



Introduction
Aging is a complex biological process marked by the gradual decline of a multitude of physiological processes [1-5]. Some functional changes, such as decreased muscular strength, have a tremendous impact on the quality of life. Normal aging involves sarcopenia, a combination of atrophy and decreased muscular strength that develops despite dietary interventions and increased physical activity [6, 7]. The physical, psychological and socio-economic impact of sarcopenia is largely underestimated despite the fact that it is a leading contributor to debilitating injuries due to repetitive falls, loss of independence, and a reduced quality of life in the elderly population [4]. Understanding the cellular mechanisms that contribute to sarcopenia is essential for the development of effective treatments and improved care for the elderly.
While many cellular modifications [8-13] may contribute to muscle aging, excitation-contraction (E-C) coupling is an elemental process that must be considered because it is so integral to the control of muscle contractility. E-C coupling requires the close coordination of extracellular Ca2+ ([Ca2+]o) entry or voltage-sensing changes with Ca2+ release from the principal intracellular storage organelle, the sarcoplasmic reticulum (SR). Changes in E-C coupling machinery may act as causative factors for, or adaptive mechanisms in, muscle aging that directly contribute to muscle weakness. Decreased muscle function during aging has been associated with muscle fiber denervation, loss of motor units, and motor unit remodeling. Moreover, altered function of several triad junction proteins involved in the regulation and transduction of E-C coupling has been shown to contribute to disrupted Ca2+ homeostasis in aged skeletal muscle [14-17].
Thus, reduced homeostatic capacity for intracellular Ca2+ movement may underlie the progression of sarcopenia and contractile dysfunction during muscle aging. We have previously demonstrated that changes in aged skeletal muscle Ca2+ homeostasis are accompanied by distinct modification of the structure and protein composition of triad junctional complexes. We found that expression of mitsugumin 29 (MG29), a triad junction protein, drastically decreases with aging and that skeletal muscle from adolescent MG29 knockout mice contain many of the phenotypic changes we observe in aged skeletal muscle, including defective store-operated Ca2+ entry (SOCE) [16-18]. SOCE is an important mechanism linking [Ca2+]o entry and intracellular Ca2+ storage, in particular during the repetitive cycles of E-C coupling when a reduced SR Ca2+ store necessitates the activation of SOCE. Several previous studies have demonstrated that SOCE plays a role in muscle fatigue [19-22] as part of a functional stress response in skeletal muscle [18].
However, to date, to the best of our knowledge, no systematic studies have yet been performed to test if a reduced SOCE function contributes to the decreased contractile capacity of aged skeletal muscles. Furthermore, it is not known if and how SOCE contributes to normal muscle contractility, two essential physiological questions that we aimed to answer in these novel studies.
To examine the contribution of SOCE to muscle contractile function during aging, we made use of ex vivo contractility assays, in which the components of the extracellular milieu surrounding isolated anatomical muscles can be precisely manipulated. This allowed use of pharmacological and experimental manipulations to alter the function of SOCE in order to define its physiological role in young and aged skeletal muscle. We found that reagents that prevent [Ca2+]o entry reduce contractile force in skeletal muscle, an effect that was more prominent at high frequency stimulation as compared to lower frequencies of stimulation, and that SOCE is a robust mechanism of [Ca2+]o entry in young, healthy skeletal muscle. Of utmost importance, we also demonstrate that SOCE is compromised in aged skeletal muscle, and that loss of this physiological component contributes to at least a part of the decreased contractile force generation typical of aged muscle.
Our novel studies that now link SOCE with muscle contractile function, suggest that manipulation of SOCE may present a therapeutically valid target for improvement of contractile function and, potentially the amelioration of muscle weakness seen during aging.
Results
Extracellular Ca2+ contributes to tetanic contractile force in skeletal muscle
In the first series of ex vivo contractility experiments, it was critical to validate that muscle preparation deterioration or run-down did not play any role in our results. Fig. 1 shows an example that is representative of the muscle preparations used in our studies. Fig. 1A demonstrates that our preparations can undergo very lengthy protocols without any detectable run-down of contractile force. When a single muscle fiber is dissected from the intact muscle, it produces a maximal tetanic force that is equivalent to the amount force produced by the same fiber after it is permeabilized with Triton X-100 and maximally activated with calcium (Figs. 1B, C). These results illustrate the robustness of our preparations and the validity of our force calibrations.
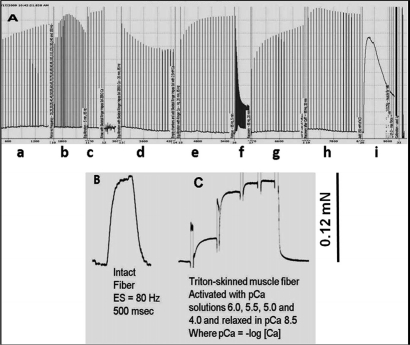
Figure 1. Original and unfiltered recording of a SOL muscle from Wt mice illustrates robustness of our preparations as it can undergo a multitude of experimental manipulations during a prolonged protocol without signs of deterioration. (A) An entire protocol of more than 6h in duration in a soleus (SOL) muscle is shown. The experimental points shown in this record are: a) Initial equilibration period where muscle is carefully stretched; b) Force vs. frequency relationship, c) Equilibration, followed by solution change (from 2.5 mM Ca2+ to zero Ca2+ + 0.1 mM EGTA), d) Equilibration in Zero Ca2+, followed by change of solution back to 2.5 mM Ca2+, e) Recovery in 2.5 mM Ca2+, f) Fatiguing stimulation, g) Recovery from fatigue in the absence of caffeine, h) Recovery in the presence of 20 mM caffeine, i) After stimulation being stopped, the muscle is treated with 80 mM KCl and still produces a very large response to KCl that approximates the maximal tetanic force. Preparation produced 11 g (~107 mN) of relative force. (B) A single muscle fiber was dissected from the same intact SOL muscle shown in panel A. Single muscle fiber was the electrically stimulated with 80 Hz. and produced ~ 0.12 mN of force. The same fiber was then chemically skinned with Triton X-100 and produced a maximal contractile force of ~ 0.12 mN. From our own observations, we estimate that a SOL muscle has ~925 muscle fibers; thus, the predicted contractile force for this preparation is ~111 mN (0.12 mN/fiber × 925 = 111 mN), which is very similar to the relative force of 107 mN determined by the force calibration in our intact muscle system.
