



Introduction
Adenosine deaminase (ADA) is a 41 kDa monomer protein whose functions included not only catalyst activity, co-stimulatory, allosteric modifications, and cell-cell communication, but it also plays a vital role in purine metabolism [1]. The enzyme is generally distributed in human tissues such as the thymus and spleen, with the highest levels found in the gastrointestinal tract and moderate activity found in the brain [2]. Deficiency of ADA activity can lead to severe combined immunodeficiency which manifested as liver disease, tuberculosis, infectious mononucleosis, HIV infection, and reperfusion injury of the infracted myocardium [3, 4].
With the development of medical technology, the survival expectancy of patients with ACI has greatly improved, but it is still a major threat to human health [5]. Adenosine has been known as a neuroprotective agent for more than 30 years. Phillis and his colleagues [6] showed that the neuroprotective mechanisms include inhibition of neuronal excitability through adenosine, reducing intracellular calcium levels, and reducing nerve damage. Notably, adenosine may be an important endogenous neuroprotectant [6–8]. Extracellular concentrations of adenosine can increase from normal baseline levels of approximately 1 mM to 100 mM or more during and after hypoxic or ischemic attacks [9]. Moreover, elevated adenosine levels after ischemic stroke has been reported [10, 11]. However, there are few reports on the association between ADA activity and ACI occurrence. This study aim was to explore this relationship.
Methods
Study population
The study enrolled a total of 7913 participants in the Affiliated Hospital of Qingdao University from December 2012 to June 2019, including 3968 subjects who met the diagnostic criteria for ACI and 3945 controls. Patients with a diagnosis of ACI were supported by magnetic resonance imaging (MRI) findings. The study excluded (1) individuals who underwent incomplete laboratory tests; (2) individuals with autoimmune diseases, such as systemic lupus erythematosus, liver disease, blood diseases, tuberculosis, and other serious illnesses; and (3) individuals with hemorrhagic stroke. The 3945 participants in the control group had no signs and symptoms of ACI, and MRI did not support the diagnosis. The written consent of all subjects or their legal representatives has been obtained. The study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University.
Clinical parameters
A detailed medical history and risk factors for ACI were recorded for all participants. Height and weight were recorded in centimeters (cm) and kilograms (Kg) respectively. Body mass index (BMI) was calculated as weight (kg) divided by the square of height (m). Systolic and diastolic blood pressure were measured twice every 30 minutes with an automatic oscilloscope device, and the mean values of systolic and diastolic blood pressure were taken respectively. Hypertension was diagnosed when systolic blood pressure (≥140 mmHg) and/or diastolic blood pressure (≥90 mmHg), or using antihypertensive medications. Diabetes mellitus (DM) was diagnosed if hypoglycemic drugs were used or fasting blood glucose (FBG) level ≥7.0 mmol/L or glycosylated hemoglobin (HbA1c) concentration ≥6.5%.
Evaluation of intracranial arterial stenosis
All subjects with ACI in the study underwent three-dimensional time-of-flight (3D TOF) magnetic resonance angiography (MRA) (3D- TOF MRA) with 3.0 T magnetic resonance scans. We defined intracranial vascular stenosis using the Warfarin-Aspirin Symptomatic Intracranial Disease (WASID) test criteria [12]. Intracranial artery stenosis (ICAS) was diagnosed when MRA showed occlusion or 50% to 99% stenosis. The following vessels were evaluated: bilateral internal carotid artery (ICA), bilateral middle cerebral artery (MCA, M1/M2), anterior cerebral artery (ACA, A1/A2), posterior cerebral artery (PCA, P1/P2), vertebral artery (VA), or basilar artery (BA).
Biochemical measurements
Blood samples from all participants were taken after overnight fasting of at least 8 hours. Whole blood samples from participants were collected by vacuum tube in the absence of anticoagulant and centrifuged at 1500 × g for 15 min. Serum concentrations of alanine aminotransferase (ALT), total cholesterol (TC), low--density lipoprotein cholesterol (LDL-C), serum creatinine (SCr), triglycerides (TG), high-density lipoprotein cholesterol (HDL-C), fasting blood glucose (FBG), UA, and ADA was measured with an automatic biochemistry analyzer (Hitachi HCP-7600, Hitachi, Japan).
Adenosine deaminase assay
ADA concentrations were tested at 37°C based on Giusti and Galanti [13]. ADA activity was measured by the peroxidase method using a commercial kit (Beijing Leadman Biochemistry Co., Ltd; China). ADA enzyme deaminates adenosine to generate inosine. Inosine generates hypoxanthine through purine nucleoside phosphorylase (PNP). The latter yielded UA and hydrogen peroxide (H2O2) under xanthine oxidase (XOD). Finally, H2O2 was also reacted with N-Ethyl-N- (2-hydroxy-3-sulfopropyl) -3-methylaniline and 4-aminoantipyrine in the presence of peroxidase to generate quinone dye that is monitored kinetically. Under standard assay conditions, the amount of enzyme required for adenosine to release 1 mmol of ammonia per minute was defined as 1 unit (1U) of ADA. Figure 1 shows the process of the enzymatic reaction.
