



Introduction
Variability in the trajectory of age-related cognitive decline has stimulated research into the relationship between systemic inflammation and cognitive function. Polymorphisms in immune response-related genes [1–6] and elevated markers of systemic inflammation [7–11] have been associated with an increased rate of cognitive decline. In addition, severe acute systemic inflammation can result in long-lasting cognitive impairments, including increased susceptibility to neurodegeneration after resolution of the infection [12–14]. Concern about the history of inflammation has increased due to the COVID-19 pandemic and neurological features of the disease, which suggest possible long-term effects [15–19]. The results from studies in humans suggest that in order to understand the relationship between severe infections that arise in adulthood and cognitive impairment with advanced age, it is important to distinguish between changes in baseline verses a difference in the trajectory of cognitive decline [10, 11, 20–26].
Animal models indicate that inflammation induced by lipopolysaccharide (LPS) treatment, during a period of neurodevelopment or in young adulthood, may increase vulnerability to cognitive impairment with age or due to a subsequent occurrence of systemic inflammation through altered synaptic plasticity mechanisms [27–29]. One possibility is that early inflammation primes or trains the brain to increase responsiveness to a secondary immune stimulation by either a subsequent systemic inflammation or chronic low-level systemic inflammation associated with aging. In contrast, other studies indicate that LPS treatment in young adults result in immune tolerance in the brain due to long-lasting epigenetic changes, resulting in decreased expression of LPS-induced pro-inflammatory genes, cytokines, chemokines, and neurotrophic factors, and increased phagocytosis [30, 31]. The goals of this study was to longitudinally examine the effect of systemic inflammation, administered in young adult (6 months) male rats, on subsequent cognitive decline during aging, and related treatment effects on synaptic function and gene expression in the hippocampus.
Results
Behavioral characterization at 12 months of age
At 6 months of age, rats were injected with vehicle (n = 12) or LPS (n = 12) once a week for 7 weeks. Cognitive function was first assessed at 12 months of age (6 months after the onset of injections). For the cue discrimination task, there was an effect of training [F(4, 80) = 18.76, p < 0.0001] in which animals swam less distance to find the platform over the blocks of training (Figure 1A), in the absence of a treatment effect. Similarly, for spatial discrimination training, there was an effect of training [F(4, 80) = 12.2, p < 0.0001] on the distance to find the platform (Figure 1B), in the absence of a treatment effect. A repeated measures ANOVA performed on the DI scores for the acquisition, 2-hr, and 24-hr memory probe trials indicated no effect of treatment and an effect of repeated testing [F(2, 44) = 3.848, p < 0.05]. Post hoc analysis indicated that the DI scores for the 24-hr probe trial were reduced relative to the acquisition and 2-hr retention probe trials (Figure 1C). Finally, one group t-tests, on the DI scores within each treatment group and probe trial, indicated performance was above chance for each group for each probe trial. The results indicate that, at middle-age, no group effects were observed for acquisition or retention of a spatial episodic memory, examined 6 months after the LPS injections.
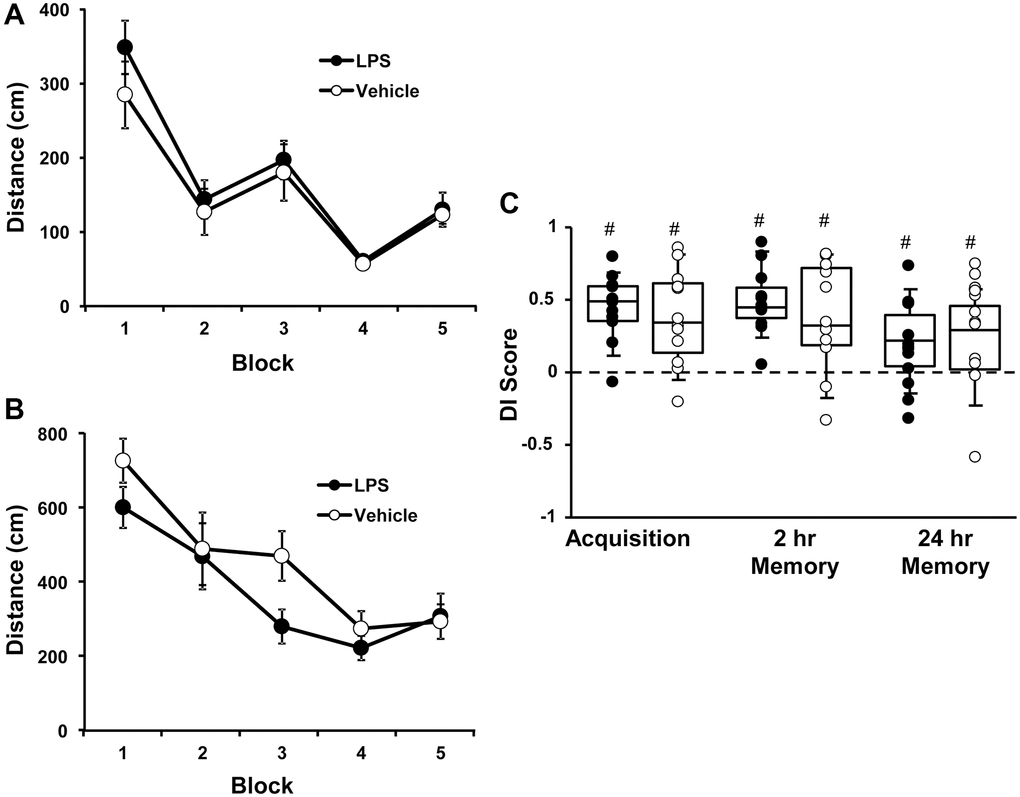
Figure 1. Vehicle and LPS treated animals learn cue and spatial discrimination with no difference in learning or memory at 12 months, 6 months after treatment. Symbols represent mean escape path length (± SEM) for vehicle (open circles) and LPS treated (filled circles) animals over the training blocks for (A) cue and (B) spatial discrimination. (C) Box and whisker plots and individual DI scores from the acquisition, 2 hr and 24 hr probe trials. Pound sign indicates significant difference from chance (p < 0.05).
Behavioral characterization at 18 months of age
At 18 months of age (12 months after the injections), the escape platform location was moved to a different location and rats were again assessed for acquisition and retention of a spatial episodic memory. A repeated measures ANOVA indicated an effect of training [F(4, 80) = 12.31, p < 0.0001], such that the distance to find the platform decreased over the training blocks, in the absence of a treatment effect (Figure 2A).
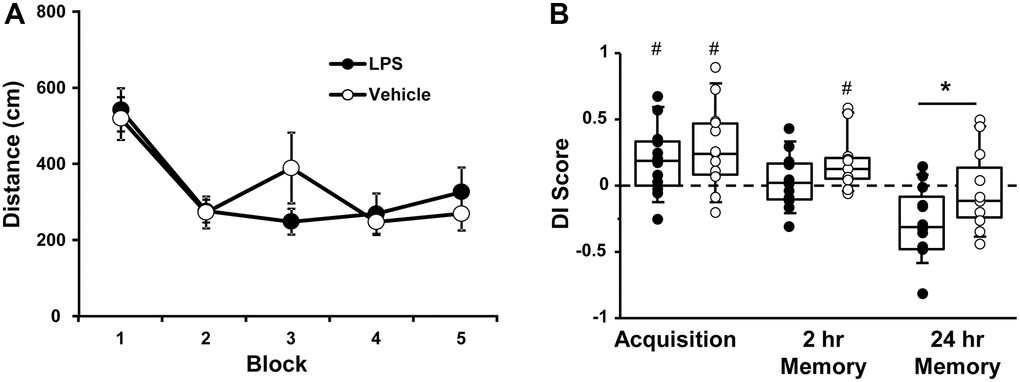
Figure 2. Vehicle and LPS treated animals learn the spatial discrimination and differences in memory emerge at 18 months, 12 months after treatment. Symbols represent mean escape path length (± SEM) for vehicle (open circles) and LPS treated (filled circles) animals over the training blocks for (A) spatial discrimination. (B) Box and whisker plots and individual DI scores from the acquisition, 2 hr and 24 hr probe trials. Pound sign indicates significant difference from chance (p < 0.05). Asterisk indicates significant treatment difference (p < 0.05).
A repeated measures ANOVA on the DI scores indicated an effect of repeated testing [F(2, 44) = 17.65, p < 0.0001] and treatment [F(1, 22) = 5.0, p < 0.05]. Post hoc tests for the repeated testing indicated that the 24-hr DI scores were reduced compared to the acquisition and the 2-hr probe trials and the 2-hr probe trial was reduced relative to the acquisition probe. Post hoc tests for treatment effects on each probe trial indicated that LPS-treated animals performed poorly compared to vehicle treated for the 24-hr probe [F(1, 22] = 4.65, p < 0.05]. One-tailed t-test indicated that both groups performed above chance during the acquisition and only vehicle treated animals were above chance for the 2-hr probe trail (Figure 2B).