SOCE is dependent upon extracellular calcium. To begin testing the role of extracellular Ca2+ in skeletal muscle contractility, Ca2+ was removed from the extracellular solution bathing intact, isolated extensor digitorum longus (EDL, glycolitic mostly ast-twitch) muscle fibers by chelation with 0.1 mM EGTA. Under these conditions, we observed a decrease in the contractile force following high frequency stimulation, while the low frequency component is not significantly altered (Fig. 2A). This suggests that during high frequency tetanic contractions, repetitive cycles of SR Ca2+ release results in loss of Ca2+ from intracellular SR stores to trigger entry of Ca2+ into muscle fibers. In multiple experiments, we observed full recovery of contractile force upon restoration of 2.5 mM [Ca2+]o by washout of EGTA-containing solution (n=18/20) (Fig. 2B). Note that both the initial loss of force and recovery after washout are rapid, suggesting that the effect of [Ca]o may be related to direct shuttling of Ca2+ across the sarcolemmal membrane through mechanisms that these type of experiments are not able to reveal. Our next step was to investigate if the extracellular Ca2+ dependence observed in EDL muscles was also present in the soleus (SOL, oxidative mostly slow-twitch) muscle. We found that removal of extracellular Ca2+ actually produced a larger drop in contractile force in young SOL as compared to young EDL muscles (Fig. 2C-D). Further evidence of the necessity of [Ca2+]o entry in the maintenance of skeletal muscle contractility is provided by a series of experiments that inhibit [Ca2+]o entry through inclusion of NiCl2 in the extra-cellular solution, which has been shown to specifically inhibit a component of Ca2+ entry in smooth muscle cells that is not affected by either nifedipine or verapamil, which suggest its selectivity to block the SOCE machinery [23, 24]. Since the experiments shown in Fig. 2 were performed at ambient temperature (~25°C) and physiological mechanisms may display dependence on temperature, we conducted these contractility experiments involving NiCl2 at the 37°C to establish the extent to which Ca2+ entry effects contraction at physiologically normal temperature (Fig. 3). These experiments revealed that a similar effect of [Ca2+]o entry on contractility can be observed at physiological temperatures. As shown in Fig. 3A, addition of 1 mM Ni2+ to the extracellular solution results in a nearly instantaneous drop in the contractile force of isolated EDL muscles. The effect of Ni2+ was completely reversible after washout in the majority of the experiments (Fig. 3B, n=9/11). Similarly, the SOL muscles also displayed a component of contractile force that is sensitive to inhibition by NiCl2 (Fig. 3C). As summarized in Figs. 2D and 3D, SOL muscles demonstrate a larger Ca2+ dependence component in both Ca removal and SOCE blockade with Ni2+. As with chelation of [Ca2+]o by EGTA we found that Ni2+ has more pronounced effects on the contractile force developed during high frequency stimulation than during low frequency stimulation, further suggesting that under conditions of high Ca2+ demand, [Ca2+]o exerts a more significant role in skeletal muscle contractility. An intriguing observation during the NiCl2 experiments was the transient increase in the low frequency force (Fig. 3A). It is possible that at these lower frequencies, the addition of Cl- induced Ca2+ release from the SR, a phenomenon previously reported by Stephenson et al [25]. This result actually provides very strong evidence that a drop in force at higher frequencies of stimulation in response to NiCl2 is not artifactual. From the measurement of force-frequency relationship, one can clearly see that Ni2+ effects were more pronounced at higher frequencies of stimulation (Fig. 4A) supporting the concept that SOCE may contribute to DPH muscle contractility under conditions of higher demand, which might have important clinical implications, since DPH fatigue can lead to serious disease and death. In Fig. 4B, DPH muscles were contracted with a high frequency of 200Hz. Clearly, a fast initial drop in force within 1min is easily detected. This initial drop in force is followed by an additional time-dependent force reduction component, since a further reduction in force is observed in the following 8min of exposure to Ni2+ (Fig. 4B). These data further confirm our findings in EDL and SOL muscles, and suggest that SOCE response is a universal phenomenon in different muscle types of young animals.
![Removal of [Ca2+]o reduces tetanic contractile force in young skeletal muscle.](https://cdn.aging-us.com/article/100335/figure/F2/large.png)
Figure 2. Removal of [Ca2+]o reduces tetanic contractile force in young skeletal muscle. (A) An Intact EDL muscle was electrically stimulated with low and high frequency in a bath solution with either 2.5 mM [Ca]o (control) or 0 [Ca]o+0.1 mM EGTA. (B) An Intact SOL muscle was electrically stimulated with high frequency in a bath solution with either 2.5 mM [Ca]o (control) or 0 [Ca]o+0.1 mM EGTA. Fast recovery upon return of preparations to 2.5 mM [Ca]o is observed. (C) Data summary for the effects of 0 [Ca]o on EDL muscle at T = 25° C (n = 12, p < 0.01). (D) Data summary for the effects of 0 [Ca]o on SOL muscle at T = 25° C (n = 10, p < 0.01).
