



Introduction
Acute cardiac injury caused by septic shock is a common cardiovascular complication in critically ill patients [1]. In septic shock, the myocardium is injured due to a variety of causes, including severe infection, anoxia, ischemia, trauma, and surgery, resulting in high morbidity and mortality [2]. Unfortunately, no effective treatment exists for septic cardiomyopathy. Thus, studying the molecular pathogenesis of acute myocardial injury may provide an effective target for the early diagnosis and treatment of septic cardiomyopathy.
Recently, studies of sepsis-induced myocardial injury have found that excessive reactive oxygen species (ROS) production caused by mitochondrial damage results in increased oxidative stress, leading to cell death and tissue damage [3]. Studies have also found that early treatment with ROS scavengers is an effective therapy for septic cardiomyopathy [4, 5]. At the molecular level, ROS-induced cardiomyocyte oxidative stress promotes protein oxidation and lipid peroxidation [6], which reduce cardiomyocyte contractility and thus decrease heart pump function, leading to low perfusion of distant organs or tissues [7]. More severely, oxidative stress triggers cardiomyocyte death through apoptosis, necroptosis, and necrosis [8]. Mitochondria are the main source of ROS and are vulnerable to oxidative stress [9]. Therefore, protection of mitochondria is vital to suppress oxidative stress.
Mitochondrial biogenesis is the process of mitochondrial degradation and regeneration [10]. It is regulated by several nuclear genes that control mitochondrial DNA synthesis and protein expression, such as peroxisome proliferator-activated receptorγcoactivator-1α (PGC1α) [11]. PGC1α, a member of the transcriptional coactivator family PGC-1, primarily exists in high-energy-demand tissues and organs, including the heart, controlling both mitochondrial biogenesis and energy metabolism [12, 13]. PGC-1α expression is activated by a variety of stresses such as oxidative stress, exercise training, myocardial ischemia-reperfusion injury, myocardial infarction, and cardiac fibrosis [14, 15]. Elevated PGC-1α expression is correlated with increased mitochondrial biogenesis, which sustains mitochondrial quality and quantity [16]. Conversely, knockout of PGC-1α decreases cardiac function, possibly due to mitochondrial turnover arrest and cardiomyocyte mitochondrial death [17]. At the molecular level, PGC-1α transcription and activity are controlled by the AMPK pathway [18]. The protective function of AMPK on diabetic cardiomyopathy, cardiac ischemia-reperfusion injury, cardiac remodeling, and inflammation-related myocardial damage has been widely studied [19, 20]. Considering the beneficial effects exerted by AMPK and PGC-1α on mitochondrial homeostasis and cardioprotection, determining their effect on sepsis-related myocardial injury and mitochondrial damage is important.
Sirtuin-3 (SirT3) is a nuclear NAD+-dependent histone deacetylase that regulates mitochondrial oxidative stress and bioenergetics [21]. SirT3 increases the expression of genes related to mitochondrial DNA repair through deacetylation, including NEIL1, NEIL2, OGG1, MUTYH, APE1, and LIG3 [22]. SirT3 also inhibits mitochondria-mediated apoptosis in cardiomyocyte ischemia-reperfusion injury, and this effect has been associated with activation of the AMPK pathway [23]. Anti-senescence [24], anti-fibrosis [25], and anti-inflammation [26] effects of SirT3 have also been demonstrated. Increased SirT3 has been shown to prevent apoptosis [23], oxidative stress [27], mitochondrial fission [28], unfolded protein response [29], and metabolic reprogramming [30] in many cardiovascular disorders. However, there is a lack of evidence demonstrating the effect of SirT3 on septic cardiomyopathy. The aim of our study is to determine whether SirT3 protects cardiomyocytes against septic shock by sustaining mitochondrial biogenesis via the AMPK pathway.
Results
SirT3 and AMPK are downregulated in LPS-induced septic cardiomyopathy
Lipopolysaccharide (LPS) was used to establish the septic cardiomyocyte model in vitro. Then, RNA was isolated to analyze the alterations of SirT3 and AMPK in cardiomyocytes. As shown in Figure 1A, 1B, compared to the control group, SirT3 was significantly downregulated. A decrease in AMPK transcription was also seen. These data indicate that LPS suppresses SirT3 and AMPK transcription. In addition, ELISA assay demonstrated that SirT3 and AMPK activity was significantly downregulated in cardiomyocytes after exposure to LPS (Figure 1C, 1D), suggesting that LPS may inhibit SirT3 and AMPK protein expression. Cardiomyocyte viability was also reduced in response to LPS treatment (Figure 1E). To determine whether SirT3 and AMPK inactivation contributes to LPS-mediated cardiomyocyte death, lentivirus-loaded SirT3 (SirT3-OE) and AMPK agonist (AICAR) were incubated with cardiomyocytes in the presence of LPS. Then, cardiomyocyte viability was measured using the TUNEL assay. As shown in Figure 1F, 1G, approximately 3% of cardiomyocytes were TUNEL positive under normal condition, whereas this rate increased to approximately 41% after exposure to LPS. Treatment with either Sirt3-OE or AICAR drastically suppressed the rate of TUNEL-positive cardiomyocytes. Together, our results indicate that SirT3 and AMPK are inhibited by LPS, leading to cardiomyocyte damage.
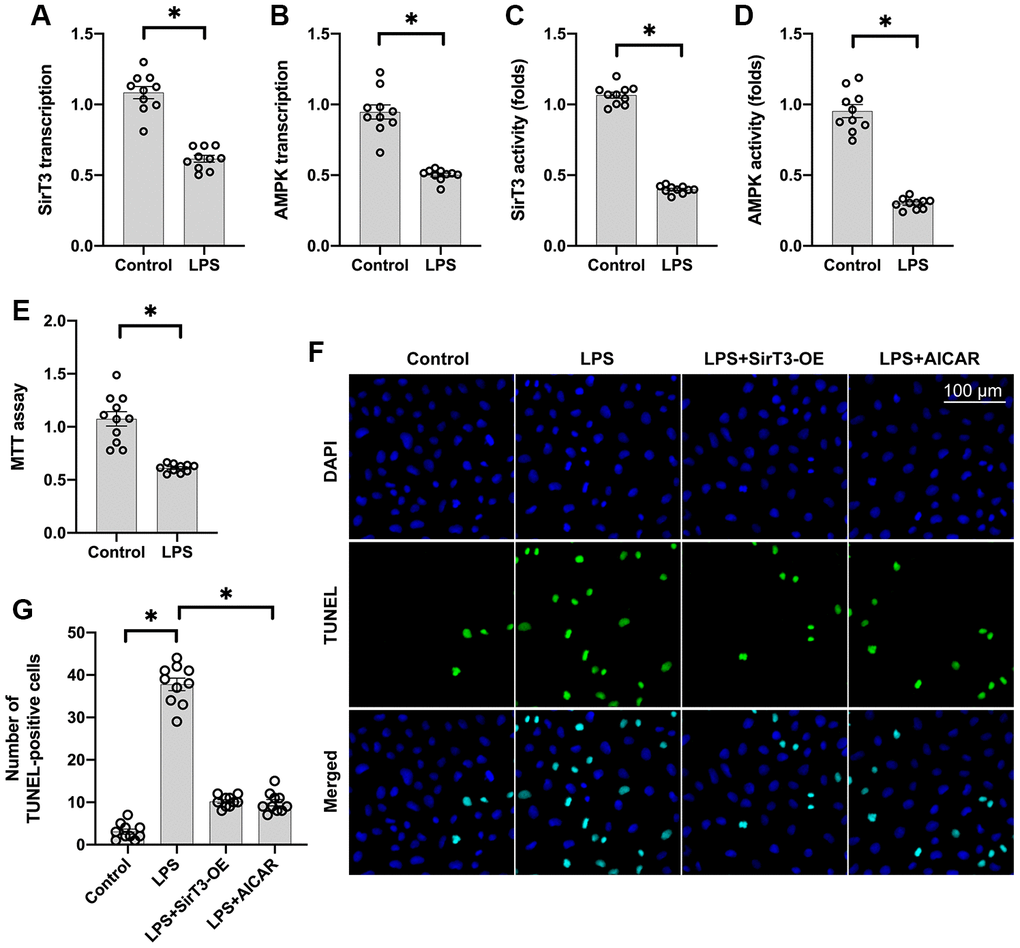
Figure 1. SirT3 and AMPK are downregulated in response to LPS-induced septic cardiomyopathy. (A, B) RNA was isolated, and the transcription of SirT3 and AMPK was remeasured. (C, D) ELISA was used to analyze the activities of SirT3 and AMPK in response to LPS treatment. (E) MTT assay was used to detect cardiomyocyte viability under LPS treatment. (F, G). TUNEL staining for apoptotic cardiomyocytes. Lentivirus-loaded SirT3 (SirT3-OE) and AMPK agonist (AICAR) were incubated with cardiomyocytes in the presence of LPS. Then, the number of TUNEL-positive cardiomyocytes was determined. *P < 0.05.
