



Introduction
Vascular endothelium releases several factors involved in the local regulation of vascular tone and modulation of pro-inflammatory molecule production [1]. Among them are endothelium-derived relaxing factors in particular nitric oxide (NO) as well as endothelium-derived contracting factors such as prostaglandins and superoxide anion [2, 3]. Constitutive expression of endothelial nitric oxide synthase (eNOS) is mainly responsible for NO production in endothelial cells of conduit arteries [4, 5]. On the other hand, cyclooxygenase (COX) is a rate-limiting enzyme in the biosynthesis of prostaglandins. COX1 is constitutively expressed and plays an important role in vascular homeostasis, while COX2 is considered an inducible enzyme in mouse conduit arteries [6].
Aging of blood vessels are major contributors to the development of cardiovascular disease [7]. Existing evidence obtained in human and animal studies suggest that the earliest detectable vascular phenotype in aging involves the development of endothelial dysfunction, which is commonly characterized by the reduced endothelial production of NO and by the increased production of COX-derived vasoconstrictor factors [8–10]. Moreover, clinical studies have reported that endothelium-dependent vasodilatation progressively declines with age and occurs earlier in men than in women [11, 12].
Amyloid-β precursor protein (APP) is an evolutionarily conserved protein and is implicated in the development of Alzheimer's disease [13–15]. However, the exact function of APP outside the central nervous system, particularly in peripheral tissues, remains poorly understood [16–18]. We and others have shown that APP is expressed in cultured endothelial cells as well as in vascular wall of wild-type mice [19–24]. Under physiological conditions, APP is constitutively cleaved at the cell surface by α-secretase via non-amyloidogenic processing pathway. Alpha-processing of APP causes release of soluble APP alpha (sAPPα) ectodomain in the lumen of blood vessel wall [25–30]. Recently, we showed that endothelium-specific inactivation of APP (eAPP−/−) caused endothelial dysfunction in mice cerebral arteries [29]. However, no previous studies tested the effects of aging on vascular APP expression in systemic large conduit arteries. In the present study, we advance the hypothesis that APP exerts vascular protective effects on endothelial function during aging.
Results
Characterization of mice
Body weight was increased in both aged wild-type (WT) and eAPP−/− mice (P<0.05 vs. respective young mice; Table 1). Blood pressure measurements revealed that systolic blood pressure (SBP), mean blood pressure (MBP), and diastolic blood pressure (DBP) were unchanged in young and aged WT and eAPP−/− mice (P>0.05; Table 1). Blood glucose was also not different between eAPP−/− mice and their WT irrespective of age (Table 1). Lipid profile was unchanged in plasma of young WT and eAPP−/− mice (P>0.05; Table 1). Aging caused a slight but significant increase in circulating levels of cholesterol in WT mice (P<0.05 vs. young WT; Table 1). In contrast, levels of triglycerides were decreased in aged eAPP−/− mice (P<0.05 vs. young eAPP−/− mice; Table 1). Plasma levels of norepinephrine were significantly increased in aged WT and eAPP−/− mice (P<0.05 vs. respective young mice; Table 1). Aging significantly increased plasma levels of sAPPα in both WT and eAPP−/− mice however, levels of sAPPα remained significantly decreased in eAPP−/− mice as compared to age-matched WT mice (P<0.05; Table 1). However, plasma levels of amyloid-β 1-40 (Aβ1-40) were unaltered in young and aged WT and eAPP−/− mice (P>0.05; Table 1).
Table 1. Characteristics of young and aged eAPP−/− mice.
| Parameters | Young | Aged | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| WT littermates | eAPP−/− | WT littermates | eAPP−/− | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Body weight (g) | 30±1 (10) | 29±1 (10) | 32±1 (12) * | 33±1 (12) * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SBP (mmHg) | 121±2 (9) | 119±2 (9) | 120±2 (9) | 121±2 (11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MBP (mmHg) | 93±1 (9) | 92±1 (9) | 94±2 (9) | 95±2 (11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DBP (mmHg) | 79±1 (9) | 78±1 (9) | 81±2 (9) | 82±2 (11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Glucose (mg/dL) | 147±8 (10) | 156±11 (10) | 133±7 (12) | 155±10 (12) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cholesterol (mg/dL) | 63±2 (11) | 63±3 (11) | 88±14 (11) * | 72±3 (11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HDL (mg/dL) | 54± 2 (11) | 52± 3 (11) | 68±13 (11) | 54±4 (11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Triglycerides (mg/dL) | 85± 5 (11) | 91± 6 (11) | 74±8 (11) | 63±5 (11) * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Norepinephrine (pg/mL) | 1599±222 (16) | 1898±280 (16) | 3601±1019 (9) * | 3371±399 (7) * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| sAPPα (pg/mL) | 549±39 (10) | 244±27 (10) † | 734±72 (12) * | 428±55 (12) *† | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Aβ1-40 (pg/mL) | 90±8 (10) | 88±10 (10) | 93±11 (8) | 92±20 (8) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SBP indicates systolic blood pressure; MBP, mean blood pressure; DBP, diastolic blood pressure; HDL, high-density lipoprotein; WT, wild-type. Data are means ± SEM and the numbers of mice are indicated in the parentheses. * P<0.05 vs. young mice of same strain; † P<0.05 vs. age-matched WT littermates (two-way ANOVA followed by Tukey’s HSD test). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
APP expression and processing in aorta
Western blot analyses revealed that APP expression was significantly lower (by 27%) in the aorta of young eAPP−/− mice (P<0.05 vs. young WT; Supplementary Figure 1). Aging caused increase in APP expression in WT mice aortas (P<0.05 vs. young WT; Figure 1A) while APP expression was unchanged in aged eAPP−/− mice aortas (P>0.05 vs. young eAPP−/− mice; Figure 1A). Release of sAPPα from the aorta was significantly increased in aged WT mice (P<0.05 vs. young WT mice; Figure 1B). In contrast, sAPPα release was not significantly affected by aging of eAPP−/− mice (P>0.05 vs. young eAPP−/− mice; Figure 1B). Release of Aβ1-40 from the aorta was not altered in young and aged WT and eAPP−/− mice (P>0.05; Supplementary Figure 2). Moreover, protein expressions of a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and beta-site APP cleaving enzyme 1 (BACE1) were unaltered in WT and eAPP−/− mice irrespective of age (P>0.05; Figures 1C, 1D, respectively).
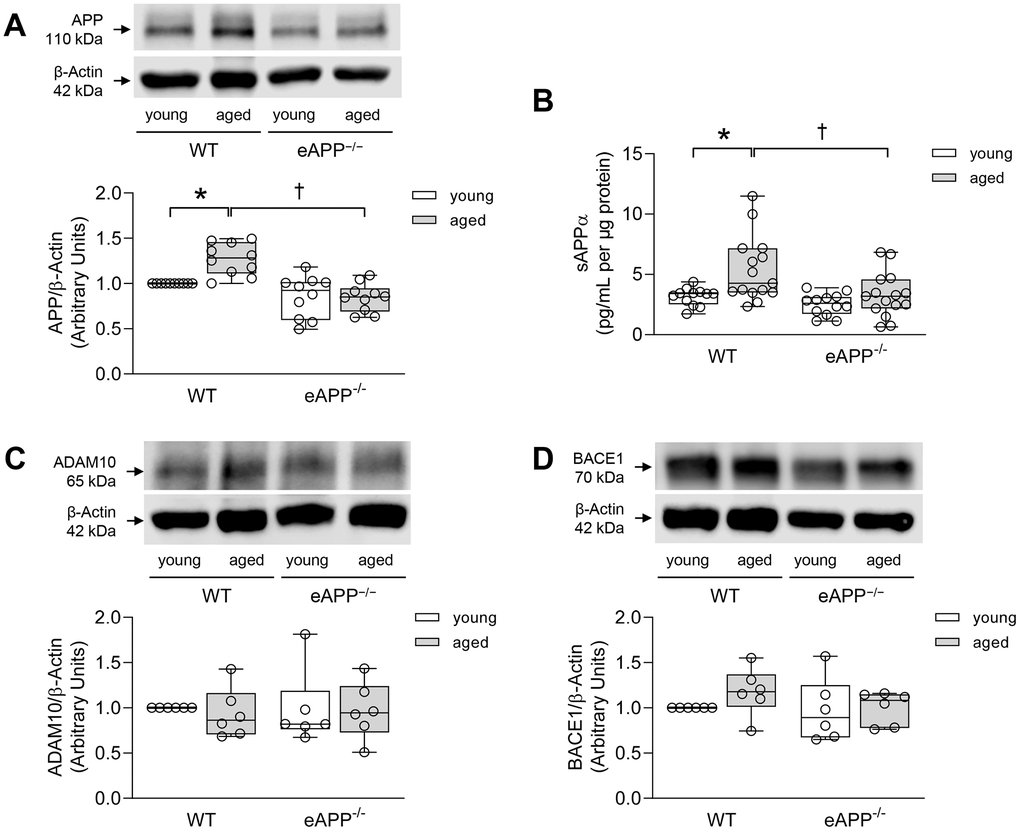
Figure 1. (A) Effects of aging on protein expression of APP in the aortas of wild-type (WT) littermates and eAPP−/− mice (n=10 per group). (B) Effects of aging on ex-vivo sAPPα secretion from wild-type (WT) littermates and eAPP−/− mice aortas. The supernatants were collected and analyzed for sAPPα levels. Results were normalized against tissue protein levels (n=12 per group for young WT littermates and eAPP−/− mice and n=15 per group for aged WT littermates and eAPP−/− mice). (C) Effects of aging on protein expression of ADAM10 in the aortas of wild-type (WT) littermates and eAPP−/− mice (n=6 per group). (D) Effects of aging on protein expression of BACE1 in the aortas of wild-type (WT) littermates and eAPP−/− mice (n=6 per group). Western blot results are the relative densitometry compared with β-actin protein. All results are representing box plots with whiskers showing the median, 25th to 75th percentiles, and min-max range. * P<0.05 versus young mice of same strain; † P<0.05 versus age-matched WT littermates (two-way ANOVA followed by Tukey’s HSD test).