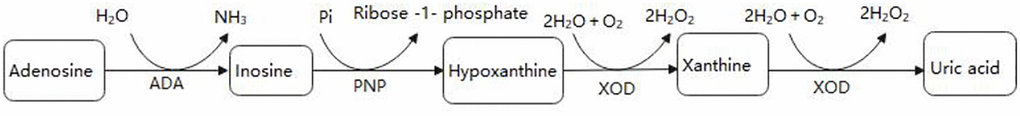
Figure 1. The enzymatic reaction scheme.
Statistical analyses
SPSS statistical software was used for statistical analysis (version 25.0; SPSS Inc., Chicago, Illinois, USA). The mean ± standard deviation (SD) was used to describe continuous data, while frequency and percentage were used to describe categorical variables. For comparisons in categorical variables, we used the Chi-square test. The study variables were compared between the patient group and the control group using the unpaired student’s t-test, and Spearman’s correlation coefficients was used to assess interrelationships. To investigate the interaction of other variables between ADA concentrations and ACI, logistic regression was used in this study. A two-sided test was used for statistical analysis, and P < 0.05 were considered statistically significant.
Results
A total of 7913 participants were enrolled including 3968 patients with ACI (68.33 ± 11.08 years) and 3945 controls without ACI (60.79 ± 14.51years). Of the 3968 ACI patients, 536 patients had single-diseased blood vessels, 313 patients presented two-diseased blood vessels and 1290 had three-diseased blood vessels. The concentrations of TG, BMI, FBG, TC, LDL-C, and ALT in the ACI group were higher than those in the control group. Patients with ACI were significantly older than those in the control group. Furthermore, the rates of hypertension, DM, smoking, and alcohol consumption were higher in the patient group than in the control group. We observed no statistical difference in SCr between the control group and the patients. There was no significant difference in ADA activity between different ICAS groups classified by the number of stenotic arteries (Figure 2). The information and clinical characteristics of the participants are shown in Table 1.
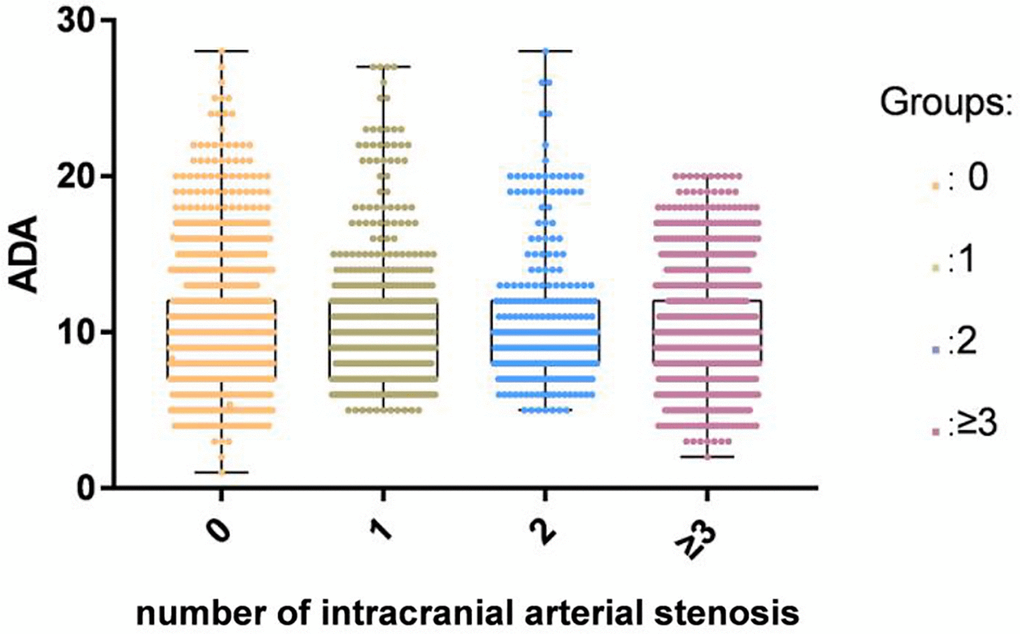
Figure 2. There was no significant difference in ADA activity among different ICAS groups classified by the number of stenotic arteries in the ACI group.
Table 1. Demographic and clinical characteristics of ACI patients and controls.
