



Introduction
Cell death is an essential cellular process that plays crucial roles in development and immune responses. Apoptosis is a well-known example of cell death. Cytotoxic T cells and natural killer cells can utilize the apoptosis mechanism to kill virus-infected host cells. During apoptosis, DNA fragmentation can destroy the viral genome, eliminating viral pathogens. After the discovery of apoptosis, other cell death pathways have been discovered, including autophagy, ferroptosis, pyroptosis, necroptosis, and NETosis [1]. We have proposed a framework encompassing all discovered host immunological pathways, such as TH1, TH2a, TH2b, TH3, TH9, TH17, TH22, TH1-like, and THαβ immune reactions [2–4]. These immune responses combat different types of pathogens and are linked to four types of hypersensitivities. TH1 and TH1-like immune responses fight against intracellular microorganisms, including intracellular bacteria, protozoa, and fungi. TH1 immunity is an eradicable immune reaction, while TH1-like is a tolerable immune reaction. TH1 and TH1-like immune responses are associated with type 4 delayed-type hypersensitivities. TH2 and TH9 immune responses combat parasites, including ectoparasites (insects) and endoparasites (helminths). TH2 immunity is an eradicable immune reaction, and TH9 is a tolerable immune reaction. TH2 and TH9 immune responses are associated with type 1 allergic hypersensitivities. TH22 and TH17 immune responses fight against extracellular microorganisms, including extracellular bacteria, protozoa, and fungi. TH22 immunity is an eradicable immune reaction, and TH17 is a tolerable immune reaction. TH22 and TH17 immune responses are associated with type 3 immune complex-related hypersensitivities. THαβ and TH3 immune responses combat infectious particles, including viruses and prions. THαβ immunity is an eradicable immune reaction, and TH3 is a tolerable immune reaction. THαβ and TH3 immune responses are associated with type 2 antibody-dependent cytotoxic hypersensitivities. Programmed cell death is a crucial component of the host defense mechanism. Thus, different types of host immunological reactions can be related to different types of programmed cell death to defend against different pathogens. Here, we will review these cell death pathways associated with the host immunological pathways.
Overview of cell death pathways
Apoptosis
Apoptosis, the earliest discovered cell death pathway, stands in contrast to necrosis, an unprogrammed form of cell death induced by pathogens or various external factors. Unlike necrosis, apoptosis is a tightly regulated and programmed cell death pathway governed by genetic machinery. During embryonic development, apoptosis plays a crucial role as embryonic cells utilize this mechanism to eliminate unwanted cells. Moreover, the apoptosis mechanism is employed in host immune responses to combat pathogenic infections. For instance, the natural killer cell antibody-dependent cellular cytotoxic reaction uses apoptosis to eliminate virus-infected cells. Consequently, apoptosis emerges as a vital component in the body’s self-defense reactions.
Apoptosis can be categorized into two main pathways: the extrinsic pathway and the intrinsic pathway. The extrinsic pathway is activated by external signal molecules, initiating the apoptosis machinery. A classic example of the extrinsic apoptosis pathway involves the interaction between Fas and Fas ligand. On the other hand, the intrinsic pathway is activated by internal cellular signal molecules, with the release of cytochrome c from mitochondria being a characteristic event. Cytosolic cytochrome c serves as a trigger for cell apoptosis. Notably, there is an interconnection between the extrinsic and intrinsic pathways, converging into a common cell death pathway. The apoptosis machinery comprises initiator and executor caspases responsible for breaking down intracellular DNA and proteins. Initiator caspases include caspase 2, 8, 9, and 10, while executor caspases include caspase 3, 6, and 7. This intricate system underscores the convergence of both extrinsic and intrinsic apoptosis pathways into a unified mechanism of cell death.
Autophagy
Autophagy is a natural, conserved cellular process that digests unwanted, damaged, or old organelles through a lysosome-dependent regulated mechanism. It is referred to as type 2 cell death. Autophagy can be initiated during starvation or other cellular stress situations [5]. It is a process that recycles cell contents to maintain the required metabolism of cells. Special organelles involved in autophagy include mitophagy, the autophagy of mitochondria, and others. Autophagy is an essential cellular process. Autophagic death is a cellular process involving autophagy-induced programmed cell death. This machinery also plays a crucial role in the cell’s defense mechanism [6]. Although initially recognized as a principal degradation pathway to protect against starvation, it is now evident that autophagy also plays a vital role in the homeostasis of non-starved cells. Defects in autophagy are associated with various human diseases, especially neurodegenerative disorders, and modulating autophagy becomes a potential treatment for these detrimental illnesses.
Four types of autophagy have been classified: macroautophagy, microautophagy, chaperone-mediated autophagy (CMA), and crinophagy. In macroautophagy, cytoplasmic components such as mitochondria are targeted and isolated from the main part of the cell within a double-membrane autophagosome. Then, it fuses with a lysosome to become an autolysosome, and eventually, the contents of the vesicle are degraded and recycled. Compared to crinophagy, unnecessary secretory granules are degraded and recycled. In disease, autophagy has been seen as an adaptive response to stress, promoting cell survival; but in other conditions, it could promote cell death and morbidity. In extreme hunger, the breakdown of cellular components promotes cell survival by maintaining cellular energy.
Pyroptosis
Pyroptosis is another form of programmed cell death [7]. It is related to interleukin-1 and interleukin-18. It is associated with programmed cell death of macrophages. This process can help rapidly clear intracellular pathogens. Pyroptosis usually occurs in immune cells, keratinocytes, and sometimes epithelial cells. This process is triggered by the formation of an inflammasome complex (pyroptosome complex) via the stimulation of intracellular danger signals. The pyroptosome complex is related to the activation of caspases 1/4/5 in humans, which are different caspase sets compared to apoptosis. Caspases 1/4/5 cause the maturation of pro-inflammatory cytokines interleukin-1β and interleukin-18. These caspases also activate the pore-forming protein gasdermin D (GSDMD). Gasdermin D is the key effector molecule of pyroptosis. The inflammasome pathway can be canonical or noncanonical. The canonical pathway involves the activation of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) recognized by several endogenous pattern recognition receptors (PRRs). For example, NLRP3 or NLRC4 protein is activated by different PAMPs and DAMPs. These receptors can upregulate pro-inflammatory cytokines, including interleukin-12, via the NFkB and MAPK signaling mechanisms. Then, pro-IL-1β and pro-IL-18 are released to be activated via the action of cysteine-regulated caspase-1. Both NLRC4 and procaspase-1 contain a caspase activation and recruitment domain (CARD). After NLRC4 recruits pro-caspase-1, the homotypic CARD-CARD interaction will induce an autocatalytic reaction, allowing pro-caspase-1 to become active caspase-1. Activated caspase-1 cleaves pro-IL-1β and pro-IL-18, enabling these two cytokines to become activated forms. Besides, caspase-1 also cleaves the intracellular gasdermin D. GSDMD will be cleaved into two fragments: the N-terminal GSDMD-N and the C-terminal GSDMD-C. GSDMD-N can form transmembrane pores. These transmembrane pores allow the secretion of IL-1β and IL-18 into extracellular spaces. These pores also impair the extracellular-intracellular ion gradients, causing an increase in osmotic pressure with the influx of water, leading to cell swelling and bursting, resulting in pyroptosis. It is worth noting that GSDMD-N can only insert itself into the inner membrane of specific lipid compositions. And, without cleavage, GSDMD-N is autoinhibited by GSDMD-C. The noncanonical pathway involves the interaction of bacterial lipopolysaccharide and human caspase 4/5. Binding LPS to these caspases induces oligomerization and activation. These caspases also cleave GSDMD to become GSDMD-N, promoting pyroptosis.