Overexpression of SirT3 or activation of AMPK reduces LPS-induced cardiomyocyte dysfunction
Cardiomyocyte contractile properties determine cardiac function [31]. Previous studies have reported that septic cardiomyopathy is characterized by depressed myocardial ejection function, which results in lower organ blood perfusion [32, 33]. In our study, a single cardiomyocyte was isolated, and cardiomyocyte mechanical features were observed. As shown in Figure 2A–2F, the resting cell length of the cardiomyocyte was similar in the presence or absence of LPS. In addition, SirT3-OE or AICAR treatment did not affect resting cell length. However, LPS treatment significantly reduced peak shortening in the cardiomyocyte, and this effect was improved by the addition of SirT3-OE or AICAR (Figure 2A–2F). Similarly, the cardiomyocyte maximal velocity of shortening (+dL/dt) was also suppressed by LPS, whereas SirT3-OE or AICAR treatment restored the +dL/dt (Figure 2A–2F). In addition to the maximal velocity of shortening, the maximal velocity of relengthening (−dL/dt), which is used to evaluate cardiomyocyte relaxation, was also compromised by LPS treatment and reversed to near-normal levels with Sirt3-OE or AICAR treatment (Figure 2A–2F). To quantify cardiomyocyte contractile and diastolic function, time to peak shortening (TPS) and time to 90% relengthening (TR90) were measured [34], as previously described. As shown in Figure 2A–2F, compared to the control group, both TPS and TR90 increased after LPS treatment, suggesting impaired cardiomyocyte contractile and diastolic capacities. However, SirT3-OE or AICAR treatment improved TPS and TR90 in cardiomyocytes that had been treated with LPS (Figure 2A–2F). Our results indicate that SirT3 and AMPK are important in sustaining cardiomyocyte mechanical properties in the presence of LPS.
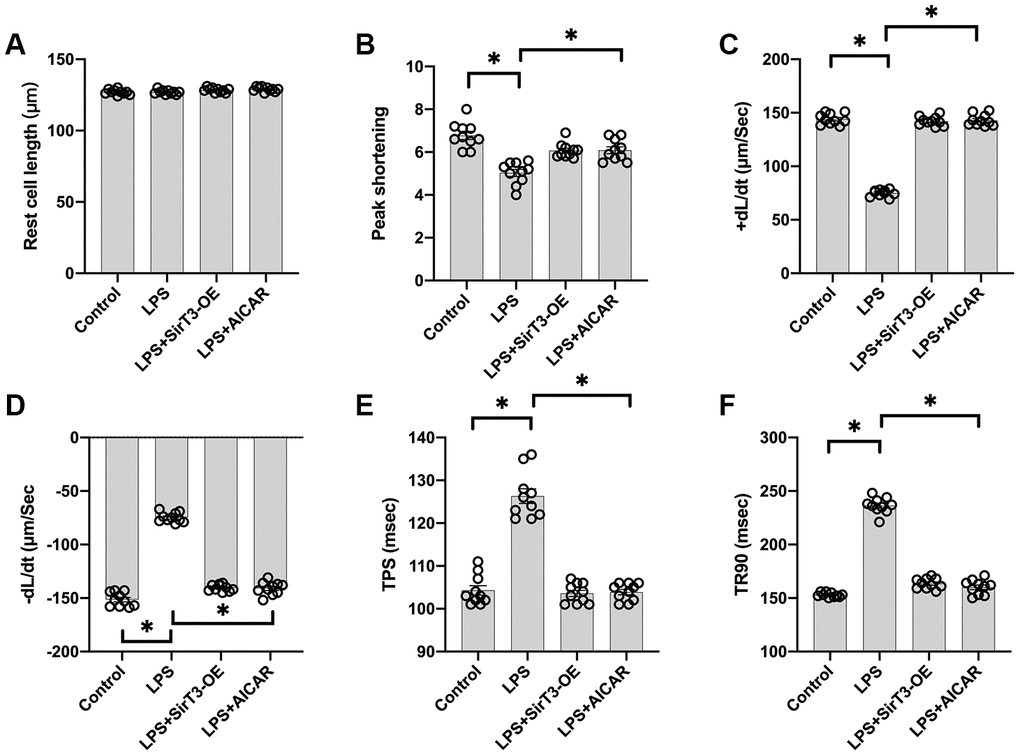
Figure 2. Overexpression of SirT3 or activation of AMPK attenuates LPS-mediated cardiomyocyte dysfunction. (A–F) Cardiomyocyte contractility in response to LPS treatment. Lentivirus-loaded SirT3 (SirT3-OE) and AMPK agonist (AICAR) were incubated with cardiomyocyte in the presence of LPS. +dL/dt is the maximal velocity of shortening. −dL/dt is the maximal velocity of relengthening. TPS, time to peak shortening; TR90, time to 90% relengthening. *P < 0.05.
SirT3 and AMPK sustain mitochondrial function
At the subcellular level, SirT3 is primarily localized in mitochondria and sustains mitochondrial bioenergetics [35]. In addition, mitochondria-mediated ATP production is vital for cardiomyocyte contraction and relaxation [36]. Thus, we wanted to determine whether mitochondrial function is sustained by SirT3 and AMPK in LPS-treated cardiomyocytes. First, mitochondrial metabolism was determine by analyzing mitochondrial respiration. Compared to the control group, the activity of mitochondrial electron transport chain complexes was significantly downregulated by LPS treatment (Figure 3A–3C); this effect was reversed by SirT3-OE or AICAR treatment. Subsequently, due to impaired mitochondrial respiration, mitochondrial ROS production was significantly elevated in LPS-treated cardiomyocytes, whereas SirT3-OE or AICAR decreased mitochondrial ROS accumulation (Figure 3D–3E). The levels of cellular anti-oxidants, including GSH and SOD, were decreased by LPS, which may prevent mitochondrial ROS clearance (Figure 3F–3G). However, SirT3-OE or AICAR increased the levels of GSH and SOD and thus reduced intracellular mitochondrial ROS (Figure 3D–3G).