| Variable | ACI (3968) | Non-ACI (3945) | P value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years* | 68.33 ± 11.08 | 60.79 ± 14.51 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender, male n (%) # | 2779 (70.04%) | 2161 (54.78%) | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMI (kg/m2) * | 25.36 ± 5.69 | 24.99 ± 8.49 | 0.023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hypertension, n (%) # | 2573 (64.84%) | 879 (22.28%) | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diabetes, n (%) # | 1087 (27.39%) | 128 (3.24%) | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Smoking, n (%) # | 1608 (40.52%) | 685 (17.36%) | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Drinking, n (%) # | 1393 (35.11%) | 651 (16.50) | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FBG, mmol/L* | 13.34 ± 3.92 | 6.27 ± 2.41 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TG, mmol/L* | 1.57 ± 0.97 | 1.50 ± 1.14 | 0.005 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TC, mmol/L* | 6.26 ± 3.63 | 4.47 ± 1.17 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| UA, μmol/L* | 286.95 ± 72.11 | 276.05 ± 100.72 | 0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HDL-C, mmol/L* | 2.00 ± 1.36 | 2.08 ± 1.44 | 0.009 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LDL-C, mmol/L* | 3.33 ± 0.98 | 2.76 ± 0.97 | 0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SCr, μmol/L | 83.42 ± 19.26 | 82.65 ± 28.73 | 0.165 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ALT, U/L* | 18.84 ± 8.99 | 18.33 ± 7.57 | 0.007 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ADA, U/L* | 10.10 ± 3.72 | 11.07 ± 2.85 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Male, U/L* | 9.74 ± 3.65 | 10.85 ± 2.80 | <0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Female, U/L* | 10.92 ± 3.74 | 11.33 ± 2.89 | 0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Single-diseased vessels, n (%) # | 536 (13.51%) | − | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Double-diseased vessels, n (%) # | 313 (7.89%) | − | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Triple-diseased vessels, n (%) # | 1290 (32.51%) | − | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| #Categorical variables are expressed as percentages. p values of the categorical variables were calculated by χ2 test. *Continuous variables are expressed as the mean ± SD. p values of the continuous variables were calculated using unpaired t test. Abbreviations: ADA: adenosine deaminase; ALT: alanine aminotransferase; BMI: body mass index; FBG: fasting blood glucose; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; SCr: Serum creatinine; SD: standard deviation; TC: total cholesterol; TG: triglyceride; UA: uric acid. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
We found that serum ADA activity was positively correlated with age (r = 0.173, P < 0.001), FBG (r = 0.142, P < 0.001), TG (r = 0.038, P = 0.001), LDL-C (r = 0.065, P < 0.001), TC (r = 0.075, P < 0.001) and ALT (r = 0.034, P = 0.003) levels in patients with ACI. Furthermore, we observed a negative relationship in UA (r = −0.038, P < 0.001), and HDL-C (r = −0.075, P < 0.001) with serum ADA. These results are shown in Figure 3.
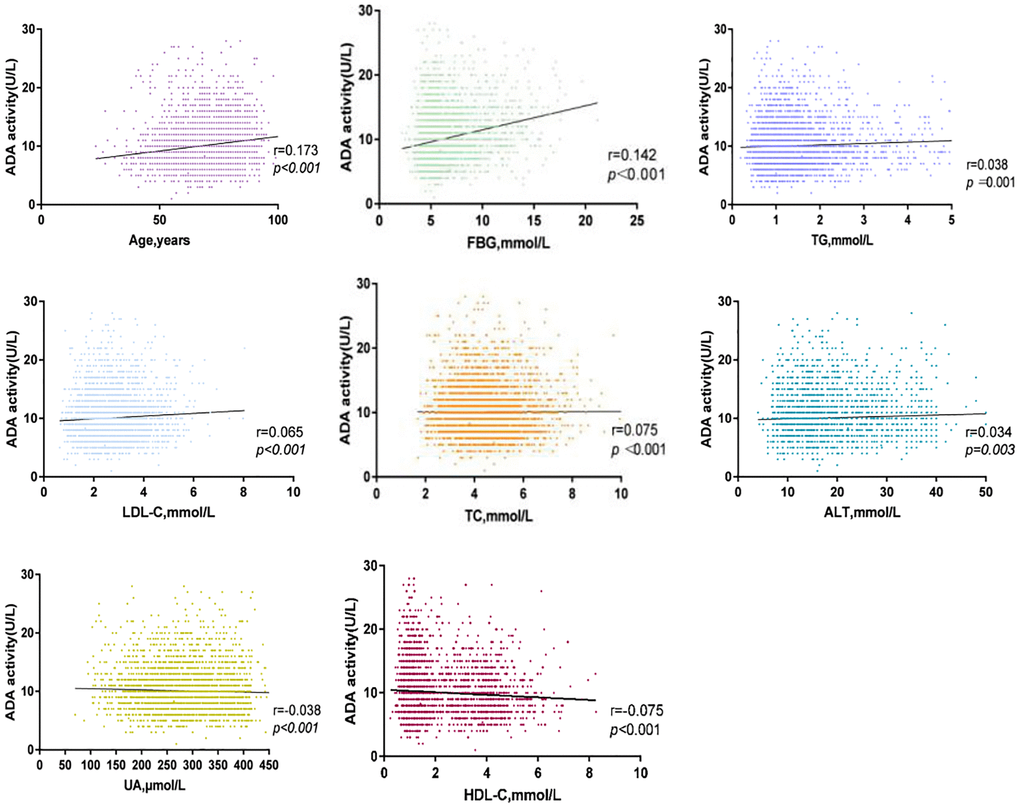
Figure 3. Correlation between serum ADA and TC, ALT, FBG, LDL-C, Age, UA, HDL-C, TG in ACI group.