Ferroptosis
Ferroptosis is a type of programmed cell death triggered by excess iron intracellularly. It is characterized by the accumulation of lipid peroxides. Its term is oxytosis. Ferroptosis is triggered by the failure of glutathione-mediated antioxidant defenses. The overall pattern of ferroptosis is the iron-mediated accumulation of oxidatively damaged phospholipids, especially lipid peroxides. When free radicals abstract electrons from a phospholipid, oxidation of phospholipids will occur. Typically, it affects polyunsaturated fatty acids. The main cellular defense mechanism against ferroptosis is mediated by glutathione peroxidase 4 (GPX4). GPX4 can convert lipid peroxides into non-toxic lipid alcohol molecules. Iron is vital and necessary to generate reactive oxygen species to initiate ferroptosis. Thus, treating cells with iron chelators can stop the occurrence of ferroptosis. Additionally, intracellular glutathione (GSH) levels are key to the function of GPX4, so depletion of GSH will lead to ferroptotic cell death. Besides, ferroptosis causes phenotypic changes in mitochondria.
Necroptosis
Necroptosis is a programmed form of cell death compared to necrosis. The key cytokine mediating necroptosis is TNFα. Binding of TNFα leads to the activation of its receptor TNFR1. TNFR1 receptor binds to TNFR-associated death protein (TRADD) and TNF receptor-associated factor 2 (TRAF2) to activate RIPK1, which recruits RIPK3 to form the necrosome (ripoptosome). During the necroptosis process, the anti-apoptotic protein cFLIP can inactivate caspase 8, facilitating necroptosis. In the absence of caspase 8, RIPK1 and RIPK3 can autophosphorylate and transphosphorylate each other to form a microfilament-like complex named the necrosome. The necrosome phosphorylates the pro-necroptotic protein MLKL, which causes MLKL oligomerization. The oligomerized MLKL will insert into plasma and organelle membranes to induce permeability. Besides, MLKL insertion will induce the leakage of cellular contents of the damage-associated molecular patterns (DAMPs) to trigger inflammation. The necrosome also inhibits the adenine nucleotide translocase in mitochondria, lowering intracellular ATP concentrations. Furthermore, uncoupling of the mitochondrial electron transport chain will lead to mitochondrial damage and open the mitochondrial permeability transition pores, allowing mitochondrial proteins to move into the cytoplasm. The necrosome can additionally cause leaks of lysosomal enzymes into the cytosol via the induction of reactive oxygen radicals by JNK, calpain activation by calcium release, and sphingosine formation. In contrast to apoptosis, the process of necroptosis does not relate to caspase activation. No apoptotic body formation is seen in necroptosis. Cells undergo necroptotic rupture, leaking cellular contents into intercellular spaces.
NETosis
Neutrophil extracellular traps (NETs) are networks of neutrophil-derived extracellular fibers binding to extracellular pathogens [8]. NETs allow neutrophils to kill extracellular microorganisms with minimal damage to the body [9]. NETs consist of DNA stretches and proteins, including azurophilic granules (neutrophil elastase, cathepsin G, and myeloperoxidase), tertiary granules (gelatinase), and specific granules (lactoferrin). NETs can also form intravascularly via the regulation of platelets. Platelet TLR4 can bind to extracellular microorganisms and activate neutrophils to initiate NETs. Thus, NETs can capture bacteria in blood vessels, stopping their migration via blood circulation. NETs activation and release are usually associated with neutrophil programmed cell death, suicidal NETosis. The NETosis pathway typically begins with NADPH oxidase activation of arginine deiminase 4 (PAD4) via reactive oxygen radicals. PAD4 will induce the citrullination of histones in the neutrophil cell nuclei, resulting in chromatin decondensation. Azurophilic granules (neutrophil elastase, cathepsin G, and myeloperoxidase) enter the neutrophil nucleus and cause the rupture of the nuclear envelope. Then, the decondensed chromatin enters the cytoplasm, where it combines with other cellular granules to form the early-stage NET. NETosis is a double-edged sword, which may cause complications. There is a report suggesting a relationship between NETosis and organ injury [10].
Overview of host immunological pathways
The immune system is a marvelously complex network, where host immunological pathways play a pivotal role in defending against diverse pathogens. These pathways are categorized based on the dominance of certain immunoglobulins, predominantly into IgG-dominant eradicable immune responses and IgA-dominant tolerable immune responses [2–4, 11]. Eradicable immune responses are initiated by follicular helper T cells (Tfh) via interleukin-21, and tolerable immune responses are initiated by regulatory T cells (Treg) via TGF-β. Understanding the intricacies of these pathways is crucial in comprehending how the immune system combats various threats.
In the realm of eradicable immune responses, the action primarily revolves around combating different types of pathogens through specialized immune mechanisms. The TH1 immunity, for instance, stands guard against intracellular microorganisms such as bacteria, protozoa, and fungi. This branch mobilizes an array of defenders including M1 macrophages, IFNγ-producing CD4 T cells, iNKT1 cells, CD8 T cells (Tc1, EM4), and IgG3 B cells, forming a formidable defense line against these intruders. TH1 immunity is also intricately linked to type 4 delayed type hypersensitivity reactions, highlighting its role in specific immune responses.
In contrast, TH2 immunity gears up against parasites, presenting two distinct subtypes: TH2a and TH2b. TH2a tackles endoparasites (helminths) with its lineup of inflammatory eosinophils (iEOS), interleukin-4/interleukin-5 producing CD4 T cells, mast cells-tryptase (MCt), iNKT2 cells, and IgG4 B cells. On the other hand, TH2b focuses on combating ectoparasites (insects), marshaling basophils, interleukin-13/interleukin-4 producing CD4 T cells, mast cells-tryptase/chymase (MCtc), iNKT2 cells, and IgE B cells. These branches of TH2 immunity are instrumental in addressing parasitic threats and are associated with type 1 allergic hypersensitivity responses.
Expanding further, TH22 immunity is dedicated to countering extracellular microorganisms such as bacteria, protozoa, and fungi. Neutrophils (N1), interleukin-22 producing CD4 T cells, iNKT17 cells, and IgG2 B cells collaboratively orchestrate the defense in this domain. TH22 immunity plays a significant role in type 3 immune complex mediated hypersensitivity reactions, showcasing its specialized function in immune responses.
Moreover, THαβ immunity is specifically tailored to combat infectious particles like viruses and prions [12–15]. This immune pathway employs NK cells (NK1), interleukin-10 producing CD4 T cells, iNKT10 cells, CD8 T cells (Tc2, EM1), and IgG1 B cells to combat these minute yet potent adversaries. Its connection to type 2 antibody-dependent cytotoxic hypersensitivity underscores its significance in addressing infectious threats.
Transitioning to tolerable immune responses dominated by IgA, these pathways exemplify the system’s ability to mount defenses without causing excessive damage to the host. Regulatory T cells play a crucial role in steering these responses, facilitating the switch to IgA, thereby establishing a more tolerable immune milieu.
TH1-like immunity within the tolerable response framework mirrors TH1 immunity but in a more regulated manner. It safeguards against intracellular microorganisms through M2 macrophages, TGFβ/IFNγ-producing CD4 T cells, iNKT1 cells, CD8 T cells (EM3), and IgA1 B cells, while maintaining a balance to prevent hyperactive responses that might harm the host.