Vascular contraction responses
Contractions to KCl were significantly increased in aged WT and eAPP−/− mice aortas as compared to young mice (P<0.05; Table 2) while there were no differences between age-matched WT and eAPP−/− mice (P>0.05; Table 2).
Table 2. Efficacy and potency of vasoconstrictors in young and aged eAPP−/− mice aortas.
| Parameters | Young | Aged | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| WT littermates | eAPP−/− | WT littermates | eAPP−/− | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KCl (g): | 1.95±0.06 (18) | 1.87±0.04 (18) | 2.53±0.11 (16) * | 2.38±0.08 (16) * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PGF2α: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Emax (%) | 139±4 (9) | 137±3 (9) | 123±3 (8) * | 124±4 (8) * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pEC50 (−log mol/L) | 5.49±0.03 (9) | 5.34±0.04 (9) † | 5.69±0.03 (8) * | 5.57±0.04 (8) * † | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phenylephrine: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Emax (%) | 63±6 (9) | 50±5 (9) | 41±8 (8) * | 33±9 (8) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pEC50 (−log mol/L) | 7.03±0.02 (9) | 6.98±0.02 (9) | 6.74±0.07 (8) * | 6.68±0.05 (8) * | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Emax indicates maximal efficacy; pEC50, potency; WT, wild-type. Data are means ± SEM and the numbers of mice are indicated in the parentheses. * P<0.05 vs. young mice of same strain; † P<0.05 vs. WT littermates of same age (two-way ANOVA followed by Tukey’s HSD test). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sensitivity to prostaglandin F2α (PGF2α)-induced contractions was slightly lower in aortas of young and aged eAPP−/− mice (P<0.05 as compared to their respective WT mice; Figures 2A, 2B, respectively; Table 2). In contrast, PGF2α potency was significantly increased in aged WT and eAPP−/− mice (P<0.05 vs. respective young mice; Table 2), and efficacy of PGF2α was significantly decreased in aged WT and eAPP−/− mice (P<0.05 vs. respective young mice; Table 2).
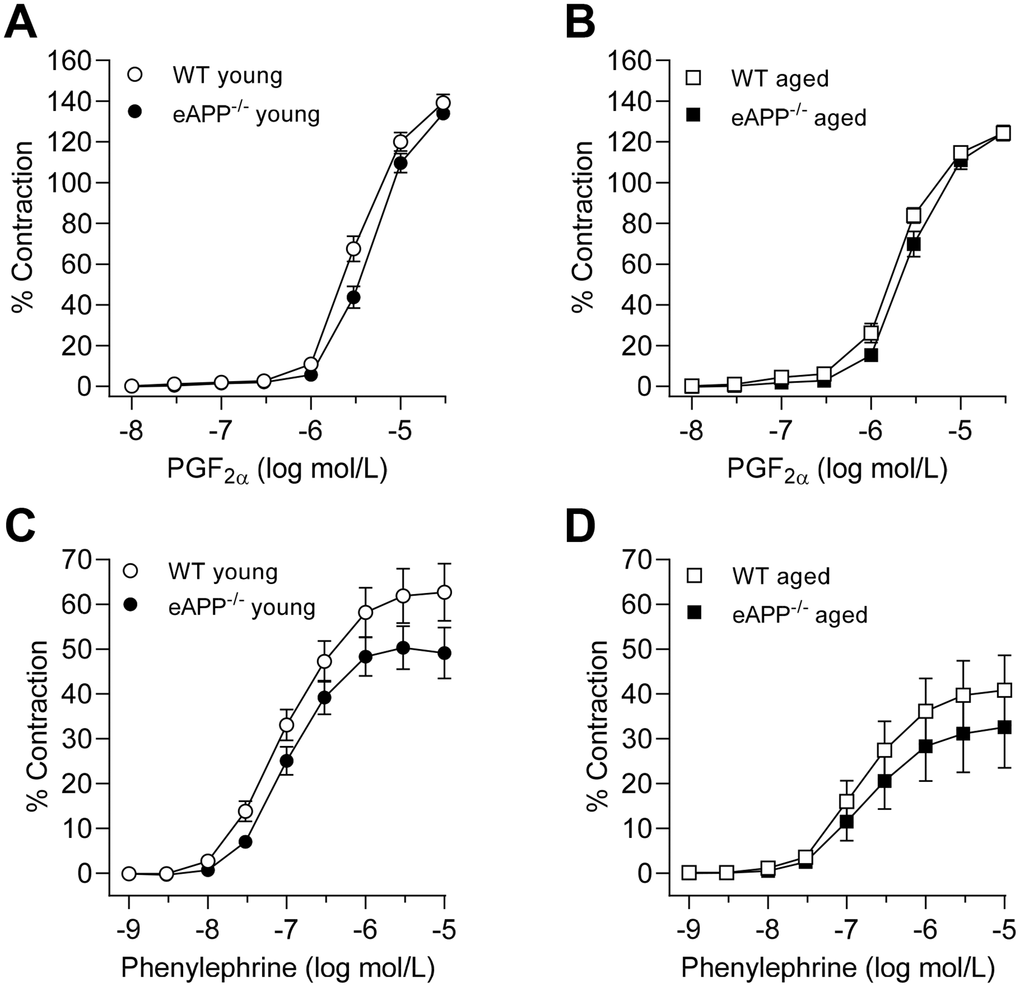
Figure 2. Concentration-dependent contractions to PGF2α (A, B) and phenylephrine (C, D) in isolated aortic rings derived from young and aged eAPP−/− mice and their respective wild-type (WT) littermates. Results are shown as mean ± SEM (n=9 per group for young WT littermates and eAPP−/− mice and n=8 per group for aged WT littermates and eAPP−/− mice) and contractions are expressed as percentage of response to a second KCl (80 mmol/L). No significant differences were detected between WT littermates and eAPP−/− mice at same age (ANOVA with Bonferroni's correction).
While no significant differences between age-matched WT and eAPP−/− mice were observed, the potency of phenylephrine was reduced in aged WT and eAPP−/− mice (P<0.05 vs. respective young mice; Table 2; Figure 2C, 2D). Furthermore, the efficacy was significantly reduced in aged WT mice (P<0.05 vs. young WT mice; Table 2) while it tended to be reduced in aged eAPP−/− mice (P=0.088; Table 2).
Vascular endothelial function
Acetylcholine (ACh)-induced endothelium-dependent relaxations were unaltered in young mice with endothelial-specific APP deletion (P>0.05 vs. young WT littermates; Figure 3A and Table 3). Aging caused a significant impairment to ACh in WT mice (P<0.05 vs. young WT mice; Table 3) while the efficacy was unaltered (Table 3). Furthermore, relaxations to ACh were further decreased in aged eAPP−/− mice (P<0.05 vs. aged WT mice; Figure 3B and Table 3).
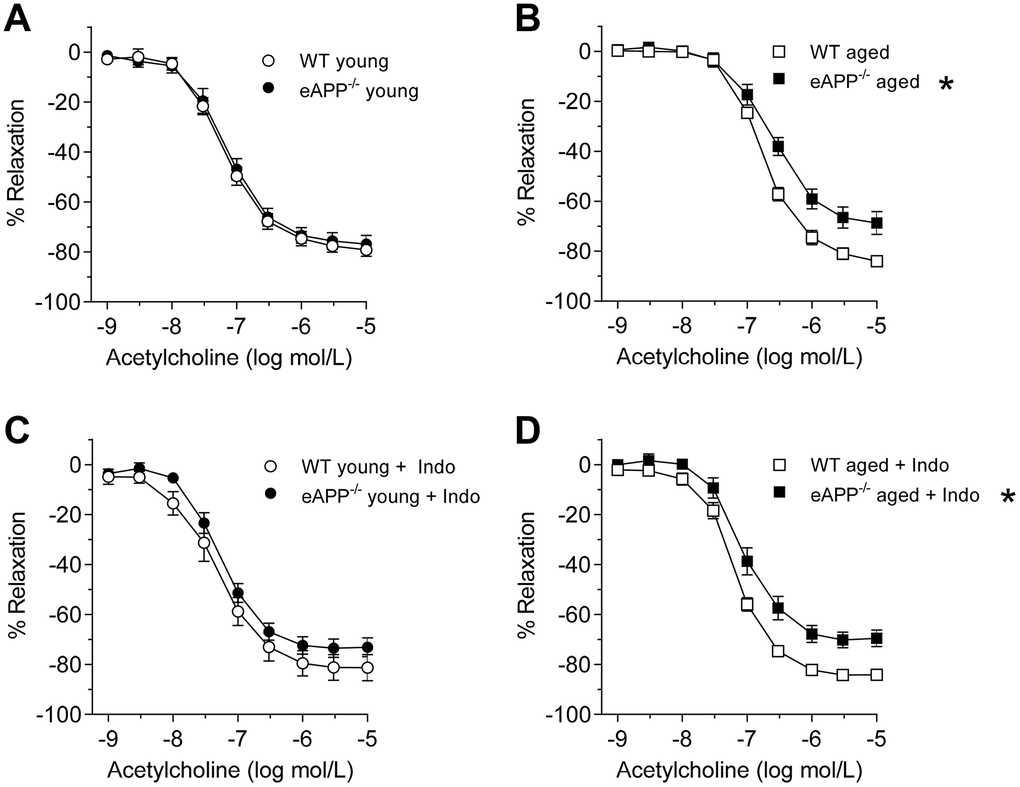
Figure 3. Endothelium-dependent relaxations to ACh in isolated aorta from young (A; n=9 per group) and aged (B; n=8 per group) wild-type (WT) littermates and eAPP−/− mice aortas. Effects of indomethacin (Indo; 10-5 mol/L) on responses to ACh in young (C; n=9 per group) and aged (D; n=7 per group) WT littermates and eAPP−/− mice aortas. Results are shown as mean ± SEM and expressed as percent relaxation from submaximal contractions induced by PGF2α. * P>0.05 versus wild-type littermates (ANOVA with Bonferroni's correction).