We observed that ADA concentrations of ACI patients were affected by DM, hypertension, high alcohol consumption and smoking status. This study showed that in the experimental group, DM and hypertension markedly elevated serum ADA concentrations, whereas alcohol consumption, and smoking decreased ADA activity (Table 2).
Table 2. Clinical parameters and ADA activity.
| ACI patients with | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Smoking | Non-smoking | Drinking | Non-drinking | Hypertension | Non-hypertension | Diabetes | Non-diabetes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Patients number, n | 1608 | 2360 | 1393 | 2575 | 2570 | 1395 | 1087 | 2880 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ADA activity, U/L | 9.68 ± 3.57 | 10.39 ± 3.79 | 9.61 ± 3.75 | 10.56 ± 3.85 | 10.11 ± 3.62 | 10.08 ± 3.90 | 10.95 ± 3.96 | 9.78 ± 3.57 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0.037 | 0.043 | 0.007 | >0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ADA is expressed as the mean ± SD. ADA: adenosine deaminase; ACI: acute cerebral infarction; SD: standard deviation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In our retrospective study, serum ADA concentrations of ACI patients were markedly lower compared to that in the control group (10.10 ± 3.72 vs. 11.07 ± 2.85 U/L, P < 0.001) (Table 1). After adjusting the multivariate logistic regression model adjusted for potential risk factors, such as BMI, FBG, TG, TC, HDL-C, LDL-C, UA, ALT, smoking, alcohol consumption, hypertension, and DM status, serum ADA concentrations were significantly associated with the presence of ACI (OR = 1.161, 95% CI: 1.140–1.183, p < 0.001) (Table 3).
Table 3. Associations between serum ADA activity and presence of stroke.
| Adjustment | Models | OR 95% CI | P | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Model 1 | Crude, no adjustment | − | P < 0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Model 2 | Adjusting for age, sex, BMI, smoking, drinking, hypertension and diabetes statues | 1.144 (1.126–1.162) | P < 0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Model 3 | Adjusting for FBG, TG, TC, HDL-C, LDL-C, UA, SCr and ALT | 1.165 (1.144–1.187) | P < 0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Model 4 | Adjusting for FBG, TG, TC, HDL-C, LDL-C, UA, ALT, age, sex, BMI, smoking, drinking, hypertension and diabetes statues | 1.161 (1.140–1.183) | P < 0.001 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: ADA: adenosine deaminase; ALT: alanine aminotransferase; BMI: body mass index; ACI: acute cerebral infarct; CI: confidence interval; FBG: fasting blood glucose; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; OR: odds ratio; SCr: serum creatinine; TC: total cholesterol; TG: triglyceride; UA: uric acid. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Discussion
The present study was the first to identify an independent correlation between the attenuated concentrations of serum ADA activity and the occurrence of ACI. However, ADA activity across ICAS groups defined by the number of stenotic arteries did not show significant differences.
Our results showed that serum ADA concentrations were positively correlated with TG, LDL-C, and TC levels in the ACI group. Atherosclerosis is the most common risk factor for ACI. It has been reported that purinergic signal transduction is to participate in the regulatory process of vascular inflammation and atherosclerosis and plays a key role [14]. Increasing ADA concentrations have been reported to precede macrophage accumulation of macrophages and vascular lipid deposition, and then increased with higher plaque formation in a mouse model of atherosclerosis [15]. Therefore, increased vascular ADA activity has been proposed as an early marker and trigger of atherosclerosis. Kutryb Zajac et al. [16] revealed there was a positively correlated that markers of endothelial activation, vascular lipid content, plasma triglycerides, and LDL-C.
HDL-C has long been considered good cholesterol and epidemiological studies have shown that its plasma level is negatively correlated with cardiovascular and cerebrovascular risk [17]. We also observed an inverse correlation between HDL-C levels and ADA. ADA is involved in the atherosclerosis process, and HDL-C acts as a protective factor in atherosclerosis. However, how HDL-C acts on ADA remain unclear, and further research is needed.
In this work, DM was also observed to significantly increase ADA activity in stroke patients. A study showed that ADA concentrations of subjects with DM were significantly elevated than that in healthy controls and ADA concentrations of DM subjects were positively correlated with FBG levels [18]. Previous studies have shown that diabetes is a chronic low-grade inflammatory disease in which T lymphocytes are to participate in the immune response, characterized by islet β-cell dysfunction [19, 20]. ADA not only acts as a key factor in the proliferation, differentiation, and maturation of T lymphocytes [21, 22], but it is also an important enzyme that regulates the adenosine concentrations inactivation—which also acts as an important factor in glucose and insulin homeostasis and the pathophysiology of diabetes [23–25]. Elevated ADA activity in diabetic patients accelerates adenosine decomposition, thereby influencing blood glucose homeostasis [20]. On the other hand, ADA binds to DDPIV/CD26, a transmembrane glycoprotein on the surface of T lymphocytes, via the A2b receptor, thus inhibiting glucagon-like peptide-1 (GLP-1) [26]. It acts as a key factor in promoting insulin secretion, inhibiting glucagon secretion, and promoting islet cell proliferation and differentiation of islet cells [27].