While LPS treated animals exhibited impaired retention relative to controls, it is clear that age also influenced retention over the 24-hr delay. The interaction of age (12 and 18 months) and treatment was examined for probe trial performance using repeated measures ANOVAs. For the acquisition probe trial, a repeated measures ANOVA indicated decreased performance with age [F(1, 22] = 4.5, p < 0.05] in the absence of a treatment effect. However, planned repeated measures ANOVAs within each group indicated an age effect for the LPS group [F(1, 11] = 6.23, p < 0.05], but not the vehicle group (p = 0.52). Similarly, for the 2-hr probe trial a repeated measures ANOVA indicated a significant effect of age [F(1, 22] = 17.46, p < 0.0005] in the absence of a treatment effect and planned repeated measures ANOVAs within each group indicated a significant effect of age for the LPS group [F(1, 11] = 20.19, p < 0.001], but not the vehicle group (p = 0.12). Finally, for the 24-hr retention probe trial, a significant effect of age [F(1, 22] = 16.86, p < 0.0005], was observed in the absence of a treatment effect and planned repeated measures ANOVAs within each group indicated a significant effect of age for the LPS group [F(1, 11] = 14.36, p < 0.005] and a trend for an age effect in the vehicle group (p = 0.07). Thus, while age-related memory deficits over 24-hrs were variable in vehicle treated animals, a robust decline in memory was observed over the course of aging for LPS treated animals.
Hippocampal synaptic function
The effect of LPS exposure at 6 months of age on hippocampal synaptic transmission examined at 18 months of age was assessed in animals that were not behaviorally characterized. Hippocampal CA1-CA3 synaptic strength was examined by recording total-fEPSP and generating input-output curves and plotting the slope of total synaptic response across the different stimulation intensities for vehicle (n = 8/4 slices/animals) and LPS (n = 8/4 slices/animals) treated animals. A repeated-measures ANOVA performed across the stimulation intensities indicated an effect of stimulation intensity [F(7, 98) = 44.281, p < 0.0001]. Despite a general decrease in the synaptic response for LPS treated animals, no effect of treatment or interaction of treatment and stimulation intensity was observed for total synaptic response (Figure 3A). After assessing the total synaptic response, the NMDAR-mediated synaptic component was pharmacologically isolated. Again, we generated input-output curves for vehicle and LPS treated animals. For the NMDAR-mediated synaptic response, a repeated measures ANOVA indicated an effect of stimulation intensity [F(7, 98) = 42.774 p < 0.0001], treatment [F(1, 14) = 39.439, p < 0.0001], and an interaction of intensity X treatment [F(7, 98) = 26.346, p < 0.0001] due to a robust decrease in the NMDAR-mediated synaptic response in LPS treated animals compared to vehicle (Figure 3B).
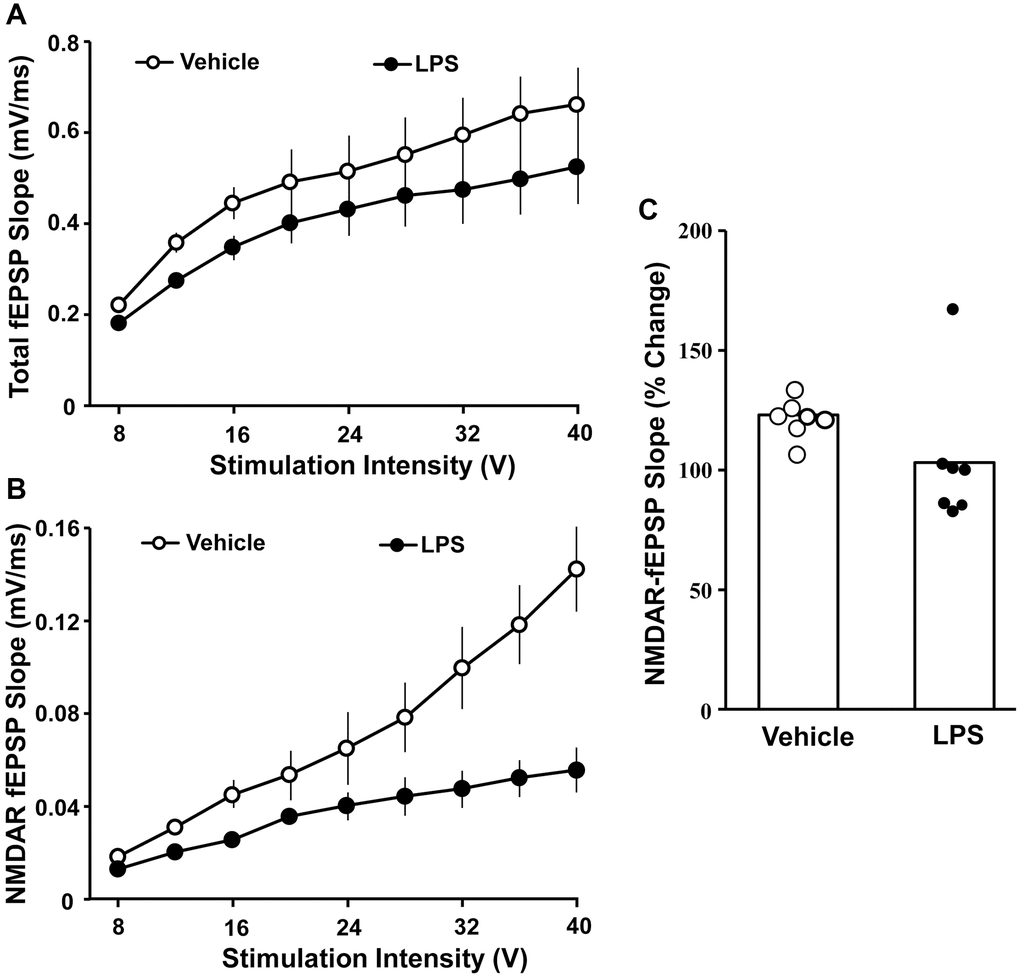
Figure 3. Decreased NMDAR-mediated synaptic responses associated with prior LPS treatment. Input-output curves for the mean slope (± SEM) of the total fEPSP (A) and NMDAR-fEPSP (B) evoked by increasing stimulation voltage (V). Data is presented for the vehicle (open circles) and LPS treatment (filled circles) recorded at 18 months, 12 months after the final LPS or vehicle injection. (C) Bars illustrating mean percentage change in NMDAR fEPSP slope induced by bath application of DTT in slices obtained from LPS (n = 7/4 slices/animal) or vehicle (n = 7/4 slices/animal)-treated animals. The distribution of individual responses is also depicted.
The decline in the NMDAR-mediated component of synaptic transmission with age can result from a decline in receptors or a redox mediated hypofunction [32–34]. To determined possible differences in NMDAR function due to redox state, the reducing agent, DTT, was added to the bath after collection of baseline NMDAR-mediated synaptic responses and the response was collected for 1 hr in slices obtained from LPS (7/4 slices/animal) and vehicle (7/4 slices/animal) treated animals (Figure 3C). An ANOVA indicated no difference between groups; however, t-tests indicated that DTT application increased the synaptic response relative to baseline in slices from vehicle control animals [t (6) = 6.26, p < 0.001], while the post DTT response was not different from baseline in LPS treated animals (Figure 3C).
RNA expression
For behaviorally characterized LPS (n = 8) and vehicle (n = 8) treated rats, RNA-sequencing was performed on the CA1 region of the hippocampus collected at 18 months of age. Differential expression filtering resulted in 157 genes that increased and 286 genes that decreased with treatment. For the genes that increased with LPS treatment, unsupervised functional annotation analysis indicated that these genes were enriched for lipid biosynthetic process (Figure 4, Table 1). Functional annotation clustering analysis for genes that were downregulated with LPS treatment were related to excitatory synapse, postsynaptic density, dendrite, synapse, neuron projection development, and synapse organization (Figure 4, Table 1). In order to examine possible candidate genes related to inflammation, we examined genes within the clusters response to lipopolysaccharide (GO:0032496), regulation of immune system process (GO:0002682), and inflammatory response (GO:0006954). The LPS treatment group exhibited decreased expression of genes that are normally upregulated by LPS, involved in the production of cytokines (Adam8, Tnfrsf25, Comt, Bcr, Pml, Mavs, Nectin2, Dapk1), or linked to regulation of the immune response (Asb2, Cdkn1a, Cyp4f5, Dnaaf2, Mtor, Nthl1), including transcription regulators (Kdm6b, Tfe3, Tead4). In contrast, upregulated genes were associated with phagocytosis and toxic effects of microglial activation (Ripk1, Kcnn4, Snx4, Fcgr3a). Interestingly, Gng12, a negative regulator of the LPS response was increased. Interestingly, no differences were observed for markers of activated microglia (Aif1, Cd68) and astrocytes (Gfap). The results confirm other studies that find memory impairment linked to aging is associated with a decrease in synaptic genes [35–37] and suggest that prior LPS treatment was associated with an altered expression of genes linked to inflammatory/immune response.
