



Introduction
Ischemic stroke is one of the leading causes of death and long-term disability in the world. Revascularization therapy via intravenous recombinant tissue plasminogen activator [1, 2] or endovascular mechanical thrombectomy [3, 4] is the mainstay and most effective strategy for acute ischemic stroke treatment. Even though the restoration of blood flow and reoxygenation is essential in preventing neuronal injury, it may give rise to ischemia-reperfusion injury and augment the neuronal damage partly through an enhanced oxidative stress and neuroinflammation [5, 6], which largely limits the efficacy of revascularization treatment. However, effective therapy for cerebral ischemia-reperfusion injury remains elusive.
Mitochondria are the powerhouses of the cell and play a pivotal role in energy metabolism, cell signaling, redox homeostasis and regulation of cellular life and death [7]. Cerebral ischemia-reperfusion injury induces mitochondrial fragmentation and dysfunction, which plays a key role in initiating the cellular apoptotic cascades and subsequent neuronal death. Maintaining the structural and functional integrity of the mitochondria is crucial in increasing neuronal survival and promoting neurological improvement. Targeting mitochondria has been proven to be a potential promising therapeutic strategy for cerebral ischemia-reperfusion injury [8, 9]. Amlodipine camsylate, a L-type calcium channel blocker, protects against oxygen-glucose deprivation (OGD) induced neural stem cell damage via maintaining mitochondrial structure and function [10]. However, more mitochondria-targeted approaches are urgently needed to improve the outcome of cerebral ischemia-reperfusion injury.
Mitofusin 2 (MFN2) is a key protein involved in mitochondrial fusion, mitophagy and interorganellar communication. It also participates in regulation of cell proliferation, apoptosis and many other diverse biological processes. Mutations in MFN2 lead to the development of neurodegeneration [11]. MFN2 mutations are associated with Charcot-Marie-Tooth disease type 2A (CMT2A). MFN2 agonists ameliorate dominant mitochondrial defects and normalize axonal mitochondrial trafficking in preclinical models of CMT2A [12]. MFN2 is a potential therapeutic target for disorders with impairment of mitochondrial dynamism and trafficking.
USP30 is a mitochondrion-localized deubiquitylase that removes ubiquitin moieties from target proteins. USP30 protects mitochondria and peroxisomes from damage [13]. USP30 regulates pexophagy and protects basal peroxisome from degradation [14]. USP30 also plays a key role in maintaining the integrity of the mitochondria. USP30 suppresses ubiquitin ligase parkin-dependent ubiquitylation of mitochondrial proteins and inhibits mitochondrial-induced cell death [15]. USP30 antagonizes mitophagy mediated by the parkin and PINK1. USP30 inhibition is beneficial for Parkinson's disease through promoting mitochondrial clearance to ensure a healthy mitochondrial network. USP30 is supposed to be a promising therapeutic target for Parkinson's disease [16–19]. However, the potential role of USP30 in cerebral ischemia-reperfusion injury remains largely unknown.
Therefore, on the basis of previous findings, the aims of this study were to determine: (1) whether oxygen-glucose deprivation/reperfusion (OGDR) induced mitochondrial proteins and MFN2 ubiquitination and degradation in SK-N-BE(2) cells; (2) whether overexpression of MFN2 and USP30 protected against OGDR induced mitochondrial fragmentation; and (3) whether USP30 regulated ubiquitination and degradation of MFN2 after OGDR insult. In this study, we confirmed that OGDR induced ubiquitination of mitochondrial proteins during the early stage of reperfusion. MFN2 was also rapidly ubiquitinated and degraded in the early stage of reperfusion. Overexpression of MFN2 and USP30 attenuated OGDR induced mitochondrial fragmentation, and USP30 overexpression inhibited OGDR induced ubiquitination and degradation of MFN2. Our results suggest that USP30 is a promising target for the development of innovative therapeutic regimens for cerebral ischemia-reperfusion injury.
Results
Ubiquitination of mitochondrial proteins is increased during early reperfusion after OGD
We used a ubiquitin antibody to explore the ubiquitination of mitochondrial and cytoplasmic proteins after OGDR insult (Figure 1). SK-N-BE(2) cells were cultured, and mitochondrial and cytoplasmic proteins were isolated after 45 min reperfusion following 4 h OGD. As demonstrated in Figure 1A, we found that the ubiquitination of mitochondrial proteins increased significantly after 45 min reperfusion following 4 h OGD(P=0.0052), while there was no significant change in the ubiquitination of cytoplasmic proteins (Figure 1A, 1B). These data suggest that mitochondrial proteins are rapidly ubiquitinated in the early stage of reperfusion after OGD.