In this study, we also observed a significant effect of smoking on serum ADA activity in patients with ACI. The main components of cigarettes are nicotine, tar, carbon monoxide (CO), and other substances [28]. Nicotine in cigarettes affects the cardiovascular system through sympathetic activation [28]. Studies have shown that patients who smoke can secrete more adrenaline, which induces platelet aggregation, and CO in smoke leads to tissue ischemia and hypoxia. Adenosine inhibits platelet aggregation, dilates blood vessels, and alleviates ischemia and hypoxia. Under the condition of smoking, ADA activity through feedback adjustment, reduce the decomposition of adenosine, thus maintaining high adenosine activity to protect the cardiovascular system. Alcohol consumption exerts a negative influence on serum ADA activity.
In a previous study, several metabolites were found to be risk factors for ACI such as homocysteine (Hcy), asymmetric dimethylarginine (ADMA), and UA [29–31]. However, both adenosine and NO have potential roles as an endogenous neuroprotective agent in ischemia [32–34]. Adenosine is a signaling molecule that appears in the extracellular environment and plays a key role in human physiology. It has a dual function, acting both as a homeostatic transcellular messenger and as a neuromodulator. Previous studies have indicated that extracellular adenosine concentrations can increase from approximately 1 mM at normal baseline levels to 100 mM or more during and after episodes of hypoxia or ischemia attacks [32]. It cannot pass freely through cell membranes, but it can be transported into the cell by equilibrative nucleoside transporters (ENTs) and requires the use of nucleoside transporters to facilitate this process [35]. When adenosine levels are increased rapidly, it is transported to vascular endothelial cells and red blood cells, where ADA rapidly metabolizes it to inosine [36]. ADA is a purine metabolic enzyme that irreversibly converts adenosine to inosine [37]. Adenosine can appear in the extracellular environment. Adenosine, also one of the methionine cycle products, is opposite to UA, Hcy, and ADMA in maintaining physiological homeostasis [38–40]. Furthermore, there are many enzymes, factors, and substances involved in the cycle, including L-arginine, vitamin B12, vitamin B6, and folate. Ultimately, adenosine and NO act on the cerebrovascular endothelium to induce vasodilation and also play a role in neuroprotection [33, 41, 42] (Figure 4).
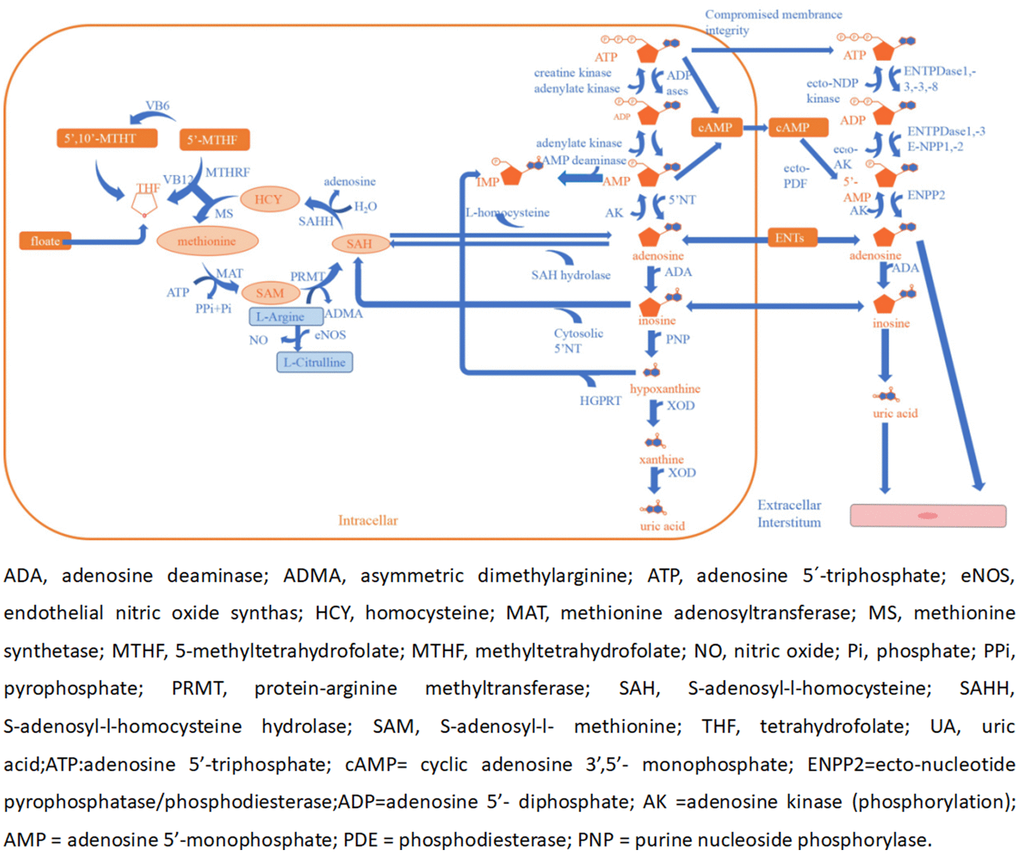
Figure 4. Adenosine and methionine metabolism pathways and endothelial dysfunction.