Table 3. Efficacy and potency of endothelium-dependent relaxations in young and aged eAPP−/− mice aortas.
| Parameters | Young | Aged | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| WT littermates | eAPP−/− | WT littermates | eAPP−/− | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Emax (%): | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ACh | 78±2 (9) | 76±4 (9) | 83±2 (8) | 69±5 (8) † | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ACh + Indomethacin | 81±6 (9) | 73±4 (9) | 84±2 (7) | 70±3 (7) † | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| pEC50 (−log mol/L): | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ACh | 7.18±0.04 (9) | 7.16±0.05 (9) | 6.75±0.01 (8) * | 6.61±0.05 (8) * † | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ACh + Indomethacin | 7.33±0.08 (9) | 7.25±0.04 (9) | 7.17±0.03 (7) # | 7.04±0.06 (7) * # | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Emax indicates maximal efficacy; pEC50, potency; WT, wild-type. Data are means ± SEM and the numbers of mice are indicated in the parentheses. * P<0.05 vs. young mice of same strain; † P<0.05 vs. WT littermates of same age (two-way ANOVA followed by Tukey’s HSD test); # P<0.05 vs. without indomethacin treatment (unpaired t-test). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Incubation of aortas with indomethacin did not affect relaxations to ACh in young mice (P>0.05; Figure 3C). In contrast, indomethacin significantly improved endothelium-dependent relaxations to ACh in both aged WT and eAPP−/− mice (P<0.05 vs. aortas without indomethacin; Table 3). However, the efficacy is still significantly reduced in aged eAPP−/− mice in the presence of indomethacin (P<0.05 vs. aged WT mice with indomethacin; Figure 3D and Table 3). Blockade of NOS with Nω-nitro L-arginine methyl ester (L-NAME) in the presence of indomethacin completely blocks relaxations to ACh in both young and aged WT and eAPP−/− aortas (Supplementary Figure 3).
Ex-vivo production of prostaglandins in aorta
We next determined whether age-related changes in vascular prostaglandins contribute to the observed decline in endothelial function of aortas, we measured release of prostaglandins in conditioned medium. Thromboxane B2 (TXB2) and prostaglandin E2 (PGE2) productions were significantly increased in both aged WT and eAPP−/− mice (P<0.05 vs. young mice; Figure 4A, 4B) while production of PGF2α was significantly increased only in aged WT mice (P<0.05 vs. young WT; Figure 4C). In contrast, production of 6-keto PGF1α (a stable metabolite of prostaglandin I2) was not different among young and aged WT and eAPP−/− mice (P>0.05; Figure 4D).
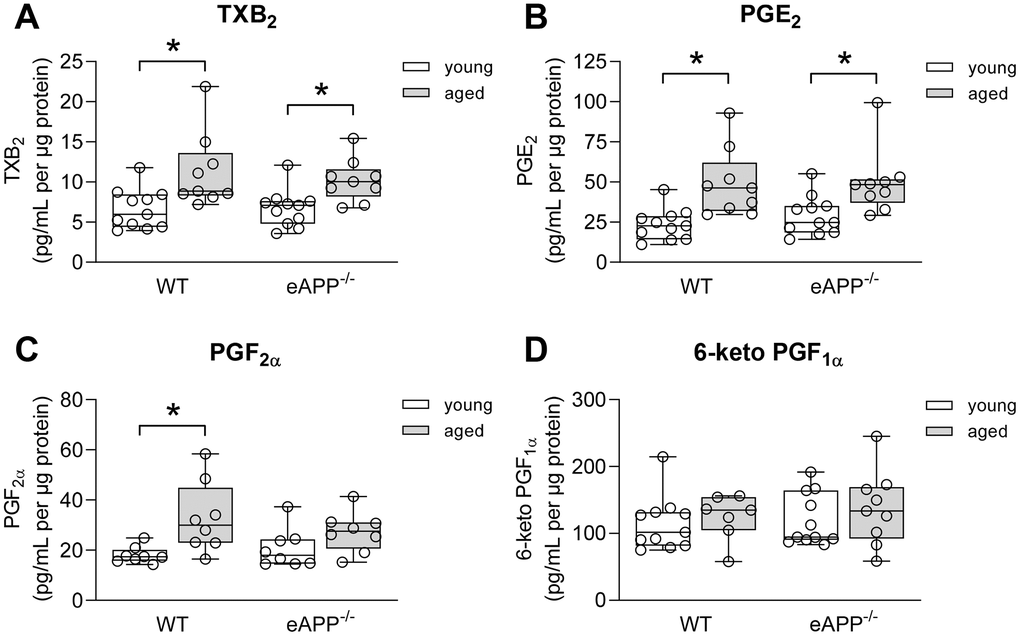
Figure 4. Effects of aging on ex-vivo secretion of prostaglandins from wild-type (WT) littermates and eAPP−/− mice aortas. The supernatants were collected and analyzed for TXB2 (A; n=11 per group for young mice and n=9 per group for aged mice), PGE2 (B; n=11 per group for young mice and n=9 per group for aged mice); PGF2α (C; n=8 per group for young mice and n=8 per group for aged mice); and 6-keto PGF1α (D; n=11 per group for young mice and n=7-9 per group for aged mice). All results were normalized against tissue protein levels and are representing box plots with whiskers showing the median, 25th to 75th percentiles, and min-max range. * P<0.05 versus young mice of same strain (two-way ANOVA followed by Tukey’s HSD test).
We also performed western blot analyses for protein expression of COX isoforms in aortas. COX1 expression was not different between WT and eAPP−/− mice irrespective of age (P>0.05; Figure 5A). In contrast, protein expression of COX2 was significantly increased in both aged WT and eAPP−/− mice (P<0.05 vs. young mice; Figure 5B). Moreover, the increase in COX2 was significantly reduced in aged eAPP−/− mice (P<0.05 vs. aged WT mice; Figure 5B).
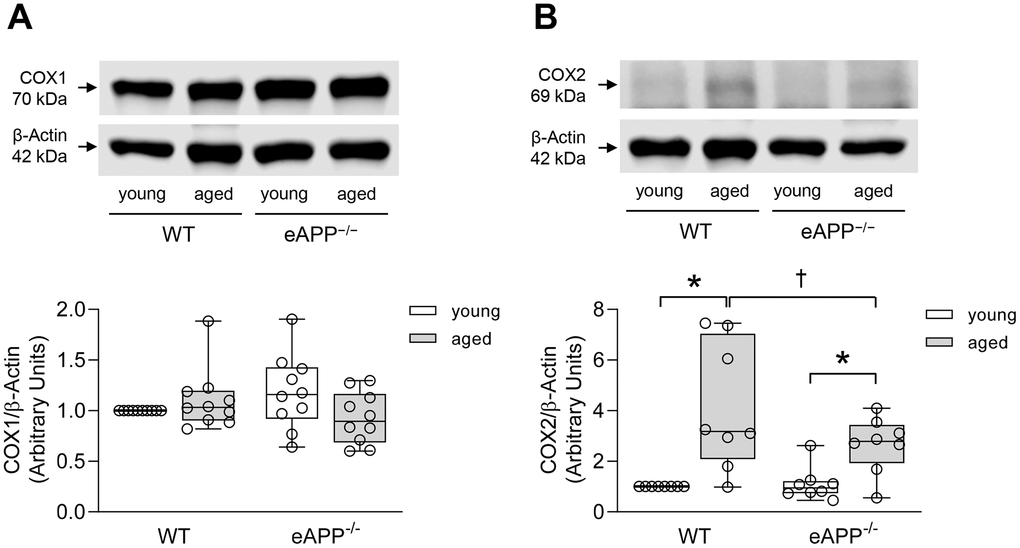
Figure 5. Protein expressions of COX1 (A) and COX2 (B) in the aortas of wild-type (WT) littermates and eAPP−/− mice. Western blot results are the relative densitometry compared with β-actin protein (n=10 per group for COX1 and n=8 per group for COX2). All results are representing box plots with whiskers showing the median, 25th to 75th percentiles, and min-max range. * P<0.05 versus young mice of same strain; † P<0.05 versus age-matched WT littermates (two-way ANOVA followed by Tukey’s HSD test).
Protein expression of eNOS
Expression of eNOS was significantly decreased in the aorta of aged eAPP−/− mice (P<0.05 vs. young eAPP−/− mice; Figure 6). Furthermore, eNOS protein tended to be reduced in aged WT mice but it did not reach statistical significance (Figure 6).