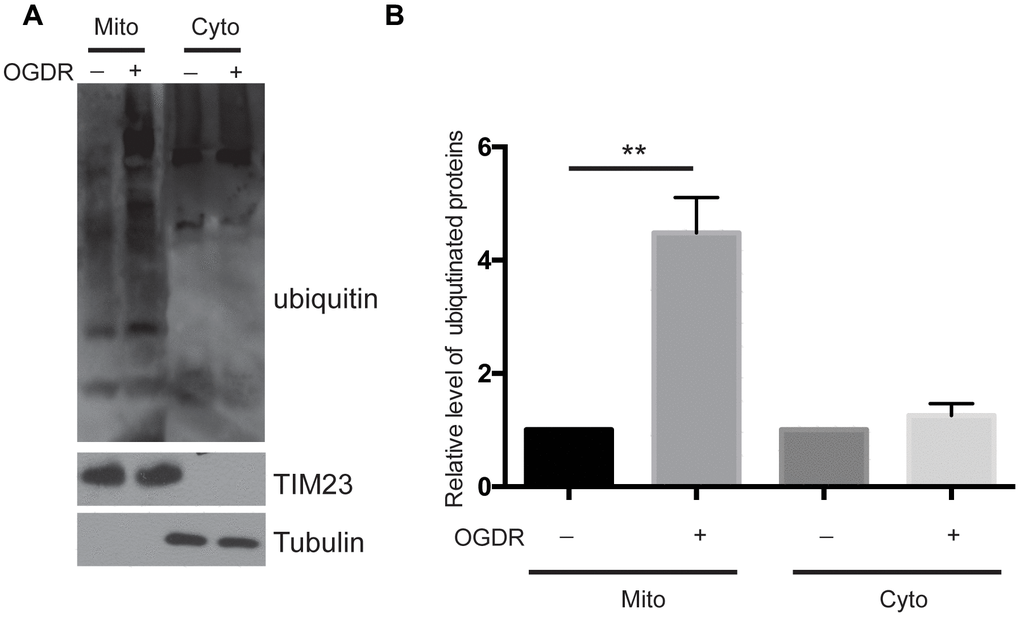
Figure 1. Ubiquitination of mitochondrial and cytoplasmic proteins in the early stage of reperfusion after 4 h OGD. (A) Western blot using an antibody against ubiquitin was performed to examine the ubiquitination of mitochondrial (Mito) and cytoplasmic (Cyto) proteins after 45 min reperfusion following 4 h OGD in SK-N-BE(2) cells. (B) Quantitation (Mean ± SEM) of A from three independent experiments. There was a significant increase in the ubiquitination of mitochondrial proteins during early reperfusion after 4 h of OGD, but not in the ubiquitination of the cytoplasmic proteins.
Ubiquitination of MFN2 is increased during early reperfusion after OGD
MFN2 is a GTPase dynamin-like protein localized to the outer mitochondrial membrane. MFN2 participates in mitochondrial clustering and fusion. We further evaluated the ubiquitination of MFN2 after OGDR insult (Figure 2). Consistent with the above data of ubiquitination of total mitochondrial proteins, the ubiquitination of MFN2 also increased significantly after 45 min reperfusion following 4 h OGD in SK-N-BE (2) cells (Figure 2A, 2B, P=0.0034). Meanwhile, the expression of total MFN2 protein decreased significantly after 45 min reperfusion following OGD insult (Figure 2A–2C, P=0.041). This data indicates that MFN2 is rapidly ubiquitinated and degraded in the early stage of reperfusion after OGD injury.
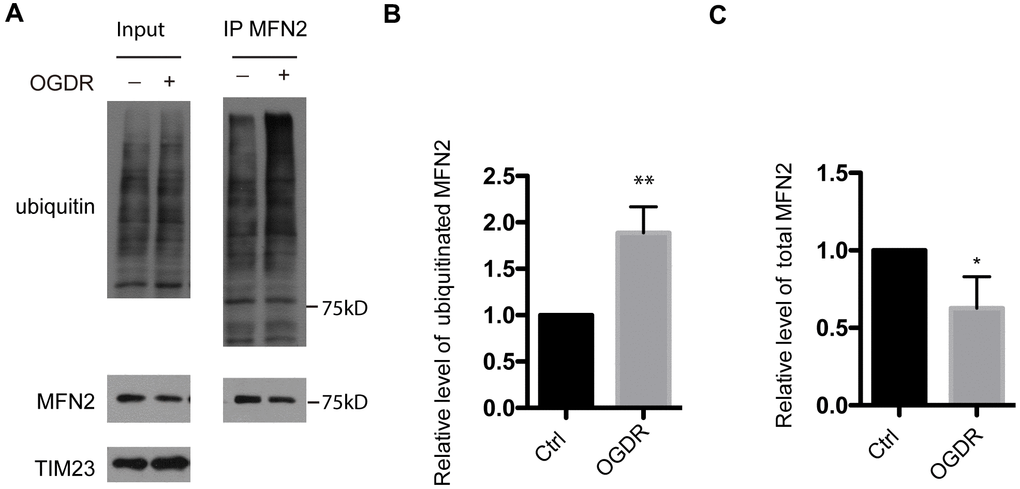
Figure 2. Ubiquitination of MFN2 in the early stage of reperfusion after 4 h OGD. (A) After immunoprecipitation of MFN2, western blot using an antibody against ubiquitin was performed to examine the ubiquitination of MFN2 after 45 min reperfusion following 4 h OGD in SK-N-BE(2) cells. (B) Quantitation (Mean ± SEM) of ubiquitinated MFN2 from three independent experiments. (C) Quantitation (Mean ± SEM) of total MFN2 from three independent experiments. There was a significant increase in the ubiquitination of MFN2 during early reperfusion after 4 h OGD.
MFN2 is rapidly degraded during early reperfusion after OGD
We have found that the ubiquitination of mitochondrial proteins and MFN2 was increased in the early stage of reperfusion after 4 h OGD, we further analyzed the expression of mitochondrial proteins after OGDR treatment. Reduced MFN2 protein expression was observed in SK-N-BE(2) cells subjected to 1.5 h (P=0.0085) and 2 h (P=0.0027) reperfusion following 4 h OGD. However, the expression of mitochondrial inner membrane protein TIM23 did not change significantly (Figure 3A, 3B). These data suggest that MFN2 is rapidly degraded in the early stage of reperfusion following OGD, which is an early event of mitochondrial damage induced by cerebral ischemia-reperfusion injury.