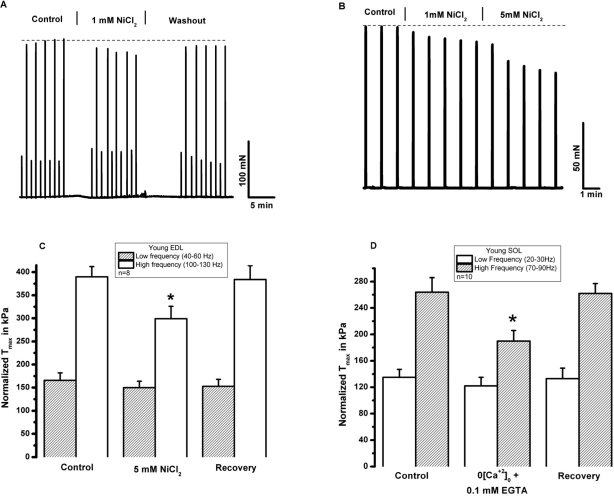
Figure 3. Extracellular Ni2+ reduces tetanic force in young skeletal muscle. (A) An intact EDL muscle was electrically stimulated at 37°C with low frequency and high-frequency in a bath solution with 2.5 mM [Ca]o (control) and following addition of 1 mM NiCl2. Ni inhibits force generated with high- but not low-frequency stimulation. (B) An intact SOL muscle was electrically stimulated at 37°C with high-frequency in a bath solution with 2.5 mM [Ca]o (control) and following addition of 1mM and 5mM NiCl2. A cumulative effect to NiCl2 is noted. (C) Data summary for the effects of NiCl2 on EDL muscle at T = 37° C (n = 8, p < 0.01). (D) Data summary for the effects of NiCl2 on SOL at T = 37° C (n = 6, p < 0.01).
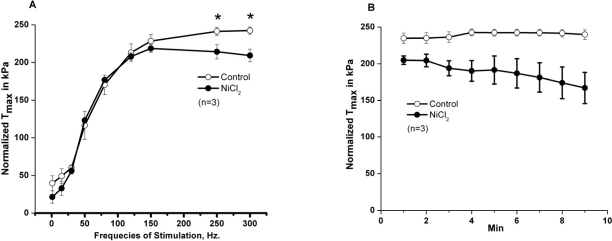
Figure 4. Effects of Ni2+ on contractility of young diaphragm muscle. (A) Force vs. frequency relationship in control diaphragm muscles (open circles) or diaphragm muscles treated with NiCl2 (solid circles). (n = 3). (B) Intact diaphragm muscle bundles from young mice were electrically stimulated at 37°C with high frequency (200 Hz/min) in the absence (open circles, control) or in the presence of 3 mM NiCl2 (solid circles, NiCl2).
Aged skeletal muscle displays reduced contractile force and negligible [Ca2+]o dependence
Extending our contractile measurements to preparations of EDL muscles from aged mice, we first confirmed our previously determined decrease of ~30% in normalized specific contractile force in this tissue (Fig. 5A). Since this reduction in force is already normalized to the different cross-sectional areas of young and aged muscles, such difference is independent of age related atrophy. These experiments revealed quite surprising phenotypic changes in aged muscle - a remarkable suppression of the sensitivity of aged muscle to inhibition of contractile force by inhibition of [Ca2+]o entry. As illustrated in Fig. 5B, there are no detectable effects of Ni2+ on the high-frequency tetanic force in aged muscles. Similarly to the young muscles, Ni2+ had a tendency to increase the low frequency force, but by the end of the recovery force the low frequency force returned to its control levels. Over the course of several experiments, we found only negligible effects of extracellular Ni2+ on muscle contractility (Fig. 5C). Additionally, we found that chelating [Ca2+]o with EGTA had no significant acute impact on the contractile function of aged EDL skeletal muscle (Fig. 5D). Complementary experiments in aged soleus muscle demonstrated similar effects of Ni2+ and 0[Ca2+]o on contractility (Fig. 5D). These results indicate that the acute functional contribution of [Ca2+]o to skeletal muscle contractility is impaired in aged skeletal muscles, and stands in stark contrast to the significant contribution of [Ca2+]o to acute contractility in young muscle.
![Reduced specific force in aged skeletal muscle associates with blunted [Ca2+]o dependence.](https://cdn.aging-us.com/article/100335/figure/F5/large.png)
Figure 5. Reduced specific force in aged skeletal muscle associates with blunted [Ca2+]o dependence. (A) Maximal tetanic force normalized to the cross sectional area in young (red) and aged (green) EDL muscles (n = 12, * p < 0.001). (B) Intact EDL muscle from aged mice subjected to the same protocol as in Figs 2-3, demonstrates that NiCl2 lacks an effect in aged muscle. (C) Summary data for the effects of NiCl2 and washout after treatment (n = 8). (D) Summary data for the effects of 0[Ca2+]o and washout after treatment (n = 6).