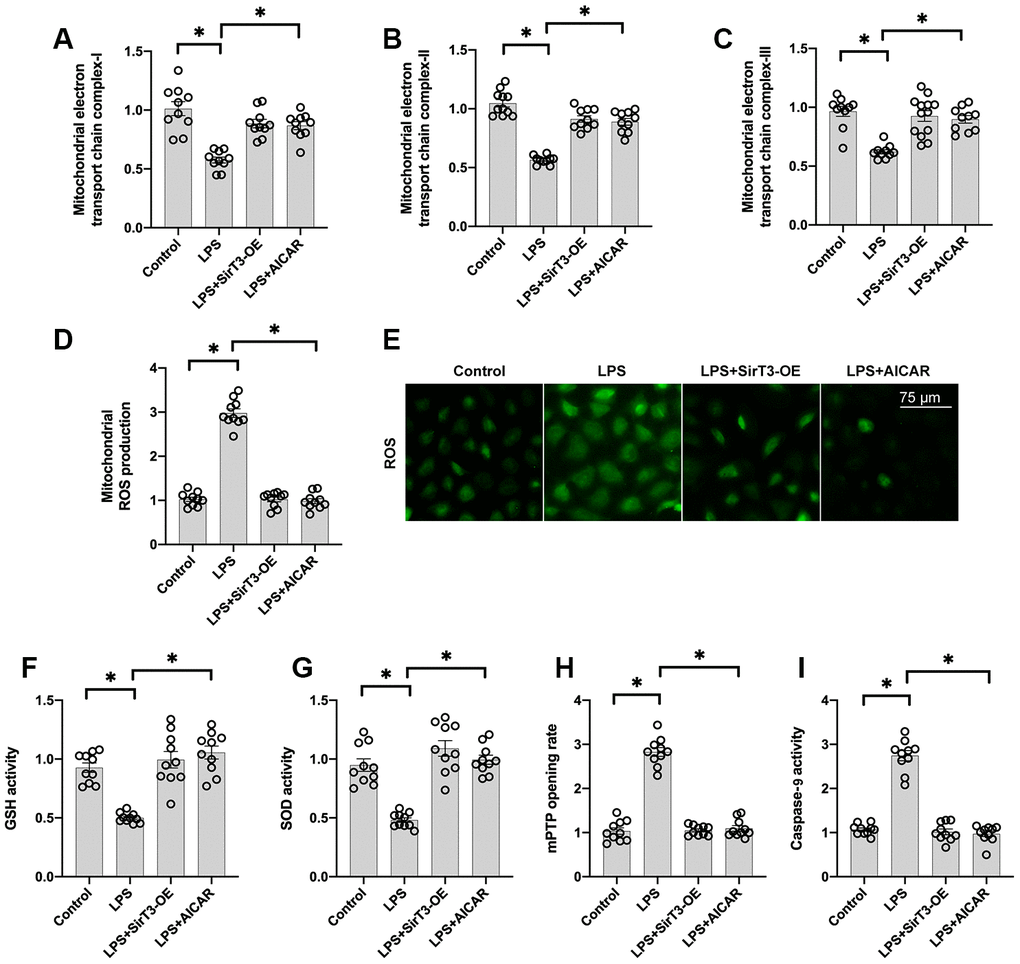
Figure 3. SirT3 and AMPK sustain mitochondrial function. (A–C) ELISA assay was used to analyze the activity of mitochondrial electron transport chain complexes in the presence of LPS. Lentivirus-loaded SirT3 (SirT3-OE) and AMPK agonist (AICAR) were incubated with cardiomyocytes before LPS treatment. (D–E) Immunofluorescence for mitochondrial ROS production. (F, G) GSH and SOD activity was measured using ELISA. (H) mPTP opening rate was measured in response to LPS treatment, SirT3-OE transfection, and AICAR supplementation. (I) ELISA was used to detect the activity of caspase-9. *P < 0.05.
Previous studies have proposed that damaged mitochondria may induce cardiomyocyte death by increasing the opening rate of mitochondrial permeability transition pore (mPTP) [37]. As shown in Figure 3H–3I, compared to the control group, LPS increased mPTP opening, which activated caspase-9. In contrast, SirT3-OE or AICAR treatment blocked mPTP opening (Figure 3H) and thus suppressed LPS-mediated caspase-9 activation (Figure 3I), suggesting that LPS may induce mitochondrial apoptosis in cardiomyocytes by inhibiting the SirT3-AMPK pathway.
Mitochondrial biogenesis is increased by SirT3 via the AMPK pathway
Recent studies have found a relationship between SirT3 overexpression and mitochondrial biogenesis activation in kidney tumor cells [38] and osteoblasts [39]. Considering the beneficial effects exerted by mitochondrial biogenesis on mitochondrial turnover, we aimed to determine whether SirT3 induces mitochondrial biogenesis to preserve mitochondrial function under LPS stress. As shown in Figure 4A–4C, compared with the control group, genes associated with mitochondrial biogenesis, such as peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC1α), transcription factor A mitochondrial (Tfam), and nuclear factor erythroid 2-related factor 2 (Nrf2), were significantly downregulated at the transcriptional level in the LPS model. Interestingly, SirT3-OE or AICAR treatment increased mitochondrial biogenesis (Figure 4A–4C). To determine whether SirT3 modulates mitochondrial biogenesis via AMPK, compound C (CC), an inhibitor of the AMPK pathway, was added to SirT3-treated cardiomyocytes before LPS treatment. Compared to the control group, although SirT3 improved mitochondrial biogenesis (as demonstrated by increased PGC1α, Tfam, and Nrf2), this effect was negated with the addition of CC (Figure 4A–4C), confirming that AMPK is required for SirT3-activated mitochondrial biogenesis. Subsequently, immunofluorescence demonstrated that PGC1α and Nrf2 levels were significantly downregulated in LPS-treated cardiomyocytes (Figure 4D–4F). Although SirT3-OE upregulated PGC1α and Nrf2 expression, this action was nullified by CC (Figure 4D–4F). Our results demonstrate that SirT3 activates mitochondrial biogenesis through AMPK.
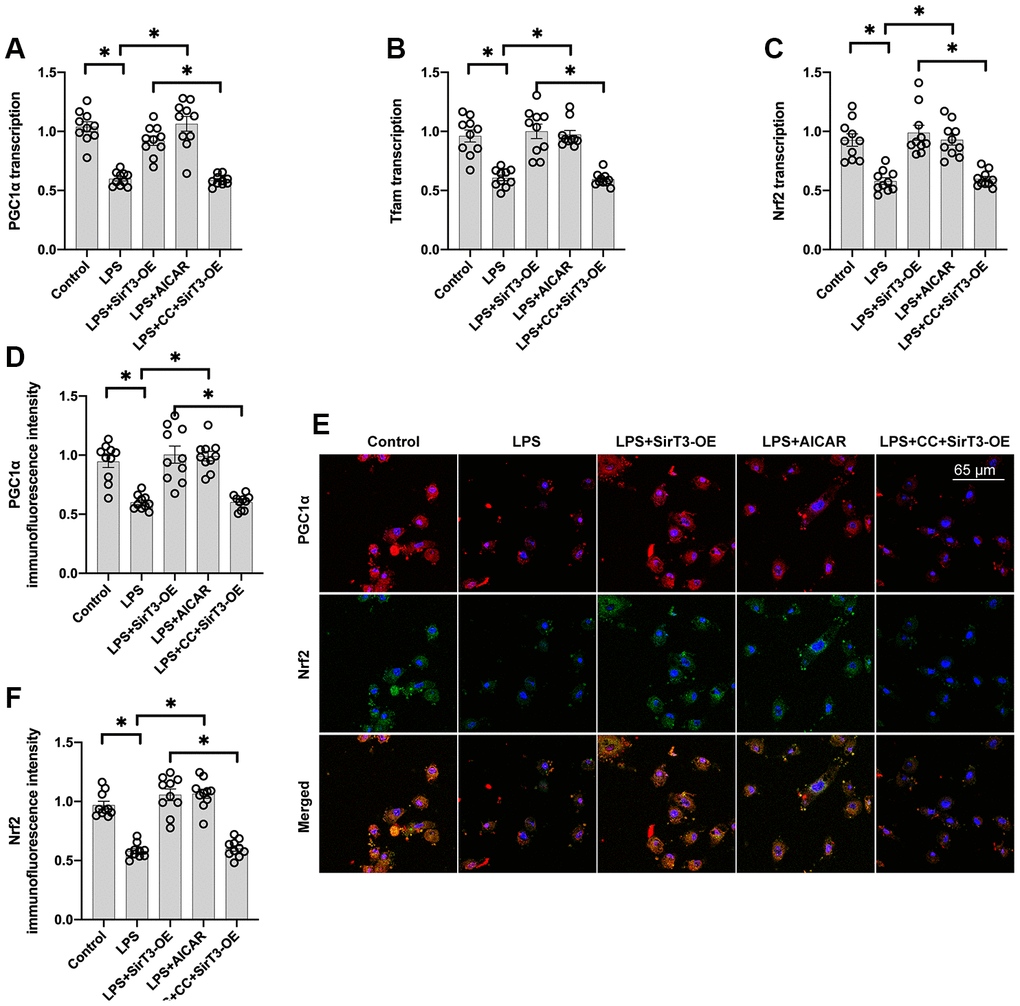
Figure 4. Mitochondrial biogenesis is increased by SirT3 via the AMPK pathway. (A–C) RNA was isolated from cardiomyocytes, and then transcription of mitochondrial biogenesis parameters was determined. Lentivirus-loaded SirT3 (SirT3-OE) and AMPK agonist (AICAR) were incubated with cardiomyocyte before LPS treatment. Compound C (CC), an antagonist of AMPK, was used to inhibit the activation of AMPK in SirT3-OE–transfected cardiomyocytes. (D–F) Immunofluorescence assay for Nrf2 and PGC1α in the presence of LPS. *P < 0.05.