TH9 immunity, targeting parasites such as insects and helminths, relies on regulatory eosinophils (rEOS), basophils, interleukin-9 producing CD4 T cells, iNKT2 cells, mast cells (MMC9), and IgA2 B cells to ensure a measured and controlled defense. This pathway, associated with type 1 allergic hypersensitivity, showcases the immune system’s ability to mount responses without tipping the balance toward excessive reactions.
Continuing within the tolerable responses, TH17 immunity is specialized in combating extracellular microorganisms. Neutrophils (N2), interleukin-17 producing CD4 T cells, iNKT17 cells, and IgA2 B cells are the primary players in this pathway, illustrating a fine-tuned defense against extracellular threats while limiting immune-mediated damage through type 3 immune complex mediated hypersensitivity.
Lastly, TH3 immunity within tolerable responses gears up against infectious particles employing NK cells (NK2), interleukin-10/TGFβ-producing CD4 T cells, iNKT10 cells, CD8 T cells (EM2), and IgA1 B cells. This pathway showcases the immune system’s adaptability, mounting responses against infectious particles while maintaining a balanced immune environment to prevent excessive host damage, closely linked to type 2 antibody-dependent cytotoxic hypersensitivity.
The intricate network of host immunological pathways, categorized into eradicable and tolerable immune responses, showcases the remarkable adaptability and specificity of the immune system in combating diverse pathogens. These pathways not only defend against various threats but also highlight the delicate balance between mounting effective responses and preventing immune-mediated damage to the host. The framework of host immunological pathways and their relations to different types of cell death is shown in Figure 1.
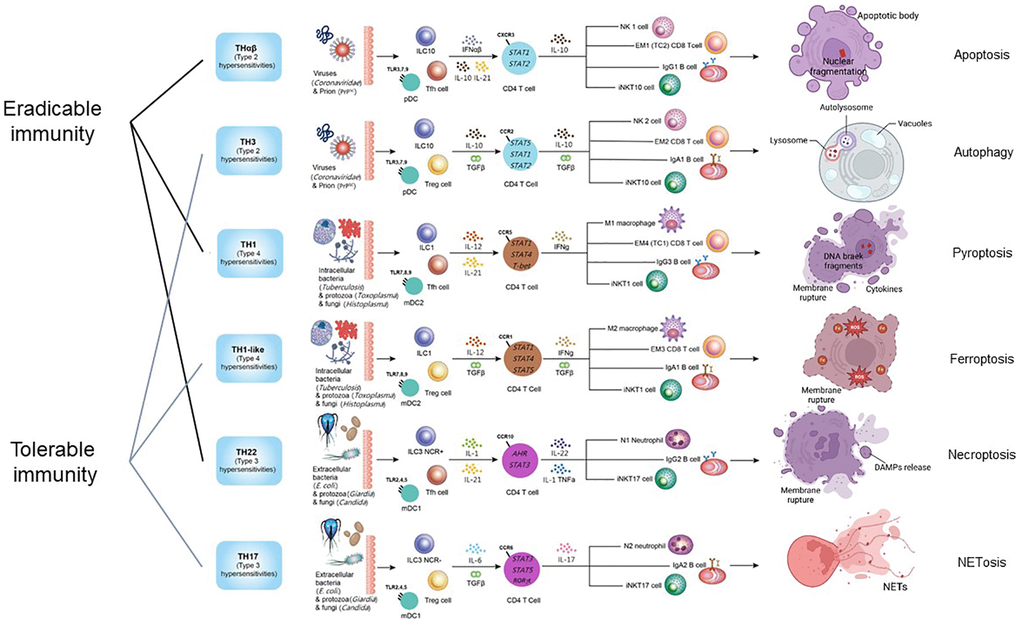
Figure 1. The framework of host immunological pathways and their relation to programmed cell death. Apoptosis is related to host anti-virus eradicable THαβ immunity. Autophagy is related to host anti-virus tolerable TH3 immunity. Pyroptosis is related to host anti-intracellular micro-organism eradicable TH1 immunity. Ferroptosis is related to host anti-intracellular micro-organism tolerable TH1-like immunity. Necroptosis is related to host anti-extracellular micro-organism eradicable TH22 immunity. NETosis is related to host anti-extracellular micro-organism tolerable TH17 immunity.
THαβ immune response and its relation to apoptosis
The host immunological THαβ pathway is the host’s immune reaction against infectious particles, including viruses and prions. Viruses and prions must live intracellularly to replicate and produce more transmissible particles. Apoptosis, the most well-studied programmed cell death pathway, is a key mediator regulating the death of virus-infected cells. During apoptosis, cell death leads to DNA or RNA fragmentation, allowing the intracellular viral DNA or RNA to be destroyed. Thus, virus particles can be eliminated by sacrificing the infected cells. Additionally, activated caspases degrade all intracellular proteins, leading to the destruction of prions, which are protein-based infectious particles, during apoptosis.
THαβ-related immune cells include natural killer (NK) cells, cytotoxic T cells, and IgG1-producing B lymphocytes. NK cells can induce antibody-dependent cellular cytotoxicity (ADCC) of virus-infected cells by binding to IgG1 antibodies [16]. ADCC is an apoptosis mechanism involving DNA and RNA fragmentation. Cytotoxic T cells can also cause apoptosis of virus-infected cells through DNA or RNA fragmentation, thereby killing the viral genomes. This process is mediated by the recognition of viral peptides presented on major histocompatibility complex (MHC) molecules by specific T cell receptors on cytotoxic T cells, which induces the apoptosis machinery. A similar mechanism can be observed in other infectious particles, like prions.
The immunosuppressive cytokine TGFβ has been found to inhibit the apoptosis process [17, 18]. Inhibition of TGFβ signaling can promote NK cell ADCC and cause target cell apoptosis [19]. Conversely, TGFβ can suppress NK cell ADCC. TGFβ-activated kinase 1 (TAK1) can antagonize apoptosis [20]. TGFβ can also inhibit Fas and caspase 8-related apoptosis [21, 22] and induce anti-apoptotic transcription factors to prevent apoptosis. Apoptosis-related protein degradation can lead to the destruction of infectious prion protein pathogens. Additionally, type 1 interferons can induce caspase cascades to trigger apoptosis in malignant cell lines [23–25]. Thus, apoptosis is a THαβ-related host defense mechanism against infectious particles, including viruses and prions.
Furthermore, the THαβ immune response is the host’s eradicable immune reaction induced by follicular helper T cells via the production of interleukin-21. Reports suggest that interleukin-21 is associated with apoptosis, including lymphocyte or cancer cell apoptosis [26–29].
TH3 immune response and its relation to autophagy
Autophagy is the type 2 programmed cell death pathway and is a milder control mechanism for virus infection of host cells [30–32]. Since type 1 interferons can help control virus infection, research has found a correlation between type 1 interferons and autophagy [24, 33]. Type 1 interferon is an inducer of autophagy [34–36]. Interferon regulatory factor 1 (IRF1), which can activate interferon beta, is also related to autophagy [37].
Autophagy is involved in the presentation of cytosolic antigens to MHC class II molecules and the digestion of intracellularly produced viral protein antigens. Autophagy is a protective mechanism against virus infection by degrading viral particles in autolysosomes. For example, autophagy has been found in liver cells to protect against hepatic virus infection [38]. Hepatitis C virus induces autophagy and interferes with the anti-viral innate eradicable immunity [39–41]. In contrast to apoptosis, autophagy with organelle degradation induces mild host inflammation.