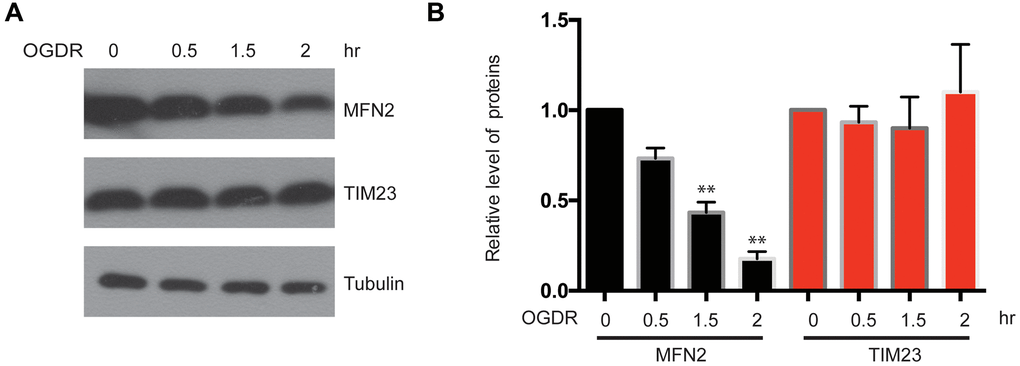
Figure 3. Expression of mitochondrial outer membrane protein MFN2 and mitochondrial inner membrane protein TIM23 in the early stage of reperfusion after 4 h OGD. (A) Western blot was performed to examine the protein level of MFN2 and TIM23 in the early stage of reperfusion following 4 h OGD in SK-N-BE(2) cells. Tubulin was as a loading control. (B) Quantitation (Mean ± SEM) of A from three independent experiments. The expression of MFN2 was reduced in a time-dependent manner during early reperfusion after OGD, whereas the expression of TIM23 was unchanged.
Overexpression of MFN2 protects against OGDR induced mitochondrial fragmentation
We overexpressed MFN2 in SK-N-BE(2) cells to determine whether intervention of mitochondrial protein degradation can play a protective role in OGDR induced mitochondrial damage. SK-N-BE(2) cells were transfected with MFN2-Myc(200ng or 500ng) and Mito-GFP plasmids. Mito-GFP was employed to indicate mitochondria. After transfection 36 h, cells were treated with OGD 4 h plus reperfusion. As demonstrated in Figure 4A, most of the cells displayed normal tubular and long mitochondria in SK-N-BE(2) cells transfected with vector without OGDR insult, while lots of mitochondria were fragmented after 4 h reperfusion following 4 h OGD in SK-N-BE(2) cells transfected with vector. However, transfection with MFN2(200ng or 500ng) significantly increased the number of SK-N-BE(2) cells with normal tubular and long mitochondria in a dose-dependent manner. MFN2(200ng or 500ng) overexpression significantly attenuated OGDR-induced mitochondrial fragmentation in SK-N-BE(2) cells in a dose-dependent manner (Figure 4A, 4B, P=0.031 and 0.0053 for 200ng and 500ng respectively). Therefore, these data strongly suggest inhibition of mitochondrial protein degradation in the early stage of reperfusion may protect mitochondria against OGDR induced fragmentation.
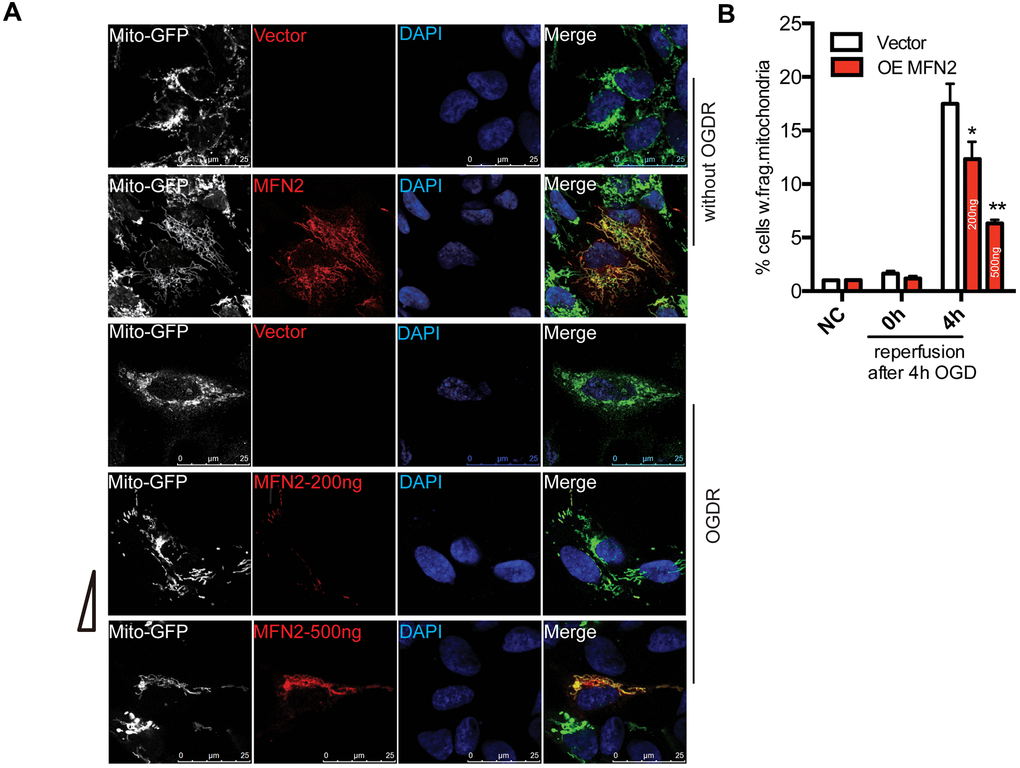
Figure 4. Effect of MFN2 overexpression on mitochondrial morphology in SK-N-BE(2) cells exposed to OGDR. SK-N-BE(2) cells were transfected with MFN2-Myc(200ng or 500ng) and Mito-GFP plasmids. After transfection 36 h, cells were treated with 4 h OGD plus reperfusion. (A) Digital photomicrograph under fluorescent illumination showed the morphology of mitochondria by mito-GFP. Most SK-N-BE(2) cells transfected with vector without OGDR displayed normal mitochondria, while fragmented mitochondria were evident in SK-N-BE(2) cells transfected with vector subjected to 4 h reperfusion after 4 h OGD. However, MFN2(200ng or 500ng) transfection significantly increased the number of SK-N-BE(2) cells with typical tubular and long mitochondria in a dose-dependent manner. (B) Quantitation (Mean ± SEM) of A from three independent experiments. Transfection with MFN2(200ng or 500ng) significantly attenuated OGDR-induced fragmentation of mitochondria in a dose-dependent manner. OE: over expression.