Inhibition of mitochondrial biogenesis decreases SirT3-induced mitochondrial protection and cardiomyocyte survival
To verify whether mitochondrial biogenesis is required for SirT3/AMPK-mediated mitochondrial protection, azithromycin, an antagonist of mitochondrial biogenesis, was used. Administration of azithromycin inhibited mitochondrial electron transport chain complex activity (Figure 5A, 5B) and induced mitochondrial ROS overload (Figure 5C, 5D) in cardiomyocytes treated with SirT3-OE or AICAR. Azithromycin was used to determine whether mitochondrial biogenesis activation is the underlying mechanism of SirT3/AMPK-induced cardioprotection, and TUNEL apoptosis staining and caspase-3 activity were measured. As shown in Figure 5E and 5F, compared to the control group, the number of TUNEL-positive cardiomyocytes was significantly increased after exposure to LPS. SirT3-OE reduced the ratio of apoptotic cardiomyocytes, but this effect was not seen in azithromycin-treated cardiomyocytes (Figure 5E, 5F). Similarly, caspase-3 activity was increased in cardiomyocytes when they were cultured with LPS (Figure 5G). Although SirT3-OE was shown to prevent caspase-3 activation, this effect was nullified by azithromycin (Figure 5G). Therefore, these results confirm that mitochondrial biogenesis, which is governed by the SirT3/AMPK pathway, protects against LPS-induced mitochondrial damage and cardiomyocyte death.
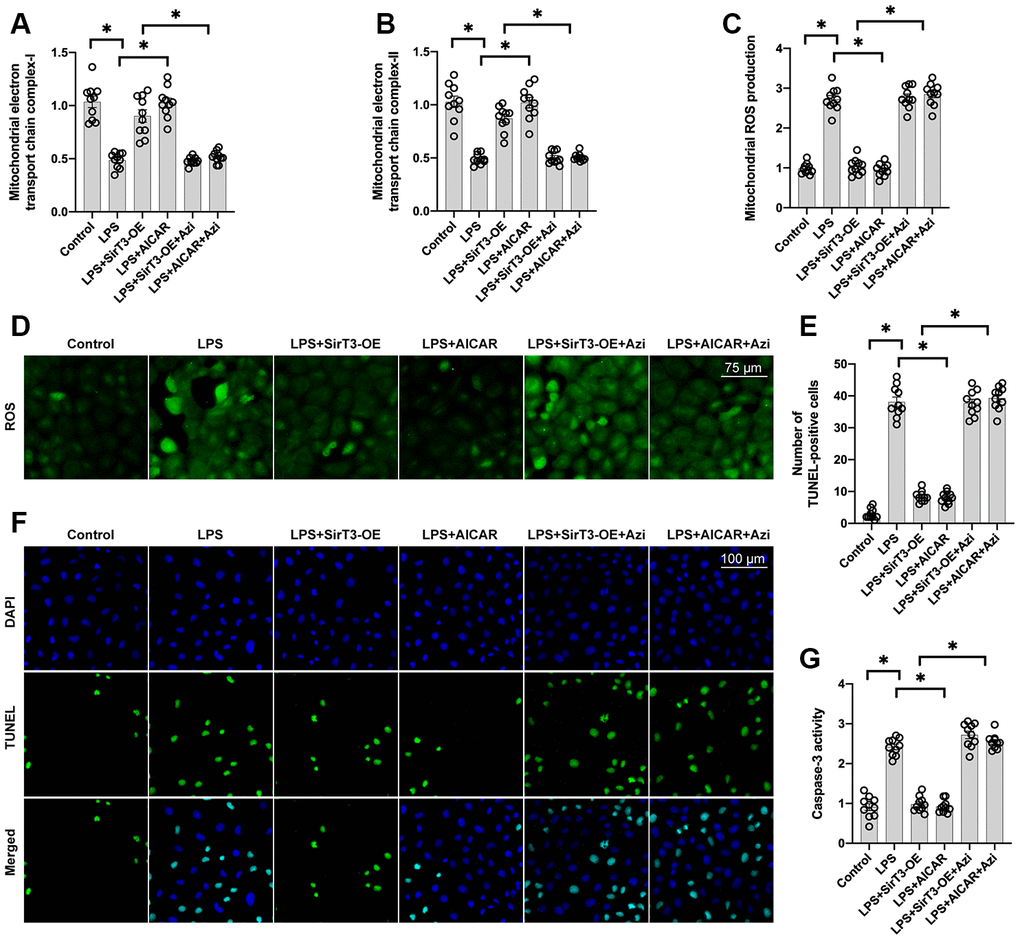
Figure 5. Inhibition of mitochondrial biogenesis decreases SirT3-mediated mitochondrial protection and cardiomyocyte survival. (A, B) ELISA assay was used to analyze mitochondrial electron transport chain complex activity in the presence of LPS. Lentivirus-loaded SirT3 (SirT3-OE) was incubated with cardiomyocytes in the presence of LPS. Compound C (CC), an antagonist of AMPK, was used to inhibit the activation of AMPK in SirT3-OE–treated cardiomyocytes. (C, D) Immunofluorescence of mitochondrial ROS production. (E, F) TUNEL staining of apoptotic cardiomyocytes. SirT3-OE was incubated with cardiomyocytes in the presence of LPS. CC was used to inhibit the activation of AMPK in SirT3-OE–treated cardiomyocytes. Then, the number of TUNEL-positive cardiomyocytes was determined. (G) ELISA was used to detect the activity of caspase-3. *P < 0.05.
Discussion
Septic cardiomyopathy is a common feature of severe sepsis syndromes and is characterized by global ventricular dysfunction with decreased ejection fraction, diffuse cardiomyocyte death, and interstitial edema. Diagnostic approaches include echocardiography, detection of biomarkers such as troponin, and measurement of pro-inflammatory factors [40]. Several molecular mechanisms have been proposed to explain the pathogenesis underlying septic cardiomyopathy including cytokine-induced cardiomyocyte apoptosis, impaired microvascular reperfusion, histone-mediated angioedema, and mitochondrial dysfunction–related energy depletion [41]. In response to these possible pathological mechanisms, fluid resuscitation, vasoactive agents, negative chronotropic agents, and supportive care have been used to treat septic cardiomyopathy [42]. In the present study, we found that SirT3 downregulation and AMPK inactivation are the primary subcellular events in the progression of sepsis-induced myocardial damage. Decreased SirT3 was followed by AMPK inactivation, resulting in blunted mitochondrial biogenesis. Subsequently, defective biogenesis in mitochondria caused mitochondrial damage, as characterized by decreased mitochondrial metabolism, increased mitochondrial ROS production, and increased mitochondrial apoptosis. Mitochondrial dysfunction impaired cardiomyocyte contractility and even triggered cardiomyocyte death. Overexpression of SirT3 sustained mitochondrial function and cardiomyocyte viability through AMPK-controlled mitochondrial biogenesis. These findings provide new insight into the subcellular molecular mechanisms underlying septic cardiomyopathy. Based on our results, drugs targeting the SirT3-AMPK-mitochondrial biogenesis axis may benefit patients suffering from sepsis-related myocardial injury.
Recently, studies have examined the role of SirT3 in cardioprotection. For example, cardiomyocyte hypertrophy is improved by SirT3 activation via reduction of cytoskeletal protein expression [43]. Cardiomyocyte-specific overexpression of SirT3 reduces myocardial infarct size by inhibiting cardiomyocyte death and preventing infarct zone extension [44, 45]. With respect to arrhythmia, SirT3 upregulation stimulates Na+/K+-ATPase and thus moderately increases intracellular K+, augmenting the stability of cardiomyocyte membrane potential [46]. In addition, activation of SirT3 accelerates glucose metabolism via upregulation of PPARα [47], and SirT3-induced metabolic reprogramming has been shown to sustain cardiomyocyte function in a mouse model of heart failure [48]. Due to the critically important role played by SirT3 in cardiomyocyte contractility, metabolism, and survival, drugs targeting SirT3 have been developed and studied. For example, exendin-4, an anti-diabetes drug that has been used to control fasting blood glucose, upregulated SirT3 activity in a mouse model of myocardial ischemia-reperfusion injury [23]. Pyridostigmine, a vasoactive agent modulating blood pressure, has been reported to affect SirT3 expression and enhances glucose metabolism in diabetic mice [49]. Exogenous H2S, an anti-oxidative gas, interacts with SirT3 and promotes transcription of mitochondrial respiratory enzymes [50]. Unfortunately, no drugs targeting SirT3 have been investigated in the setting of septic cardiomyopathy. Thus, cellular and animal studies are needed to determine how to restore SirT3 in LPS-treated cardiomyocytes.