Compared to the THαβ eradicable host immune reaction, the TH3 immunological pathway is the host’s tolerable immune response against viruses and prions. During autophagy, organelles containing virus particles are degraded. Autophagy is often observed in chronic viral infections. The key cytokines in the TH3 immunological pathway are interleukin-10 and TGF-β. However, interleukin-10 is more important for the eradicable THαβ immunity. Research has reported that interleukin-10 can prevent autophagy, and neutralization of interleukin-10 can recover the cellular machinery of autophagy [42–44].
Previous studies have found that TGF-β can promote autophagy [45]. TGF-β can prevent caspase 8-induced apoptosis and induce cell autophagy. TGF-β is mainly produced by regulatory T cells (Treg cells), and impaired Treg activity also impairs autophagy activity [46]. Follicular helper T cells (Tfh cells), which produce interleukin-21, have the opposite function of Treg cells. Previous literature reported that interleukin-21 can suppress autophagy [47].
The TH3 immune response is an IgA-dominant immune reaction, and autophagy has been found to be associated with the pathogenesis of IgA nephropathy [48]. This implies that the TH3 immune response could be related to the autophagy pathway. Interleukin-1, a key cytokine of the TH22/TH17 immunity, increases after the TH3-associated autophagy is blocked. Another THαβ/TH3 cytokine, interleukin-27, can also promote autophagy [49, 50].
TH1 immune response and its relation to pyroptosis
The TH1 immunological pathway is the host’s eradicable immunity against intracellular microorganisms, including intracellular bacteria, protozoa, and fungi. Pyroptosis, a programmed cell death mechanism, defends against intracellular pathogens [51, 52]. The major effector cells of the TH1 immune reaction are macrophages. Pyroptosis is related to the programmed cell death of macrophages. The key TH1 cytokine, interferon-gamma, is related to the activation of pyroptosis. The inflammasome complex in pyroptosis induces the activation of interleukin-1β and interleukin-18, both of which are pro-inflammatory cytokines against microorganisms. Additionally, interleukin-18 can augment the potency of interferon-gamma, which is the key immune mediator of the TH1 immunological pathway. The activation of interleukin-1β and interleukin-18, triggered by the inflammasome, further induces the production of interferon-gamma.
M1 macrophages are the key effector immune cells of TH1 immunity, and a correlation between M1 macrophage polarization and pyroptosis has been noted in previous studies [53, 54]. Interleukin-23, a vital cytokine in triggering TH1 and TH17 immune reactions, is also associated with macrophage pyroptosis [55]. The activation of the inflammasome also causes the upregulation of NF-κB, the master gene for immune activation signaling. Furthermore, the activation of the inflammasome inactivates interleukin-33, a mediator of the TH2 immunological pathway. Pyroptosis can also trigger pore-induced intracellular traps to capture intracellular bacteria, protozoa, and fungi, leading to their clearance [56]. Caspase-1-induced pyroptosis is an innate immune effector machinery fighting against intracellular microorganisms [52]. The immunosuppressive mediator TGF-β can suppress pyroptosis [57, 58].
Moreover, TH1 immunity is the host’s IgG-dominant eradicable immunity induced by follicular helper T cells via interleukin-21. Previous literature reported that interleukin-21 can cause pyroptosis of certain cells like regulatory T cells (Treg cells) [59]. Additionally, IgG immune complexes can induce macrophage pyroptosis by upregulating the expression of GSDMD [60].
TH1-like immune response and its relation to ferroptosis
Ferroptosis is a programmed cell death process triggered by intracellular iron overload. Iron is a key chemical element that helps the survival of microorganisms. According to increasing evidence, the occurrence of ferroptosis is always accompanied by inflammation. During the infection of microorganisms, including bacteria, protozoa, or fungi, higher concentrations of iron elements lead to worse infection control by the host. High intracellular iron concentrations help the survival of intracellular microorganisms. To reduce the availability of iron for intracellular microorganisms, iron-triggered cell death can sacrifice the infected cells and eliminate the microorganisms. This is the underlying logical principle of ferroptosis.
Chronic iron overload can drive macrophages to polarize into M2 macrophages, the effector cells of the TH1-like immune reaction [61, 62]. During ferroptosis, iron triggers the accumulation of lipid peroxides, causing membrane peroxidation and damage. Thus, the cell membranes of intracellular bacteria, protozoa, or fungi are damaged, leading to their death. This is why ferroptosis is a mechanism to kill and control intracellular microorganism infections. Glutathione peroxidase 4 (GPX4), which can prevent lipid peroxidation, is a protective mechanism against ferroptosis.
Regulatory T cells (Treg cells) with their key effector cytokine TGF-β can induce a tolerable immune response and tissue fibrosis. TGF-β could enhance ferroptosis via further GPX4 inhibition [63]. Additionally, GPX4 can enhance follicular helper T cells to inhibit ferroptosis [64]. There is a linkage between ferroptosis and fibrosis [65]. Chronic inflammation can be related to ferroptosis-associated tissue destruction and subsequent tissue fibrosis. TGF-β inhibitors can inhibit both ferroptosis and fibrosis [66]. Previous literature suggested an association between ferroptosis and tissue fibrosis, including renal fibrosis, pulmonary fibrosis, and liver cirrhosis [67]. For example, SARS-CoV-2 infection of lung epithelial cells can induce ferroptosis and subsequently lead to pulmonary fibrosis.
The TH1 key cytokine interferon-gamma can enhance ferroptosis in cancer cell lines and epithelial cells [68–70]. TH1-like immunity is an IgA-dominant tolerable immune reaction, and ferroptosis has been found to be related to the pathogenesis of IgA nephropathy [71].
TH22 immune response and its relation to necroptosis
The TH22 immune response is the host’s eradicable immune reaction against extracellular microorganisms, including extracellular bacteria, protozoa, and fungi. The TH22 immune response is associated with pro-inflammatory cytokines, including TNF-α. TNF-α is the key immune cytokine of the TH22 immune reaction that activates neutrophils to kill extracellular microorganisms. TNF-α is also the major mediator that induces necroptosis. The reason for triggering necroptosis, a programmed cell death pathway, can be to initiate a potent pro-inflammatory immune reaction to kill these invading extracellular microorganisms [72]. Macrophage necroptosis is observed in acute bacterial pneumonia caused by Serratia marcescens, Staphylococcus aureus, Streptococcus pneumoniae, Listeria monocytogenes, or uropathogenic Escherichia coli (UPEC) [73]. Necroptosis is a key function of neutrophils [74].
Another reason for necroptosis is to destroy potential nutrients from host cells to prevent the growth of extracellular microorganisms. TNF-α activates RIP kinases to form the necrosome. Interferon-gamma, which belongs to the TH1 immune response (different from the TH22/TH17 immunity), can downregulate necroptosis. Type 3 innate lymphoid cells, which can help trigger the TH22/TH17 immunity, are associated with necroptosis [75]. Necroptosis can also stimulate the secretion of TH22/TH17-related pro-inflammatory cytokines. A research study found that TGF-β–activated kinase 1 binding protein 2 (TAB2) deficiency causes dilated cardiomyopathy by enhancing RIPK1-dependent apoptosis and necroptosis [76]. TGF-β is the mediator of tolerable immunological pathways. Thus, eradicable immune mechanisms like apoptosis or necroptosis can be enhanced without TGF-β signaling. Another study pointed out that TGF-β-activated kinase 1 (TAK1) serves as a key survival factor in cardiac organs by directly antagonizing necroptosis [20].