The underlying mechanism of attenuated ADA activity in patients with ACI is unclear, but we believe that a complex metabolic process is involved. ADA is a key enzyme that is related to cellular metabolism and its activity, biosynthesis, and catabolism and is regulated by the neurohormone axis. When ACI occurs, adenosine activity increases rapidly, acting on endothelial cells and causing vasodilation and neuroprotection effects. Under these conditions, to maintain higher adenosine activity to protect the cerebrovascular system. ADA activity undergoes negative feedback regulation and reduces adenosine decomposition. However, this study is preliminary, and the specific mechanisms involved need to be further studied.
This study has several limitations. First, we used 3D MRA to assess intracranial arterial stenosis, but MRA is not the gold standard for assessing intracranial stenosis. Second, in this study, not all control participants underwent exhaustive MRA. Third, the ADA activity exam was a one-time exam, which may not effectively represent the fluctuation of mediator levels. Fourth, in this study, only the differences in total ADA were observed, and the differences in its isoenzymes ADA1 and ADA2 between the study group and the control group were not further evaluated. Elevated serum isoenzyme ADA2 is commonly found in viral diseases, such as immunodeficiency virus infections. Studies have detected elevated levels of tADA and its isoenzymes ADA1 and ADA2 in saliva in patients diagnosed and convalescent with the new coronavirus disease 2019 (COVID-19). Finally, our study did not evaluate interactions with oral medications that may have affected ADA levels.
Conclusions
Our study provides proof that ADA concentrations are decreased in patients with ACI. We also show that ADA is influenced by hypertension, diabetes, and lifestyle. The results suggest that the decreased ADA activity may be involved in the pathogenesis of ACI. The exact physiopathological mechanism of ADA in ACI needs further study in the further.
Author Contributions
HQZ and JLF designed this article, and revised the manuscript. YYL collected and statistically analyzed the data, prepared the charts, and drafted and revised the manuscript. CJ, ZZX, XS, QL, BJW, NNZ, MLH, and FL assisted with data acquisition, data analysis and statistical analysis, and revision of the manuscript. All the authors contributed to the writing and revision of the paper, and all the authors have read and approved the final version.
Acknowledgments
We thank all the participants and all the authors for their support and contributions to this study.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement and Consent
The study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University. The written consent of all subjects or their legal representatives has been obtained.
References
- 1. Asakura M, Asanuma H, Kim J, Liao Y, Nakamaru K, Fujita M, Komamura K, Isomura T, Furukawa H, Tomoike H, Kitakaze M. Impact of adenosine receptor signaling and metabolism on pathophysiology in patients with chronic heart failure. Hypertens Res. 2007; 30:781–7. https://doi.org/10.1291/hypres.30.781 [PubMed]
- 2. Novotný J. [Adenosine and its role in physiology]. Cesk Fysiol. 2015; 64:35–44. [PubMed]
- 3. Bradford KL, Moretti FA, Carbonaro-Sarracino DA, Gaspar HB, Kohn DB. Adenosine Deaminase (ADA)-Deficient Severe Combined Immune Deficiency (SCID): Molecular Pathogenesis and Clinical Manifestations. J Clin Immunol. 2017; 37:626–37. https://doi.org/10.1007/s10875-017-0433-3 [PubMed]
- 4. Tavilani H, Sheikh N, Vaisi-raygani A, Setarehbadi R. Sex differences in adenosine deaminase activity of stroke patients. Clin Chem Lab Med. 2008; 46:506–9. https://doi.org/10.1515/CCLM.2008.108 [PubMed]
- 5. Yang G, Wang Y, Zeng Y, Gao GF, Liang X, Zhou M, Wan X, Yu S, Jiang Y, Naghavi M, Vos T, Wang H, Lopez AD, Murray CJ. Rapid health transition in China, 1990-2010: findings from the Global Burden of Disease Study 2010. Lancet. 2013; 381:1987–2015. https://doi.org/10.1016/S0140-6736(13)61097-1 [PubMed]
- 6. Phillis JW, Goshgarian HG. Adenosine and neurotrauma: therapeutic perspectives. Neurol Res. 2001; 23:183–9. https://doi.org/10.1179/016164101101198316 [PubMed]