Mitochondrial biogenesis is significant in neonatal cardiomyocytes in order to meet the metabolic requirements of cardiomyocyte contractility [13, 51]. With increasing age, the extent and activity of mitochondrial biogenesis are slowly reduced [12, 52]. Therefore, mitochondrial biogenesis has been identified as a factor that may delay aging [53]. The goal of mitochondrial biogenesis is to produce new mitochondria through mitochondrial division, in order to sustain the mitochondrial cycle in cooperation with mitochondrial autophagy [54, 55]. Damaged or defective mitochondrial biogenesis is associated with an accumulation of old or dysfunctional mitochondria with reduced mitochondrial potential or increased ROS [56]. Subsequently, these damaged mitochondria disrupt cardiomyocyte metabolism and induce mitochondrial apoptosis or necrosis through mPTP opening [57, 58]. Therefore, activation of mitochondrial biogenesis is a protective tool to reduce cardiomyocyte vulnerability to stress. For example, increased mitochondrial biogenesis protects cardiomyocytes against ischemia-reperfusion injury by reducing mitochondrial oxidative stress and improving mitochondrial metabolism [59]. In addition, mitochondrial response sensitivity and mitochondrial senescence were shown to be normalized by mitochondrial biogenesis activation in a mouse model of myocardial hypertrophy [60, 61]. In this study, we observed the pathological alterations and protective mechanisms underlying mitochondrial biogenesis in septic cardiomyopathy. Our findings provide further evidence of the cardioprotective properties of mitochondrial biogenesis in cardiovascular disorders.
A limitation of our study is the lack of in vivo studies to support our findings. In addition, we did not evaluate the impact of the SirT3/AMPK axis on cardiomyocyte metabolism and the myocardial inflammatory response.
In conclusion, SirT3 downregulation, AMPK inactivation, and mitochondrial biogenesis inhibition are present in septic cardiomyocytes. Overexpression of SirT3 increases AMPK activity and improves mitochondrial biogenesis, which sustains mitochondrial function and reduces sepsis-related cardiomyocyte injury.
Materials and Methods
Cell culture
Neonatal mice cardiomyocytes were isolated from 1- to 2-day-old mice. In brief, hearts were removed, ventricles were pooled, and cells were dispersed by successive enzymatic digestion with collagenase A (0.4 mg/mL, Roche) and pancreatin (0.5 mg/mL, Sigma-Aldrich) [62]. Cell suspension was thereafter purified by centrifugation through a discontinuous Percoll gradient to obtain myocardial cell cultures with 99% myocytes. After seeding on either plastic dishes coated with gelatin (0.2% in PBS, Sigma-Aldrich) or, for confocal microscopy, glass coverslips coated with poly-D-lysine (0.1 mg/mL, Sigma-Aldrich) in 30-mm plastic wells, cardiomyocytes were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/medium 199 (4:1) supplemented with 10% horse serum, 5% calf serum, 1% glutamine, and antibiotics and placed in 37°C-5% CO2 atmosphere for 20 hours [63]. Approximately 95% of the cells displayed spontaneous contractile activity in culture. Then, cells, transiently transfected if necessary, were cultured in serum-free media for 24 hours before treatment. LPS was added into the medium to induce a septic cardiomyopathy model in vitro, as previously described [64].
RNA isolation and PCR
RNA isolation from cell culture was done using Trifast (Peqlab) as per the manufacturer’s instructions. Isolated RNA (500-1000 ng) was reversed transcribed with random primer using iScript Select cDNA synthesis kit (Biorad). Real-time quantitative polymerase chain reaction (PCR) was done with iQ SYBR Green mix (Biorad) on C1000 Touch Thermocycler (Biorad) using specific primer pairs [68]. For amplification of circular RNAs, divergent primers were used, whereas normal linear transcripts were amplified by convergent primers as usual [69]. For RNase R resistant assay, RNA was incubated with RNase R (Biozym Scientific) at 37°C for 10 minutes and heat activated at 95°C for 3 minutes. RNA was then reverse transcribed and amplified by specific PCR primers as mentioned previously. Validation was performed as three individual experiments with three replicates each time [70].
Mitochondrial function assay
Mitochondrial oxygen consumption and ATP synthesis rates were measured in saponin-permeabilized fibers using palmitoyl-carnitine/malate, pyruvate/malate, or glutamate/malate as substrate combinations, as described previously [71]. Mitochondria were isolated by differential centrifugation, and oxygen consumption and ATP synthesis were measured using palmitoyl-carnitine/malate, pyruvate/malate, or glutamate/malate as substrate combinations, as described previously [72]. Mitochondrial respiration complexes of isolated mitochondrial membranes were separated by blue native gel electrophoresis, and complex activities were determined by in-gel staining assays, as described previously [73]. Mitochondrial respiration rate in cardiomyocytes was assessed using a Seahorse XFp Extracellular Flux Analyzer with the XFp Cell Mito Stress Test Kit (Agilent, Santa Clara, CA) [74].
MTT assay
MTT assay was performed using Cell Proliferation Kit I (Roche) as per the manufacturer’s instruction [75]. Briefly, cells were seeded in a 96-well plate and the next day treated with LPS in normal medium for 48 hours [36]. Next, 10 μL of MTT reagent were added, and cells were incubated for 4 hours followed by addition of dissolving reagent and incubation overnight. The next day, absorbance was measured at 580 and 690 nm by an HT Synergy (Biotek) plate reader.
TUNEL staining
Cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature and then permeabilized with ice-cold 0.1% Triton-X-100 in PBS for 2 minutes at room temperature. Next, cells were incubated with enzyme labeling solution provided with In Situ Cell Death Detection Kit (Roche) for 1 hour at 37°C. For negative staining, enzyme was not added to the labeling solution [76]. Cells were then washed and incubated with DAPI for 15 minutes. Images were taken with a Nikon Eclipse Ti microscope, and images were analyzed with Nikon NIS-Elements [33]. For analysis in each case, 10 different images were analyzed from different regions, and the average value was taken. Three data points representing three individual experiments are shown.
Immunoblot analysis
Mitochondrial and cytosolic fractions were generated by homogenizing freshly excised hearts in homogenization buffer (20 mM HEPES, 140 mM KCl, 10 mM EDTA, 5 mM MgCl2, pH 7.4) with a Dounce tissue homogenizer, centrifuging the homogenate at 800 × g for 10 minutes and centrifuging the resulting supernatant at 8,000 × g for 10 minutes [77]. The supernatant is the cytosolic fraction. The pellet was washed by centrifugation at 10,000 × g and represents the mitochondrial fraction. Whole-cell extracts and mitochondrial membranes were prepared as described previously. Samples were loaded on SDS-PAGE, transferred to nitrocellulose or PVDF membranes, and incubated with specific antibodies [78]. Bands were visualized using horseradish peroxidase–conjugated secondary antibodies and the ECL detection system (GE Healthcare, Piscataway, NJ) or fluorophore-conjugated secondary antibodies and the Odyssey fluorescence detection system (Li-Cor Biosciences, Alpharetta, GA).
Statistical analysis
Quantitative data are presented as mean ± standard error. Student’s t-test (two-tailed, unpaired) was used for comparisons between two groups; one-way and two-way ANOVA with Student-Newman-Keuls post hoc tests were used as appropriate to evaluate statistically significant differences in multiple group comparisons. P < 0.05 was considered statistically significant.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Editorial Note
This corresponding author has a verified history of publications using the personal email addresses for correspondence.