Mechanisms of loss of Ca2+-dependence in aged skeletal muscle
Our results indicate that the dependence of skeletal muscle on [Ca2+]o is lost in aged muscle. Elucidation of the mechanism contributing to this loss of Ca2+-dependence may provide a therapeutic target for increasing contractile force and function in aged skeletal muscle or perhaps new insights into the prevention of muscle loss in young subjects during aging. To establish whether the effect of [Ca2+]o entry on contractility of young skeletal muscle was mediated by the L-type Ca2+ channel, we incubated the EDL or soleus muscles with nifedipine, an inhibitor of this channel, at a concentration (10 μM) previously shown to block Ca2+ entry via the L-type Ca channel during excitation-coupled Ca2+ entry (ECCE) in C2C12 myotubes [26] As shown in Fig. 6, there was no significant decrease in contractility observed with up to 13 minutes of exposure to nifedipine, a finding in agreement with previous reports from Reid et al. [27; 28]. Therefore, the mechanism that underlies [Ca2+]o entry contribution to contractility is likely to involve pathways other than those linked to the L-type channels. Our findings further suggest that this mechanism must be functional in skeletal muscle from young, but not aged, individuals. Considering our previous findings indicate compromised SOCE in aged skeletal muscle, we hypothesize that SOCE could be one such pathway. To test this hypothesis, we examined how abrogated SOCE might affect force production in young and aged skeletal muscle. First, we examined the effects of BTP-2, which has been shown to be a specific blocker of SOCE in skeletal muscles [29], on contractility of young and aged muscle. We found that BTP-2 reduce the high frequency tetanic force produced by EDL and SOL muscles from young mice (Fig. 7A and 7B), further suggesting that a component of force generation in young, healthy skeletal muscles derives from functional SOCE. In striking contrast, BTP-2 did not have noticeable effects on the contractile force generation of aged EDL and SOL muscles (Fig. 7C and 7D).
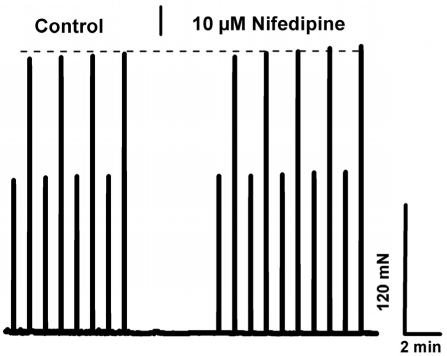
Figure 6. L-type/DHPR inhibition does not explain ex-tracellular Ca+2 dependence in young skeletal muscles. Intact EDL muscle from young mice subjected to the low/high frequency stimulation protocol demonstrates that 10 μM nifedipine lacks an effect in young muscle.
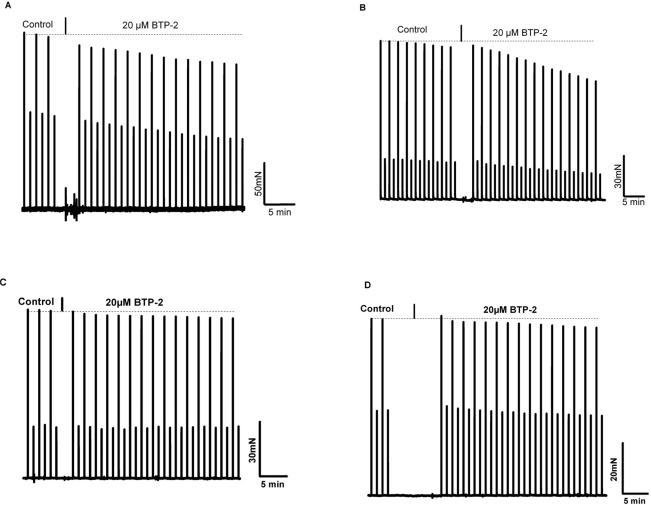
Figure 7. BTP-2, a selective and potent inhibitor of SOCE reduces contractile force in young but not in aged skeletal muscle. (A) Intact EDL muscle stimulated with low and high frequencies every minute at 37°C; in control (no drug) contractions are shown, followed by the progressive effect of 20 μM BTP-2. On average the decrease in force with 20 μM it was 19 ± 4%. (B) Same protocol as in panel A, but using young SOL muscles, representative of 3 experiments. In these 3 experiments force decreased by 31 ± 7% in 15 min in SOL muscles. (C) EDL muscles from aged Wt mice were equilibrated with 30 and 80 Hz under control condition and then were exposed to 20 μM BTP-2. (D) SOL muscles from aged WT mice tested with the same protocol as shown for aged EDL muscles in panel C. No statistical differences between control and BTP-2 were seen in aged EDL and SOL muscles.
Previous studies by Bolotina and colleagues [30, 31] showed that bromoenol lactone (BEL) is a potent inhibitor of SOCE, while studies by Gong and Reid found that BEL can reduce contractile force in skeletal muscle from young adult mice [32]. Thus, a series of studies were conducted a series of experiments to test if inhibition of SOCE by BEL produced differential effects on contractility in young or aged skeletal muscle fibers. Since BEL has not been used to inhibit SOCE in primary skeletal muscle, we first validated that BEL was effective under these conditions. We conducted Ca2+ imaging studies in individual flexor digitorum brevis (FDB) muscle cells to measure SOCE using Mn2+-quenching of Fura-2 fluorescence to measure SOCE [33]. Our data with young isolated FDB muscle fibers revealed that 25 μM BEL could almost completely inhibit SOCE elicited by depletion of the SR Ca2+ stores using thapsigargin (TG) or caffeine/ryanodine treatment (Fig. 8). Knowing that BEL acted as a very effective blocker of SOCE in young FDB muscle cells, we then used mechanically-skinned muscle fibers to monitor Ca2+ exit from the T-tubules via SOCE using the fluorescence of Rhod-5N trapped in the T-tubule space as a reporter [16, 34]. Under these conditions, BEL effectively blocked SOCE as monitored with confocal imaging of Rhod-5 fluorescence in young muscle fibers (Fig. 9A), but had negligible effect in aged ones (Fig. 9B). Our last series of experiments was to investigate the effects of BEL in contractility experiments, and we found that BEL can reduce the high-frequency force production of young EDL muscle (Fig. 10A), providing additional pharmacological evidence that extracellular Ca2+ is linked to force production in skeletal muscle. When these experiments are conducted with aged skeletal muscle there is no decrease in contractile force following BEL application (Fig. 10B). Thus, the SOCE-dependent component of skeletal muscle contractility that is present in young skeletal muscle is no longer active in aged skeletal muscle, and parallels at least part of the loss of the contractile force in this tissue [13] (see also figures in the manuscript indicating the atrophy-related and atrophy-independent components of force generation in aged muscles and accompanying Editorial Commentary).