USP30 overexpression ameliorates OGDR-induced mitochondrial fragmentation
To determine whether overexpression of USP30, a deubiquitinase localized to mitochondria, also protects against OGDR induced mitochondrial fragmentation, after transfected with USP30-Flag(200ng or 500ng) and Mito-GFP plasmids for 36 h, SK-N-BE(2) cells were subjected to OGD 4 h plus 4 h reperfusion (Figure 5). The percentage of SK-N-BE(2) cells with fragmentized mitochondria was increased after OGDR injury. However, SK-N-BE(2) cells transfected with USP30(200ng or 500ng) displayed a significant decrease in mitochondrial fragmentation after OGDR exposure in a dose-dependent manner(P=0.043 and 0.037 for 200ng and 500ng respectively). These data strongly suggest that increased USP30 expression significantly attenuates fragmentation of mitochondria after OGDR insult in a dose-dependent manner.
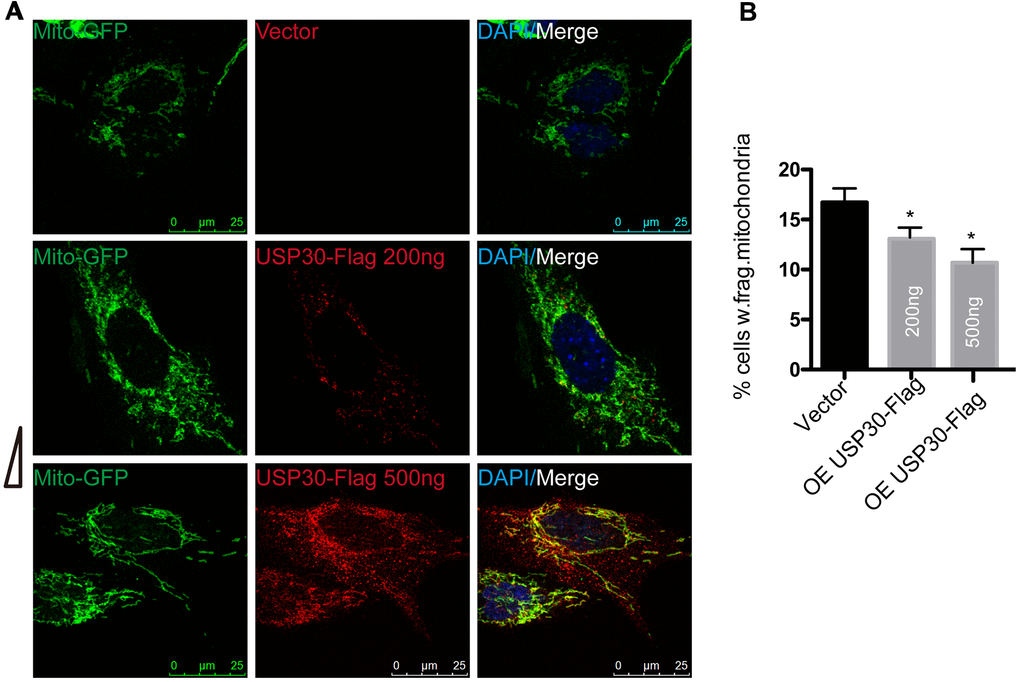
Figure 5. Effect of USP30 overexpression on mitochondrial morphology in SK-N-BE(2) cells exposed to OGDR. SK-N-BE(2) cells were transfected with indicated plasmids. After transfection 36 h, SK-N-BE(2) cells were treated with 4 h OGD plus 4 h reperfusion. (A) Digital photomicrograph under fluorescent illumination showed the morphology of mitochondria by mito-GFP. Mitochondrial fragmentation was evident in SK-N-BE(2) cells after 4 h reperfusion following 4 h OGD. However, USP30(200ng or 500ng) transfection significantly increased the number of SK-N-BE(2) cells with typical tubular and long mitochondria without fragmentation in a dose-dependent manner. (B) Quantitation (Mean ± SEM) of A from three independent experiments. Transfection with USP30(200ng or 500ng) protected against OGDR-induced mitochondrial fragmentation in a dose-dependent manner. OE: over expression.
USP30 overexpression inhibits ubiquitination and degradation of MFN2 after OGDR
To further analyze the role of USP30 in the regulation of ubiquitination and expression of MFN2 after OGDR insult, we transfected SK-N-BE(2) cells with USP30-Flag plasmids to increase USP30 expression. After transfection 36 h, SK-N-BE(2) cells were treated with OGD 4 h plus 4 h reperfusion. As demonstrated in Figure 6, overexpression of USP30 significantly inhibited OGDR induced ubiquitination of MFN2 (Figure 6A, 6B, P=0.0029) and increased the protein expression of MFN2 (Figure 6A–6C, P=0.0085). Together with the results above, our data indicate that USP30 overexpression protects against OGDR induced mitochondrial fragmentation through regulation of the ubiquitination and expression of MFN2.