The TH22 immune response is the host’s IgG-mediated eradicable immunity, which is initiated by follicular helper T cells via interleukin-21. There is no direct evidence suggesting that interleukin-21 can induce necroptosis. However, previous literature reported that interleukin-21 can cooperate with TNF-α, the key factor of necroptosis, to induce autoimmune disorders [77]. Additionally, IgG immune complexes have been found to trigger necroptosis in a previous study [78]. Interleukin-22 is the central cytokine of the TH22 immune reaction, and previous research pointed out that interleukin-22-producing type 3 innate lymphoid cells are related to necroptosis [75].
TH17 immune response and its relation to NETosis
The TH17 host immune reaction is the tolerable immune response against extracellular bacteria, fungi, or protozoa. Neutrophils play dominant roles in the TH17 tolerable immune response. In this situation, neutrophils cannot successfully kill and eradicate these extracellular microorganisms. Thus, these neutrophils sacrifice themselves to stop the progression of these extracellular microorganisms. These polymorphonuclear neutrophils (PMNs) have condensed DNA contents in their cell nuclei, and they trigger the NETosis cell programmed death pathway. Then, these extracellular microorganisms can be entrapped in the neutrophil extracellular traps (NETs), and other alive neutrophils will digest these extracellular bacteria, protozoa, or fungi. NETs can also induce TH17 immune cells [79].
The tolerable antibody IgA is found to activate NETosis. NETosis is also found to be correlated with chronic inflammation and delayed wound healing [80]. The TH17 immune reaction-related IgA immune complex formation is also associated with NETosis via the activation of the Fc-α receptor [81, 82]. IgA vasculitis has also been reported to be associated with NETs [83]. The central cytokine of the TH17 immune response, interleukin-17, can also induce NETosis [84]. Neutrophils can release IL-17 through extracellular trap formation during psoriasis [85]. IL-17A is expressed on NETs in ankylosing spondylitis [86]. The TH22/TH17 key cytokine TNF signaling can induce NETosis of CCR5+ neutrophils [87]. Thus, the TH17 host tolerable immunological pathway is associated with NETosis.
TAK1 is also required for neutrophil extracellular trap formation [88], pointing out the significance of TGF-β in NETosis. TGF-β itself can also induce NETs [89]. Regulatory T cells, characterized by the secretion of TGF-β, suggest that Treg cells are mediators of NETosis. NETs can also directly trigger epithelial and endothelial cell death [90].
Conclusions
Programmed cell death pathways are related to different host immunological pathways. Apoptosis is related to the host’s anti-viral eradicable THαβ immunity. Autophagy is related to the host’s anti-viral tolerable TH3 immunity. Pyroptosis is related to the host’s anti-intracellular microorganism eradicable TH1 immunity. Ferroptosis is related to the host’s anti-intracellular microorganism tolerable TH1-like immunity. Necroptosis is related to the host’s anti-extracellular microorganism eradicable TH22 immunity. NETosis is related to the host’s anti-extracellular microorganism tolerable TH17 immunity. These relationships can help us understand the host defense mechanisms against invading pathogens and provide new insights for developing better therapeutic strategies against infections or autoimmune disorders.
Data availability statement
This review article was performed via literature search without conducting experiments. Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Author Contributions
YKW, KWT, KCL, and WCH conducted the study and wrote the manuscript. YKW, KCL, and WCH aided in collecting reference literature and assisted in drafting the manuscript. KWT and WCH created figures. YKW, KWT, KCL, and WCH oversaw the study and edited the manuscript. WCH finally approved the manuscript.
Acknowledgments
The authors would also like to thank the Core Laboratory at the Department of Research, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, for their technical support and use of their facilities.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
This review article explored the relationship between immune pathways and cell death mechanism via literature search without conducting experiments. Sources were drawn from PubMed and Medline, eliminating the necessity for ethical statements and informed consent in the preparation and finalization of the article.
Funding
This study was supported by grants from Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (TCRD-TPE-110-02(2/3) and TCRD-TPE-111-01(3/3)).
References
- 1. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. 2022; 7:196. https://doi.org/10.1038/s41392-022-01046-3 [PubMed]
- 2. Hu WC. A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front Immunol. 2020; 11:1992. https://doi.org/10.3389/fimmu.2020.01992 [PubMed]
- 3. Lee YH, Tsai KW, Lu KC, Shih LJ, Hu WC. Cancer as a Dysfunctional Immune Disorder: Pro-Tumor TH1-like Immune Response and Anti-Tumor THαβ Immune Response Based on the Complete Updated Framework of Host Immunological Pathways. Biomedicines. 2022; 10:2497. https://doi.org/10.3390/biomedicines10102497 [PubMed]
- 4. Wen TH, Tsai KW, Wu YJ, Liao MT, Lu KC, Hu WC. The Framework for Human Host Immune Responses to Four Types of Parasitic Infections and Relevant Key JAK/STAT Signaling. Int J Mol Sci. 2021; 22:13310. https://doi.org/10.3390/ijms222413310 [PubMed]
- 5. Kriel J, Loos B. The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 2019; 26:640–52. https://doi.org/10.1038/s41418-018-0267-4 [PubMed]
- 6. Abdoli A, Alirezaei M, Mehrbod P, Forouzanfar F. Autophagy: The multi-purpose bridge in viral infections and host cells. Rev Med Virol. 2018; 28:e1973. https://doi.org/10.1002/rmv.1973 [PubMed]
- 7. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017; 277:61–75. https://doi.org/10.1111/imr.12534 [PubMed]
- 8. Branzk N, Papayannopoulos V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol. 2013; 35:513–30. https://doi.org/10.1007/s00281-013-0384-6 [PubMed]
- 9. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007; 176:231–41. https://doi.org/10.1083/jcb.200606027 [PubMed]
- 10. Cahilog Z, Zhao H, Wu L, Alam A, Eguchi S, Weng H, Ma D. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation. 2020; 43:2021–32. https://doi.org/10.1007/s10753-020-01294-x [PubMed]