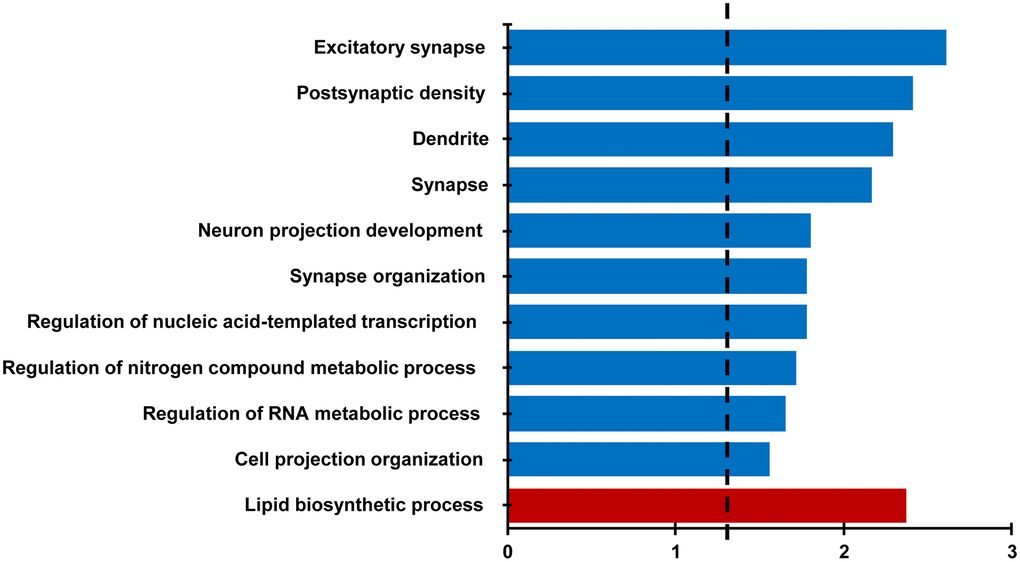
Figure 4. Differential gene expression analysis evaluating the effect of treatment in the CA1 region. Bars represent the –log (p value) for selected GO terms that were significant for down regulated (blue) and upregulated (red) genes. Dotted line is the –log (0.05).
Table 1. Gene ontology categories and lists of differentially expressed genes evaluating the effect of treatment in the CA1 region.
| Category | Term | Count | Genes | Benjamini p-value |
| GOTERM_BP | GO:0008610~lipid biosynthetic process | 17 | HMGCS1, NUS1, ST8SIA4, GPAT3, HSD17B7, INPP4B, INSIG1, IGF2, IDH1, MSMO1, PEX2, PDSS1, PTGDS, PTPMT1, SELENOI, SC5D, THRSP | 0.004 |
| GOTERM_CC | GO:0060076~excitatory synapse | 15 | BCR, MAGI2, BAIAP2, GRIK5, MINK1, BSN, PPP1R9B, SH2D5, SEMA4C, LRFN1, CAMK2B, NSMF, UNC13A, DISC1, ADD2 | 0.002 |
| GOTERM_CC | GO:0014069~postsynaptic density | 14 | BCR, MAGI2, BAIAP2, GRIK5, MINK1, BSN, PPP1R9B, SH2D5, SEMA4C, LRFN1, CAMK2B, NSMF, DISC1, ADD2 | 0.004 |
| GOTERM_CC | GO:0030425~dendrite | 25 | CRTC1, GRIK5, HCFC1, COMT, KCNJ2, ZMYND8, NUMA1, HTR1A, INPP5J, ANK3, AGO2, CAMK2B, HAP1, SLC8A2, MAGI2, BAIAP2, STRN4, MINK1, BSN, PPP1R9B, ADCY9, KHSRP, NSMF, RGS8, MTOR | 0.005 |
| GOTERM_CC | GO:0045202~synapse | 28 | CRTC1, GRIK5, COMT, KCNJ2, ZMYND8, AMPH, SH2D5, ANK3, LRFN1, CAMK2B, HAP1, DISC1, SLC8A2, MAGI2, BCR, BAIAP2, RIMBP2, STRN4, MINK1, BSN, PPP1R9B, DOK7, LRP6, SEMA4C, NSMF, DOC2B, UNC13A, ADD2 | 0.008 |
| GOTERM_BP | GO:0031175~neuron projection development | 29 | CRTC1, ZMYND8, FOXO6, IGF1R, JADE2, FOLR1, UNC5A, INPP5J, ANK3, CAMSAP1, LRFN1, OBSL1, CAMK2B, MKL1, HAP1, DISC1, MAGI2, BAIAP2, LRRN2, SDK1, MINK1, NTNG2, ARID1B, PPP1R9B, SEMA4C, NSMF, MTOR, UNC13A, KIF26B | 0.02 |
| GOTERM_BP | GO:0050808~synapse organization | 13 | SEZ6L2, HTR1A, MAGI2, ANK3, MDGA1, DOK7, BSN, CAMK2B, SEZ6L, ZMYND8, UNC13A, DISC1, ADGRB2 | 0.02 |
Western blot analysis
CA1 tissue samples from LPS- (n = 4) and vehicle-treated (n = 4) behaviorally characterized animals were prepared for Western blot analysis. Membranes were immuno-stained for GluN2B, GluN2A, PSD95, and GAPDH (Figure 5). PSD95 signal intensities, normalized to GAPDH, were not significantly different between treatment groups (p = 0.66), which suggests no difference in synapse number. In addition, we found no significant group differences in the expression of either subunit of NMDAR, GluN2B and GluN2A normalized to GAPDH (p = 0.26 and p = 0.85 respectively, not shown), or for each subunit normalized to PSD95 expression (p = 0.52 and p = 0.89, respectively) (Figure 5).
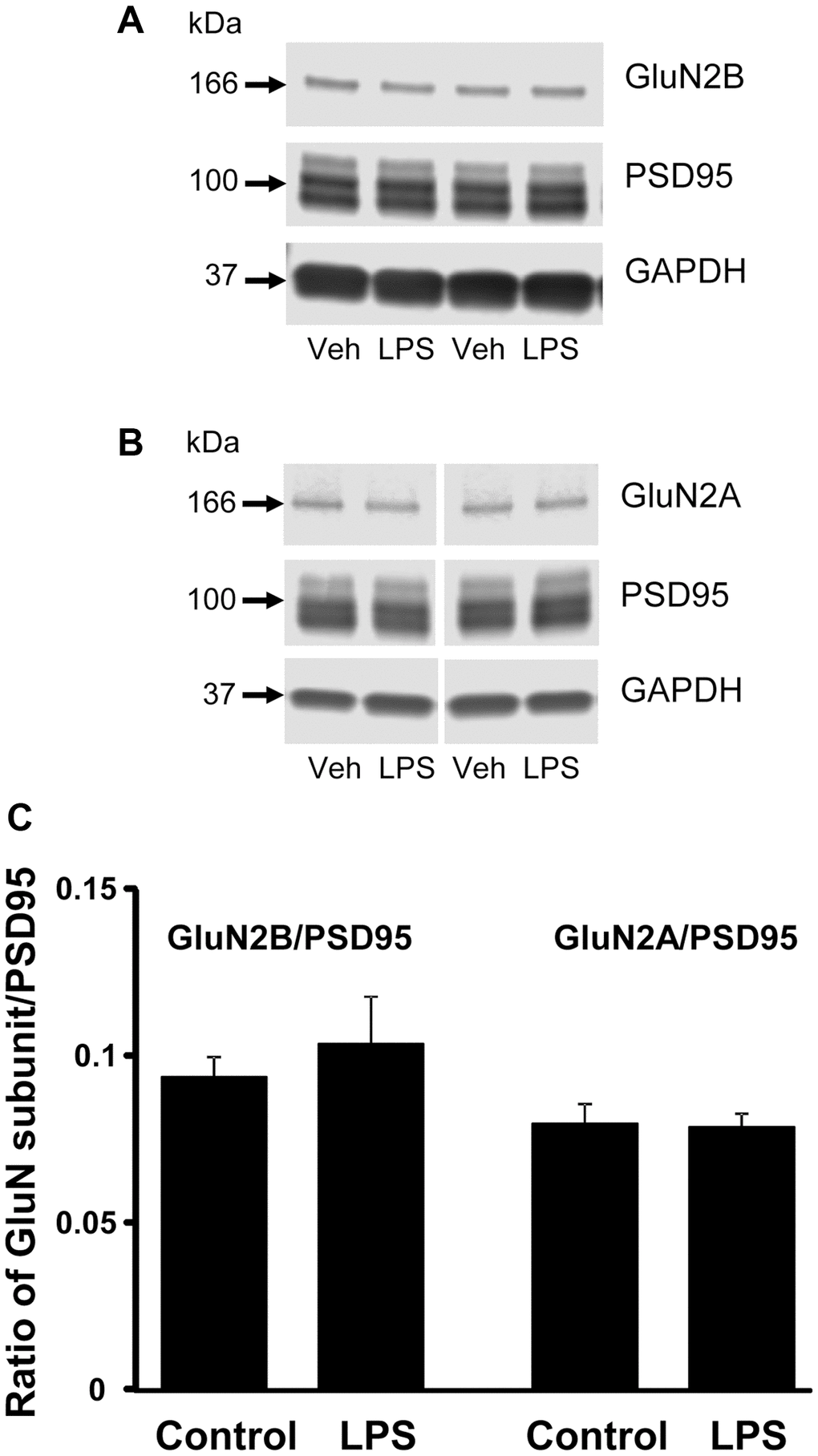
Figure 5. Western blot analysis of NMDAR subunit expression in CA1 region of hippocampus. The blots illustrate expression of (A) GluN2B, (B) GluN2A, and PSD95. (C) The bars represent the mean (± SEM) ratio of expression for vehicle (n = 4) and LPS treated (n = 4) animals.
Discussion
The current study indicates that inflammation in adulthood influences the trajectory of cognitive decline, from middle-age (12 months of age) to old age (18 months of age). At 18 months, 12 months after LPS injections, both groups exhibited a similar level of learning observed as a decrease in the distance to find the platform over the course of training. Furthermore, no difference was observed for the acquisition probe trial and both groups performed above chance indicating similar acquisition of a spatial search strategy. For retention probe trials, only the vehicle control group performed above chance for the 2-hr retention period. Also, the DI score for the 24-hr retention probe trial was decreased in LPS animals relative to vehicle controls. Furthermore, repeated measures analysis indicates that the age-related decline in cognition from 12 to 18 months was mainly due to LPS treated animals. The current study adds to the previous literature by demonstrating that inflammation initiated in adults, interacts with aging, contributing to the trajectory of cognitive decline, such that LPS treated animals exhibit greater susceptibility to memory deficits from middle-age to old age.