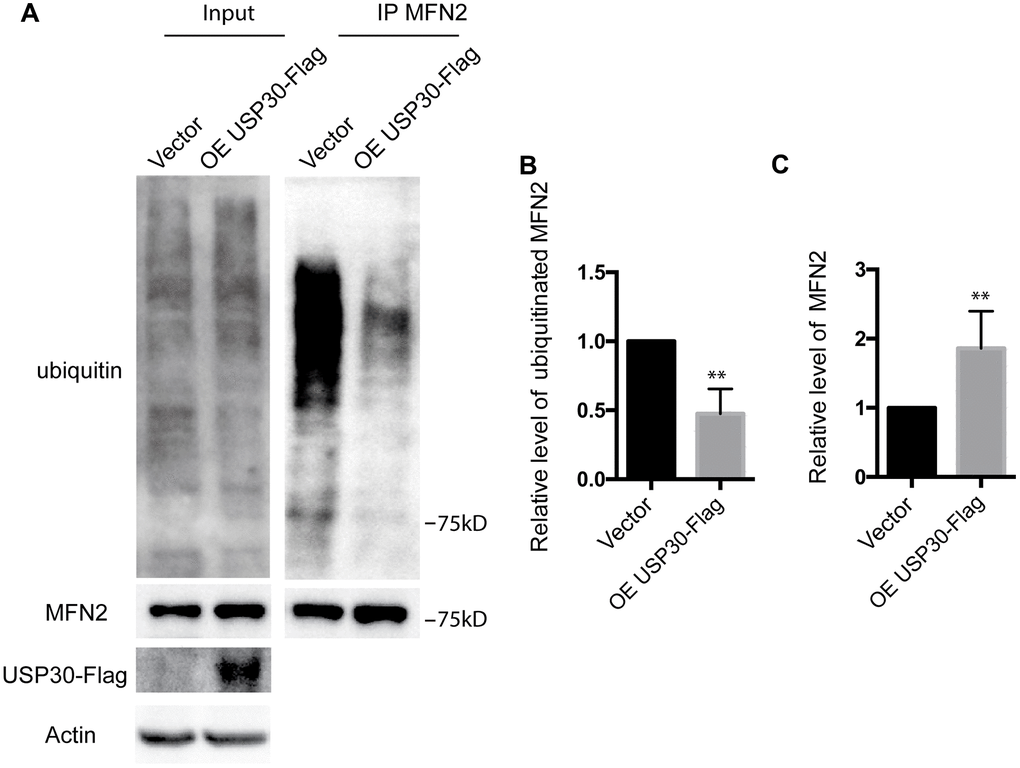
Figure 6. Effect of USP30 overexpression on the ubiquitination and protein expression of MFN2 in SK-N-BE(2) cells exposed to OGDR. SK-N-BE(2) cells were transfected with indicated plasmid. After transfection 36 h, SK-N-BE(2) cells were treated with 4 h OGD plus 4 h reperfusion. (A) After immunoprecipitation of MFN2, western blot was performed to examine the ubiquitination and protein level of MFN2 after 4 h reperfusion following 4 h OGD in SK-N-BE(2) cells. Transfection with USP30-Flag significantly attenuated OGDR induced ubiquitination of MFN2 and increased protein expression of MFN2. (B) Quantitation (Mean ± SEM) of ubiquitination of MFN2 from three independent experiments. (C) Quantitation (Mean ± SEM) of protein expression of MFN2 from three independent experiments. OE: over expression.
Discussion
The ubiquitin-proteasome system (UPS) and autophagy are the two complicated tightly controlled protein degradation pathways that are essential for maintaining cellular protein homeostasis. They prevent accumulation of potentially toxic proteins within the neuron and play crucial roles in neuronal survival after cerebral ischemia and many other neurological disorders [20]. Ubiquitylation serves as a degradation signal in both the UPS and autophagy systems. Cerebral ischemia-reperfusion injury induces accumulation of ubiquitin, proteasome activity impairment, and neuronal cell death [21]. Cerebral ischemia increases the expression of ubiquitin-protein conjugates in the brain. Ubiquitination level is immediately increased during reperfusion after cerebral ischemia. Elevated levels of ubiquitination is found in neurons in the cerebral ischemic penumbra with survival potential [22]. Regulation of ubiquitination may confer neuroprotection in cerebral ischemia-reperfusion injury. GS Rg1 protects against cerebral ischemia-reperfusion induced injury through suppressing proteasomal activity and ubiquitinated protein aggregation in brain [23]. The ubiquitin E3 ligase TRAF6 promotes cerebral ischemia-reperfusion injury through increased ubiquitination and activation of Rac1 [24]. However, despite recent advances, the exact role of the ubiquitylation in cerebral ischemia-reperfusion injury and the factors contributing to ubiquitinated protein aggregation still need further exploration [25]. In the study, we found that the mitochondrial proteins are immediately ubiquitinated during reperfusion after OGD. Ubiquitination of mitochondrial proteins increased as early as 45 min reperfusion following 4 h OGD. Ubiquitylation plays a key role in the maintenance of mitochondrial integrity. Therefore, increased ubiquitination of mitochondrial proteins after OGDR would lead to impairment of mitochondrial structure via continuous ubiquitination and degradation.
Cerebral ischemia-reperfusion injury decreases the protein expression of MFN2 in mice [26]. Mitochondrial release and decreased expression of MFN2 after cerebral ischemia-reperfusion injury was found in mitochondria of the cerebral cortex, but not in hippocampal mitochondria [27]. Consistent with previous studies, we found that MFN2 was immediately ubiquitinated and degraded in the early stage of reperfusion after 4h OGD. Therefore, the increased ubiquitination and degradation of MFN2 may represent an important mechanism of mitochondrial damage and neuronal death after OGDR injury.
Mitochondria play a pivotal role in neuronal survival and death via their functions in ATP production, apoptosis, reactive oxygen species generation and Ca2+ homeostasis. Cerebral ischemia-reperfusion induces mitochondrial fragmentation and dysfunction, which will aggravate neuronal death and is supposed to be the key pathogenic mechanism. Mitochondria play diverse roles in cerebral ischemia and are promising targets for the clinical treatment of cerebral ischemia-reperfusion injury [28]. However, the mechanisms underlying mitochondria in ischemic neuronal death and protection still need further exploration. In our study, we found that overexpression of MFN2 protected against OGDR induced mitochondrial fragmentation. MFN2 is a potential target for CMT2A and MFN2 agonists attenuate MFN2 mutations induced mitochondrial defects in CMT2A [12]. Nuclear receptor subfamily 4 group A member 1 (NR4A1) inhibition protects against cerebral ischemia-reperfusion induced mitochondrial damage through upregulation expression of MFN2 and reversing MFN2-mediated mitophagy [29]. HDAC2 knockout enhances neuronal expression of MFN2 and promotes neuronal survival in mice model of cerebral ischemia-reperfusion injury [30]. Thus, targeting MFN2 may open avenues for discovering innovative therapeutic regimens for cerebral ischemia-reperfusion injury.