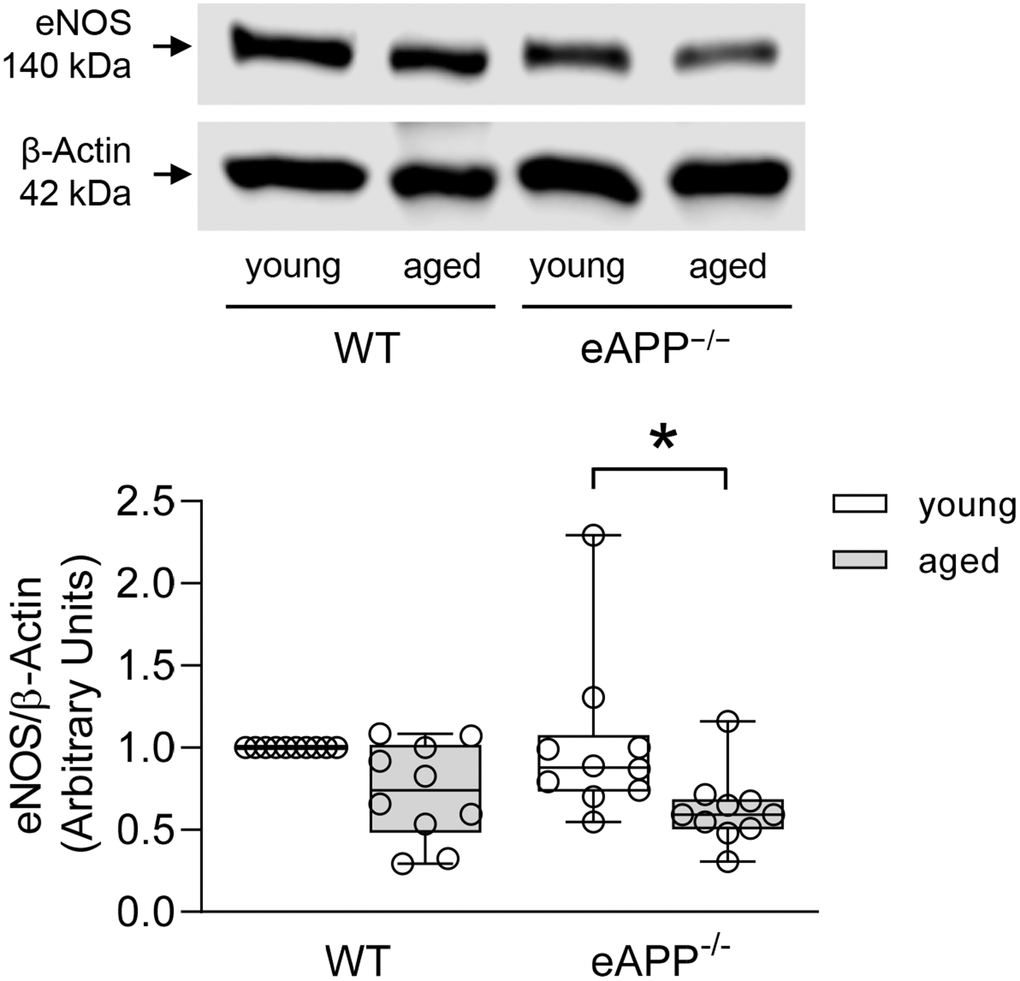
Figure 6. Effects of aging on protein expression of eNOS in the aortas of wild-type (WT) littermates and eAPP−/− mice. Western blot results are the relative densitometry compared with β-actin protein (n=10 per group). All results are representing box plots with whiskers showing the median, 25th to 75th percentiles, and min-max range. * P<0.05 versus young mice of same strain (two-way ANOVA followed by Tukey’s HSD test).
Superoxide anion
To investigate whether endothelial dysfunction in aging was caused by increased superoxide anion production in aorta, we performed HPLC-based assay of 2-hydroxyethidium. Levels of superoxide anion were significantly enhanced in aged eAPP−/− mice (P<0.05 vs. young eAPP−/− mice; Figure 7) while superoxide anion levels were unaltered in aged WT mice (P>0.05 vs. young WT mice; Figure 7).
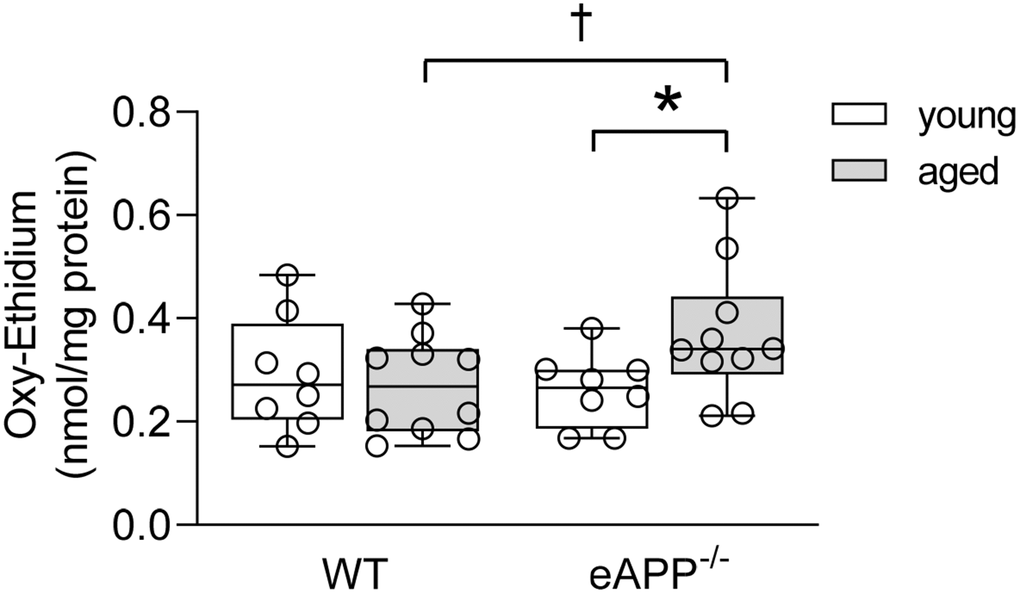
Figure 7. Quantitative HPLC analysis of superoxide anion in aortas from young and aged wild-type (WT) littermates and eAPP−/− mice aortas. All results were normalized against tissue protein levels and are representing box plots with whiskers showing the median, 25th to 75th percentiles, and min-max range (n=8 per group for young WT littermates and eAPP−/− mice and n=10 per group for aged WT littermates and eAPP−/− mice). * P<0.05 versus young mice of same strain; † P<0.05 versus age-matched WT littermates (two-way ANOVA followed by Tukey’s HSD test).
Intracellular cGMP levels
Under basal conditions, cyclic guanosine 3',5'-monophosphate (cGMP) were unchanged in aortas of WT and eAPP−/− mice irrespective of age (P>0.05; Figure 8). ACh stimulated cGMP levels were significantly increased in young and aged WT and eAPP−/− mice (P<0.05 vs. basal levels; Figure 8). However, ACh stimulated cGMP levels were significantly decreased in aged WT and eAPP−/− mice (P<0.05 young mice in the presence of ACh; Figure 8). In contrast, cGMP levels did not differ between WT and eAPP−/− mice groups under basal and stimulated conditions (P>0.05; Figure 8).
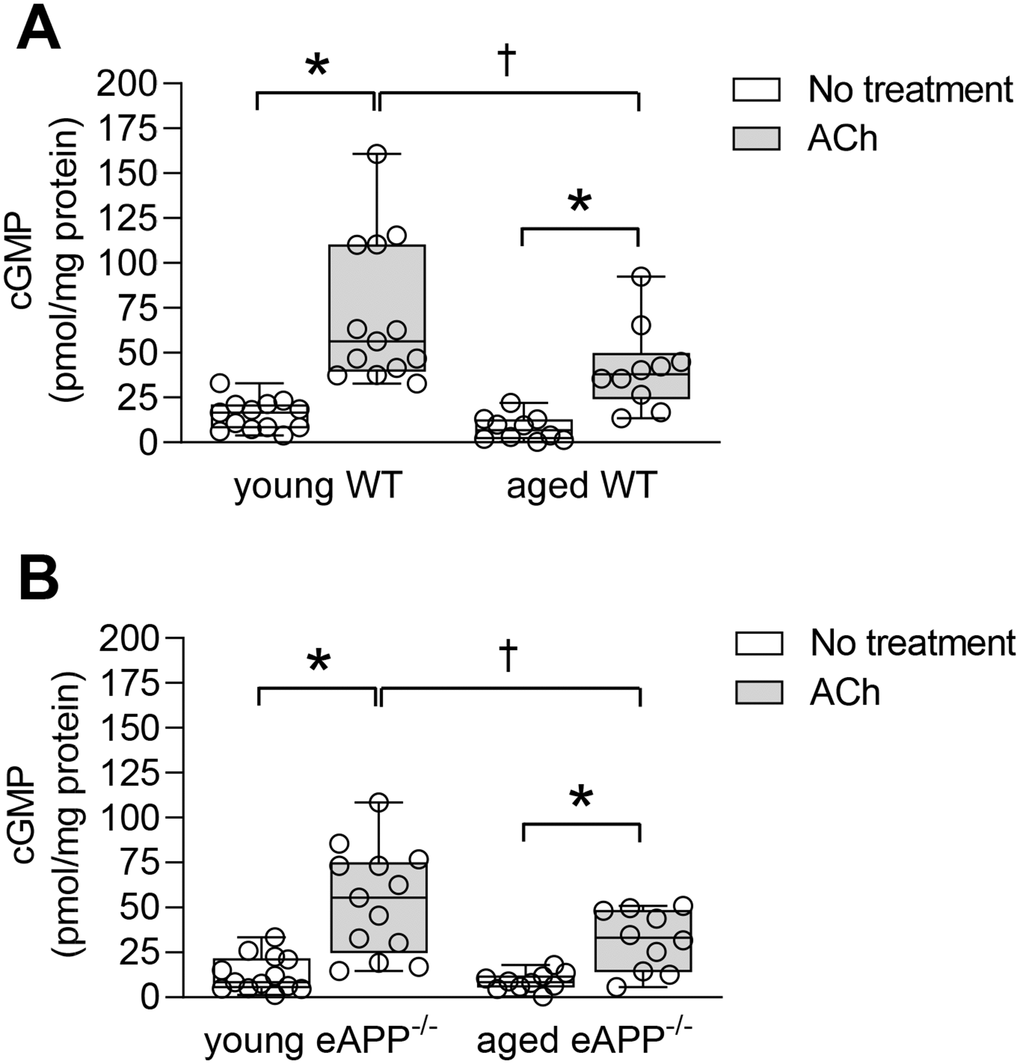
Figure 8. Quantitative analysis of cGMP in aortas from young and aged wild-type (WT) littermates aortas (A) and eAPP−/− mice aortas (B) under basal and ACh (10 μM) stimulated conditions. All results were normalized against tissue protein levels and are representing box plots with whiskers showing the median, 25th to 75th percentiles, and min-max range (n=13 per group for young WT littermates and eAPP−/− mice and n=10 per group for aged WT littermates and eAPP−/− mice). * P<0.05 versus basal levels; † P<0.05 versus young WT mice (two-way ANOVA followed by Tukey’s HSD test).