- 11. Chu YT, Liao MT, Tsai KW, Lu KC, Hu WC. Interplay of Chemokines Receptors, Toll-like Receptors, and Host Immunological Pathways. Biomedicines. 2023; 11:2384. https://doi.org/10.3390/biomedicines11092384 [PubMed]
- 12. Hu WC. The Central THαβ Immunity Associated Cytokine: IL-10 Has a Strong Anti-Tumor Ability Toward Established Cancer Models In Vivo and Toward Cancer Cells In Vitro. Front Oncol. 2021; 11:655554. https://doi.org/10.3389/fonc.2021.655554 [PubMed]
- 13. Hu WC. Human immune responses to Plasmodium falciparum infection: molecular evidence for a suboptimal THαβ and TH17 bias over ideal and effective traditional TH1 immune response. Malar J. 2013; 12:392. https://doi.org/10.1186/1475-2875-12-392 [PubMed]
- 14. Tsou A, Chen PJ, Tsai KW, Hu WC, Lu KC. THαβ Immunological Pathway as Protective Immune Response against Prion Diseases: An Insight for Prion Infection Therapy. Viruses. 2022; 14:408. https://doi.org/10.3390/v14020408 [PubMed]
- 15. Shih LJ, Yang CC, Liao MT, Lu KC, Hu WC, Lin CP. An important call: Suggestion of using IL-10 as therapeutic agent for COVID-19 with ARDS and other complications. Virulence. 2023; 14:2190650. https://doi.org/10.1080/21505594.2023.2190650 [PubMed]
- 16. Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, Inogés S, Melero I, Berraondo P. Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol Cell Biol. 2017; 95:347–55. https://doi.org/10.1038/icb.2017.6 [PubMed]
- 17. Cano-González A, López-Rivas A. Opposing roles of TGF-β and EGF in the regulation of TRAIL-induced apoptosis in human breast epithelial cells. Biochim Biophys Acta. 2016; 1863:2104–14. https://doi.org/10.1016/j.bbamcr.2016.05.011 [PubMed]
- 18. Gal A, Sjöblom T, Fedorova L, Imreh S, Beug H, Moustakas A. Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene. 2008; 27:1218–30. https://doi.org/10.1038/sj.onc.1210741 [PubMed]
- 19. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, Wald DN. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS One. 2018; 13:e0191358. https://doi.org/10.1371/journal.pone.0191358 [PubMed]
- 20. Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG. TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol. 2013; 33:582–95. https://doi.org/10.1128/MCB.01225-12 [PubMed]
- 21. Park SM, Kim S, Choi JS, Hur DY, Lee WJ, Lee MS, Choe J, Lee TH. TGF-beta inhibits Fas-mediated apoptosis of a follicular dendritic cell line by down-regulating the expression of Fas and caspase-8: counteracting role of TGF-beta on TNF sensitization of Fas-mediated apoptosis. J Immunol. 2005; 174:6169–75. https://doi.org/10.4049/jimmunol.174.10.6169 [PubMed]
- 22. Schlapbach R, Spanaus KS, Malipiero U, Lens S, Tasinato A, Tschopp J, Fontana A. TGF-beta induces the expression of the FLICE-inhibitory protein and inhibits Fas-mediated apoptosis of microglia. Eur J Immunol. 2000; 30:3680–8. https://doi.org/10.1002/1521-4141(200012)30:12%3c3680::AID-IMMU3680%3e3.0.CO;2-L [PubMed]
- 23. Kotredes KP, Gamero AM. Interferons as inducers of apoptosis in malignant cells. J Interferon Cytokine Res. 2013; 33:162–70. https://doi.org/10.1089/jir.2012.0110 [PubMed]
- 24. Makita K, Hara H, Sano E, Okamoto Y, Ochiai Y, Harada T, Ueda T, Nakayama T, Aizawa S, Yoshino A. Interferon-β sensitizes human malignant melanoma cells to temozolomide-induced apoptosis and autophagy. Int J Oncol. 2019; 54:1864–74. https://doi.org/10.3892/ijo.2019.4743 [PubMed]
- 25. Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, Einhorn S, Grandér D. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene. 2002; 21:1251–62. https://doi.org/10.1038/sj.onc.1205179 [PubMed]
- 26. Akamatsu N, Yamada Y, Hasegawa H, Makabe K, Asano R, Kumagai I, Murata K, Imaizumi Y, Tsukasaki K, Tsuruda K, Sugahara K, Atogami S, Yanagihara K, Kamihira S. High IL-21 receptor expression and apoptosis induction by IL-21 in follicular lymphoma. Cancer Lett. 2007; 256:196–206. https://doi.org/10.1016/j.canlet.2007.06.001 [PubMed]
- 27. Gelebart P, Zak Z, Anand M, Dien-Bard J, Amin HM, Lai R. Interleukin-21 effectively induces apoptosis in mantle cell lymphoma through a STAT1-dependent mechanism. Leukemia. 2009; 23:1836–46. https://doi.org/10.1038/leu.2009.100 [PubMed]
- 28. Mehta DS, Wurster AL, Whitters MJ, Young DA, Collins M, Grusby MJ. IL-21 induces the apoptosis of resting and activated primary B cells. J Immunol. 2003; 170:4111–8. https://doi.org/10.4049/jimmunol.170.8.4111 [PubMed]
- 29. Sarosiek KA, Malumbres R, Nechushtan H, Gentles AJ, Avisar E, Lossos IS. Novel IL-21 signaling pathway up-regulates c-Myc and induces apoptosis of diffuse large B-cell lymphomas. Blood. 2010; 115:570–80. https://doi.org/10.1182/blood-2009-08-239996 [PubMed]
- 30. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018; 16:341–54. https://doi.org/10.1038/s41579-018-0003-6 [PubMed]
- 31. Datan E, Salman S. Autophagic cell death in viral infection: Do TAM receptors play a role? Int Rev Cell Mol Biol. 2020; 357:123–68. https://doi.org/10.1016/bs.ircmb.2020.10.001 [PubMed]
- 32. Espert L, Codogno P, Biard-Piechaczyk M. Involvement of autophagy in viral infections: antiviral function and subversion by viruses. J Mol Med (Berl). 2007; 85:811–23. https://doi.org/10.1007/s00109-007-0173-6 [PubMed]
- 33. Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, Coyle AJ, Kolbeck R, Green DR, Sanjuan MA. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012; 37:986–97. https://doi.org/10.1016/j.immuni.2012.09.014 [PubMed]
- 34. Li J, Kemper T, Broering R, Chen J, Yuan Z, Wang X, Lu M. Interferon Alpha Induces Cellular Autophagy and Modulates Hepatitis B Virus Replication. Front Cell Infect Microbiol. 2022; 12:804011. https://doi.org/10.3389/fcimb.2022.804011 [PubMed]
- 35. Schmeisser H, Bekisz J, Zoon KC. New function of type I IFN: induction of autophagy. J Interferon Cytokine Res. 2014; 34:71–8. https://doi.org/10.1089/jir.2013.0128 [PubMed]
- 36. Tian Y, Wang ML, Zhao J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses. 2019; 11:132. https://doi.org/10.3390/v11020132 [PubMed]
- 37. Cui Z, Li S, Liu Z, Zhang Y, Zhang H. Interferon Regulatory Factor 1 Activates Autophagy to Aggravate Hepatic Ischemia-Reperfusion Injury by Increasing High Mobility Group Box 1 Release. Cell Physiol Biochem. 2018; 48:328–38. https://doi.org/10.1159/000491732 [PubMed]