References
- 1. Chang W, Xie JF, Xu JY, Yang Y. Effect of levosimendan on mortality in severe sepsis and septic shock: a meta-analysis of randomised trials. BMJ Open. 2018; 8:e019338. https://doi.org/10.1136/bmjopen-2017-019338 [PubMed]
- 2. Honda T, He Q, Wang F, Redington AN. Acute and chronic remote ischemic conditioning attenuate septic cardiomyopathy, improve cardiac output, protect systemic organs, and improve mortality in a lipopolysaccharide-induced sepsis model. Basic Res Cardiol. 2019; 114:15. https://doi.org/10.1007/s00395-019-0724-3 [PubMed]
- 3. Tan Y, Ouyang H, Xiao X, Zhong J, Dong M. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones. 2019; 24:595–608. https://doi.org/10.1007/s12192-019-00992-2 [PubMed]
- 4. Yu W, Mei X, Zhang Q, Zhang H, Zhang T, Zou C. Yap overexpression attenuates septic cardiomyopathy by inhibiting DRP1-related mitochondrial fission and activating the ERK signaling pathway. J Recept Signal Transduct Res. 2019; 39:175–86. https://doi.org/10.1080/10799893.2019.1641822 [PubMed]
- 5. Falk L, Hultman J, Broman LM. Extracorporeal membrane oxygenation for septic shock. Crit Care Med. 2019; 47:1097–105. https://doi.org/10.1097/CCM.0000000000003819 [PubMed]
- 6. Haileselassie B, Mukherjee R, Joshi AU, Napier BA, Massis LM, Ostberg NP, Queliconi BB, Monack D, Bernstein D, Mochly-Rosen D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol. 2019; 130:160–69. https://doi.org/10.1016/j.yjmcc.2019.04.006 [PubMed]
- 7. Chang W, Feng M, Li Y, Sun Y, Sun L. MKP1 overexpression reduces TNF-α-induced cardiac injury via suppressing mitochondrial fragmentation and inhibiting the JNK-MIEF1 pathways. J Cell Physiol. 2019. [Epub ahead of print]. https://doi.org/10.1002/jcp.28273 [PubMed]
- 8. Xiao Y, Zhang Q, Wang Y, Wang B, Sun F, Han Z, Feng Y, Yang H, Meng S, Wang Z. Dual-functional protein for one-step production of a soluble and targeted fluorescent dye. Theranostics. 2018; 8:3111–25. https://doi.org/10.7150/thno.24613 [PubMed]
- 9. Shang X, Li J, Yu R, Zhu P, Zhang Y, Xu J, Chen K, Li M. Sepsis-related myocardial injury is associated with Mst1 upregulation, mitochondrial dysfunction and the Drp1/f-actin signaling pathway. J Mol Histol. 2019; 50:91–103. https://doi.org/10.1007/s10735-018-09809-5 [PubMed]
- 10. Liu H, He Z, Germič N, Ademi H, Frangež Ž, Felser A, Peng S, Riether C, Djonov V, Nuoffer JM, Bovet C, Mlinarič-Raščan I, Zlobec I, et al. ATG12 deficiency leads to tumor cell oncosis owing to diminished mitochondrial biogenesis and reduced cellular bioenergetics. Cell Death Differ. 2020; 27:1965–80. https://doi.org/10.1038/s41418-019-0476-5 [PubMed]
- 11. Bhargava P, Janda J, Schnellmann RG. Elucidation of cGMP-dependent induction of mitochondrial biogenesis through PKG and p38 MAPK in the kidney. Am J Physiol Renal Physiol. 2020; 318:F322–28. https://doi.org/10.1152/ajprenal.00533.2019 [PubMed]
- 12. Wang J, Toan S, Zhou H. Mitochondrial quality control in cardiac microvascular ischemia-reperfusion injury: new insights into the mechanisms and therapeutic potentials. Pharmacol Res. 2020; 156:104771. https://doi.org/10.1016/j.phrs.2020.104771 [PubMed]
- 13. Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020; 23:299–314. https://doi.org/10.1007/s10456-020-09720-2 [PubMed]
- 14. Ojo B, Simenson AJ, O’Hara C, Wu L, Gou X, Peterson SK, Lin D, Smith BJ, Lucas EA. Wheat germ supplementation alleviates insulin resistance and cardiac mitochondrial dysfunction in an animal model of diet-induced obesity. Br J Nutr. 2017; 118:241–49. https://doi.org/10.1017/S0007114517002082 [PubMed]
- 15. Rahn JJ, Stackley KD, Chan SS. Opa1 is required for proper mitochondrial metabolism in early development. PLoS One. 2013; 8:e59218. https://doi.org/10.1371/journal.pone.0059218 [PubMed]
- 16. Zu YX, Lu HY, Liu WW, Jiang XW, Huang Y, Li X, Zhao QC, Xu ZH. Jiang gui fang activated interscapular brown adipose tissue and induced epididymal white adipose tissue browning through the PPARγ/SIRT1-PGC1α pathway. J Ethnopharmacol. 2020; 248:112271. https://doi.org/10.1016/j.jep.2019.112271 [PubMed]
- 17. Kalliora C, Kyriazis ID, Oka SI, Lieu MJ, Yue Y, Area-Gomez E, Pol CJ, Tian Y, Mizushima W, Chin A, Scerbo D, Schulze PC, Civelek M, et al. Dual peroxisome-proliferator-activated-receptor-α/γ activation inhibits SIRT1-PGC1α axis and causes cardiac dysfunction. JCI Insight. 2019; 5:e129556. https://doi.org/10.1172/jci.insight.129556 [PubMed]
- 18. Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L, Reiter RJ, Qiao S, Yuan J. AMPK/PGC1α activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. 2018; 129:59–72. https://doi.org/10.1016/j.freeradbiomed.2018.08.032 [PubMed]
- 19. Zhu J, Wang YF, Chai XM, Qian K, Zhang LW, Peng P, Chen PM, Cao JF, Qin ZH, Sheng R, Xie H. Exogenous NADPH ameliorates myocardial ischemia-reperfusion injury in rats through activating AMPK/mTOR pathway. Acta Pharmacol Sin. 2020; 41:535–45. https://doi.org/10.1038/s41401-019-0301-1 [PubMed]
- 20. Wang ZP, Shen D, Che Y, Jin YG, Wang SS, Wu QQ, Zhou H, Meng YY, Yuan Y. Corosolic acid ameliorates cardiac hypertrophy via regulating autophagy. Biosci Rep. 2019; 39:BSR20191860. https://doi.org/10.1042/BSR20191860 [PubMed]
- 21. Li R, Xin T, Li D, Wang C, Zhu H, Zhou H. Therapeutic effect of sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018; 18:229–43. https://doi.org/10.1016/j.redox.2018.07.011 [PubMed]
- 22. Kabziński J, Walczak A, Mik M, Majsterek I. Sirt3 regulates the level of mitochondrial DNA repair activity through deacetylation of NEIL1, NEIL2, OGG1, MUTYH, APE1 and LIG3 in colorectal cancer. Pol Przegl Chir. 2019; 92:1–4. https://doi.org/10.5604/01.3001.0013.5539 [PubMed]
- 23. Eid RA, Bin-Meferij MM, El-Kott AF, Eleawa SM, Zaki MS, Al-Shraim M, El-Sayed F, Eldeen MA, Alkhateeb MA, Alharbi SA, Aldera H, Khalil MA. Exendin-4 protects against myocardial ischemia-reperfusion injury by upregulation of SIRT1 and SIRT3 and activation of AMPK. J Cardiovasc Transl Res. 2020. [Epub ahead of print]. https://doi.org/10.1007/s12265-020-09984-5 [PubMed]
- 24. Xiang X, Huang J, Song S, Wang Y, Zeng Y, Wu S, Ruan Y. 17β-estradiol inhibits H2O2-induced senescence in HUVEC cells through upregulating SIRT3 expression and promoting autophagy. Biogerontology. 2020. [Epub ahead of print]. https://doi.org/10.1007/s10522-020-09868-w [PubMed]
- 25. Gao J, Zhang K, Wang Y, Guo R, Liu H, Jia C, Sun X, Wu C, Wang W, Du J, Chen J. A machine learning-driven study indicates emodin improves cardiac hypertrophy by modulation of mitochondrial SIRT3 signaling. Pharmacol Res. 2020; 155:104739. https://doi.org/10.1016/j.phrs.2020.104739 [PubMed]
- 26. Dikalova AE, Pandey A, Xiao L, Arslanbaeva L, Sidorova T, Lopez MG, Billings FT