Ex-vivo studies with cytokines
We next attempted in different experimental conditions to provide insight into the effects of APP deletion on eNOS expression in vascular endothelium. Young WT and eAPP−/− mice aortas were used to examine the effects of pro-inflammatory cytokines cocktail on expression of APP and eNOS. Western blot analysis revealed that APP protein was significantly increased in young WT mice treated with cytokines (P<0.05 vs. PBS treatment; Figure 9A) while APP expression was not significantly affected in young eAPP−/− mice (P>0.05; Figure 9A). In contrast, eNOS expression was significantly reduced in young eAPP−/− but not in young WT aortas after cytokines treatment (P<0.05 vs. PBS treatment; Figure 9B). Expressions of iNOS and COX2 were equally increased in both groups of mice (P<0.05 vs. PBS treatment; Supplementary Figure 4).
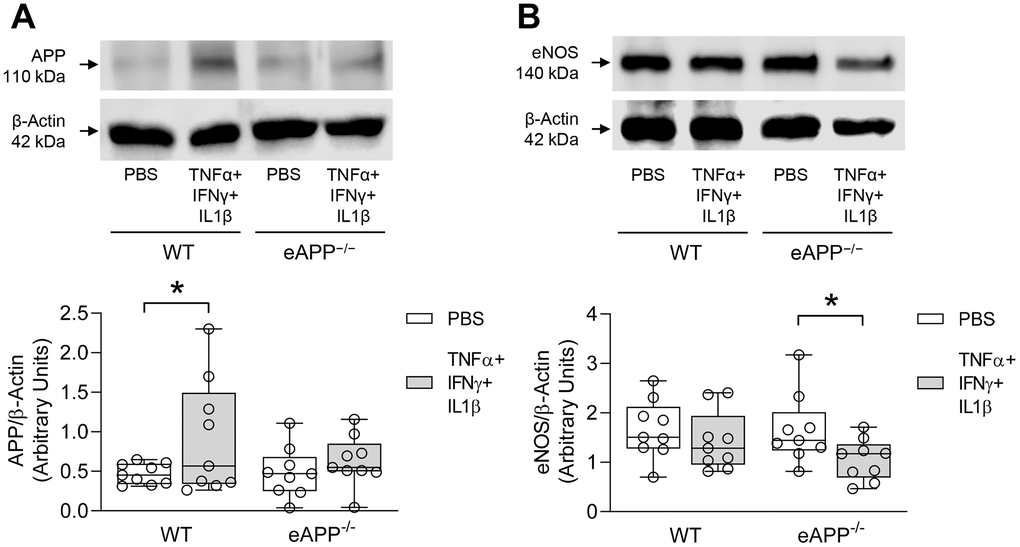
Figure 9. Effects of ex-vivo treatment for 24 hours with cytokines cocktail on protein expressions of APP (A) and eNOS (B) in the aortas of young wild-type (WT) littermates and eAPP−/− mice. Western blot results are the relative densitometry compared with β-actin protein (n=9 per group). All results are representing box plots with whiskers showing the median, 25th to 75th percentiles, and min-max range. * P<0.05 versus control PBS treatment (two-way ANOVA followed by Tukey’s HSD test).
Discussion
There are several major findings in this study. First, protein expression of endothelial APP and production of sAPPα in endothelium of aged WT mice are increased as compared to aged eAPP−/− mice. Second, impairment of endothelium-dependent relaxations to acetylcholine induced by aging is exacerbated in eAPP−/− mice aortas. Third, eNOS expression was decreased whereas levels of superoxide anion were enhanced in aortas of aged eAPP−/− mice. Fourth, pro-inflammatory cytokines increased APP protein expression only in isolated aortas of young WT mice. In contrast, pro-inflammatory cytokines decreased expression of eNOS exclusively in young eAPP−/− mice. Taken together, the results suggest that during aging endothelial APP exerts vascular protective effects. These effects appear to be mediated by preservation of eNOS and endothelium-dependent vasodilator function.
In the present study we demonstrated for the first time that APP expression and sAPPα release were increased in aged wild-type mice aortas. Importantly, these age-induced changes in expression and processing of APP were not observed in the aorta of eAPP−/− mice thus demonstrating that endothelial cells are a major site of increased APP expression and production of sAPPα. Of note, Austin and colleagues reported that APP expression is increased mainly in intimal layer of both human and apolipoprotein E-deficient mice atherosclerotic aortas [21]. Importantly, protein expression of mature α-secretase ADAM10 was unchanged in aged aortas. Whether enhanced α-processing of APP could be explained by the increased enzymatic activity of ADAM10, or some other α-secretases, remains to be determined [27].
Prior study reported that hypertension causes shift towards amyloidogenic APP β-processing and development of cerebral amyloid angiopathy [31]. However, we detect neither blood pressure increases nor changes of aortic protein expression of β-secretase, BACE1, and production of Aβ1-40 in aged wild-type and eAPP−/− mice indicating that amyloidogenic processing of APP in aorta is not affected by aging. Thus, our study demonstrates that upregulation of APP expression in aged wild-type mice is not induced by alterations of arterial blood pressure.
It is not clear what are the exact mechanisms responsible for increased expression and α-processing of APP in large conduit arteries of aged WT mice. Relevant to our observations, it is well known that aging is characterized by a state of chronic, low-grade inflammation [32, 33]. Moreover, expression of inflammatory cytokines such as TNFα, INFγ, and/or IL-1ß have been reported to be elevated in arteries of aged mice [34–37]. We speculate that the presence of proinflammatory stimuli is the most likely explanation for increased expression and non-amyloidogenic processing of APP. Consistent with this hypothesis, TNFα and IL-1ß increase APP expression and secretion of sAPPα from endothelial cells [19–21, 38]. In the present study, we also demonstrated that ex-vivo treatment of isolated aortas with cocktail of inflammatory cytokines led to increased APP expression in young WT mice but not in eAPP−/− mice again indicating that endothelium is a major source of APP. The concentrations of cytokines used for ex-vivo studies were higher than published serum cytokines levels detected in-vivo in young control mice [39]. Whether aging significantly increases circulating levels of cytokines tested in our study remains to be determined. Nevertheless, it is conceivable that the observed upregulation of APP represents an adaptative/protective response of endothelial cells to aging-induced inflammation [40, 41].
Although plasma levels of sAPPα were lower in eAPP−/− mice as compared to WT mice, aging increased sAPPα levels not only in WT mice but also in eAPP−/− mice. This indicates that other cell types besides endothelial cells must contribute to elevation of circulating sAPPα. It is well known that APP is also expressed in platelets and cleaved by non-amyloidogenic APP processing thereby releasing sAPPα upon activation [38, 42, 43]. Notably, platelets count is significantly increased in aged wild-type C57BL/6J mice [44]. Increased release of sAPPα from platelets may help explain elevated circulating levels of sAPPα in aged eAPP−/− mice.
Circulating levels norepinephrine were increased in both aged WT and eAPP−/− mice despite normal blood pressure. This may explain the observed reduced responsiveness to phenylephrine in aging. Chronic exposure of the peripheral vasculature to high circulating levels of norepinephrine causes compensatory downregulation of reactivity to phenylephrine. Indeed, similar observations were made in healthy human subjects showing that α-adrenergic vasocontraction responsiveness to increased circulating norepinephrine levels is reduced with age in healthy men [45].
Aging also alters arachidonic acid metabolism in endothelial cells and these alterations can profoundly affect vasomotor function [46, 47]. Human and animal studies have established that enhanced production of vasoconstrictor prostaglandins can impair endothelium-dependent relaxations during aging [8, 10]. In subjects older than 60 years, the treatment with COX-inhibitor, indomethacin, potentiates endothelium-dependent vasodilation to ACh thus demonstrating that vasoconstrictor prostaglandins contribute to impairment of endothelium-dependent relaxations in humans [10]. In line with these studies, we presented evidence that indomethacin significantly improved endothelium-dependent relaxations in response to ACh in both aged WT and eAPP−/− mice aortas. Consistent with these observations, production of TXA2 and PGE2 was significantly increased in both aged WT and eAPP−/− mice. The reason why the production of PGF2α was increased only in aged WT littermates is unclear and remains to be determined. However, this could be related to different expression levels of COX2 between aged WT and eAPP−/− mice (see below). In contrast, the production of 6-keto PGF1α remained unchanged in aged conduit arteries demonstrating that the metabolism of prostaglandin I2, which causes direct vasodilation in smooth muscle cells, is unaffected.
Interestingly, our study revealed that protein expression of COX2 was significantly increased in both aged wild-type and eAPP−/− mice while protein expression of COX1 remained unchanged. Although COX1 primarily contributes to basal vascular production of prostaglandins, COX2 also contributes to the production of prostaglandins [48]. Indeed, it has been reported that COX2 is expressed in endothelial cells and that inhibition of eNOS unmasks the ability of ACh to elicit endothelium-dependent contractions which are sensitive to inhibition of COX2 [49]. Thus, increased production of vasoconstrictor prostaglandins by COX2 appears to be responsible for reduced endothelium-dependent relaxations in aged mice. Notably, this phenomenon was not affected by genetic deletion of APP in endothelium.
To determine the reason why endothelium-dependent relaxations to ACh were still impaired in aged eAPP−/− mice despite treatment with indomethacin, we studied eNOS protein expression. Interestingly, eNOS protein was decreased in the aorta of aged eAPP−/− mice but not in young eAPP−/− mice. In contrast, expression of eNOS is unchanged in aged WT mice aortas and this is in line with the previous reports [50, 51]. Moreover, eNOS expression is not altered in conduit and resistance arteries of different mouse models of cardiovascular disease despite presence of inflammatory cytokines [52–54]. These observations are confirmed and further extended by our ex-vivo studies of isolated aortas treated with cytokines cocktail showing that eNOS expression was downregulated in young eAPP−/− mice, but not in young WT mice. From these findings we concluded that during aging, the presence of APP in aortic vascular endothelium is essential for normal expression and function of eNOS.