- 38. Tian Z, Wang M, Yao N, Yang S, Liu J, Yang Y, Chen T, Zhao Y, He Y. Expression of autophagy-modulating genes in peripheral blood mononuclear cells from familial clustering patients with chronic hepatitis B virus infection. Arch Virol. 2019; 164:2005–13. https://doi.org/10.1007/s00705-019-04248-3 [PubMed]
- 39. Dreux M, Chisari FV. Impact of the autophagy machinery on hepatitis C virus infection. Viruses. 2011; 3:1342–57. https://doi.org/10.3390/v3081342 [PubMed]
- 40. Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011; 121:37–56. https://doi.org/10.1172/JCI41474 [PubMed]
- 41. Rautou PE, Cazals-Hatem D, Feldmann G, Mansouri A, Grodet A, Barge S, Martinot-Peignoux M, Duces A, Bièche I, Lebrec D, Bedossa P, Paradis V, Marcellin P, et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am J Pathol. 2011; 178:2708–15. https://doi.org/10.1016/j.ajpath.2011.02.021 [PubMed]
- 42. Chen J, Guo Q, Chen Q, Chen Y, Chen D, Chen Z, Wang X, Huang Y. Interleukin 10 inhibits oxidative stress-induced autophagosome formation in hepatic stellate cells by activating the mTOR-STAT3 pathway. Exp Cell Res. 2022; 411:113001. https://doi.org/10.1016/j.yexcr.2021.113001 [PubMed]
- 43. Maneechotesuwan K, Kasetsinsombat K, Wongkajornsilp A, Barnes PJ. Role of autophagy in regulating interleukin-10 and the responses to corticosteroids and statins in asthma. Clin Exp Allergy. 2021; 51:1553–65. https://doi.org/10.1111/cea.13825 [PubMed]
- 44. Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, Park CG, Chun T. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol. 2011; 48:720–7. https://doi.org/10.1016/j.molimm.2010.10.020 [PubMed]
- 45. Mandatori S, Pacella I, Marzolla V, Mammi C, Starace D, Padula F, Vitiello L, Armani A, Savoia C, Taurino M, De Zio D, Giampietri C, Piconese S, et al. Altered Tregs Differentiation and Impaired Autophagy Correlate to Atherosclerotic Disease. Front Immunol. 2020; 11:350. https://doi.org/10.3389/fimmu.2020.00350 [PubMed]
- 46. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, Wu C, Vogel P, Neale G, Green DR, Chi H. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016; 17:277–85. https://doi.org/10.1038/ni.3365 [PubMed]
- 47. Kato H, Perl A. Blockade of Treg Cell Differentiation and Function by the Interleukin-21-Mechanistic Target of Rapamycin Axis Via Suppression of Autophagy in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018; 70:427–38. https://doi.org/10.1002/art.40380 [PubMed]
- 48. Chang M, Shi X, Ma S, Zhao M, Fan J, Pan Z, Xue S, Zhang Z, Shi Z, Yang B, Zhang Y. Inhibition of excessive autophagy alleviates renal injury and inflammation in a rat model of immunoglobulin A nephropathy. Eur J Pharmacol. 2023; 961:176198. https://doi.org/10.1016/j.ejphar.2023.176198 [PubMed]
- 49. Laverdure S, Wang Z, Yang J, Yamamoto T, Thomas T, Sato T, Nagashima K, Imamichi T. Interleukin-27 promotes autophagy in human serum-induced primary macrophages via an mTOR- and LC3-independent pathway. Sci Rep. 2021; 11:14898. https://doi.org/10.1038/s41598-021-94061-3 [PubMed]
- 50. Ting L, Feng Y, Zhou Y, Tong Z, Dong Z. IL-27 induces autophagy through regulation of the DNMT1/lncRNA MEG3/ERK/p38 axis to reduce pulmonary fibrosis. Respir Res. 2023; 24:67. https://doi.org/10.1186/s12931-023-02373-x [PubMed]
- 51. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015; 265:130–42. https://doi.org/10.1111/imr.12287 [PubMed]
- 52. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010; 11:1136–42. https://doi.org/10.1038/ni.1960 [PubMed]
- 53. Li N, Chen J, Geng C, Wang X, Wang Y, Sun N, Wang P, Han L, Li Z, Fan H, Hou S, Gong Y. Myoglobin promotes macrophage polarization to M1 type and pyroptosis via the RIG-I/Caspase1/GSDMD signaling pathway in CS-AKI. Cell Death Discov. 2022; 8:90. https://doi.org/10.1038/s41420-022-00894-w [PubMed]
- 54. Xia W, Lu Z, Chen W, Zhou J, Zhao Y. Excess fatty acids induce pancreatic acinar cell pyroptosis through macrophage M1 polarization. BMC Gastroenterol. 2022; 22:72. https://doi.org/10.1186/s12876-022-02146-8 [PubMed]
- 55. Wang C, Liu T, Wang Z, Li W, Zhao Q, Mi Z, Xue X, Shi P, Sun Y, Zhang Y, Wang N, Bao F, Chen W, et al. IL-23/IL-23R Promote Macrophage Pyroptosis and T Helper 1/T Helper 17 Cell Differentiation in Mycobacterial Infection. J Invest Dermatol. 2023; 143:2264–74.e18. https://doi.org/10.1016/j.jid.2023.04.019 [PubMed]
- 56. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016; 213:2113–28. https://doi.org/10.1084/jem.20151613 [PubMed]
- 57. Xie Y, Chen X, Li Y, Chen S, Liu S, Yu Z, Wang W. Transforming growth factor-β1 protects against LPC-induced cognitive deficit by attenuating pyroptosis of microglia via NF-κB/ERK1/2 pathways. J Neuroinflammation. 2022; 19:194. https://doi.org/10.1186/s12974-022-02557-0 [PubMed]
- 58. Tamura Y, Morikawa M, Tanabe R, Miyazono K, Koinuma D. Anti-pyroptotic function of TGF-β is suppressed by a synthetic dsRNA analogue in triple negative breast cancer cells. Mol Oncol. 2021; 15:1289–307. https://doi.org/10.1002/1878-0261.12890 [PubMed]
- 59. Chang L, Wu H, Huang W, Li Y, Chen Y, Li X, Yao Z, Chen X, Lai X, Zheng R, Huang Z, Wu X, Zhang G. IL-21 induces pyroptosis of Treg cells via Akt-mTOR-NLRP3-caspase 1 axis in eosinophilic chronic rhinosinusitis. J Allergy Clin Immunol. 2023; 152:641–55.e14. https://doi.org/10.1016/j.jaci.2023.04.013 [PubMed]
- 60. Yang L, Liu T, Zhuo Y, Li D, Li D, Liu J, Gao H, Zhang L, Lin J, Wang X. Verbenalin alleviates acute lung injury induced by sepsis and IgG immune complex through GPR18 receptor. Cell Signal. 2023; 109:110768. https://doi.org/10.1016/j.cellsig.2023.110768 [PubMed]
- 61. Yang Y, Wang Y, Guo L, Gao W, Tang TL, Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis. 2022; 13:355. https://doi.org/10.1038/s41419-022-04775-z [PubMed]
- 62. Yang M, Shen Z, Zhang X, Song Z, Zhang Y, Lin Z, Chen L. Ferroptosis of macrophages facilitates bone loss in apical periodontitis via NRF2/FSP1/ROS pathway. Free Radic Biol Med. 2023; 208:334–47. https://doi.org/10.1016/j.freeradbiomed.2023.08.020 [PubMed]
- 63. Kim DH, Kim WD, Kim SK, Moon DH, Lee SJ. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 2020; 11:406. https://doi.org/10.1038/s41419-020-2618-6 [PubMed]
- 64. Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, Wei Y, Wan Y, Wang N, Zhou M, Qiu C, Zeng Q, Ong HS, et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol. 2021; 22:1127–39. https://doi.org/10.1038/s41590-021-00996-0 [PubMed]