4th , Verdin E, Auwerx J, Harrison DG, Dikalov SI. Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ Res. 2020; 126:439–52. https://doi.org/10.1161/CIRCRESAHA.119.315767 [PubMed] - 27. Zhu Z, Li H, Chen W, Cui Y, Huang A, Qi X. Perindopril improves cardiac function by enhancing the expression of SIRT3 and PGC-1α in a rat model of isoproterenol-induced cardiomyopathy. Front Pharmacol. 2020; 11:94. https://doi.org/10.3389/fphar.2020.00094 [PubMed]
- 28. Wang Q, Xu J, Li X, Liu Z, Han Y, Xu X, Li X, Tang Y, Liu Y, Yu T, Li X. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J Cell Physiol. 2019; 234:23495–506. https://doi.org/10.1002/jcp.28918 [PubMed]
- 29. Parodi-Rullán RM, Chapa-Dubocq XR, Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Front Physiol. 2018; 9:1094. https://doi.org/10.3389/fphys.2018.01094 [PubMed]
- 30. Wang G, Fu XL, Wang JJ, Guan R, Sun Y, Tony To SS. Inhibition of glycolytic metabolism in glioblastoma cells by Pt3glc combinated with PI3K inhibitor via SIRT3-mediated mitochondrial and PI3K/Akt-MAPK pathway. J Cell Physiol. 2019; 234:5888–903. https://doi.org/10.1002/jcp.26474 [PubMed]
- 31. Heusch G. 25 years of remote ischemic conditioning: from laboratory curiosity to clinical outcome. Basic Res Cardiol. 2018; 113:15. https://doi.org/10.1007/s00395-018-0673-2 [PubMed]
- 32. Bøtker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femminò S, García-Dorado D, et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. 2018; 113:39. https://doi.org/10.1007/s00395-018-0696-8 [PubMed]
- 33. Davidson SM, Arjun S, Basalay MV, Bell RM, Bromage DI, Bøtker HE, Carr RD, Cunningham J, Ghosh AK, Heusch G, Ibanez B, Kleinbongard P, Lecour S, et al. The 10th biennial hatter cardiovascular institute workshop: cellular protection-evaluating new directions in the setting of myocardial infarction, ischaemic stroke, and cardio-oncology. Basic Res Cardiol. 2018; 113:43. https://doi.org/10.1007/s00395-018-0704-z [PubMed]
- 34. Zhou H, Yue Y, Wang J, Ma Q, Chen Y. Melatonin therapy for diabetic cardiomyopathy: a mechanism involving syk-mitochondrial complex I-SERCA pathway. Cell Signal. 2018; 47:88–100. https://doi.org/10.1016/j.cellsig.2018.03.012 [PubMed]
- 35. Carrico C, Meyer JG, He W, Gibson BW, Verdin E. The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 2018; 27:497–512. https://doi.org/10.1016/j.cmet.2018.01.016 [PubMed]
- 36. Nwadozi E, Ng A, Strömberg A, Liu HY, Olsson K, Gustafsson T, Haas TL. Leptin is a physiological regulator of skeletal muscle angiogenesis and is locally produced by PDGFRα and PDGFRβ expressing perivascular cells. Angiogenesis. 2019; 22:103–15. https://doi.org/10.1007/s10456-018-9641-6 [PubMed]
- 37. Kim S, Jin H, Seo HR, Lee HJ, Lee YS. Regulating BRCA1 protein stability by cathepsin s-mediated ubiquitin degradation. Cell Death Differ. 2019; 26:812–25. https://doi.org/10.1038/s41418-018-0153-0 [PubMed]
- 38. Liu H, Li S, Liu X, Chen Y, Deng H. SIRT3 overexpression inhibits growth of kidney tumor cells and enhances mitochondrial biogenesis. J Proteome Res. 2018; 17:3143–52. https://doi.org/10.1021/acs.jproteome.8b00260 [PubMed]
- 39. Gao J, Feng Z, Wang X, Zeng M, Liu J, Han S, Xu J, Chen L, Cao K, Long J, Li Z, Shen W, Liu J. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018; 25:229–40. https://doi.org/10.1038/cdd.2017.144 [PubMed]
- 40. Cao Y, Han X, Pan H, Jiang Y, Peng X, Xiao W, Rong J, Chen F, He J, Zou L, Tang Y, Pei Y, Zheng J, et al. Emerging protective roles of shengmai injection in septic cardiomyopathy in mice by inducing myocardial mitochondrial autophagy via caspase-3/Beclin-1 axis. Inflamm Res. 2020; 69:41–50. https://doi.org/10.1007/s00011-019-01292-2 [PubMed]
- 41. Durand A, Duburcq T, Dekeyser T, Neviere R, Howsam M, Favory R, Preau S. Involvement of mitochondrial disorders in septic cardiomyopathy. Oxid Med Cell Longev. 2017; 2017:4076348. https://doi.org/10.1155/2017/4076348 [PubMed]
- 42. Fattahi F, Frydrych LM, Bian G, Kalbitz M, Herron TJ, Malan EA, Delano MJ, Ward PA. Role of complement C5a and histones in septic cardiomyopathy. Mol Immunol. 2018; 102:32–41. https://doi.org/10.1016/j.molimm.2018.06.006 [PubMed]
- 43. Guo R, Liu N, Liu H, Zhang J, Zhang H, Wang Y, Baruscotti M, Zhao L, Wang Y. High content screening identifies licoisoflavone a as a bioactive compound of tongmaiyangxin pills to restrain cardiomyocyte hypertrophy via activating Sirt3. Phytomedicine. 2020; 68:153171. https://doi.org/10.1016/j.phymed.2020.153171 [PubMed]
- 44. Akhmedov A, Montecucco F, Costantino S, Vdovenko D, Schaub Clerigué A, Gaul DS, Burger F, Roth A, Carbone F, Liberale L, Amrollahi-Sharifabadi M, Vellone VG, Eriksson U, et al. Cardiomyocyte-specific JunD overexpression increases infarct size following ischemia/reperfusion cardiac injury by downregulating Sirt3. Thromb Haemost. 2020; 120:168–80. https://doi.org/10.1055/s-0039-3400299 [PubMed]
- 45. Liu J, Yan W, Zhao X, Jia Q, Wang J, Zhang H, Liu C, He K, Sun Z. Sirt3 attenuates post-infarction cardiac injury via inhibiting mitochondrial fission and normalization of AMPK-Drp1 pathways. Cell Signal. 2019; 53:1–13. https://doi.org/10.1016/j.cellsig.2018.09.009 [PubMed]
- 46. Xiong S, Sun HJ, Cao L, Zhu M, Liu T, Wu Z, Bian JS. Stimulation of Na+/K+-ATPase with an antibody against its 4th extracellular region attenuates angiotensin II-induced H9c2 cardiomyocyte hypertrophy via an AMPK/SIRT3/PPAR γ signaling pathway. Oxid Med Cell Longev. 2019; 2019:4616034. https://doi.org/10.1155/2019/4616034 [PubMed]
- 47. Chen X, Wang Q, Shao M, Ma L, Guo D, Wu Y, Gao P, Wang X, Li W, Li C, Wang Y. Ginsenoside Rb3 regulates energy metabolism and apoptosis in cardiomyocytes via activating PPARα pathway. Biomed Pharmacother. 2019; 120:109487. https://doi.org/10.1016/j.biopha.2019.109487 [PubMed]
- 48. Zeng H, Chen JX. Sirtuin 3, endothelial metabolic reprogramming, and heart failure with preserved ejection fraction. J Cardiovasc Pharmacol. 2019; 74:315–23. https://doi.org/10.1097/FJC.0000000000000719 [PubMed]
- 49. Yang Y, Zhao M, Yu XJ, Liu LZ, He X, Deng J, Zang WJ. Pyridostigmine regulates glucose metabolism and mitochondrial homeostasis to reduce myocardial vulnerability to injury in diabetic mice. Am J Physiol Endocrinol Metab. 2019; 317:E312–26. https://doi.org/10.1152/ajpendo.00569.2018 [PubMed]
- 50. Sun Y, Teng Z, Sun X, Zhang L, Chen J, Wang B, Lu F, Liu N, Yu M, Peng S, Wang Y, Zhao D, Zhao Y, et al. Exogenous H2S reduces the acetylation levels of mitochondrial respiratory enzymes via regulating the NAD+-SIRT3 pathway in cardiac tissues of db/db mice. Am J Physiol Endocrinol Metab. 2019; 317:E284–97. https://doi.org/10.1152/ajpendo.00326.2018 [PubMed]