Superoxide anion levels were increased in aged eAPP-/- aortas but not in aged WT aortas. Increased ROS generation and/or reduced antioxidant capacity could contribute to the increased levels of superoxide anion. However, insufficient NO production in aged eAPP-/- mice can also lead to enhanced production of superoxide anion thereby further exacerbating impairment of endothelium-dependent relaxations (Figure 10). Indeed, several studies provided evidence that NO inactivated superoxide anion by rapid nonenzymatic reaction with superoxide anion [55–58].
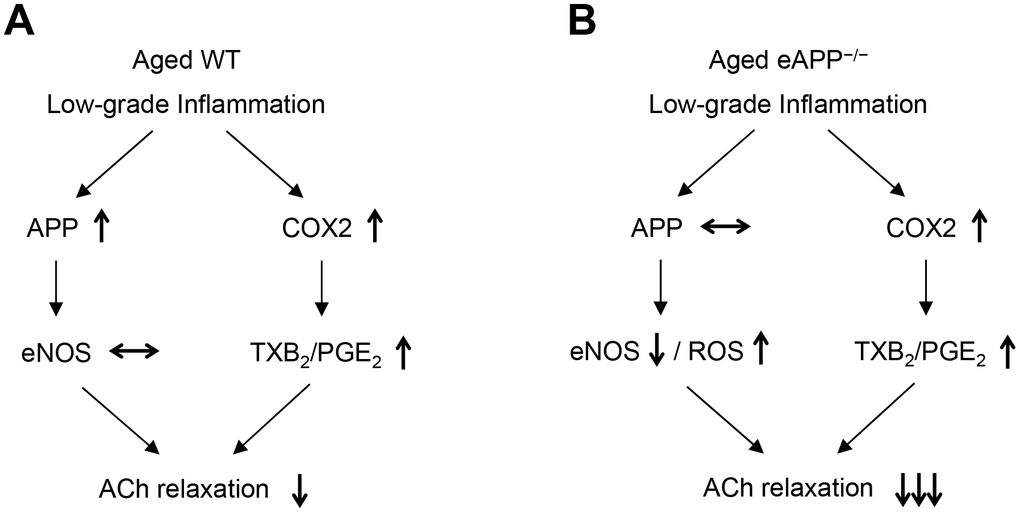
Figure 10. Schematic summary of the effects of aging on endothelial function in wild-type (WT) mice (A) and endothelium-specific amyloid precursor protein-deficient (eAPP−/−) mice (B). Please note that expression of APP is increased in aged wild-type mice (A) but not in aged eAPP−/− mice (B) aortas. ↑ = increase; ↓ = decrease; ↔ = no change; eNOS = endothelial nitric oxide synthase; ROS = reactive oxygen species; COX2 = cyclooxygenase 2; TXB2 = thromboxane B2; PGE2 = prostaglandin E2; ACh = acetylcholine.
We were unable to observe any changes in superoxide anion production in conduit arteries derived from aged WT mice under basal conditions and this is in line with existing literature [37, 51, 59]. In contrast, several studies reported increased levels of superoxide anion in aged wild-type mice arteries [50, 60, 61]. The reason for this discrepancy is unclear. One possible explanation is that vascular superoxide production is increased in very old (31 months old) wild-type mice [37, 61].
Elevation of aortic cGMP levels induced by ACh are decreased in both aged WT and eAPP−/− mice as compared to young mice aortas while basal cGMP levels were not different between WT and eAPP−/− mice. As mentioned earlier, the endothelium is a major source not only of NO but of prostaglandins as well [3]. Relevant to our observations, endogenous prostaglandins have been shown to decrease cGMP levels and to induce contractions in response to ACh in the aorta [62, 63] thus suggesting that exaggerated production of prostaglandins during aging can impair ACh-stimulated production of cGMP. At the present time we do not have an explanation as to why levels of cGMP were not decreased in aged eAPP−/− mice.
In aggregate, the present study demonstrates that endothelial dysfunction (loss of NO) is more pronounced in aged eAPP−/− mice than in aged WT littermates. We also provide evidence that the absence of APP in endothelial cells causes downregulation of eNOS expression in eAPP−/− mice. Our findings support the concept that up-regulation of endothelial APP and production of sAPPα are adaptive mechanisms designed to protect blood vessel wall from detrimental effects of inflammation associated with aging. In this regard, preservation of NO production in endothelium of aged aortas appears to be critically important function of endothelial APP.
Materials and Methods
Experimental animals
APPflox/flox;Tie2-Cre− mice (wild-type [WT] littermates) and APPflox/flox;Tie2-Cre+ (eAPP−/−) mice were generated and genotyped in our laboratory as described previously [29]. All mice were maintained on 12h/12h light/dark cycle and on standard chow with free access to drinking water. Institutional Animal Care and Use Committee of the Mayo Clinic reviewed and approved the experimental design. In addition, the protocols complied with the guidelines of ARRIVE and NIH for care and use of laboratory animals. Young male mice (4-6 months old) and aged male mice (23-26 months old) were anaesthetized with overdose of pentobarbital (200-250 mg/kg BW, i.p.) and were exsanguinated using cardiac puncture method for collection of blood samples. The aortas were carefully harvested and were thereafter dissected free from surrounding connective tissues in cold (4° C) Krebs solution [composition (in mmol/L): NaCl 118.6; KCl 4.7; CaCl2 2.5; MgSO4 1.2; KH2PO4 1.2; NaHCO3 25.1; glucose 10.1; EDTA 0.026]. Age-matched WT littermate mice served as controls in studies designed to determine vascular phenotype characteristics of eAPP−/− mice.
Blood pressure
A tail-cuff technique (Harvard Apparatus Ltd., Kent, England) was used to monitor SBP and MBP [64]. DBP was calculated through the formula: DBP = [(3xMBP) - SBP]/2.
Glucose and lipid levels
Blood was collected through cardiac puncture and was transferred to a tube containing EDTA. Glucose was determined immediately using Accu Check® (Roche Diagnostics, Indianapolis, IN). Thereafter, blood samples were centrifuged for 10 min at 2000 rpm (4° C) and plasma was obtained and stored at -80° C. Plasma levels of cholesterol, HDL, and triglyceride were measured as described previously [24].
Determination of plasma norepinephrine levels
Blood was placed in a tube containing EGTA with reduced glutathione. After centrifugation, plasma was stored at -80° C until assayed. After extraction with activated alumina norepinephrine levels were determined by HPLC with electrochemical detection as described [65].
Determination of plasma sAPPα and Aβ1-40 levels
A highly sensitive sAPPα mouse/rat ELISA kit (Immuno-Biological Laboratories America, Minneapolis, MN) and Aβ1-40 mouse ELISA kit (Invitrogen, Camarillo, CA) were used to determine sAPPα and Aβ1-40 levels, respectively.
Determination of ex-vivo release of sAPPα and Aβ1-40
Ten millimeters long aortas were opened longitudinally and were incubated in minimal essential medium (MEM) in the presence of bovine serum albumin (0.1%), penicillin (100 U/mL) and streptomycin (100 μg/mL; GIBCO® Thermo Fischer Scientific Inc., Waltham, MA) for 24 hours at 37° C in CO2 incubator. Thereafter, the cultured medium was collected and stored at -80° C until assayed. Levels sAPPα and Aβ1-40 in conditioned medium were determined using the ELISA kits as described above and all results were normalized against protein levels of aortas.
Organ chamber studies in isolated aortas
An isometric force measurement was used to study vasomotor function of isolated thoracic aortic rings as previously described [66]. Aortic rings were stretched to optimal force of 1.5 grams. Thereafter, the rings were contracted twice with KCl (80 mmol/L) and washed out. Concentration-dependent response curves to PGF2α (10-8−3x10-5 mol/L; Cayman, Ann Arbor, MI) and L-phenylephrine (10-9−10-5 mol/L; Sigma, St. Louis, MO) were cumulatively obtained. After washing out, concentration-dependent responses to ACh (10-9−10-5 mol/L; Sigma) were recorded in parallel in the absence and in the presence of indomethacin (10-5 mol/L, 30 minutes; Sigma) and indomethacin + L-NAME (3x10-4 mol/L, 30 minutes; Sigma). Various concentrations of PGF2α (3x10-6–8x10-6 mol/L) were used in order to achieve similar submaximal contractions in young WT and eAPP−/− mice (57±5% and 61±5%, respectively; P>0.05; n=9) as well as in aged WT and eAPP−/− mice (66±4% and 69±3%, respectively; P>0.05; n=8).
Determination of ex-vivo release of prostaglandins
Ten millimeters long thoracic aortas were opened longitudinally and were incubated in modified MEM in the presence of 0.1% BSA, 100 U/mL penicillin and 100 μg/mL streptomycin for 24 hours (37° C in CO2 incubator). Thereafter, the cultured medium was collected and stored at -80° C. Commercially available colorimetric ELISA kits (Cayman, Ann Arbor, MI) were used for measurements of TXB2 (a stable metabolite of TXA2), PGE2, PGF2α, and 6-keto PGF1α concentrations in the medium. All results were normalized against aortic protein levels.
Determination of intracellular superoxide anions
Intracellular levels of superoxide anion were quantified in fresh aortic rings using 2-hydroxyethidium standard by HPLC-based fluorescence detection and normalized against tissue protein levels as described in detail in previous studies [67, 68].
Determination of cGMP
Twelve millimeters long aortas were incubated for 2 hours at 37° C in MEM containing bovine serum albumin (0.1%), penicillin (100 U/mL) and streptomycin (100 μg/mL). A phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (100 μmol/L; Sigma) was also added to prevent degradation of cyclic nucleotides. At the end of incubation time, ACh (10 μM) was added for 2 minutes. Thereafter, all samples were immediately placed in liquid N2 and stored at -80° C until assayed. Colorimetric cGMP ELISA immunoassay (Cell Biolabs Inc., San Diego, CA) was used to determine cGMP levels and the results were normalized against aortic protein concentrations.