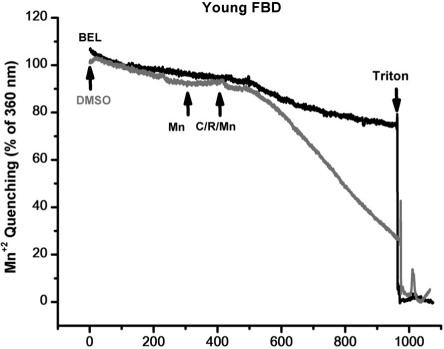
Figure 8. Mn+2 quenching of Fura-2 in FDB muscles of young muscle validates the use of BEL as an effective SOCE blocker. Enzymatically dissociated FDB muscle fibers were tested for their SOCE activity using the Mn+2 quenching assay. BEL was able to block most 90% of the SOCE response under these experimental conditions.
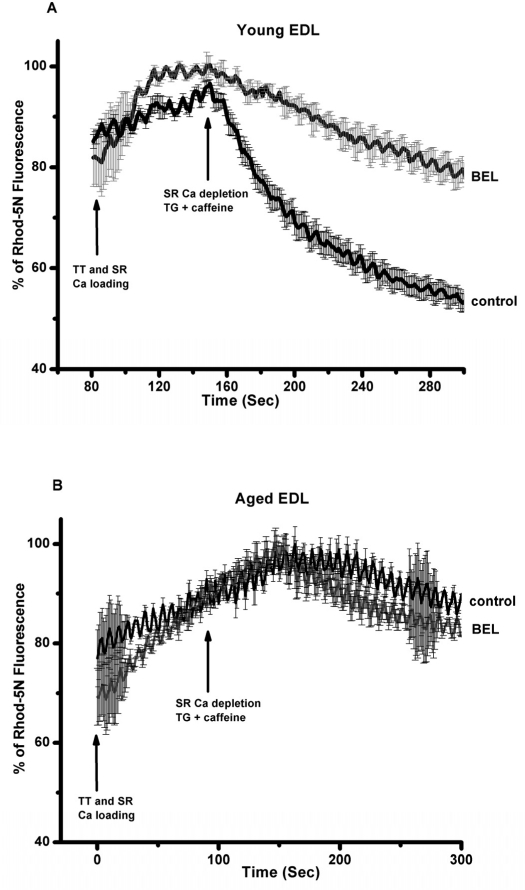
Figure 9. Direct visualization of SOCE in mechanically EDL skinned muscle fibers of young and old muscles with BEL reveals robust SOCE in young but compromised SOCE in aged muscle. Confocal imaging of Ca movement from the sealed T-tubules of mechanically skinned muscle fibers from young and aged mice demonstrates that SOCE is fully function in young, but severely ablated in aged muscle fibers.
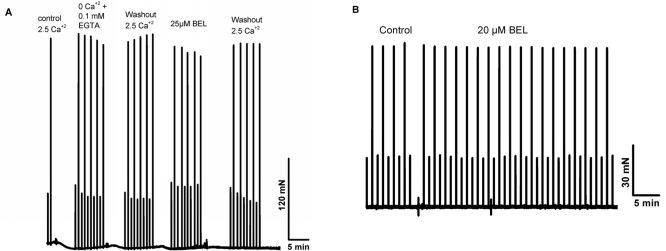
Figure 10. BEL reduces contractile force in young but not in aged skeletal muscle. Intact EDL and SOL muscles from young and aged muscles reveal that this agent is able to reduce the contractile force in only young muscles.
Discussion
As aging populations grow world-wide (It has been estimated that by 2050 more than 200 million humans will have sarcopenia), the aging-related decline in muscle function becomes a more pressing public health concern that requires more effective therapeutic approaches, which in turn requires an understanding of sarcopenia at a mechanistic level. While a portion of the decrease in muscular strength during aging can be explained by muscle atrophy, it is clear that other components contribute to reduced force production during aging. Furthermore, it is unquestionable that an array of cellular modifications [8-13] may contribute to muscle aging, and the pleiotropic nature of muscle aging muscle be recognized and accepted if we are to make significant progress towards its prevention and management.
While the role of SOCE during aging is largely unexplored, we recently had established the novel finding that SOCE is compromised in aged skeletal muscle [16], but our inaugural studies failed to directly link reduced SOCE in aged muscles with decreased contractile force in aged muscles. Now, our new data reveals that a functionally significant, SOCE-dependent component of skeletal muscle contractility in young muscle is lost in aged skeletal muscle. A viable hypothesis for atrophy-independent muscle weakness in aging, therefore, involves substantial abrogation of SOCE, a process vital to the full functionality of E-C coupling.
Our studies indicate that in young skeletal muscle, SOCE contributes to maintenance of contractile force under high frequency stimulation, where Ca2+ requirements may exceed the capacity of the intracellular store to provide for sustained and repetitive contractions, almost as if there is a fatigue component to the SOCE regulation of contractile force. The fact that force generation in skeletal muscle from aged animals is significantly reduced under conditions of high-frequency stimulation suggest that this tissue has lost its ability to use [Ca2+]o during periods of increased demand, a phenomenon we recently introduced [18]. Our data now suggests that SOCE is actually required for repetitive stimulation of skeletal muscles, and its loss in aging significantly contributes to the loss of contractile function in the skeletal muscle.