- 51. Zhou H, Toan S. Pathological Roles of Mitochondrial Oxidative Stress and Mitochondrial Dynamics in Cardiac Microvascular Ischemia/Reperfusion Injury. Biomolecules. 2020; 10:85. https://doi.org/10.3390/biom10010085 [PubMed]
- 52. Li R, Toan S, Zhou H. Role of mitochondrial quality control in the pathogenesis of nonalcoholic fatty liver disease. Aging (Albany NY). 2020; 12:6467–85. https://doi.org/10.18632/aging.102972 [PubMed]
- 53. Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S, Li Y, Zhou H, Chen Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018; 16:157–68. https://doi.org/10.1016/j.redox.2018.02.019 [PubMed]
- 54. Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017; 63:e12413. https://doi.org/10.1111/jpi.12413 [PubMed]
- 55. Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S, Zhang Y, Han T, Ren J, Cao F, Chen Y. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J Pineal Res. 2017; 63. https://doi.org/10.1111/jpi.12438 [PubMed]
- 56. Delmotte P, Sieck GC. Endoplasmic reticulum stress and mitochondrial function in airway smooth muscle. Front Cell Dev Biol. 2020; 7:374. https://doi.org/10.3389/fcell.2019.00374 [PubMed]
- 57. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019; 26:605–16. https://doi.org/10.1038/s41418-018-0252-y [PubMed]
- 58. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018; 25:486–541. https://doi.org/10.1038/s41418-017-0012-4 [PubMed]
- 59. Qi X, Wang J. Melatonin improves mitochondrial biogenesis through the AMPK/PGC1α pathway to attenuate ischemia/reperfusion-induced myocardial damage. Aging (Albany NY). 2020; 12:7299–312. https://doi.org/10.18632/aging.103078 [PubMed]
- 60. Weng X, Zhang X, Lu X, Wu J, Li S. Reduced mitochondrial response sensitivity is involved in the anti-apoptotic effect of dexmedetomidine pretreatment in cardiomyocytes. Int J Mol Med. 2018; 41:2328–38. https://doi.org/10.3892/ijmm.2018.3384 [PubMed]
- 61. Song M, Franco A, Fleischer JA, Zhang L, Dorn GW
2nd . Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 2017; 26:872–83.e5. https://doi.org/10.1016/j.cmet.2017.09.023 [PubMed] - 62. Kraft L, Erdenesukh T, Sauter M, Tschöpe C, Klingel K. Blocking the IL-1β signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res Cardiol. 2019; 114:11. https://doi.org/10.1007/s00395-019-0719-0 [PubMed]
- 63. Ndongson-Dongmo B, Lang GP, Mece O, Hechaichi N, Lajqi T, Hoyer D, Brodhun M, Heller R, Wetzker R, Franz M, Levy FO, Bauer R. Reduced ambient temperature exacerbates SIRS-induced cardiac autonomic dysregulation and myocardial dysfunction in mice. Basic Res Cardiol. 2019; 114:26. https://doi.org/10.1007/s00395-019-0734-1 [PubMed]
- 64. Zhong J, Tan Y, Lu J, Liu J, Xiao X, Zhu P, Chen S, Zheng S, Chen Y, Hu Y, Guo Z. Therapeutic contribution of melatonin to the treatment of septic cardiomyopathy: a novel mechanism linking Ripk3-modified mitochondrial performance and endoplasmic reticulum function. Redox Biol. 2019; 26:101287. https://doi.org/10.1016/j.redox.2019.101287 [PubMed]
- 65. Aalto AL, Mohan AK, Schwintzer L, Kupka S, Kietz C, Walczak H, Broemer M, Meinander A. M1-linked ubiquitination by LUBEL is required for inflammatory responses to oral infection in drosophila. Cell Death Differ. 2019; 26:860–76. https://doi.org/10.1038/s41418-018-0164-x [PubMed]
- 66. Ansari D, Torén W, Zhou Q, Hu D, Andersson R. Proteomic and genomic profiling of pancreatic cancer. Cell Biol Toxicol. 2019; 35:333–43. https://doi.org/10.1007/s10565-019-09465-9 [PubMed]
- 67. Golovynska I, Golovynskyi S, Stepanov YV, Garmanchuk LV, Stepanova LI, Qu J, Ohulchanskyy TY. Red and near-infrared light induces intracellular Ca2+ flux via the activation of glutamate N-methyl-D-aspartate receptors. J Cell Physiol. 2019. [Epub ahead of print]. https://doi.org/10.1002/jcp.28257 [PubMed]
- 68. Amin AH, El-Missiry MA, Othman AI, Ali DA, Gouida MS, Ismail AH. Ameliorative effects of melatonin against solid ehrlich carcinoma progression in female mice. J Pineal Res. 2019; 67:e12585. https://doi.org/10.1111/jpi.12585 [PubMed]
- 69. Bowman CM, Benet LZ. Interlaboratory variability in human hepatocyte intrinsic clearance values and trends with physicochemical properties. Pharm Res. 2019; 36:113. https://doi.org/10.1007/s11095-019-2645-0 [PubMed]
- 70. Arun KG, Sharanya CS, Sadasivan C. Computational and experimental validation of morin as adenosine deaminase inhibitor. J Recept Signal Transduct Res. 2018; 38:240–45. https://doi.org/10.1080/10799893.2018.1476543 [PubMed]
- 71. Chandrasekharan A, Varadarajan SN, Lekshmi A, Lupitha SS, Darvin P, Chandrasekhar L, Pillai PR, Santhoshkumar TR, Pillai MR. A high-throughput real-time in vitro assay using mitochondrial targeted roGFP for screening of drugs targeting mitochondria. Redox Biol. 2019; 20:379–89. https://doi.org/10.1016/j.redox.2018.10.013 [PubMed]
- 72. Zarfati M, Avivi I, Brenner B, Katz T, Aharon A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis. 2019; 22:185–96. https://doi.org/10.1007/s10456-018-9649-y [PubMed]
- 73. Zhang S, Huang F, Tian W, Lai J, Qian L, Hong W, Chen H, Li LC. Andrographolide promotes pancreatic duct cells differentiation into insulin-producing cells by targeting PDX-1. Biochem Pharmacol. 2020; 174:113785. https://doi.org/10.1016/j.bcp.2019.113785 [PubMed]
- 74. Zeng X, Zhang X, Li C, Wang X, Jerwick J, Xu T, Ning Y, Wang Y, Zhang L, Zhang Z, Ma Y, Zhou C. Ultrahigh-resolution optical coherence microscopy accurately classifies precancerous and cancerous human cervix free of labeling. Theranostics. 2018; 8:3099–110. https://doi.org/10.7150/thno.24599 [PubMed]
- 75. Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D, Zhou H, Chen Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones. 2018; 23:101–13. https://doi.org/10.1007/s12192-017-0827-4 [PubMed]
- 76. Lu J, Li J, Hu Y, Guo Z, Sun D, Wang P, Guo K, Duan DD, Gao S, Jiang J, Wang J, Liu P. Chrysophanol protects against doxorubicin-induced cardiotoxicity by suppressing cellular PARylation. Acta Pharm Sin B. 2019; 9:782–93. https://doi.org/10.1016/j.apsb.2018.10.008 [PubMed]
- 77. Chou LF, Chen CY, Yang WH, Chen CC, Chang JL, Leu YL, Liou MJ, Wang TH. Suppression of hepatocellular carcinoma progression through FOXM1 and EMT inhibition via hydroxygenkwanin-induced miR-320a expression. Biomolecules. 2019; 10:20. https://doi.org/10.3390/biom10010020 [PubMed]
- 78. Chrifi I, Louzao-Martinez L, Brandt MM, van Dijk CG, Bürgisser PE, Zhu C, Kros JM, Verhaar MC, Duncker DJ, Cheng C. CMTM4 regulates angiogenesis by promoting cell surface recycling of VE-cadherin to endothelial adherens junctions. Angiogenesis. 2019; 22:75–93. https://doi.org/10.1007/s10456-018-9638-1 [PubMed]