Ex-vivo studies with cytokines
Ten millimeters long thoracic aortas were opened longitudinally and were incubated with control solution (PBS) or cytokines cocktail composing 20 ng/mL recombinant mouse tumor necrosis factor α (TNFα; R&D Systems, Minneapolis, MN), 50 ng/mL recombinant mouse interferon γ (INFγ; R&D Systems), and 1 ng/mL recombinant mouse interleukin-1ß (IL-1ß; R&D Systems) in MEM in the presence of bovine serum albumin (0.1%), penicillin (100 U/mL) and streptomycin (100 μg/mL) for 24 hours at 37° C in CO2 incubator [69]. Thereafter, the aortas were homogenized in lysis buffer for western blot analysis (see below).
Western blot analysis
Dissected aortas were homogenized in lysis buffer using glass grinder and pestle 20 as previously described [68]. After centrifugation, protein levels were determined in supernatants using DC protein assay kit (BioRad, Hercules, CA). For each sample, 50 μg protein was loaded on 7.5% or 10% TGX SDS-PAGE gels (BioRad). Antibodies against APP (1:250; cat# 51-2700, Invitrogen, Carlsbad, CA), BACE1 (1:250; cat# S5606, Cell signaling, Danvers, MA), ADAM10 (1:500; cat# AB19026, Millipore, Burlington, MA), eNOS (1:250, cat# 610297, BD Biosciences, San Jose, CA), inducible nitric oxide synthase (iNOS; 1:250, cat# 610333, BD Biosciences), COX1 (1:500; cat# 35-8100, Invitrogen), and COX2 (1:250; cat# 610204, BD Biosciences) were used. The antibody specificities for APP, BACE1, and eNOS were verified in knockout mice obtained from The Jackson Laboratory (stock #004133, stock #004714, and stock #002684, respectively; Supplementary Figure 5). In addition, C57BL/6J mouse heart was used as positive control for COX1, and whole cell lysate RAW 264.7 (cat# ab7187, Abcam) was used to identify COX2 and ADAM10 bands (Supplementary Figure 6). All blots were reprobed with β-actin antibody (1:50,000, A5316, Sigma). Densitometry analyses were performed in Odyssey Fc imaging system with Image Studio™ 5.2 software (Li-Cor Biotechnology, Lincoln, NE).
Statistical analysis
All results are presented as mean ± standard error of mean (SEM; n indicates the number of mice used per experiment). The efficacy and potency of the drugs were determined using LabChart Pro Dose Response Module (ADInstruments Inc., Colorado Springs, CO). Concentration-response curves were compared by analysis of variance (ANOVA) for repeated measurements followed by Bonferroni's correction as described [29]. Two-way ANOVA followed by Tukey’s HSD test and unpaired t-test were used for multiple and single comparison, respectively (JMP Pro 14.1 software; SAS Institute, Cary, NC). Differences of P<0.05 were considered statistically significant.
Supplementary Materials
Author Contributions
Livius d’Uscio: experimental design, data collection and analysis, and manuscript writing. Zvonimir Katusic: experimental design, data interpretation, and manuscript writing.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by National Institutes of Health R01 grant HL-131515 and by the Mayo Foundation.
References
- 1. Lüscher TF, Vanhoutte PM. The Endothelium: Modulator of Cardiovascular Function. Boca Raton, Fla: CRC Press. 1990.
- 2. Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988; 333:664–66. https://doi.org/10.1038/333664a0 [PubMed]
- 3. Katusić ZS, Shepherd JT. Endothelium-derived vasoactive factors: II. Endothelium-dependent contraction. Hypertension. 1991; 18:III86–92. https://doi.org/10.1161/01.hyp.18.5_suppl.iii86 [PubMed]
- 4. Pollock JS, Förstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, Murad F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci USA. 1991; 88:10480–84. https://doi.org/10.1073/pnas.88.23.10480 [PubMed]
- 5. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995; 377:239–42. https://doi.org/10.1038/377239a0 [PubMed]
- 6. Qi Z, Cai H, Morrow JD, Breyer MD. Differentiation of cyclooxygenase 1- and 2-derived prostanoids in mouse kidney and aorta. Hypertension. 2006; 48:323–28. https://doi.org/10.1161/01.HYP.0000231934.67549.b7 [PubMed]
- 7. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003; 107:139–46. https://doi.org/10.1161/01.cir.0000048892.83521.58 [PubMed]
- 8. Küng CF, Lüscher TF. Different mechanisms of endothelial dysfunction with aging and hypertension in rat aorta. Hypertension. 1995; 25:194–200. https://doi.org/10.1161/01.hyp.25.2.194 [PubMed]
- 9. Wong MS, Vanhoutte PM. COX-mediated endothelium-dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin. 2010; 31:1095–102. https://doi.org/10.1038/aps.2010.127 [PubMed]
- 10. Taddei S, Virdis A, Mattei P, Ghiadoni L, Fasolo CB, Sudano I, Salvetti A. Hypertension causes premature aging of endothelial function in humans. Hypertension. 1997; 29:736–43. https://doi.org/10.1161/01.hyp.29.3.736 [PubMed]
- 11. Zeiher AM, Drexler H, Saurbier B, Just H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest. 1993; 92:652–62. https://doi.org/10.1172/JCI116634 [PubMed]
- 12. Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Coll Cardiol. 1994; 24:471–76. https://doi.org/10.1016/0735-1097(94)90305-0 [PubMed]
- 13. Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987; 325:733–36. https://doi.org/10.1038/325733a0 [PubMed]
- 14. Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA. 1988; 85:7341–45. https://doi.org/10.1073/pnas.85.19.7341 [PubMed]
- 15. Nicolas M, Hassan BA. Amyloid precursor protein and neural development. Development. 2014; 141:2543–48. https://doi.org/10.1242/dev.108712 [PubMed]
- 16. Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature. 1988; 331:528–30. https://doi.org/10.1038/331528a0 [PubMed]
- 17. Golde TE, Estus S, Usiak M, Younkin LH, Younkin SG. Expression of beta amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron. 1990; 4:253–67. https://doi.org/10.1016/0896-6273(90)90100-t [PubMed]
- 18. d’Uscio LV, He T, Katusic ZS. Expression and Processing of Amyloid Precursor Protein in Vascular Endothelium. Physiology (Bethesda). 2017; 32:20–32. https://doi.org/10.1152/physiol.00021.2016 [PubMed]
- 19. Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, Gajdusek DC. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci USA. 1989; 86:7606–10. https://doi.org/10.1073/pnas.86.19.7606 [PubMed]
- 20. Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain Res Mol Brain Res. 1992; 16:128–34. https://doi.org/10.1016/0169-328x(92)90202-m [PubMed]
- 21. Austin SA, Sens MA, Combs CK. Amyloid precursor protein mediates a tyrosine kinase-dependent activation response in endothelial cells. J Neurosci. 2009; 29:14451–62. https://doi.org/10.1523/JNEUROSCI.3107-09.2009 [PubMed]
- 22. Kitazume S, Tachida Y, Kato M, Yamaguchi Y, Honda T, Hashimoto Y, Wada Y, Saito T, Iwata N, Saido T, Taniguchi N. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J Biol Chem. 2010; 285:40097–103. https://doi.org/10.1074/jbc.M110.144626 [PubMed]
- 23. Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res. 2010; 107:1498–502. https://doi.org/10.1161/CIRCRESAHA.110.233080 [PubMed]
- 24. d’Uscio LV, Das P, Santhanam AV, He T, Younkin SG, Katusic ZS. Activation of PPARδ prevents endothelial dysfunction induced by overexpression of amyloid-β precursor protein. Cardiovasc Res. 2012; 96:504–12. https://doi.org/10.1093/cvr/cvs266 [PubMed]
- 25. Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992; 357:500–03. https://doi.org/10.1038/357500a0 [PubMed]
- 26. Sisodia SS. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci USA. 1992; 89:6075–79. https://doi.org/10.1073/pnas.89.13.6075 [PubMed]
- 27. Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999; 96:3922–27. https://doi.org/10.1073/pnas.96.7.3922 [PubMed]
- 28. He T, Santhanam AV, Lu T, d’Uscio LV, Katusic ZS. Role of prostacyclin signaling in endothelial production of soluble amyloid precursor protein-α in cerebral microvessels. J Cereb Blood Flow Metab. 2017; 37:106–22. https://doi.org/10.1177/0271678X15618977 [PubMed]
- 29. d’Uscio LV, He T, Santhanam AV, Katusic ZS. Endothelium-specific amyloid precursor protein deficiency causes endothelial dysfunction in cerebral arteries. J Cereb Blood Flow Metab. 2018; 38:1715–26. https://doi.org/10.1177/0271678X17735418 [PubMed]
- 30. Austin SA, Katusic ZS. Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. J Cereb Blood Flow Metab. 2020; 40:392–403. https://doi.org/10.1177/0271678X18822474 [PubMed]
- 31. Cifuentes D, Poittevin M, Bonnin P, Ngkelo A, Kubis N, Merkulova-Rainon T, Lévy BI. Inactivation of Nitric Oxide Synthesis Exacerbates the Development of Alzheimer Disease Pathology in APPPS1 Mice (Amyloid Precursor Protein/Presenilin-1). Hypertension. 2017; 70:613–23. https://doi.org/10.1161/HYPERTENSIONAHA.117.09742 [PubMed]