USP30, a deubiquitinase localized to the mitochondrial outer membrane, is essential in maintaining the integrity of mitochondria. USP30 depletion leads to elongated and interconnected mitochondria, which is rescued by ectopic expression of USP30 [31]. USP30 removes Lys 6- and Lys 11-linked multimers from intact ubiquitylated mitochondria and regulates atypical ubiquitin chains on mitochondria [32]. USP30 inhibition confers neuroprotection in Parkinson's disease through rescuing the defective parkin-mediated mitophagy, promoting mitochondrial degradation and maintenance the integrity of mitochondria [16]. USP30 also play a key role in the regulation of BAX/BAK-dependent apoptosis and USP30 inhibition promotes mitochondrial cell death [15]. However, the potential role of USP30 in cerebral ischemia-reperfusion injury is still largely unknown. Therefore, in this study, we investigated the role of USP30 in SK-N-BE(2) cells exposed to OGDR. We demonstrated that overexpression of USP30 attenuated OGDR-induced mitochondrial fragmentation, suggesting that USP30 overexpression is beneficial for cerebral ischemia-reperfusion injury through maintaining mitochondrial structure integrity.
The ubiquitin-proteasome system plays a pivotal role in the maintenance of mitochondrial homeostasis through regulating the ubiquitination of MFN2. MITOL, a mitochondrial ubiquitin ligase, regulates mitochondrial dynamics by activating MFN2 via K192 ubiquitination [33]. Parkin, an E3 ubiquitin ligase, induces a rapid ubiquitination and degradation of MFN2 in a proteasome- and p97-dependent manner [34, 35]. Therefore, we further studied the neuroprotective mechanisms of USP30 in cerebral ischemia-reperfusion injury. We found that USP30 overexpression inhibited OGDR induced MFN2 ubiquitination and degradation. MFN2 is supposed to be a potential target for USP30. USP30 is essential for the maintenance of mitochondrial homeostasis through regulating the deubiquitination of ubiquitylated MFN2 and promoting mitochondrial fusion [36]. Therefore, the neuroprotective effect of USP30 against OGDR induced mitochondria damage may be relate to its inhibition of ubiquitination and degradation of MFN2.
In conclusion, our study demonstrates that in the early stage of reperfusion after OGD, the mitochondrial proteins are immediately ubiquitinated. The ubiquitination and degradation of MFN2 is also increased during early reperfusion after OGD, while overexpression of MFN2 ameliorates OGDR induced mitochondrial fragmentation. USP30 overexpression inhibits OGDR-induced ubiquitination and degradation of MFN2, and protects against mitochondrial fragmentation. Our results suggest that overexpression of USP30 may be a promising strategy for the treatment of disorders whose etiology is based upon cerebral ischemia-reperfusion injury.
Materials and Methods
Antibodies, cells and reagents
SK-N-BE(2) cell was obtained from ATCC. Ubiquitin (#3933, #3936), MFN2(#11925), Tom20(#42406), β-tubulin and β-actin antibodies were purchased from Cell Signaling Tech. TIM23 (sc-514463) antibody was purchased from Santa Cruz Biotechnology. USP30-Flag (Addgene, #22578) and Mito-GFP (Addgene, #44385) were obtained from Addgene.
Cell culture and transfection
SK-N-BE(2) cell line was cultured in Eagle's Minimum Essential Medium and F12 Medium (Invitrogen Life Technologies) supplemented with 10% fetal bovine serum(Sigma-Aldrich), 50 μg/mL penicillin, and streptomycin in a 5% CO2 incubator. Cells were transiently transfected using Lipofectamine 2000 reagent (Invitrogen Life Technologies) with the indicated plasmids MFN2-Myc(200ng or 500ng per well for 24-well plate), USP30-Flag(200ng or 500ng per well for 24-well plate) or Mito-GFP following the protocol.
OGDR
SK-N-BE(2) cells were transferred into a temperature controlled (37° C) anaerobic chamber (Forma Scientific) containing a gas mixture composed of 5% CO2, 95% N2. The culture medium was replaced with deoxygenated glucose-free Hanks' Balanced Salt Solution (Invitrogen) and cells were maintained in the chamber for 4 h. After OGD, SK-N-BE(2) cells were maintained in culture medium supplemented with 10% FBS under normoxic culture conditions for different times.
Mitochondrial fractionation
Cells were washed twice with ice-cold PBS, and then scraped into ice-cold PBS followed by centrifugation at 1,000 g for 5 min at 4° C. Cell pellets were resuspended in mitochondrial isolation buffer (5 mM Hepes pH 7.4, 3 mM MgCl2, 1 mM EGTA, and 250 mM sucrose) containing protease and phosphatase inhibitors. Lysates were passed through a 5/8-inch 25-gauge needle 20 times using a 1-mL syringe and centrifuged at 1,000 g, 4° C for 20 min. Supernatants were collected, and cytosolic extracts were recovered by centrifugation at 10,000 g, 4° C for 15 min to obtain crude mitochondrial pellets.
Immunoprecipitation
For immunoprecipitation, cell lysis buffer included 1% Triton, 10 mM HEPES, pH7.5, 142.5 mM KCl, 5 mM MgCl2,1 mM EDTA, 10% glycerol, and a protease inhibitor cocktail. MFN2 antibody and Protein G agarose beads (Sigma-Aldrich) were used for immunoprecipitation. Total protein was analysed by SDS-PAGE, and a rabbit anti-ubiquitin antibody (Sigma-Aldrich) was used for immunoblotting.