- 65. Ling H, Xiao H, Luo T, Lin H, Deng J. Role of Ferroptosis in Regulating the Epithelial-Mesenchymal Transition in Pulmonary Fibrosis. Biomedicines. 2023; 11:163. https://doi.org/10.3390/biomedicines11010163 [PubMed]
- 66. Bao R, Wang Q, Yu M, Zeng Y, Wen S, Liu T, Wang M, Li Y, Chang S, Chi H, Ma S, Wang K, Yang A, et al. AAV9-HGF cooperating with TGF-beta/Smad inhibitor attenuates silicosis fibrosis via inhibiting ferroptosis. Biomed Pharmacother. 2023; 161:114537. https://doi.org/10.1016/j.biopha.2023.114537 [PubMed]
- 67. Du X, Dong R, Wu Y, Ni B. Physiological Effects of Ferroptosis on Organ Fibrosis. Oxid Med Cell Longev. 2022; 2022:5295434. https://doi.org/10.1155/2022/5295434 [PubMed]
- 68. Wei TT, Zhang MY, Zheng XH, Xie TH, Wang W, Zou J, Li Y, Li HY, Cai J, Wang X, Tan J, Yang X, Yao Y, Zhu L. Interferon-γ induces retinal pigment epithelial cell Ferroptosis by a JAK1-2/STAT1/SLC7A11 signaling pathway in Age-related Macular Degeneration. FEBS J. 2022; 289:1968–83. https://doi.org/10.1111/febs.16272 [PubMed]
- 69. Yu X, Zhu D, Luo B, Kou W, Cheng Y, Zhu Y. IFNγ enhances ferroptosis by increasing JAK-STAT pathway activation to suppress SLCA711 expression in adrenocortical carcinoma. Oncol Rep. 2022; 47:97. https://doi.org/10.3892/or.2022.8308 [PubMed]
- 70. Zitvogel L, Kroemer G. Interferon-γ induces cancer cell ferroptosis. Cell Res. 2019; 29:692–3. https://doi.org/10.1038/s41422-019-0186-z [PubMed]
- 71. Wu J, Shao X, Shen J, Lin Q, Zhu X, Li S, Li J, Zhou W, Qi C, Ni Z. Downregulation of PPARα mediates FABP1 expression, contributing to IgA nephropathy by stimulating ferroptosis in human mesangial cells. Int J Biol Sci. 2022; 18:5438–58. https://doi.org/10.7150/ijbs.74675 [PubMed]
- 72. Zhu K, Liang W, Ma Z, Xu D, Cao S, Lu X, Liu N, Shan B, Qian L, Yuan J. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018; 9:500. https://doi.org/10.1038/s41419-018-0524-y [PubMed]
- 73. González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015; 11:e1005337. https://doi.org/10.1371/journal.ppat.1005337 [PubMed]
- 74. Wang X, Yousefi S, Simon HU. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 2018; 9:111. https://doi.org/10.1038/s41419-017-0058-8 [PubMed]
- 75. Shindo R, Ohmuraya M, Komazawa-Sakon S, Miyake S, Deguchi Y, Yamazaki S, Nishina T, Yoshimoto T, Kakuta S, Koike M, Uchiyama Y, Konishi H, Kiyama H, et al. Necroptosis of Intestinal Epithelial Cells Induces Type 3 Innate Lymphoid Cell-Dependent Lethal Ileitis. iScience. 2019; 15:536–51. https://doi.org/10.1016/j.isci.2019.05.011 [PubMed]
- 76. Yin H, Guo X, Chen Y, Zeng Y, Mo X, Hong S, He H, Li J, Steinmetz R, Liu Q. TAB2 deficiency induces dilated cardiomyopathy by promoting RIPK1-dependent apoptosis and necroptosis. J Clin Invest. 2022; 132:e152297. https://doi.org/10.1172/JCI152297 [PubMed]
- 77. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, Heuts F, Kogimtzis A, Patel S, Rosenthal M, Ono M, Sansom DM, Narendran P, Walker LS. Follicular helper T cell signature in type 1 diabetes. J Clin Invest. 2015; 125:292–303. https://doi.org/10.1172/JCI76238 [PubMed]
- 78. Santos LD, Antunes KH, Cassão G, Gonçalves JI, Abbadi BL, Bizarro CV, Basso LA, Machado P, de Souza APD, Porto BN. SARS-CoV-2 immune complex triggers human monocyte necroptosis. Int Immunopharmacol. 2023; 117:109954. https://doi.org/10.1016/j.intimp.2023.109954 [PubMed]
- 79. Lambert S, Hambro CA, Johnston A, Stuart PE, Tsoi LC, Nair RP, Elder JT. Neutrophil Extracellular Traps Induce Human Th17 Cells: Effect of Psoriasis-Associated TRAF3IP2 Genotype. J Invest Dermatol. 2019; 139:1245–53. https://doi.org/10.1016/j.jid.2018.11.021 [PubMed]
- 80. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015; 21:815–9. https://doi.org/10.1038/nm.3887 [PubMed]
- 81. Aleyd E, Al M, Tuk CW, van der Laken CJ, van Egmond M. IgA Complexes in Plasma and Synovial Fluid of Patients with Rheumatoid Arthritis Induce Neutrophil Extracellular Traps via FcαRI. J Immunol. 2016; 197:4552–9. https://doi.org/10.4049/jimmunol.1502353 [PubMed]
- 82. Aleyd E, van Hout MW, Ganzevles SH, Hoeben KA, Everts V, Bakema JE, van Egmond M. IgA enhances NETosis and release of neutrophil extracellular traps by polymorphonuclear cells via Fcα receptor I. J Immunol. 2014; 192:2374–83. https://doi.org/10.4049/jimmunol.1300261 [PubMed]
- 83. Chen XQ, Tu L, Zou JS, Zhu SQ, Zhao YJ, Qin YH. The Involvement of Neutrophil Extracellular Traps in Disease Activity Associated With IgA Vasculitis. Front Immunol. 2021; 12:668974. https://doi.org/10.3389/fimmu.2021.668974 [PubMed]
- 84. Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, Zoltan M, Arora N, Baydogan S, Horne W, Burks J, Xu H, Hussain P, et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. 2020; 217:e20190354. https://doi.org/10.1084/jem.20190354 [PubMed]
- 85. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011; 187:490–500. https://doi.org/10.4049/jimmunol.1100123 [PubMed]
- 86. Papagoras C, Chrysanthopoulou A, Mitsios A, Ntinopoulou M, Tsironidou V, Batsali AK, Papadaki HA, Skendros P, Ritis K. IL-17A expressed on neutrophil extracellular traps promotes mesenchymal stem cell differentiation toward bone-forming cells in ankylosing spondylitis. Eur J Immunol. 2021; 51:930–42. https://doi.org/10.1002/eji.202048878 [PubMed]
- 87. Neuenfeldt F, Schumacher JC, Grieshaber-Bouyer R, Habicht J, Schröder-Braunstein J, Gauss A, Merle U, Niesler B, Heineken N, Dalpke A, Gaida MM, Giese T, Meuer S, et al. Inflammation induces pro-NETotic neutrophils via TNFR2 signaling. Cell Rep. 2022; 39:110710. https://doi.org/10.1016/j.celrep.2022.110710 [PubMed]
- 88. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Transforming Growth Factor-β-Activated Kinase 1 Is Required for Human FcγRIIIb-Induced Neutrophil Extracellular Trap Formation. Front Immunol. 2016; 7:277. https://doi.org/10.3389/fimmu.2016.00277 [PubMed]
- 89. Jablonska E, Garley M, Surazynski A, Grubczak K, Iwaniuk A, Borys J, Moniuszko M, Ratajczak-Wrona W. Neutrophil extracellular traps (NETs) formation induced by TGF-β in oral lichen planus - Possible implications for the development of oral cancer. Immunobiology. 2020; 225:151901. https://doi.org/10.1016/j.imbio.2019.151901 [PubMed]
- 90. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012; 7:e32366. https://doi.org/10.1371/journal.pone.0032366 [PubMed]