- 7. Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005; 63:191–270. https://doi.org/10.1016/S0074-7742(05)63007-3 [PubMed]
- 8. von Lubitz DK. Adenosine in the treatment of stroke: yes, maybe, or absolutely not? Expert Opin Investig Drugs. 2001; 10:619–32. https://doi.org/10.1517/13543784.10.4.619 [PubMed]
- 9. Picano E, Abbracchio MP. Adenosine, the imperfect endogenous anti-ischemic cardio-neuroprotector. Brain Res Bull. 2000; 52:75–82. https://doi.org/10.1016/s0361-9230(00)00249-5 [PubMed]
- 10. Newby AC. Adenosine: origin and clinical roles. Adv Exp Med Biol. 1991; 309A:265–70. https://doi.org/10.1007/978-1-4899-2638-8_60 [PubMed]
- 11. Heistad DD, Marcus ML, Gourley JK, Busija DW. Effect of adenosine and dipyridamole on cerebral blood flow. Am J Physiol. 1981; 240:H775–80. https://doi.org/10.1152/ajpheart.1981.240.5.H775 [PubMed]
- 12. Samuels OB, Joseph GJ, Lynn MJ, Smith HA, Chimowitz MI. A standardized method for measuring intracranial arterial stenosis. AJNR Am J Neuroradiol. 2000; 21:643–6. [PubMed]
- 13. Zanini D, Schmatz R, Pelinson LP, Pimentel VC, da Costa P, Cardoso AM, Martins CC, Schetinger CC, Baldissareli J, do Carmo Araújo M, Oliveira L, Chiesa J, Morsch VM, et al. Ectoenzymes and cholinesterase activity and biomarkers of oxidative stress in patients with lung cancer. Mol Cell Biochem. 2013; 374:137–48. https://doi.org/10.1007/s11010-012-1513-6 [PubMed]
- 14. Jalkanen J, Yegutkin GG, Hollmén M, Aalto K, Kiviniemi T, Salomaa V, Jalkanen S, Hakovirta H. Aberrant circulating levels of purinergic signaling markers are associated with several key aspects of peripheral atherosclerosis and thrombosis. Circ Res. 2015; 116:1206–15. https://doi.org/10.1161/CIRCRESAHA.116.305715 [PubMed]
- 15. Kutryb-Zajac B, Mateuszuk L, Zukowska P, Jasztal A, Zabielska MA, Toczek M, Jablonska P, Zakrzewska A, Sitek B, Rogowski J, Lango R, Slominska EM, Chlopicki S, Smolenski RT. Increased activity of vascular adenosine deaminase in atherosclerosis and therapeutic potential of its inhibition. Cardiovasc Res. 2016; 112:590–605. https://doi.org/10.1093/cvr/cvw203 [PubMed]
- 16. Kutryb-Zajac B, Mierzejewska P, Sucajtys-Szulc E, Bulinska A, Zabielska MA, Jablonska P, Serocki M, Koszalka P, Milczarek R, Jasztal A, Bartoszewski R, Chlopicki S, Slominska EM, Smolenski RT. Inhibition of LPS-stimulated ecto-adenosine deaminase attenuates endothelial cell activation. J Mol Cell Cardiol. 2019; 128:62–76. https://doi.org/10.1016/j.yjmcc.2019.01.004 [PubMed]
- 17. Adorni MP, Ronda N, Bernini F, Zimetti F. High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells. 2021; 10:574. https://doi.org/10.3390/cells10030574 [PubMed]
- 18. Khemka VK, Bagchi D, Ghosh A, Sen O, Bir A, Chakrabarti S, Banerjee A. Raised serum adenosine deaminase level in nonobese type 2 diabetes mellitus. ScientificWorldJournal. 2013; 2013:404320. https://doi.org/10.1155/2013/404320 [PubMed]
- 19. Stentz FB, Kitabchi AE. Activated T lymphocytes in Type 2 diabetes: implications from in vitro studies. Curr Drug Targets. 2003; 4:493–503. https://doi.org/10.2174/1389450033490966 [PubMed]
- 20. Cao J, Wang H, Su JB, Wang XQ, Zhang DM, Wang XH, Liu WS, Ge XQ. Inverse relationship between serum adenosine deaminase levels and islet beta cell function in patients with type 2 diabetes. Diabetol Metab Syndr. 2021; 13:54. https://doi.org/10.1186/s13098-021-00671-2 [PubMed]
- 21. Yordanova M, Gerova D, Atanassova A, Galunska B. Adenosine Deaminase as a Useful Biomarker for Diagnosis and Monitoring of Inflammatory Bowel Disease. Clin Lab. 2020; 66. https://doi.org/10.7754/Clin.Lab.2019.191124 [PubMed]
- 22. Ebrahimi-Rad M, Khatami S, Ansari S, Jalylfar S, Valadbeigi S, Saghiri R. Adenosine Deaminase 1 as a Biomarker for Diagnosis and Monitoring of Patients with Acute Lymphoblastic Leukemia. J Med Biochem. 2018; 37:128–33. https://doi.org/10.1515/jomb-2017-0042 [PubMed]
- 23. Singh A, Gibert Y, Dwyer KM. The adenosine, adrenergic and opioid pathways in the regulation of insulin secretion, beta cell proliferation and regeneration. Pancreatology. 2018; 18:615–23. https://doi.org/10.1016/j.pan.2018.06.006 [PubMed]