Immunoblotting
Cells were lysed with lysis buffer containing 1% Triton X-100. Samples were subsequently separated by SDS–PAGE and transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore). Immunoblot analysis was performed and visualized with Super-Signal West Pico Chemiluminescent substrate (Pierce) or Immobilon Western Chemiluminescent HRP substrate (Millipore). Signal intensities were analyzed using a LAS-3000 mini imaging analyzer and Multi Gauge software, version 3.0 (Fujifilm).
Immunofluorescence
Cells grown on coverslips were washed with PBS and fixed in 4% paraformaldehyde in PBS for 10 min at 4° C. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min, blocked with 3% bovine serum albumin in PBS for 30 min, and incubated with primary antibodies for 1 h. After washing, cells were incubated with secondary antibodies for 30 min. Images were acquired on a confocal laser microscope (FV1000D IX81, Olympus).
Quantitative and statistical analysis
Mitochondrial fragmentation was assessed as described before [19]. Cells with shortened, punctate, and sometimes rounded mitochondria were classified as fragmented. Quantification was performed using more than 300 cells per experiment.
The data are presented as mean ± Standard Error (SEM). Paired Student’s t-test was used for comparison between pre- and post-treatment values in each group, while unpaired t-test was used for comparison between two groups. One-way ANOVA followed by Tukey multiple comparisons test was used to compare values among three or more groups. P < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism (La Jolla, CA, USA).
Author Contributions
Chunli Chen, Haiyun Qin and Jiayu Tang conducted the experiments. Zhiping Hu and Jieqiong Tan supervised the experiments. Liuwang Zeng supervised the experiments and wrote the manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This article has been supported by National Natural Science Foundation of China (Grant no. 81771423 and 81974212), Natural Science Foundation of Hunan Province (Grant no.2020JJ4822 and 2018JJ2496) and Hunan Province Science and Technology Grant (No. 2019GK5010).
References
- 1. Ma H, Campbell BC, Parsons MW, Churilov L, Levi CR, Hsu C, Kleinig TJ, Wijeratne T, Curtze S, Dewey HM, Miteff F, Tsai CH, Lee JT, et al, and EXTEND Investigators. Thrombolysis guided by perfusion imaging up to 9 hours after onset of stroke. N Engl J Med. 2019; 380:1795–803. https://doi.org/10.1056/NEJMoa1813046 [PubMed]
- 2. Thomalla G, Simonsen CZ, Boutitie F, Andersen G, Berthezene Y, Cheng B, Cheripelli B, Cho TH, Fazekas F, Fiehler J, Ford I, Galinovic I, Gellissen S, et al, and WAKE-UP Investigators. MRI-guided thrombolysis for stroke with unknown time of onset. N Engl J Med. 2018; 379:611–22. https://doi.org/10.1056/NEJMoa1804355 [PubMed]
- 3. Nogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, Yavagal DR, Ribo M, Cognard C, Hanel RA, Sila CA, Hassan AE, Millan M, et al, and DAWN Trial Investigators. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med. 2018; 378:11–21. https://doi.org/10.1056/NEJMoa1706442 [PubMed]
- 4. Albers GW, Marks MP, Kemp S, Christensen S, Tsai JP, Ortega-Gutierrez S, McTaggart RA, Torbey MT, Kim-Tenser M, Leslie-Mazwi T, Sarraj A, Kasner SE, Ansari SA, et al, and DEFUSE 3 Investigators. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med. 2018; 378:708–18. https://doi.org/10.1056/NEJMoa1713973 [PubMed]
- 5. Tu R, Armstrong J, Lee KS, Hammock BD, Sapirstein A, Koehler RC. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral Ischemia. Sci Rep. 2018; 8:5279. https://doi.org/10.1038/s41598-018-23504-1 [PubMed]
- 6. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015; 6:524–51. https://doi.org/10.1016/j.redox.2015.08.020 [PubMed]
- 7. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. 2017; 1863:1066–77. https://doi.org/10.1016/j.bbadis.2016.11.010 [PubMed]
- 8. Russo E, Nguyen H, Lippert T, Tuazon J, Borlongan CV, Napoli E. Mitochondrial targeting as a novel therapy for stroke. Brain Circ. 2018; 4:84–94. https://doi.org/10.4103/bc.bc_14_18 [PubMed]
- 9. Liu F, Lu J, Manaenko A, Tang J, Hu Q. Mitochondria in ischemic stroke: new insight and implications. Aging Dis. 2018; 9:924–37. https://doi.org/10.14336/AD.2017.1126 [PubMed]
- 10. Park HH, Han MH, Choi H, Lee YJ, Kim JM, Cheong JH, Ryu JI, Lee KY, Koh SH. Mitochondria damaged by oxygen glucose deprivation can be restored through activation of the PI3K/Akt pathway and inhibition of calcium influx by amlodipine camsylate. Sci Rep. 2019; 9:15717. https://doi.org/10.1038/s41598-019-52083-y [PubMed]
- 11. Stuppia G, Rizzo F, Riboldi G, Del Bo R, Nizzardo M, Simone C, Comi GP, Bresolin N, Corti S. MFN2-related neuropathies: clinical features, molecular pathogenesis and therapeutic perspectives. J Neurol Sci. 2015; 356:7–18. https://doi.org/10.1016/j.jns.2015.05.033 [PubMed]
- 12. Rocha AG, Franco A, Krezel AM, Rumsey JM, Alberti JM, Knight WC, Biris N, Zacharioudakis E, Janetka JW, Baloh RH, Kitsis RN, Mochly-Rosen D, Townsend RR, et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science. 2018; 360:336–41. https://doi.org/10.1126/science.aao1785 [PubMed]