Intriguingly, reduced SOCE during the aging process is not a phenomenon limited to skeletal muscles, but is also present in other tissues, such as fibroblasts and neuronal cells [35, 36]. In addition, it is clear from the recent report by Stiber, that STIM1, and hence SOCE, is required for the normal development of skeletal muscle [22]. Our findings that [Ca2+]o contributes directly to contractility in young skeletal muscles is different than previous findings - that [Ca2+]o does not normally contribute to skeletal muscle contractility, except in a subpopulation of aged muscle fibers [37]. This may be due to the fact that most of the earlier studies suggesting a relative lack of effect of [Ca2+]o on muscle contractility were not performed under conditions that could allow detection of the effect that we observe, including the fact that many of these studies were performed in amphibian muscle, and also in some cases, these studies were conducted in single muscle fibers that might behave in ways that are different from the intact, anatomical muscle. Also, since Delbono and collaborators showed that a subpopulation of aged muscle fibers were dependent on extracellular calcium entry, while another subpopulation was not dependent on extracellular calcium entry, it does make sense that in a intact muscles, where all muscle fibers are combined, such difference is not observed. Furthermore, our results come from use of a previously unexploited high-frequency stimulation (using 80-200 Hz of 500-1000ms duration, with single pulses of 1ms duration) paradigm that mimics states when demand for Ca2+is high to elucidate the functional effects of SOCE on the contractility of three distinct intact muscle types (EDL, soleus and diaphragm) from mice. In addition, more potent SOCE inhibitors are now available and utilization of a small but effective concentration of 0.1 mM EGTA in the zero Ca+2 solution was critical for full observation of these effects, likely required to buffer the large amount of calcium in the T-tubular system. One of our most significant findings is that skeletal muscle contractile function under conditions of low-frequency stimulation remains mostly [Ca2+]o-independent, while significant contribution of SOCE to contractility is more prominent under high frequency stimulation. In line with our hypothesis that there is a loss of functional SOCE in aged muscle, there was no effect on force by the application of EGTA, NiCl2, BTP-2, and BEL in aged muscle. The utilization of ex vivo, intact isolated muscles offers the advantage of robust preparations, as evidenced by our previous report that LDH is essentially undetectable in the extracellular bathing solution of our preparations [38, 39]. Our findings indicate that the SOCE-dependent component of contractility we observed in young muscle is lost in aged muscle, which also display reduced SOCE activity (See Figs 8-9 and [16]). Such findings provide the possibility that increasing SOCE activity in aged muscle may restore this lost component of contractile force and increase force generation. In light of our present results and our recent work with MG29 null mice [16, 17], the parallels between defective SOCE and reduced MG29 levels in aged skeletal muscle may be of significant physiological import. Indeed, these findings suggest that modulation of MG29 function in aged skeletal muscle provides a potential therapeutic target to enhance SOCE and increase muscle contractility in aged patients. This finding is further supported by the recent association of mitsugumins in human muscle diseases [40]. Yet, another molecule of interest might be MIP/MTMR14, since we recently showed that mice lacking this molecule display phenotypic muscle changes that mirror muscle wasting and muscle weakness during aging [13].
In summary, we show for the first time that SOCE plays a significant role in normal contractility in young, healthy skeletal muscle. In striking contrast, in aged skeletal muscle, we now show that compromised SOCE is linked to decreased contractile force. We postulate that a chronically compromised SOCE leads to an equally chronic reduction in the amount of Ca2+ stored in the SR, creating a negative feedback loop, less Ca2+ is available, less is released, with less force being produced. Interestingly, the absolute force produced by young, healthy muscles when SOCE is inhibited approximates the amount of force produced by aged muscles. Thus, SOCE should be seen as an important functional biomarker of muscle function during aging, and it may provide a suitable target for therapeutic intervention for muscle diseases. Whether the loss of SOCE contributes to muscle atrophy during sarcopenia, or whether it is the reflection of the pleiotropic adaptive response during aging, is still very much an open question.
Materials and Methods
Muscle Preparation
[17, 34, 41]. Intact extensor digitorum longus (EDL), diaphragm (DPH) and soleus (SOL) muscles of wild type (Wt) male mice, 5 and 24 month old, were surgically removed from tendon to tendon by blunt dissection and immediately placed in a dissecting dish containing a modified bicarbonate Ringer solution with the following compositions (mM). In the case of the DPH, small bundles were prepared in such a way that they could be mounted from the rib cage and the central tendon. The pH was adjusted to between 7.4 with NaHCO3, followed by the addition of fetal bovine serum (to 0.2%) to increase viability of the dissected muscle. The solution was continuously aerated with a gas mixture consisting of 95% O2 and 5% CO2. EDL and SOL muscles were mounted vertically between two Radnoti (Monrovea, CA, USA) stimulating platinum electrodes and immersed in a 20 mL bathing chamber containing the incubation medium. The muscles were suspended from movable isometric force transducers above the chambers via the tendons, and secured to the base of the tissue support within the chambers. The analog output of the force transducer was digitized, stored and analyzed with PowerLab Software (Colorado Springs, CO, USA). For each muscle, the resting tension and the stimulatory voltage were provided by a Grass S8800 digital stimulator (West Warwick, RI, USA) and both parameters were adjusted to produce a maximal isometric tetanic force (Tmax).
| NaCl | KCl | CaCl2 | NaH2PO4 | MgCl2 | Glucose | EGTA | |
|---|---|---|---|---|---|---|---|
| Ca | 135 | 5 | 2.5 | 0.4 | 0.5 | 10 | — |
| 0 Ca | 135 | 5 | — | 0.4 | 0.5 | 10 | 0.1 |
Intact Muscle Protocol
After initial Tmax determination, the muscles were subjected to a protocol that mimics normal muscle activity (No fatigue, stimulation trains triggered at every minute, duty cycle of 2%). Muscles were placed in 2.5 mM Ca or zero Ca2+ and/or 5 mM NiCl2, 1 mm NiCl2, 10 μM nifedipine, or 25 μM bromoenol lactone (BEL). The intact muscles were allowed a 30-minute equilibration, during which time they were stimulated with pairs of alternating high (that produced Tmax) and low (that produced 1/2 Tmax) frequency pulse-trains administered with a periodicity of 1 minute, before any pharmacological intervention was attempted. The results indicate the relative contributions from the contractile proteins (Tmax) and from the SR (1/2 Tmax) to the stimulation [41].