Age-related memory impairment is associated with a decline in NMDAR function [38] and impaired memory following LPS treatment in adults is also associated with decreased NMDAR function [35]. The current study provides the first evidence that systemic inflammation can have long-term effects on NMDAR synaptic function. Previous research in neonates suggest a shift in NMDAR subunit expression and the direction is dependent on when systemic inflammation occurred during development/maturation. Systemic inflammation initiated in neonates increased NMDAR subunit expression, particularly in the dentate gyrus, while inflammation initiated in adults resulted in a decrease in hippocampal NMDAR subunits [29, 39]. We did not observe a change in expression of GluN2B or GluN2A protein. However, we cannot rule out that the decrease in the NMDAR synaptic response was due to a decrease in the GluN1 subunit or altered plasticity processes that regulate the localization of NMDARs to the synapse.
Work from several labs indicates that the age-related NMDAR hypofunction is due to redox regulation of NMDARs [32, 40–44] involving translocation of NMDARs into the synapse [34]. We observed that addition of the reducing agent, DTT, increased the NMDAR synaptic response in aged, vehicle-treated animals. The inability to increase the NMDAR response of LPS treated animals under reducing conditions suggests alterations in the molecular machinery for NMDAR plasticity relative to that observed for normal aging [35].
For genes that passed our statistical filter, unsupervised analysis of gene enrichment indicated that LPS-treatment was associated with decreased expression of genes linked to synaptic function (excitatory synapse, postsynaptic density, dendrite, synapse, and synapse organization). The decrease in synaptic gene expression is consistent with work indicating that age-related cognitive decline is associated with decreased expression of synaptic genes [36, 37, 45]. However, it is unclear if the gene changes contribute to the decline in NMDAR synaptic transmission. It is also possible that altered expression represents an attempt to compensate for the decrease in NMDAR function/plasticity [38]. In previous studies examining the transcriptional profile of young adults treated with LPS, an increase in expression of synaptic genes was observed, suggesting a recovery/resilience process for young animals in response to the decline in synaptic transmission [35, 46]. Several of the recovery/resilience synaptic genes (Sema4c, Sez6) were down regulated in the current aged-LPS treated group and other down regulated synaptic genes (Camk2b, Crtc1, Mink1) were reported to decrease in aged cognitively impaired animals [35]. Taken together, the results suggest that the history of neuroinflammation contributes to the loss of synaptic function and decreased synaptic gene transcription during aging, possibly augmenting a loss of resilience in the face of aging stressors.
LPS treatments can have long-term effects on the brain’s response to inflammatory signals, possibly through epigenetic regulation of transcription. Acute peripheral inflammation results in immune priming or training, increasing the brains inflammatory response to a subsequent bout of systemic inflammation. In contrast, repeated LPS injections induce immune tolerance of microglia, which can be detected weeks or months following LPS injections [30, 31]. In the current study, several of the transcriptional changes associated with repeated LPS injections are consistent with an immune tolerance. Relative to vehicle controls, animals previously treated with LPS exhibited decreased expression of genes that are normally upregulated by LPS or the production of cytokines including those involved in signaling in response to LPS, the Tumor necrosis factor (TNF) family receptor, Tnfrsf25, and immunoreceptor (Bcr). Similarly, down regulation was observed for genes that promote cytokine production (Pml, and Dapk1, Mavs) [47, 48], regulate the cytokine response (Comt) [49–51], and mediate signaling downstream of cytokine activation (Cdkn1a, Adam8, Ripk1) [52–54]. Moreover, upregulated genes included Gng12, a negative regulator of the LPS response [55]. Immune tolerance of the brain might be beneficial in the face of chronic or repeated inflammation-inducing conditions. However, cytokines have a biphasic function on cognition, wherein low-levels improve and high-levels impair memory function [56–59]. Thus, a shift in mechanisms for cytokine signaling, either increasing or decreasing signaling, could impair memory.
Other upregulated genes suggest an increase in phagocytosis, potassium currents, and release of nitric oxide, which are observed following an inflammatory challenge in immune tolerant microglia [31]. In particular, there was an increase in expression of the potassium channel Kcnn4, which regulates the release of nitric oxide [60–64]. Increased expression was also observed for the cell-surface phagocytosis receptor, Fcgr3a, and genes linked to autophagy (Atg12, Snx4), antigen presentation pathway (RT1-CE6), Itm2a, involved in immunoglobulin production, and Oas1g, an immune response protein against viral infection. Finally, decreased expression was observed for epigenetic and transcription factor regulators, including those involved in inflammation (Kdm6b, Tfe3, Tead4) and DNA base excision repair (Nthl1), suggesting that inflammation in adulthood may have triggered epigenetic mechanism resulting in long-term changes in responsiveness to cytokines and inflammatory signals. Finally, it is possible that behavioral and brain differences were due to LPS induced cell senescence and the release of metabolic waste and toxic factors from other tissues.
Finally, it is possible that behavioral and brain differences were due to the history of systemic inflammation interacting with the age-related increase in chronic low level systemic inflammation, sometimes referred to as “inflammaging”. A bout of severe systemic inflammation can induce senescence of peripheral cells, including cells of the immune system [65–69]. In turn, senescent cells exhibit a senescence associated secretory phenotype (SASP), releasing toxic factors: pro-inflammatory cytokines, chemokines, extracellular matrix proteases, and microRNA in extracellular vesicles, that contribute to age-related diseases. Furthermore, senescent cells exhibit hyper-activation in response to a variety of inflammatory mediators, increasing the release of pro-inflammatory cytokines and chemokines [70]. If the history of infection alters the number or responsiveness of peripheral senescent cells, this could influence the level of inflammaging, which has been linked to cognitive impairment in humans [7–11] and animal models [35, 71, 72]. Future studies should consider examining the relationship between the history of infection on measures of senescent cells or markers of systemic inflammation, as a measure of biological aging that influences the trajectory of cognitive decline.
In summary, repeated LPS treatment in adults was associated with long-term effects on the trajectory of age-related cognitive decline, NMDAR function, and expression of genes linked to the synapse and regulation of the response to inflammatory signals. The results of the current study suggest that the history of systemic inflammation is one component of environmental factors that contribute to the resilience or susceptibility to age-related brain changes and associated trajectory of cognitive decline.
Materials and Methods
Animals
Young (n = 32, 6 months) male Fischer 344 X Brown Norway hybrid rats were obtained from the National Institute on Aging colony through the University of Florida Animal Care and Service facility. Animals were housed in pairs on a 12:12 light/dark cycle (lights on at 6 PM). All procedures involving animals were approved by Institutional Animal Care and Use Committee at the University of Florida and were in agreement with guidelines recognized by the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Experimental paradigm
Figure 6 illustrates the experimental timeline used to assess the longitudinal effect of LPS on spatial learning and memory. Following arrival, animals were acclimated to the new environment for one week, then administered intraperitoneal injections of LPS (1 mg/kg; n = 16) or vehicle (n = 16) once a week for 7 weeks. Our previous work indicates that this procedure induces a cognitive impairment, reduces the hippocampal CA3-CA1 synaptic response, and alters transcription examined days after the last injection [35]. A subset of these animals (LPS n = 12, vehicle n = 12) were cognitively assessed on the cue and spatial versions of the water maze tasks at 12 months of age (6 months after the onset of injections). The cue version of the water maze task was performed only at 12 months, since procedural memory for how to perform the swim task (i.e., how to swim and that the pool wall is not an escape route) is retained across the lifespan [73–75]. These same animals were again assessed on the spatial version of the water maze task at 18 months of age (12 months after the injections). Another subset of animals (LPS n = 4, vehicle n = 4), were not behaviorally characterized and were used for electrophysiological experiments at 18 months of age (12 months after the injections).
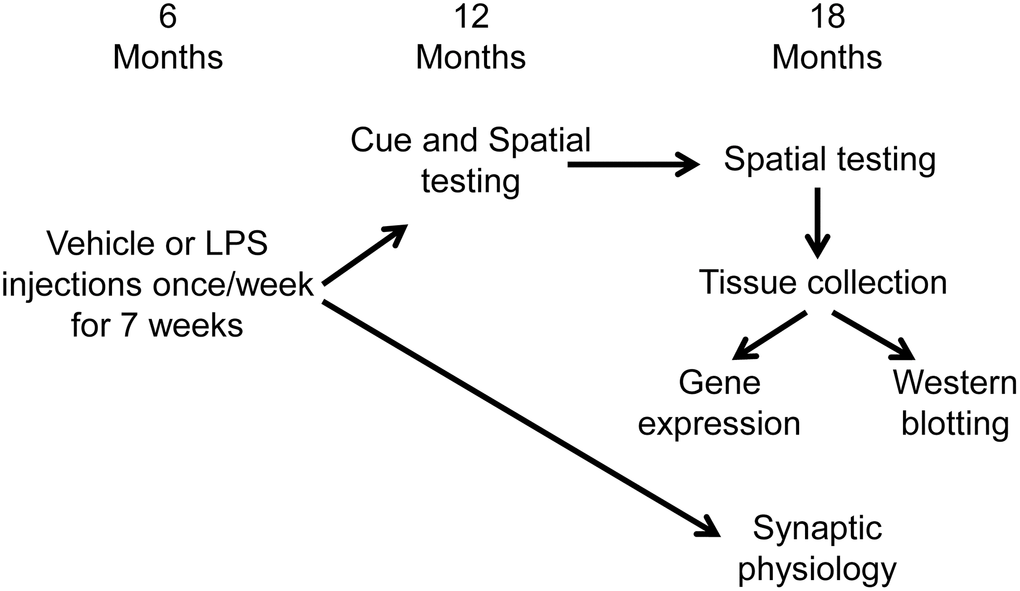
Figure 6. Schematic representing the experimental paradigm for the longitudinal effect of systemic inflammation on cognition. Young (6 months) male Fischer 344 X Brown Norway hybrid rats were either injected with vehicle (n = 16) or LPS (n = 16) once a week for 7 weeks. A subset of animals (vehicle n = 12; LPS n = 12) were cognitively assessed on the spatial discrimination water maze task at 12 and 18 months of age. Hippocampal tissue from behaviorally characterized rats was collected one week after completion of behavioral testing, at 18 months of age, and RNA sequencing was performed on the CA1 region of the hippocampus. The other group of animals (vehicle n = 4; LPS n = 4) were not behaviorally characterized and were used for electrophysiological experiments at 18 months of age (12 months after the injections).