- 13. Riccio V, McQuibban GA, Kim PK. USP30: protector of peroxisomes and mitochondria. Mol Cell Oncol. 2019; 6:1600350. https://doi.org/10.1080/23723556.2019.1600350 [PubMed]
- 14. Riccio V, Demers N, Hua R, Vissa M, Cheng DT, Strilchuk AW, Wang Y, McQuibban GA, Kim PK. Deubiquitinating enzyme USP30 maintains basal peroxisome abundance by regulating pexophagy. J Cell Biol. 2019; 218:798–807. https://doi.org/10.1083/jcb.201804172 [PubMed]
- 15. Liang JR, Martinez A, Lane JD, Mayor U, Clague MJ, Urbé S. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015; 16:618–27. https://doi.org/10.15252/embr.201439820 [PubMed]
- 16. Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014; 510:370–75. https://doi.org/10.1038/nature13418 [PubMed]
- 17. Wang Y, Serricchio M, Jauregui M, Shanbhag R, Stoltz T, Di Paolo CT, Kim PK, McQuibban GA. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy. 2015; 11:595–606. https://doi.org/10.1080/15548627.2015.1034408 [PubMed]
- 18. Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016; 100:210–22. https://doi.org/10.1016/j.freeradbiomed.2016.04.015 [PubMed]
- 19. Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017; 119:315–26. https://doi.org/10.1016/j.acthis.2017.03.001 [PubMed]
- 20. Graham SH, Liu H. Life and death in the trash heap: the ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral ischemia. Ageing Res Rev. 2017; 34:30–38. https://doi.org/10.1016/j.arr.2016.09.011 [PubMed]
- 21. Jeong EI, Chung HW, Lee WJ, Kim SH, Kim H, Choi SG, Jung YK. E2-25K SUMOylation inhibits proteasome for cell death during cerebral ischemia/reperfusion. Cell Death Dis. 2016; 7:e2573. https://doi.org/10.1038/cddis.2016.428 [PubMed]
- 22. Hochrainer K. Protein modifications with ubiquitin as response to cerebral ischemia-reperfusion injury. Transl Stroke Res. 2018; 9:157–73. https://doi.org/10.1007/s12975-017-0567-x [PubMed]
- 23. Zheng T, Jiang H, Jin R, Zhao Y, Bai Y, Xu H, Chen Y. Ginsenoside Rg1 attenuates protein aggregation and inflammatory response following cerebral ischemia and reperfusion injury. Eur J Pharmacol. 2019; 853:65–73. https://doi.org/10.1016/j.ejphar.2019.02.018 [PubMed]
- 24. Li T, Qin JJ, Yang X, Ji YX, Guo F, Cheng WL, Wu X, Gong FH, Hong Y, Zhu XY, Gong J, Wang Z, Huang Z, et al. The ubiquitin E3 ligase TRAF6 exacerbates ischemic stroke by ubiquitinating and activating Rac1. J Neurosci. 2017; 37:12123–40. https://doi.org/10.1523/JNEUROSCI.1751-17.2017 [PubMed]
- 25. Caldeira MV, Salazar IL, Curcio M, Canzoniero LM, Duarte CB. Role of the ubiquitin-proteasome system in brain ischemia: friend or foe? Prog Neurobiol. 2014; 112:50–69. https://doi.org/10.1016/j.pneurobio.2013.10.003 [PubMed]
- 26. Kumari S, Anderson L, Farmer S, Mehta SL, Li PA. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res. 2012; 3:296–304. https://doi.org/10.1007/s12975-012-0158-9 [PubMed]
- 27. Klacanova K, Kovalska M, Chomova M, Pilchova I, Tatarkova Z, Kaplan P, Racay P. Global brain ischemia in rats is associated with mitochondrial release and downregulation of Mfn2 in the cerebral cortex, but not the hippocampus. Int J Mol Med. 2019; 43:2420–28. https://doi.org/10.3892/ijmm.2019.4168 [PubMed]
- 28. Yang JL, Mukda S, Chen SD. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018; 16:263–75. https://doi.org/10.1016/j.redox.2018.03.002 [PubMed]
- 29. Zhang Z, Yu J. NR4A1 promotes cerebral ischemia reperfusion injury by repressing Mfn2-mediated mitophagy and inactivating the MAPK-ERK-CREB signaling pathway. Neurochem Res. 2018; 43:1963–77. https://doi.org/10.1007/s11064-018-2618-4 [PubMed]
- 30. Wang DB, Kinoshita C, Kinoshita Y, Sopher BL, Uo T, Lee RJ, Kim JK, Murphy SP, Dirk Keene C, Garden GA, Morrison RS. Neuronal susceptibility to beta-amyloid toxicity and ischemic injury involves histone deacetylase-2 regulation of endophilin-B1. Brain Pathol. 2019; 29:164–75. https://doi.org/10.1111/bpa.12647 [PubMed]
- 31. Nakamura N, Hirose S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol Biol Cell. 2008; 19:1903–11. https://doi.org/10.1091/mbc.e07-11-1103 [PubMed]
- 32. Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol. 2015; 17:160–69. https://doi.org/10.1038/ncb3097 [PubMed]
- 33. Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, Inatome R, Yanagi S. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. 2013; 51:20–34. https://doi.org/10.1016/j.molcel.2013.04.023 [PubMed]
- 34. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010; 191:1367–80. https://doi.org/10.1083/jcb.201007013 [PubMed]
- 35. McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I, Durcan TM, Trempe JF, Fon EA. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife. 2018; 7:e32866. https://doi.org/10.7554/eLife.32866 [PubMed]
- 36. Hou J, Eldeeb M, Wang X. Beyond deubiquitylation: USP30-mediated regulation of mitochondrial homeostasis. Adv Exp Med Biol. 2017; 1038:133–48. https://doi.org/10.1007/978-981-10-6674-0_10 [PubMed]