Intracellular Ca2+ Measurement and Mn2+ Quenching
[33]. For quantitative measurements of intracellular Ca2+], FDB muscle fibers were enzymatically isolated in a 0 Ca2+ Tyrode solution containing 2 mg/mL type I collagenase for 2 hours in a shaking bath at 37 °C, before being transferred to a 0 Ca2+ Tyrode solution without collagenase and gently triturated with a pipette. The fibers were then loaded with 5 μM Fura-2-AM for 40 minutes, after which the Fura-2 AM was washed off. As fiber motion artifacts are associated with intracellular Ca release, 20 μM N-benzyl-p-toluene sulfonamide (Sigma, St. Louis, MO), a specific myosin II inhibitor, was then applied for 20 minutes. A dual-wavelength (excitation at 340 nm and 380 nm) PTI spectrofluorometer (Photon Technology International, Birmingham, NJ) was used to determine the kinetic changes of caffeine and ryanodine (C/R)-induced intracellular Ca2+ transients.
Mn2+ quenching experiment were performed as previously described with minor modifications. Briefly, FDB fibers were loaded with Fura-2/AM fluorescent Ca2+ indicator [16]. SR Ca2+ depletion with C/R (with and without 25 μM BEL) was followed by the addition of 0.5 mM MnCl2 (with and without 25 μM BEL) to the extracellular solution. The Mn2+ quenching of Fura-2 fluorescence was measured at the Ca2+-independent wavelength of Fura-2AM excitation (360 nm). The decay of Fura-2AM fluorescence upon Mn2+ addition was expressed as percent decrease in Fura-2 fluorescence per unit time.
Confocal Imaging of Store-operated calcium entry in mechanically-skinned muscle skinned muscle fiber
These studies followed our own protocols, which are based on our original and first ever methodology to directly monitor the spatial and temporal resolution of SOCE in mammalian skeletal muscle fibers [16, 34]. Briefly, intact muscle fibers are carefully skinned in the present of 0.5mM CaCl2 and Rhod-5N, forcing the trapping of the dye and Ca+2 in the re-sealed T-tubules. Using IDL driven sub-routines, areas of interest comprising either the T-tubules or background areas can be drawn and the relative fluorescence upon activation of SOCE can be quantified. A minimum of 6 ROIs from each muscle fiber from 9 muscle fibers from 3 mice was employed to obtain the average SOCE activity in young and aged mice.
Acknowledgments
This work was supported by a NIAMS 1RC2AR058962-01 Grant to M.B., a Missouri Life Sciences Research Board Grant to M.B., a Conselho Nacional de Pesquisa-Brazil (CNPq) Grant to M.B., an AHA Scientist Development Grant to X.Z., a NIAMS AR054793 to N.W., a NIA AG028614-02 Grant and a NHLBI RO1-HL069000-02 Grant to J.M., a NIAMS AR055974 Grant to M.R. and B.H., and grants from the Department of Anesthesiology, University of Pittsburgh School of Medicine and UPMC to J.P.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare. Authors have no financial interests to disclose.
References
- 1. Faulkner JA, Brooks SV, Zerba E. Muscle atrophy and weakness with aging: Contration-induced injury as an underlying mechanism. J Gerontol. 1995; 50A: 124 -129. .
- 2. Gonzalez E, Messi ML, Delbono O. The Specific Force of Single Intact Extensor Digitorum Longus and Soleus Mouse Muscle Fibers Declines with Aging. J Membr Biol. 2000; 178: 175 -183. [PubMed] .
- 3. Faulkner JA, Brooks SV, Zerba E. Muscle atrophy and weakness with aging: contraction-induced injury as an underlying mechanism. J Gerontol A Biol Sci Med Sci. 1995; 50: Spec No 124 -129. [PubMed] .
- 4. Delbono O. Molecular mechanisms and therapeutics of the deficit in specific force in ageing skeletal muscle. Biogerontology. 2002; 3: 265 -270. [PubMed] .
- 5. Zahn JM, Sonu R, Vogel H, et al. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006; 2: e115 [PubMed] .
- 6. Bouchard C, Shephard RJ, Stephens T, Sutton JR, McPherson BD. Exercise, fitness, and health: A consensus statement. Bouchard C, Shephard RJ, Stephens T, Sutton JR, McPherson BD. Exercise, Fitness, and Health: A Consensus of Current Knowledge. Champaign Human Kinetics Books 1990; 3 -28. .
- 7. Bortz WM. How fast do we age? Exercise performance over time as a biomarker. J Gerontol A Biol Sci Med Sci. 1996; 51: M223 -M225. [PubMed] .
- 8. Pardo PS and Boriek AM. The physiological roles of Sirt1 in skeletal muscle. Aging (Albany NY). 2011; 3: 430 -437. [PubMed] .
- 9. Flach RJ and Bennett AM. MAP kinase phosphatase-1–a new player at the nexus between sarcopenia and metabolic disease. Aging (Albany NY). 2010; 2: 170 -176. [PubMed] .
- 10. Nuss JE, Amaning JK, Bailey CE, et al. Oxidative modification and aggregation of creatine kinase from aged mouse skeletal muscle. Aging (Albany NY). 2009; 1: 557 -572. [PubMed] .
- 11. Ljubicic V, Joseph AM, Adhihetty PJ, et al. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany NY). 2009; 1: 818 -830. [PubMed] .