Electrophysiology
Methods for collection of hippocampal slices and recordings have been published previously [34, 40, 76]. Briefly, rats were anesthetized with isoflurane (Halocarbon Laboratories, River Edge, NJ, USA) and swiftly decapitated. The brains were rapidly removed and the hippocampi were dissected. Hippocampal slices (~400 μm) were cut parallel to the Alvear fibers using a tissue chopper. The slices were incubated in a holding chamber (room temperature) containing standard artificial cerebrospinal fluid (aCSF) (in mM): NaCl 124, KCl 2, KH2PO4 1.25, MgSO4 2, CaCl2 2, NaHCO3 26, and glucose 10. Thirty to sixty minutes before recording, 2–3 slices were transferred to a standard interface recording chamber (Harvard Apparatus, Boston, MA, USA). The chamber was continuously perfused with standard oxygenated (95% O2, 5% CO2) aCSF at a flow rate of 2 ml/min. The pH and temperature were maintained at 7.4 and 30 ± 0.5°C, respectively. Humidified air (95% O2, 5% CO2) was continuously blown over the slices.
The total extracellular synaptic field potential (total-fEPSP) from CA3-CA1 hippocampal synaptic contacts were recorded with a glass micropipette (4–6 MΩ) filled with aCSF. Concentric bipolar stimulating electrodes (outer pole: stainless steel, 200 μm diameter; inner pole: platinum/iridium, 25 μm diameter, Fredrick Haer and Co, Bowdoinham, ME, USA) were positioned on approximately 1 mm of a recording electrode localized to the middle of stratum radiatum to stimulate CA3 inputs onto CA1. Using an SD9 stimulator (Grass Instruments, Braintree MA, USA), field potentials were induced by single diphasic stimulus pulses (100 μs). The signals were amplified, filtered between 1 Hz and 1 kHz, and stored on computer disk for off-line analysis (Data Wave Technologies, Longmont, CO, USA). The N-methyl-D-aspartate receptor (NMDAR)-mediated component of synaptic transmission (NMDAR-fEPSP) was obtained by incubating the slices in aCSF that contained low magnesium (Mg2+) (0.5 mM), 6,7-dinitroquinoxaline-2,3-dione (DNQX, 30 μM), and picrotoxin (PTX, 10 μM) [40, 71, 77]. Input-output curves for the total and NMDAR fEPSP (mV/ms) were constructed for increasing stimulation intensities.
The reducing effect of dithiothreitol (DTT) (0.5 mM) on NMDAR-fEPSP was performed by setting a baseline response at 50% of the maximum and the responses were collected for at least 10 min before and 60 min after drug application.
Spatial water maze
For behaviorally characterized animals, one week before behavioral testing, animals were handled to allow for acclimation to the new environment. The water maze tasks were tracked on the Ethovision computer software (Noldus Information Technology, Leesburg, VA, USA). The water was dyed white (Rich Art-Tempera Paint), which allowed for the animals performance to be tracked. The pool was surrounded by a black curtain.
Cue discrimination testing
The animals were first habituated to the pool by freely swimming for 30 seconds followed by a gentle guidance to the platform. After habituation, the cue version of the water maze task was performed, which assesses sensory-motor performance and permits animals to learn the procedural aspects of the task, minimizing thigmotaxis [78]. During the cue task, the platform was topped with a white flag and was 1 cm above the water level. For cue training, animals completed five blocks of three trials (15 total trials) massed into a single day. For each trial, the animal was placed into the water at a randomly assigned release point. The animal was given 60 seconds to find the randomly assigned platform. If the animal did not locate the platform within 60 seconds, the animal was gently guided to the platform.
Spatial discrimination testing
Three days after cue discrimination testing, animals were tested on the one-day version of the spatial water maze, in accordance with previously described methods [35]. Briefly, objects were attached to the black curtain surrounding the pool to act as extra-maze cues. The platform location remained the same across all trials and was submerged 1.5 cm under the water level. For each trial, the animal was placed into the water from a randomly assigned release point and given 60 seconds to find the platform. If the animal could not locate the platform, the animal was gently guided to the platform. Spatial training consisted of 4 training blocks of three trials (12 total trials). An acquisition probe trial was performed between blocks 4 and 5 to assess if animals had learned the platform location. For a probe trial, the platform was removed and the animal was released from the quadrant opposite of the platform (goal) location and allowed to swim for 60 seconds. Following the acquisition probe trial, another training block was performed (block 5). In addition, memory probe trials were delivered 2 and 24 hours after the acquisition probe trial. When animals performed the spatial discrimination task for the second time, at 18 months, the escape platform was moved to a new quadrant. To analyze performance on the probe trials, a discrimination index (DI) score was calculated. A DI score measures the time spent in the goal quadrant (contains platform) compared to the opposite quadrant [(Goal Quadrant-Opposite Quadrant)/(Goal Quadrant + Opposite Quadrant)].
Tissue collection
One week after completion of behavioral testing rats were removed from the home cage, anesthetized with isoflurane (Halocarbon Laboratories, River Edge, NJ, USA) and swiftly decapitated. The brains were rapidly removed and the subregions of the hippocampi were dissected. Samples were flash-frozen in liquid nitrogen.
RNA, library preparation, and sequencing
RNA-sequencing was performed on the hippocampal subregion CA1 from behaviorally characterized LPS (n = 8) and vehicle (n = 8) animals using the Ion Proton system. RNA was isolated using the RNeasy Lipid Tissue Mini kit (Qiagen, Hilden, Germany, catalog number 74804) with DNase digestion with RNase-Free DNase Set (Qiagen, catalog number 79254). A NanoDrop 2000 spectrophotometer was used to measure RNA concentration and a High Sensitivity (HS) RNA Screen Tape in an Agilent 2200 Tapestation system was used to quantify the RNA integrity number (RIN). As a control for library preparation, External RNA Controls Consortium (ERCC) spike-in controls (Thermo Fisher Scientific, catalog number 4456740) were added to samples. To select for poly-(A) mRNA, the Dynabead mRNA DIRECT Micro kit (Thermo Fisher Scientific, catalog number 61021) was used. Ion Total RNA-seq Kit v2 (Thermo Fisher Scientific, catalog number 4475936) was used to prepare libraries. For multiplex sequencing, Ion Xpress barcodes (Thermo Fisher Scientific, catalog number 4475485) were added. The concentration and size distribution of the libraries were assessed using the Qubit double-stranded DNA HS Assay (Thermo Fisher Scientific, catalog number 32851) and HS D1000 Screen Tape in a Tapestation system. An Ion Chef system was used for template preparation and sequenced on an Ion Proton system. Using the ERCC analysis plugin on the Torrent Server, the ERCC analysis was performed and found that samples contained at least 40 transcripts with an R2 of above 0.9. Each sample contained about 30 million reads of 145 base-pair length.
Bioinformatics and statistical analyses
Data analysis was performed using the Partek Flow server. FASTQ files were trimmed based on quality score and then aligned to the rat genome (rn6) using STAR. In Partek, gene-level counts were generated and annotated. DESeq2 was used to normalize genes. Genes for cluster analysis were filtered in two steps. First, genes with an average of less than 5 reads were removed [45, 79–81]. Second, statistical filter was used to generate a list of genes for cluster analysis. A p < 0.05 was used as a statistical filter to select differentially expressed genes (DEGs) between LPS treated animals compared to control. To assess the relationship of gene expression and cognitive performance, Spearman correlations were calculated between normalized counts of gene expression and the DI score measured during the 2-hr probe task. The criterion for a significant correlation was set at p < 0.05, consistent with our previous work [80].
Confidence in the significance of individual genes is low due to false positives associated with multiple comparisons across all genes. Gene enrichment analysis was performed under the assumption that changes in biological processes with treatment or cognition would result in a shift in the expression of clusters of genes related to the biological process [36]. Therefore, filtered genes were separated based on the direction of change and submitted to the National Institute of Health (NIH) Database for Annotation, Visualization, and Integrated Discovery (DAVID) for gene enrichment and functional annotation clustering analysis [36, 79, 80, 82, 83]. For unsupervised analysis, we report clusters for Gene Ontology (GO) terms for Cellular Components (CC), Biological Process (BP), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that exhibited a Benjamini False Discovery Rate (FDR) p < 0.05. In addition, we conducted directed analysis of several GO terms based on previous research examining age-related cognitive impairment and the hypothesis that prior LPS treatment would influence neuroinflammation responsiveness. We focused specifically on the synapse (GO:0045202), response to lipopolysaccharide (GO:0032496), regulation of immune system process (GO:0002682), and inflammatory response (GO:0006954).