- 24. Antonioli L, Blandizzi C, Csóka B, Pacher P, Haskó G. Adenosine signalling in diabetes mellitus--pathophysiology and therapeutic considerations. Nat Rev Endocrinol. 2015; 11:228–41. https://doi.org/10.1038/nrendo.2015.10 [PubMed]
- 25. Peleli M, Carlstrom M. Adenosine signaling in diabetes mellitus and associated cardiovascular and renal complications. Mol Aspects Med. 2017; 55:62–74. https://doi.org/10.1016/j.mam.2016.12.001 [PubMed]
- 26. Larijani B, Heshmat R, Ebrahimi-Rad M, Khatami S, Valadbeigi S, Saghiri R. Diagnostic Value of Adenosine Deaminase and Its Isoforms in Type II Diabetes Mellitus. Enzyme Res. 2016; 2016:9526593. https://doi.org/10.1155/2016/9526593 [PubMed]
- 27. Gracia E, Pérez-Capote K, Moreno E, Barkešová J, Mallol J, Lluís C, Franco R, Cortés A, Casadó V, Canela EI. A2A adenosine receptor ligand binding and signalling is allosterically modulated by adenosine deaminase. Biochem J. 2011; 435:701–9. https://doi.org/10.1042/BJ20101749 [PubMed]
- 28. Thomé GR, Mazzanti CM, Ahmed M, Corrêa M, Spanevello RM, Maldonado PA, Luchese C, Cargnelutti D, Morsch VM, Duarte MM, Fiorenza AM, Nogueira CW, De Bona KS, et al. Activity of ectonucleotidases and adenosine deaminase in rats exposed to cigarette smoke. Inhal Toxicol. 2009; 21:906–12. https://doi.org/10.1080/08958370802632267 [PubMed]
- 29. Chen S, Li N, Deb-Chatterji M, Dong Q, Kielstein JT, Weissenborn K, Worthmann H. Asymmetric dimethyarginine as marker and mediator in ischemic stroke. Int J Mol Sci. 2012; 13:15983–6004. https://doi.org/10.3390/ijms131215983 [PubMed]
- 30. Song Q, Wang Y, Cheng Y, Liu J, Wei C, Liu M. Serum Uric Acid and Risk of Hemorrhagic Transformation in Patients with Acute Ischemic Stroke. J Mol Neurosci. 2020; 70:94–101. https://doi.org/10.1007/s12031-019-01404-x [PubMed]
- 31. Wang M, Liang X, Cheng M, Yang L, Liu H, Wang X, Sai N, Zhang X. Homocysteine enhances neural stem cell autophagy in in vivo and in vitro model of ischemic stroke. Cell Death Dis. 2019; 10:561. https://doi.org/10.1038/s41419-019-1798-4 [PubMed]
- 32. Stone TW. Adenosine, neurodegeneration and neuroprotection. Neurol Res. 2005; 27:161–8. https://doi.org/10.1179/016164105X21896 [PubMed]
- 33. Chin-Dusting JP, Willems L, Kaye DM. L-arginine transporters in cardiovascular disease: a novel therapeutic target. Pharmacol Ther. 2007; 116:428–36. https://doi.org/10.1016/j.pharmthera.2007.08.001 [PubMed]
- 34. Bladowski M, Gawrys J, Gajecki D, Szahidewicz-Krupska E, Sawicz-Bladowska A, Doroszko A. Role of the Platelets and Nitric Oxide Biotransformation in Ischemic Stroke: A Translative Review from Bench to Bedside. Oxid Med Cell Longev. 2020; 2020:2979260. https://doi.org/10.1155/2020/2979260 [PubMed]
- 35. Camici M, Garcia-Gil M, Tozzi MG. The Inside Story of Adenosine. Int J Mol Sci. 2018; 19:784. https://doi.org/10.3390/ijms19030784 [PubMed]
- 36. Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009; 111:904–15. https://doi.org/10.1097/ALN.0b013e3181b060f2 [PubMed]
- 37. Cristalli G, Costanzi S, Lambertucci C, Lupidi G, Vittori S, Volpini R, Camaioni E. Adenosine deaminase: functional implications and different classes of inhibitors. Med Res Rev. 2001; 21:105–28. https://doi.org/10.1002/1098-1128(200103)21:2<105::aid-med1002>3.0.co;2-u [PubMed]
- 38. Rochette L, Lorin J, Zeller M, Guilland JC, Lorgis L, Cottin Y, Vergely C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: possible therapeutic targets? Pharmacol Ther. 2013; 140:239–57. https://doi.org/10.1016/j.pharmthera.2013.07.004 [PubMed]
- 39. Maxwell AJ, Bruinsma KA. Uric acid is closely linked to vascular nitric oxide activity. Evidence for mechanism of association with cardiovascular disease. J Am Coll Cardiol. 2001; 38:1850–8. https://doi.org/10.1016/s0735-1097(01)01643-6 [PubMed]
- 40. Moselhy SS, Demerdash SH. Plasma homocysteine and oxidative stress in cardiovascular disease. Dis Markers. 2003; 19:27–31. https://doi.org/10.1155/2003/137047 [PubMed]
- 41. Moat SJ, Lang D, McDowell IF, Clarke ZL, Madhavan AK, Lewis MJ, Goodfellow J. Folate, homocysteine, endothelial function and cardiovascular disease. J Nutr Biochem. 2004; 15:64–79. https://doi.org/10.1016/j.jnutbio.2003.08.010 [PubMed]
- 42. SoRelle R. Fortification of food with vitamin B12 in addition to folic acid might reduce cardiovascular disease risk. Circulation. 2002; 105:E9070. [PubMed]