- 12. Scicchitano BM, Rizzuto E, Musaro A. Counteracting muscle wasting in aging and neuromuscular diseases: the critical role of IGF-1. Aging (Albany NY). 2009; 1: 451 -457. [PubMed] .
- 13. Romero-Suarez S, Shen J, Brotto L, et al. Muscle-specific inositide phosphatase (MIP/MTMR14) is reduced with age and its loss accelerates skeletal muscle aging process by altering calcium homeostasis. Aging (Albany NY). 2010; 2: 504 -513. [PubMed] .
- 14. Delbono O, O'Rourke KS, Ettinger WH. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J Membr Biol. 1995; 148: 211 -222. [PubMed] .
- 15. Renganathan M, Messi ML, Delbono O. Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J Membr Biol. 1997; 157: 247 -253. [PubMed] .
- 16. Zhao X, Weisleder N, Thornton A, et al. Compromised store-operated Ca(2+) entry in aged skeletal muscle. Aging Cell. 8 7: 561 -568. [PubMed] .
- 17. Weisleder N, Brotto M, Komazaki S, et al. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J Cell Biol. 2006; 174: 639 -645. [PubMed] .
- 18. Brotto M, Weisleder N, Ma J. Store-operated Ca2+ entry in muscle physioloy. Current Chemical Biology. 2007; 1: 87 -95. .
- 19. Kurebayashi N and Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J Physiol. 2001; 533: 185 -199. [PubMed] .
- 20. Pan Z, Yang D, Nagaraj RY, et al. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat Cell Biol. 2002; 4: 379 -383. [PubMed] .
- 21. Senderek J, Bergmann C, Weber S, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003; 12: 349 -356. [PubMed] .
- 22. Stiber J, Hawkins A, Zhang ZS, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008; .
- 23. McElroy SP, Drummond RM, Gurney AM. Regulation of store-operated Ca2+ entry in pulmonary artery smooth muscle cells. CC. 2009; 46: 99 -106. .
- 24. McElroy SP, Gurney AM, Drummond RM. Pharmacological profile of store-operated Ca(2+) entry in intrapulmonary artery smooth muscle cells. Eur J Pharmacol. 2008; 584: 10 -20. [PubMed] .
- 25. Stephenson EW. Properties of chloride-stimulated 45Ca flux in skinned muscle fibers. J Gen Physiol. 1978; 71: 411 -430. [PubMed] .
- 26. Bannister RA, Pessah IN, Beam KG. The skeletal L-type Ca(2+) current is a major contributor to excitation-coupled Ca(2+) entry. J Gen Physiol. 2009; 133: 79 -91. [PubMed] .
- 27. Miller MJ, Shannon K, Reid MB. Effect of nifedipine on the contractile function of the rat diaphragm in vitro. Life Sci. 1989; 45: 2419 -2428. [PubMed] .
- 28. Miller MJ, Shannon K, Reid MB. Inhibition by nifedipine of the indirectly induced contractile response of the rat diaphragm. Life Sci. 1989; 45: 2429 -2435. [PubMed] .
- 29. Boittin FX, Petermann O, Hirn C, et al. Ca2+-independent phospholipase A2 enhances store-operated Ca2+ entry in dystrophic skeletal muscle fibers. J Cell Sci. 2006; 119: 3733 -3742. [PubMed] .
- 30. Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004; 6: 113 -120. [PubMed] .
- 31. Smani T, Zakharov SI, Leno E, Csutora P, Trepakova ES, Bolotina VM. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J Biol Chem. 2003; 278: 11909 -11915. [PubMed] .
- 32. Gong MC, Arbogast S, Guo Z, Mathenia J, Su W, Reid MB. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J Appl Physiol. 2006; 100: 399 -405. [PubMed] .
- 33. Zhao X, Weisleder N, Han X, et al. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J Biol Chem. 2006; 281: 33477 -33486. [PubMed] .
- 34. Zhao X, Yoshida M, Brotto L, et al. Enhanced resistance to fatigue and altered calcium handling properties of sarcalumenin knockout mice. Physiol Genomics. 2005; 23: 72 -78. [PubMed] .
- 35. Papazafiri P and Kletsas D. Developmental and age-related alterations of calcium homeostasis in human fibroblasts. Exp Gerontol. 2003; 38: 307 -311. [PubMed] .
- 36. Wojda U, Salinska E, Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008; 60: 575 -590. [PubMed] .
- 37. Payne AM, Zheng Z, Gonzalez E, Wang ZM, Messi ML, Delbono O. External Ca(2+)-dependent excitation–contraction coupling in a population of ageing mouse skeletal muscle fibres. J Physiol. 2004; 560: 137 -155. [PubMed] .
- 38. Brotto MAP and Nosek TM. Hydrogen Peroxide Disrupts Ca2+ Release from the Sarcoplasmic Reticulum of Rat Skeletal Muscle Fibers. FASEB J. 1996; .
- 39. Brotto MAP, van Leyen SA, Brotto LS, Jin JP, Nosek CM, Nosek TM. Hypoxia/fatigue-induced degradation of troponin I and troponin C: new insights into physiologic muscle fatigue. Pflugers Arch. 2001; 442: 738 -744. [PubMed] .
- 40. Waddell LB, Lemckert FA, Zheng XF, et al. Dysferlin, Annexin A1, and Mitsugumin 53 Are Upregulated in Muscular Dystrophy and Localize to Longitudinal Tubules of the T-System With Stretch. J Neuropathol Exp Neurol. 2011; 70: 302 -313. [PubMed] .
- 41. Brotto MA, Nosek TM, Kolbeck RC. Influence of ageing on the fatigability of isolated mouse skeletal muscles from mature and aged mice. Exp Physiol. 2002; 87: 77 -82. [PubMed] .