Western blot analysis
The CA1 region of the dorsal hippocampus from behaviorally characterized LPS (n = 4) and vehicle (n = 4) animals were isolated, flash frozen in liquid nitrogen, and stored at −80°C. Tissue samples were sonicated and lysed in radio-immunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific cat#89900) supplemented with phosphatase inhibitors, protease inhibitors and EDTA (Thermo Fisher Scientific, Waltham, MA, USA) and centrifuged at 20,000 × g for 10 minutes at 4°C. Protein concentrations were measured using Pierce BCA protein assay (Thermo Fisher Scientific cat#23227). Lysates were combined with 2×-Laemmli sample buffer (Bio-Rad cat#1610737) containing 2-mercaptoethanol (Sigma cat#M3148) and boiled at 97–98°C for 5 minutes prior to electrophoresis. Samples (10 μg per well) and Chameleon Duo protein ladder (Li-Cor cat#928-60000) were separated on 4%–15% Criterion TGX stain-free gels (Bio-Rad cat#5678085) in running buffer (Tris/Glycine/SDS, Bio-Rad cat#1610732) at 80V/20 min, 100V/45 min, and 120V/30 min. Prior to transfer, filter papers, membranes, and gels were equilibrated in ice-cold transfer buffer (10× Tris/Glycine, Bio-Rad cat #1610771) containing 20% methanol. Proteins were transferred to low fluorescent polyvinylidene fluoride membranes (LF-PVDF, Bio-Rad cat#1620262) using a Criterion Blotter with plate electrodes (Bio-Rad, Hercules, CA, USA) at 100V/30 minutes. The immunoblots were washed in TBS and blocked with Intercept (TBS) Blocking Buffer (Li-Cor P/N 927-60001) for 1 hour at room temperature followed by overnight incubation at 4°C with primary antibodies diluted with Intercept T20 (TBS) Antibody Diluent (Li-Cor P/N 927-65001). Antibodies used were anti-GluN2B, mouse monoclonal (Millipore/Sigma, 05-920) 1:1000; anti-GluN2A, rabbit polyclonal (Invitrogen, A-6473) 1:500; anti-PSD95, mouse monoclonal (Thermo Fisher Scientific, MA1-045) 1:1000 and anti-GAPDH, mouse monoclonal (Encor Biotechnology, MCA1D4) 1:10,000. Membranes were washed 3 times in TBST prior to a one hour room temperature incubation with IRDye 800CW and 600LT secondary antibodies (1:20,000) diluted with Intercept T20 (TBS) Antibody Diluent. Membranes were washed with TBST 3 times for 10 minutes each, and then rinsed 3 times with TBS before scanning on Li-Cor Odyssey CLx Imaging System. The data were analyzed with Image Studio Lite Ver 5.2. Target protein signals were normalized to the expression of the house-keeping protein, GAPDH. Technical replicates (×2) for signal intensity were averaged across blots.
Statistical analysis for electrophysiology, behavior, and western blot data
ANOVAs were employed to examine input/output curves and treatment effects for synaptic responses. Similarly, repeated measures ANOVAs were used to examine treatment differences across training blocks for cue or spatial discrimination testing and across probe trial DI scores within each test period, 12 and 18 months. In addition, repeated measures ANOVAs were used to examine aging and LPS treatment on behavioral measures across the 12 and 18 month time points. Significant differences were localized using Fischer’s PLSD post hoc comparisons (p < 0.05). In addition, due to the prediction that LPS-treatment would impair cognition, post hoc ANOVAs within each treatment group were employed to determine if significant effects were driven by LPS or vehicle control animals. One-tailed one-group t-tests (p < 0.05) were performed to determine if the DI scores were above that expected by chance (i.e., DI score = 0) and for DTT studies to determine if the synaptic response increased above baseline.
Author Contributions
JB, designed and performed experiments, analyzed data, constructed illustrations, and wrote the manuscript, AK, designed and performed experiments, analyze data, constructed illustrations, wrote manuscript, LB, performed experiments and constructed illustrations, MC, performed experiments, TCF, designed the experiments, analyzed data, wrote the manuscript, and constructed illustrations.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
Financial support by National Institutes of Aging Grant R01 AG052258, R01 AG037984, and R21 AG068205, P30 AG028740, and the Evelyn F. McKnight Brain Research Foundation is highly appreciated. Special thanks to Nick Sarantos, Valentina Lavieri-Sosa, and Sophia Eikenberry, for their assistance in animal handling and behavior assessment.
References
- 1. Benke KS, Carlson MC, Doan BQ, Walston JD, Xue QL, Reiner AP, Fried LP, Arking DE, Chakravarti A, Fallin MD. The association of genetic variants in interleukin-1 genes with cognition: findings from the cardiovascular health study. Exp Gerontol. 2011; 46:1010–19. https://doi.org/10.1016/j.exger.2011.09.005 [PubMed]
- 2. Mooijaart SP, Sattar N, Trompet S, Polisecki E, de Craen AJ, Schaefer EJ, Jahn SE, van Himbergen T, Welsh P, Ford I, Stott DJ, Westendorp RG, and PROSPER Study Group. C-reactive protein and genetic variants and cognitive decline in old age: the PROSPER study. PLoS One. 2011; 6:e23890. https://doi.org/10.1371/journal.pone.0023890 [PubMed]
- 3. Trompet S, de Craen AJ, Slagboom P, Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Ford I, Gaw A, Macfarlane PW, Packard CJ, Stott DJ, et al, and PROSPER Group. Genetic variation in the interleukin-1 beta-converting enzyme associates with cognitive function. The PROSPER study. Brain. 2008; 131:1069–77. https://doi.org/10.1093/brain/awn023 [PubMed]
- 4. Ma SL, Huang W, Tang NL, Lam LC. MxA polymorphisms are associated with risk and age-at-onset in Alzheimer disease and accelerated cognitive decline in Chinese elders. Rejuvenation Res. 2012; 15:516–22. https://doi.org/10.1089/rej.2012.1328 [PubMed]
- 5. Beeri MS, Moshier E, Schmeidler J, Godbold J, Uribarri J, Reddy S, Sano M, Grossman HT, Cai W, Vlassara H, Silverman JM. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech Ageing Dev. 2011; 132:583–87. https://doi.org/10.1016/j.mad.2011.10.007 [PubMed]
- 6. Sala-Llonch R, Idland AV, Borza T, Watne LO, Wyller TB, Brækhus A, Zetterberg H, Blennow K, Walhovd KB, Fjell AM. Inflammation, Amyloid, and Atrophy in The Aging Brain: Relationships with Longitudinal Changes in Cognition. J Alzheimers Dis. 2017; 58:829–40. https://doi.org/10.3233/JAD-161146 [PubMed]
- 7. Tilvis RS, Kähönen-Väre MH, Jolkkonen J, Valvanne J, Pitkala KH, Strandberg TE. Predictors of cognitive decline and mortality of aged people over a 10-year period. J Gerontol A Biol Sci Med Sci. 2004; 59:268–74. https://doi.org/10.1093/gerona/59.3.m268 [PubMed]
- 8. Weaver JD, Huang MH, Albert M, Harris T, Rowe JW, Seeman TE. Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology. 2002; 59:371–78. https://doi.org/10.1212/wnl.59.3.371 [PubMed]
- 9. Yaffe K, Lindquist K, Penninx BW, Simonsick EM, Pahor M, Kritchevsky S, Launer L, Kuller L, Rubin S, Harris T. Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology. 2003; 61:76–80. https://doi.org/10.1212/01.wnl.0000073620.42047.d7 [PubMed]
- 10. Beydoun MA, Dore GA, Canas JA, Liang H, Beydoun HA, Evans MK, Zonderman AB. Systemic Inflammation Is Associated With Longitudinal Changes in Cognitive Performance Among Urban Adults. Front Aging Neurosci. 2018; 10:313. https://doi.org/10.3389/fnagi.2018.00313 [PubMed]
- 11. Walker KA, Gottesman RF, Wu A, Knopman DS, Gross AL, Mosley TH
Jr , Selvin E, Windham BG. Systemic inflammation during midlife and cognitive change over 20 years: The ARIC Study. Neurology. 2019; 92:e1256–67. https://doi.org/10.1212/WNL.0000000000007094. [PubMed] - 12. Comim CM, Constantino LC, Barichello T, Streck EL, Quevedo J, Dal-Pizzol F. Cognitive impairment in the septic brain. Curr Neurovasc Res. 2009; 6:194–203. https://doi.org/10.2174/156720209788970045 [PubMed]
- 13. Mankowski RT, Anton SD, Ghita GL, Brumback B, Cox MC, Mohr AM, Leeuwenburgh C, Moldawer LL, Efron PA, Brakenridge SC, Moore FA. Older Sepsis Survivors Suffer Persistent Disability Burden and Poor Long-Term Survival. J Am Geriatr Soc. 2020; 68:1962–69. https://doi.org/10.1111/jgs.16435 [PubMed]
