683 -691. [PubMed] .
- 67. Apetri AC, Maki K, Roder H, Surewicz WK. Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J Am Chem Soc. 2006; 128: 11673 -11678. [PubMed] .
- 68. Yankner BA, Caceres A, Duffy LK. Nerve growth factor potentiates the neurotoxicity of beta amyloid. Proc Natl Acad Sci U S A. 1990; 87: 9020 -9023. [PubMed] .
- 69. Lorenzo A and Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A. 1994; 9: 12243 -12247. [PubMed] .
- 70. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998; 95: 6448 -6453. [PubMed] .
- 71. Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999; 19: 8876 -8884. [PubMed] .
- 72. Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002; 277: 32046 -32053. [PubMed] .
- 73. Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987; 61: 3688 -3693. [PubMed] .
- 74. Goldfarb LG, Brown P, Vrbovská A, Baron H, McCombie WR, Cathala F, Gibbs CJ Jr, Gajdusek DC. An insert mutation in the chromosome 20 amyloid precursor gene in a Gerstmann-Sträussler-Scheinker family. J Neurol Sci. 1992; 111: 189 -194. [PubMed] .
- 75. Taraboulos A, Jendroska K, Serban D, Yang SL, DeArmond SJ, Prusiner SB. Regional mapping of prion proteins in brain. Proc Natl Acad Sci U S A. 1992; 89: 7620 -7624. [PubMed] .