- 32. Cesari M, Penninx BW, Newman AB, Kritchevsky SB, Nicklas BJ, Sutton-Tyrrell K, Rubin SM, Ding J, Simonsick EM, Harris TB, Pahor M. Inflammatory markers and onset of cardiovascular events: results from the Health ABC study. Circulation. 2003; 108:2317–22. https://doi.org/10.1161/01.CIR.0000097109.90783.FC [PubMed]
- 33. Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004; 39:687–99. https://doi.org/10.1016/j.exger.2004.01.009 [PubMed]
- 34. Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. 2003; 17:1183–85. https://doi.org/10.1096/fj.02-1049fje [PubMed]
- 35. Lesniewski LA, Durrant JR, Connell ML, Henson GD, Black AD, Donato AJ, Seals DR. Aerobic exercise reverses arterial inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2011; 301:H1025–32. https://doi.org/10.1152/ajpheart.01276.2010 [PubMed]
- 36. Peña Silva RA, Chu Y, Miller JD, Mitchell IJ, Penninger JM, Faraci FM, Heistad DD. Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke. 2012; 43:3358–63. https://doi.org/10.1161/STROKEAHA.112.667063 [PubMed]
- 37. Dinh QN, Drummond GR, Kemp-Harper BK, Diep H, De Silva TM, Kim HA, Vinh A, Robertson AA, Cooper MA, Mansell A, Chrissobolis S, Sobey CG. Pressor response to angiotensin II is enhanced in aged mice and associated with inflammation, vasoconstriction and oxidative stress. Aging (Albany NY). 2017; 9:1595–606. https://doi.org/10.18632/aging.101255 [PubMed]
- 38. Kitazume S, Yoshihisa A, Yamaki T, Oikawa M, Tachida Y, Ogawa K, Imamaki R, Hagiwara Y, Kinoshita N, Takeishi Y, Furukawa K, Tomita N, Arai H, et al. Soluble amyloid precursor protein 770 is released from inflamed endothelial cells and activated platelets: a novel biomarker for acute coronary syndrome. J Biol Chem. 2012; 287:40817–25. https://doi.org/10.1074/jbc.M112.398578 [PubMed]
- 39. Tracz MJ, Juncos JP, Croatt AJ, Ackerman AW, Grande JP, Knutson KL, Kane GC, Terzic A, Griffin MD, Nath KA. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007; 72:1073–80. https://doi.org/10.1038/sj.ki.5002471 [PubMed]
- 40. Chen MB, Yang AC, Yousef H, Lee D, Chen W, Schaum N, Lehallier B, Quake SR, Wyss-Coray T. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Rep. 2020; 30:4418–32.e4. https://doi.org/10.1016/j.celrep.2020.03.012 [PubMed]
- 41. Taylor RC. Aging and the UPR(ER). Brain Res. 2016; 1648:588–93. https://doi.org/10.1016/j.brainres.2016.04.017 [PubMed]
- 42. Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid beta-protein precursor): a platelet alpha-granule protein. Science. 1990; 248:745–48. https://doi.org/10.1126/science.2110384 [PubMed]
- 43. Schuck F, Wolf D, Fellgiebel A, Endres K. Increase of α-Secretase ADAM10 in Platelets Along Cognitively Healthy Aging. J Alzheimers Dis. 2016; 50:817–26. https://doi.org/10.3233/JAD-150737 [PubMed]
- 44. Dayal S, Wilson KM, Motto DG, Miller FJ
Jr , Chauhan AK, Lentz SR. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation. 2013; 127:1308–16. https://doi.org/10.1161/CIRCULATIONAHA.112.000966 [PubMed] - 45. Dinenno FA, Dietz NM, Joyner MJ. Aging and forearm postjunctional alpha-adrenergic vasoconstriction in healthy men. Circulation. 2002; 106:1349–54. https://doi.org/10.1161/01.CIR.0000028819.64790.BE [PubMed]
- 46. Tang EH, Vanhoutte PM. Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol Genomics. 2008; 32:409–18. https://doi.org/10.1152/physiolgenomics.00136.2007 [PubMed]
- 47. Qian H, Luo N, Chi Y. Aging-shifted prostaglandin profile in endothelium as a factor in cardiovascular disorders. J Aging Res. 2012; 2012:121390. https://doi.org/10.1155/2012/121390 [PubMed]
- 48. Gomez I, Foudi N, Longrois D, Norel X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fatty Acids. 2013; 89:55–63. https://doi.org/10.1016/j.plefa.2013.04.004 [PubMed]
- 49. Wong SL, Leung FP, Lau CW, Au CL, Yung LM, Yao X, Chen ZY, Vanhoutte PM, Gollasch M, Huang Y. Cyclooxygenase-2-derived prostaglandin F2alpha mediates endothelium-dependent contractions in the aortae of hamsters with increased impact during aging. Circ Res. 2009; 104:228–35. https://doi.org/10.1161/CIRCRESAHA.108.179770 [PubMed]
- 50. Francia P, delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Lüscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004; 110:2889–95. https://doi.org/10.1161/01.CIR.0000147731.24444.4D [PubMed]
- 51. Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. 2007; 27:1941–46. https://doi.org/10.1161/ATVBAHA.107.146852 [PubMed]
- 52. d’Uscio LV, Baker TA, Mantilla CB, Smith L, Weiler D, Sieck GC, Katusic ZS. Mechanism of endothelial dysfunction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001; 21:1017–22. https://doi.org/10.1161/01.atv.21.6.1017 [PubMed]
- 53. Lee J, Lee S, Zhang H, Hill MA, Zhang C, Park Y. Interaction of IL-6 and TNF-α contributes to endothelial dysfunction in type 2 diabetic mouse hearts. PLoS One. 2017; 12:e0187189. https://doi.org/10.1371/journal.pone.0187189 [PubMed]
- 54. Silva JF, Correa IC, Diniz TF, Lima PM, Santos RL, Cortes SF, Coimbra CC, Lemos VS. Obesity, Inflammation, and Exercise Training: Relative Contribution of iNOS and eNOS in the Modulation of Vascular Function in the Mouse Aorta. Front Physiol. 2016; 7:386. https://doi.org/10.3389/fphys.2016.00386 [PubMed]
- 55. Harrison CB, Drummond GR, Sobey CG, Selemidis S. Evidence that nitric oxide inhibits vascular inflammation and superoxide production via a p47phox-dependent mechanism in mice. Clin Exp Pharmacol Physiol. 2010; 37:429–34. https://doi.org/10.1111/j.1440-1681.2009.05317.x [PubMed]
- 56. Kopkan L, Hess A, Husková Z, Cervenka L, Navar LG, Majid DS. High-salt intake enhances superoxide activity in eNOS knockout mice leading to the development of salt sensitivity. Am J Physiol Renal Physiol. 2010; 299:F656–63. https://doi.org/10.1152/ajprenal.00047.2010 [PubMed]
- 57. Majid DS, Nishiyama A, Jackson KE, Castillo A. Inhibition of nitric oxide synthase enhances superoxide activity in canine kidney. Am J Physiol Regul Integr Comp Physiol. 2004; 287:R27–32. https://doi.org/10.1152/ajpregu.00073.2004 [PubMed]
- 58. Wink DA, Miranda KM, Espey MG, Pluta RM, Hewett SJ, Colton C, Vitek M, Feelisch M, Grisham MB. Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal. 2001; 3:203–13. https://doi.org/10.1089/152308601300185179 [PubMed]
- 59. Shi Y, Savarese G, Perrone-Filardi P, Lüscher TF, Camici GG. Enhanced age-dependent cerebrovascular dysfunction is mediated by adaptor protein p66Shc. Int J Cardiol. 2014; 175:446–50. https://doi.org/10.1016/j.ijcard.2014.06.025 [PubMed]
- 60. Takenouchi Y, Kobayashi T, Matsumoto T, Kamata K. Gender differences in age-related endothelial function in the murine aorta. Atherosclerosis. 2009; 206:397–404. https://doi.org/10.1016/j.atherosclerosis.2009.03.005 [PubMed]
- 61. Lesniewski LA, Connell ML, Durrant JR, Folian BJ, Anderson MC, Donato AJ, Seals DR. B6D2F1 Mice are a suitable model of oxidative stress-mediated impaired endothelium-dependent dilation with aging. J Gerontol A Biol Sci Med Sci. 2009; 64:9–20. https://doi.org/10.1093/gerona/gln049 [PubMed]
- 62. Kato T, Iwama Y, Okumura K, Hashimoto H, Ito T, Satake T. Prostaglandin H2 may be the endothelium-derived contracting factor released by acetylcholine in the aorta of the rat. Hypertension. 1990; 15:475–81. https://doi.org/10.1161/01.hyp.15.5.475 [PubMed]
- 63. Schachter D, Sang JC. Regional differentiation in the rat aorta: effects of cyclooxygenase inhibitors. Am J Physiol. 1997; 273:H1478–83. https://doi.org/10.1152/ajpheart.1997.273.3.H1478 [PubMed]
- 64. Johns C, Gavras I, Handy DE, Salomao A, Gavras H. Models of experimental hypertension in mice. Hypertension. 1996; 28:1064–69. https://doi.org/10.1161/01.hyp.28.6.1064 [PubMed]
- 65. Jiang NS, Machacek D. Measurement of catecholamines in blood and urine by liquid chromatography with amperometric detection. In: Parvez Hea, ed. In Progress in HPLC. Utrecht: V.N.U. Science Press BV. 1987; 397–426.
- 66. d’Uscio LV, He T, Santhanam AV, Tai LJ, Evans RM, Katusic ZS. Mechanisms of vascular dysfunction in mice with endothelium-specific deletion of the PPAR-δ gene. Am J Physiol Heart Circ Physiol. 2014; 306:H1001–10. https://doi.org/10.1152/ajpheart.00761.2013 [PubMed]
- 67. Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA. 2005; 102:5727–32. https://doi.org/10.1073/pnas.0501719102 [PubMed]
- 68. d'Uscio LV, Smith LA, Katusic ZS. Differential effects of eNOS uncoupling on conduit and small arteries in GTP-cyclohydrolase I-deficient hph-1 mice. Am J Physiol Heart Circ Physiol. 2011; 301:H2227–34. https://doi.org/10.1152/ajpheart.00588.2011 [PubMed]
- 69. Huang A, Zhang YY, Chen K, Hatakeyama K, Keaney JF
Jr . Cytokine-stimulated GTP cyclohydrolase I expression in endothelial cells requires coordinated activation of nuclear factor-kappaB and Stat1/Stat3. Circ Res. 2005; 96:164–71. https://doi.org/10.1161/01.RES.0000153669.24827.DF [PubMed]