- 14. Widmann CN, Heneka MT. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014; 13:630–36. https://doi.org/10.1016/S1474-4422(14)70017-1 [PubMed]
- 15. Hampshire A, Trender W, Chamberlain SR, Jolly AE, Grant JE, Patrick F, Mazibuko N, Williams SC, Barnby JM, Hellyer P, Mehta MA. Cognitive deficits in people who have recovered from COVID-19. EClinicalMedicine. 2021; 39:101044. https://doi.org/10.1016/j.eclinm.2021.101044 [PubMed]
- 16. Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C, Fafi-Kremer S, Ohana M, Anheim M, Meziani F. Neurologic Features in Severe SARS-CoV-2 Infection. N Engl J Med. 2020; 382:2268–70. https://doi.org/10.1056/NEJMc2008597 [PubMed]
- 17. Liotta EM, Batra A, Clark JR, Shlobin NA, Hoffman SC, Orban ZS, Koralnik IJ. Frequent neurologic manifestations and encephalopathy-associated morbidity in Covid-19 patients. Ann Clin Transl Neurol. 2020; 7:2221–30. https://doi.org/10.1002/acn3.51210 [PubMed]
- 18. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Li Y, Hu B. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020; 77:683–90. https://doi.org/10.1001/jamaneurol.2020.1127 [PubMed]
- 19. Sullivan BN, Fischer T. Age-Associated Neurological Complications of COVID-19: A Systematic Review and Meta-Analysis. Front Aging Neurosci. 2021; 13:653694. https://doi.org/10.3389/fnagi.2021.653694 [PubMed]
- 20. Gimeno D, Marmot MG, Singh-Manoux A. Inflammatory markers and cognitive function in middle-aged adults: the Whitehall II study. Psychoneuroendocrinology. 2008; 33:1322–34. https://doi.org/10.1016/j.psyneuen.2008.07.006 [PubMed]
- 21. Todd MA. Inflammation and Cognition in Older Adults: Evidence from Taiwan. Biodemography Soc Biol. 2017; 63:309–23. https://doi.org/10.1080/19485565.2017.1403305 [PubMed]
- 22. Beydoun MA, Weiss J, Obhi HK, Beydoun HA, Dore GA, Liang H, Evans MK, Zonderman AB. Cytokines are associated with longitudinal changes in cognitive performance among urban adults. Brain Behav Immun. 2019; 80:474–87. https://doi.org/10.1016/j.bbi.2019.04.027 [PubMed]
- 23. Dik MG, Jonker C, Hack CE, Smit JH, Comijs HC, Eikelenboom P. Serum inflammatory proteins and cognitive decline in older persons. Neurology. 2005; 64:1371–77. https://doi.org/10.1212/01.WNL.0000158281.08946.68 [PubMed]
- 24. Teunissen CE, van Boxtel MP, Bosma H, Bosmans E, Delanghe J, De Bruijn C, Wauters A, Maes M, Jolles J, Steinbusch HW, de Vente J. Inflammation markers in relation to cognition in a healthy aging population. J Neuroimmunol. 2003; 134:142–50. https://doi.org/10.1016/s0165-5728(02)00398-3 [PubMed]
- 25. Alley DE, Crimmins EM, Karlamangla A, Hu P, Seeman TE. Inflammation and rate of cognitive change in high-functioning older adults. J Gerontol A Biol Sci Med Sci. 2008; 63:50–55. https://doi.org/10.1093/gerona/63.1.50 [PubMed]
- 26. Benros ME, Sørensen HJ, Nielsen PR, Nordentoft M, Mortensen PB, Petersen L. The Association between Infections and General Cognitive Ability in Young Men - A Nationwide Study. PLoS One. 2015; 10:e0124005. https://doi.org/10.1371/journal.pone.0124005 [PubMed]
- 27. Dinel AL, Joffre C, Trifilieff P, Aubert A, Foury A, Le Ruyet P, Layé S. Inflammation early in life is a vulnerability factor for emotional behavior at adolescence and for lipopolysaccharide-induced spatial memory and neurogenesis alteration at adulthood. J Neuroinflammation. 2014; 11:155. https://doi.org/10.1186/s12974-014-0155-x [PubMed]
- 28. Bilbo SD, Rudy JW, Watkins LR, Maier SF. A behavioural characterization of neonatal infection-facilitated memory impairment in adult rats. Behav Brain Res. 2006; 169:39–47. https://doi.org/10.1016/j.bbr.2005.12.002 [PubMed]
- 29. Bilbo SD. Early-life infection is a vulnerability factor for aging-related glial alterations and cognitive decline. Neurobiol Learn Mem. 2010; 94:57–64. https://doi.org/10.1016/j.nlm.2010.04.001 [PubMed]
- 30. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A, Blank T, Staszewski O, Datta M, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018; 556:332–38. https://doi.org/10.1038/s41586-018-0023-4 [PubMed]
- 31. Schaafsma W, Zhang X, van Zomeren KC, Jacobs S, Georgieva PB, Wolf SA, Kettenmann H, Janova H, Saiepour N, Hanisch UK, Meerlo P, van den Elsen PJ, Brouwer N, et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav Immun. 2015; 48:205–21. https://doi.org/10.1016/j.bbi.2015.03.013 [PubMed]
- 32. Kumar A, Foster TC. Linking redox regulation of NMDAR synaptic function to cognitive decline during aging. J Neurosci. 2013; 33:15710–15. https://doi.org/10.1523/JNEUROSCI.2176-13.2013 [PubMed]
- 33. Kumar A, Yegla B, Foster TC. Redox Signaling in Neurotransmission and Cognition During Aging. Antioxid Redox Signal. 2018; 28:1724–45. https://doi.org/10.1089/ars.2017.7111 [PubMed]
- 34. Kumar A, Thinschmidt JS, Foster TC. Subunit contribution to NMDA receptor hypofunction and redox sensitivity of hippocampal synaptic transmission during aging. Aging (Albany NY). 2019; 11:5140–57. https://doi.org/10.18632/aging.102108 [PubMed]
- 35. Barter J, Kumar A, Rani A, Colon-Perez LM, Febo M, Foster TC. Differential Effect of Repeated Lipopolysaccharide Treatment and Aging on Hippocampal Function and Biomarkers of Hippocampal Senescence. Mol Neurobiol. 2020; 57:4045–59. https://doi.org/10.1007/s12035-020-02008-y [PubMed]
- 36. Ianov L, Rani A, Beas BS, Kumar A, Foster TC. Transcription Profile of Aging and Cognition-Related Genes in the Medial Prefrontal Cortex. Front Aging Neurosci. 2016; 8:113. https://doi.org/10.3389/fnagi.2016.00113 [PubMed]
- 37. Smith G, Rani A, Kumar A, Barter J, Foster TC. Hippocampal Subregion Transcriptomic Profiles Reflect Strategy Selection during Cognitive Aging. J Neurosci. 2020; 40:4888–99. https://doi.org/10.1523/JNEUROSCI.2944-19.2020 [PubMed]
- 38. Foster TC. Senescent neurophysiology: Ca2+ signaling from the membrane to the nucleus. Neurobiol Learn Mem. 2019; 164:107064. https://doi.org/10.1016/j.nlm.2019.107064 [PubMed]
- 39. Harré EM, Galic MA, Mouihate A, Noorbakhsh F, Pittman QJ. Neonatal inflammation produces selective behavioural deficits and alters N-methyl-D-aspartate receptor subunit mRNA in the adult rat brain. Eur J Neurosci. 2008; 27:644–53. https://doi.org/10.1111/j.1460-9568.2008.06031.x [PubMed]
- 40. Bodhinathan K, Kumar A, Foster TC. Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2010; 30:1914–24. https://doi.org/10.1523/JNEUROSCI.5485-09.2010 [PubMed]
- 41. Haxaire C, Turpin FR, Potier B, Kervern M, Sinet PM, Barbanel G, Mothet JP, Dutar P, Billard JM. Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting D-serine-dependent NMDA receptor activation. Aging Cell. 2012; 11:336–44. https://doi.org/10.1111/j.1474-9726.2012.00792.x [PubMed]
- 42. Kumar A, Foster TC. Alteration in NMDA Receptor Mediated Glutamatergic Neurotransmission in the Hippocampus During Senescence. Neurochem Res. 2019; 44:38–48. https://doi.org/10.1007/s11064-018-2634-4 [PubMed]
- 43. Robillard JM, Gordon GR, Choi HB, Christie BR, MacVicar BA. Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLoS One. 2011; 6:e20676. https://doi.org/10.1371/journal.pone.0020676 [PubMed]
- 44. Yang YJ, Wu PF, Long LH, Yu DF, Wu WN, Hu ZL, Fu H, Xie N, Jin Y, Ni L, Wang JZ, Wang F, Chen JG. Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell. 2010; 9:709–21. https://doi.org/10.1111/j.1474-9726.2010.00595.x [PubMed]
- 45. Ianov L, De Both M, Chawla MK, Rani A, Kennedy AJ, Piras I, Day JJ, Siniard A, Kumar A, Sweatt JD, Barnes CA, Huentelman MJ, Foster TC. Hippocampal Transcriptomic Profiles: Subfield Vulnerability to Age and Cognitive Impairment. Front Aging Neurosci. 2017; 9:383. https://doi.org/10.3389/fnagi.2017.00383 [PubMed]
- 46. Barter J, Kumar A, Stortz JA, Hollen M, Nacionales D, Efron PA, Moldawer LL, Foster TC. Age and Sex Influence the Hippocampal Response and Recovery Following Sepsis. Mol Neurobiol. 2019; 56:8557–72. https://doi.org/10.1007/s12035-019-01681-y [PubMed]
- 47. Palibrk V, Suganthan R, Scheffler K, Wang W, Bjørås M, Bøe SO. PML regulates neuroprotective innate immunity and neuroblast commitment in a hypoxic-ischemic encephalopathy model. Cell Death Dis. 2016; 7:e2320. https://doi.org/10.1038/cddis.2016.223 [PubMed]
- 48. Song L, Pei L, Hu L, Pan S, Xiong W, Liu M, Wu Y, Shang Y, Yao S. Death-associated protein kinase 1 mediates interleukin-1β production through regulating inlfammasome activation in Bv2 microglial cells and mice. Sci Rep. 2018; 8:9930. https://doi.org/10.1038/s41598-018-27842-y [PubMed]
- 49. Flierl MA, Rittirsch D, Nadeau BA, Sarma JV, Day DE, Lentsch AB, Huber-Lang MS, Ward PA. Upregulation of phagocyte-derived catecholamines augments the acute inflammatory response. PLoS One. 2009; 4:e4414. https://doi.org/10.1371/journal.pone.0004414 [PubMed]
- 50. Redell JB, Dash PK. Traumatic brain injury stimulates hippocampal catechol-O-methyl transferase expression in microglia. Neurosci Lett. 2007; 413:36–41. https://doi.org/10.1016/j.neulet.2006.11.060 [PubMed]
- 51. Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, McGuire SR, List RP, Day DE, Hoesel LM, Gao H, Van Rooijen N, Huber-Lang MS, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007; 449:721–25. https://doi.org/10.1038/nature06185 [PubMed]
- 52. Ring RH, Valo Z, Gao C, Barish ME, Singer-Sam J. The Cdkn1a gene (p21Waf1/Cip1) is an inflammatory response gene in the mouse central nervous system. Neurosci Lett. 2003; 350:73–76. https://doi.org/10.1016/s0304-3940(03)00883-8 [PubMed]
- 53. Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor alpha induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron-glia interactions during neurodegeneration. J Neurosci. 2000; 20:7964–71. https://doi.org/10.1523/JNEUROSCI.20-21-07964.2000 [PubMed]
- 54. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019; 20:19–33. https://doi.org/10.1038/s41583-018-0093-1 [PubMed]
- 55. Larson KC, Lipko M, Dabrowski M, Draper MP. Gng12 is a novel negative regulator of LPS-induced inflammation in the microglial cell line BV-2. Inflamm Res. 2010; 59:15–22. https://doi.org/10.1007/s00011-009-0062-2 [PubMed]
- 56. Donzis EJ, Tronson NC. Modulation of learning and memory by cytokines: signaling mechanisms and long term consequences. Neurobiol Learn Mem. 2014; 115:68–77. https://doi.org/10.1016/j.nlm.2014.08.008 [PubMed]
- 57. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011; 25:181–213. https://doi.org/10.1016/j.bbi.2010.10.015 [PubMed]
- 58. Donegan JJ, Girotti M, Weinberg MS, Morilak DA. A novel role for brain interleukin-6: facilitation of cognitive flexibility in rat orbitofrontal cortex. J Neurosci. 2014; 34:953–62. https://doi.org/10.1523/JNEUROSCI.3968-13.2014 [PubMed]
- 59. Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, Levy-Lahad E, Yirmiya R. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007; 32:1106–15. https://doi.org/10.1016/j.psyneuen.2007.09.004 [PubMed]
- 60. Bouhy D, Ghasemlou N, Lively S, Redensek A, Rathore KI, Schlichter LC, David S. Inhibition of the Ca2+-dependent K+ channel, KCNN4/KCa3.1, improves tissue protection and locomotor recovery after spinal cord injury. J Neurosci. 2011; 31:16298–308. https://doi.org/10.1523/JNEUROSCI.0047-11.2011 [PubMed]
- 61. Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007; 27:234–44. https://doi.org/10.1523/JNEUROSCI.3593-06.2007 [PubMed]
- 62. Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. 2011; 286:3693–706. https://doi.org/10.1074/jbc.M110.135244 [PubMed]
- 63. Nguyen HM, Grössinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, Wulff H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in "classically" and "alternatively" activated microglia. Glia. 2017; 65:106–21. https://doi.org/10.1002/glia.23078 [PubMed]
- 64. Staal RGW, Khayrullina T, Zhang H, Davis S, Fallon SM, Cajina M, Nattini ME, Hu A, Zhou H, Poda SB, Zorn S, Chandrasena G, Dale E, et al. Inhibition of the potassium channel KCa3.1 by senicapoc reverses tactile allodynia in rats with peripheral nerve injury. Eur J Pharmacol. 2017; 795:1–7. https://doi.org/10.1016/j.ejphar.2016.11.031 [PubMed]
- 65. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013; 15:978–90. https://doi.org/10.1038/ncb2784 [PubMed]
- 66. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012; 11:345–49. https://doi.org/10.1111/j.1474-9726.2012.00795.x [PubMed]
- 67. Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediators Inflamm. 2018; 2018:9076485. https://doi.org/10.1155/2018/9076485 [PubMed]
- 68. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018; 24:1246–56. https://doi.org/10.1038/s41591-018-0092-9 [PubMed]
- 69. Budamagunta V, Foster TC, Zhou D. Cellular senescence in lymphoid organs and immunosenescence. Aging (Albany NY). 2021; 13:19920–41. https://doi.org/10.18632/aging.203405 [PubMed]
- 70. Budamagunta V, Manohar-Sindhu S, Yang Y, He Y, Traktuev DO, Foster TC, Zhou D. Senescence-associated hyper-activation to inflammatory stimuli in vitro. Aging (Albany NY). 2021; 13:19088–107. https://doi.org/10.18632/aging.203396 [PubMed]
- 71. Kumar A, Rani A, Scheinert RB, Ormerod BK, Foster TC. Nonsteroidal anti-inflammatory drug, indomethacin improves spatial memory and NMDA receptor function in aged animals. Neurobiol Aging. 2018; 70:184–93. https://doi.org/10.1016/j.neurobiolaging.2018.06.026 [PubMed]
- 72. Scheinert RB, Asokan A, Rani A, Kumar A, Foster TC, Ormerod BK. Some hormone, cytokine and chemokine levels that change across lifespan vary by cognitive status in male Fischer 344 rats. Brain Behav Immun. 2015; 49:216–32. https://doi.org/10.1016/j.bbi.2015.06.005 [PubMed]
- 73. Guidi M, Kumar A, Rani A, Foster TC. Assessing the emergence and reliability of cognitive decline over the life span in Fisher 344 rats using the spatial water maze. Front Aging Neurosci. 2014; 6:2. https://doi.org/10.3389/fnagi.2014.00002 [PubMed]
- 74. McQuail JA, Dunn AR, Stern Y, Barnes CA, Kempermann G, Rapp PR, Kaczorowski CC, Foster TC. Cognitive Reserve in Model Systems for Mechanistic Discovery: The Importance of Longitudinal Studies. Front Aging Neurosci. 2021; 12:607685. https://doi.org/10.3389/fnagi.2020.607685 [PubMed]
- 75. Markowska AL, Savonenko AV. Protective effect of practice on cognition during aging: implications for predictive characteristics of performance and efficacy of practice. Neurobiol Learn Mem. 2002; 78:294–320. https://doi.org/10.1006/nlme.2002.4064 [PubMed]
- 76. Guidi M, Kumar A, Foster TC. Impaired attention and synaptic senescence of the prefrontal cortex involves redox regulation of NMDA receptors. J Neurosci. 2015; 35:3966–77. https://doi.org/10.1523/JNEUROSCI.3523-14.2015 [PubMed]
- 77. Lee WH, Kumar A, Rani A, Foster TC. Role of antioxidant enzymes in redox regulation of N-methyl-D-aspartate receptor function and memory in middle-aged rats. Neurobiol Aging. 2014; 35:1459–68. https://doi.org/10.1016/j.neurobiolaging.2013.12.002 [PubMed]
- 78. Guidi M, Foster TC. Behavioral model for assessing cognitive decline. Methods Mol Biol. 2012; 829:145–53. https://doi.org/10.1007/978-1-61779-458-2_8 [PubMed]
- 79. Aenlle KK, Kumar A, Cui L, Jackson TC, Foster TC. Estrogen effects on cognition and hippocampal transcription in middle-aged mice. Neurobiol Aging. 2009; 30:932–45. https://doi.org/10.1016/j.neurobiolaging.2007.09.004 [PubMed]
- 80. Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, Landfield PW. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003; 23:3807–19. https://doi.org/10.1523/JNEUROSCI.23-09-03807.2003 [PubMed]
- 81. Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, Keime C, Marot G, Castel D, Estelle J, Guernec G, Jagla B, Jouneau L, et al, and French StatOmique Consortium. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013; 14:671–83. https://doi.org/10.1093/bib/bbs046 [PubMed]
- 82. Aenlle KK, Foster TC. Aging alters the expression of genes for neuroprotection and synaptic function following acute estradiol treatment. Hippocampus. 2010; 20:1047–60. https://doi.org/10.1002/hipo.20703 [PubMed]
- 83. Zeier Z, Madorsky I, Xu Y, Ogle WO, Notterpek L, Foster TC. Gene expression in the hippocampus: regionally specific effects of aging and caloric restriction. Mech Ageing Dev. 2011; 132:8–19. https://doi.org/10.1016/j.mad.2010.10.006 [PubMed]