



Introduction
Brain ageing is associated with cognitive decline and neurodegeneration. Normal ageing often leads to levels of decline in cognition, with estimates of a fifth of people over 71 affected by impairment that is not classed as dementia [1]. Loss of mitochondrial functionality is implicated as a key factor leading to age related decline and the development of many neurodegenerative diseases. Increasing our understanding of the changes that occur in the normal process of ageing is crucial to help distinguish between the biological features of disease and that of ageing itself. Delineating the expected changes within the lifetime of a mammal provides entry points to examine endogenous protective and degenerative pathways, these lend themselves as biomarkers or can present novel treatment targets.
Ageing research has focussed on the extension of lifespan. The mitochondrial free-radical theory of ageing suggests that reactive oxygen species (ROS) produced by the mitochondria cause a decline of molecular function, resulting in ageing [2]. Critically, the balance of beneficial and deleterious effects of mitochondrial ROS within a tissue during a lifetime still needs to be determined [3]. Recent evidence supporting a role for ROS in ageing found that increasing NADPH, by upregulation of the enzyme glucose-6-phosphate dehydrogenase (G6PD) in mice increased antioxidant defences which delayed ageing [4]. Mitochondrial dysfunction is a common feature of ageing and neurodegeneration and certain mitochondrial proteins have been shown to undergo oxidative damage in both of these processes [5–7]. However, ROS are unlikely to be the only factors contributing to age-related mitochondrial dysfunction. An understanding of how mitochondrial composition changes with age can shed light on the mechanisms affecting these organelles throughout the lifetime of mammals. A complication, when looking at diseases of ageing, is the inability to separate the effect of normal ageing from disease-related changes in the mitochondrion, for example in Parkinson’s disease where mitochondrial changes are clearly important, yet not always specifically distinguishable from the effects of ageing [5,8].
Proteomics targeted specifically at the mitochondrial fraction of the cell is a powerful approach to identify the expected age-related changes in tissues. A few studies have been reported where mitochondrial proteins have been examined in normal brain tissue [9,10]. Existing studies of skeletal muscle senescence suggest that mitochondrial enzymes are largely increased in abundance, though how this information fits with decreased complex I activity in ageing mitochondria has not yet been determined [11].
Our study stems from an interest in mitochondrial proteins within tissues with a high capacity for oxidative phosphorylation. Both the brain and skeletal muscle undergo a degree of decline in advanced age. We profiled proteins of mitochondrial fractions isolated from young (~8weeks) and old (78 weeks) mouse brain and skeletal muscle. It is pertinent to point out that our samples were deliberately chosen to reflect youth versus middle-age, rather than true old-age (old mouse would be >104 weeks) [12]. Our intention was to maximise the possibility of identifying early changes that may be occurring prior to detectable functional losses rather than the ‘gravestones’ heralding end stage dysfunction. While it is illuminating to interrogate the proteome of a particular biological entity, neither of these are closed systems. Our study highlights the importance of making assessments across these groupings. We show that the pharmaceutical target carbonic anhydrase II is increased with age in mitochondria. To investigate the potential importance of changing levels of these proteins we looked to see whether carbonic anhydrases are also changed in a similar manner in the Purkinje Cell Degeneration (pcd5J) mouse model of neurodegeneration. The pcd5j mouse is an excellent model to study the effect of a pure mitochondrial neurodegenerative phenotype that occurs early in life [13–15,7]. We compared our findings in the neuronal and non-neuronal tissues with what we found in pcd5J to understand whether the levels of carbonic anhydrase found are likely to be a protective or dysfunctional alteration. We now are able to provide the molecular context of normal mitochondrial ageing which needs to be fully considered as carbonic anhydrase inhibitor therapy becomes more widely applied in diseases affecting our ageing populations.
Results
The mitochondrial proteome is different in young and old murine skeletal muscle tissue
Six proteins were selected to have changed when comparing the young (4-11 week) and old (78 week) skeletal muscle mitochondrial proteomic profiles (Figure 1A). Carbonic anhydrase III (discussed later), calsequestrin and Voltage Dependent Anion Channel 1(VDAC1) increase with age in the old skeletal muscle mitochondria (p<0.05). Calsequestrin increases with a greater than two-fold change between the young and older mitochondria. It has recently been shown that loss of calsequestrin leads to mitochondrial dysfunction and oxidative stress in skeletal muscle [16]. It could be interpreted therefore that an upregulation of calsequestrin in this case is a protective response rather than being reflective of muscle decline; a study in postmenopausal women also identifies a (smaller fold) increase in total skeletal muscle calsequestrin [17]. Overexpression of calsequestrin in cardiomyocytes suggests that endoplasmic reticulum calcium stores may be enhanced perinuclearly to provide an independent compensatory effect in the case of misregulated calcium homeostasis. Mitochondria also accumulate perinuclearly and are regulators of calcium signalling providing more evidence that the upregulation in calsequestrin that we observe is protective in ageing skeletal muscle [18]. In skeletal muscle mitochondria we can confirm definite changes in haemoglobin subunit alpha, ATP synthase and VDAC1. These mitochondrial proteins have previously been shown to be differentially regulated in ageing and our data confirm that these are likely important regulators of ageing in skeletal muscle mitochondria [5,19,20].
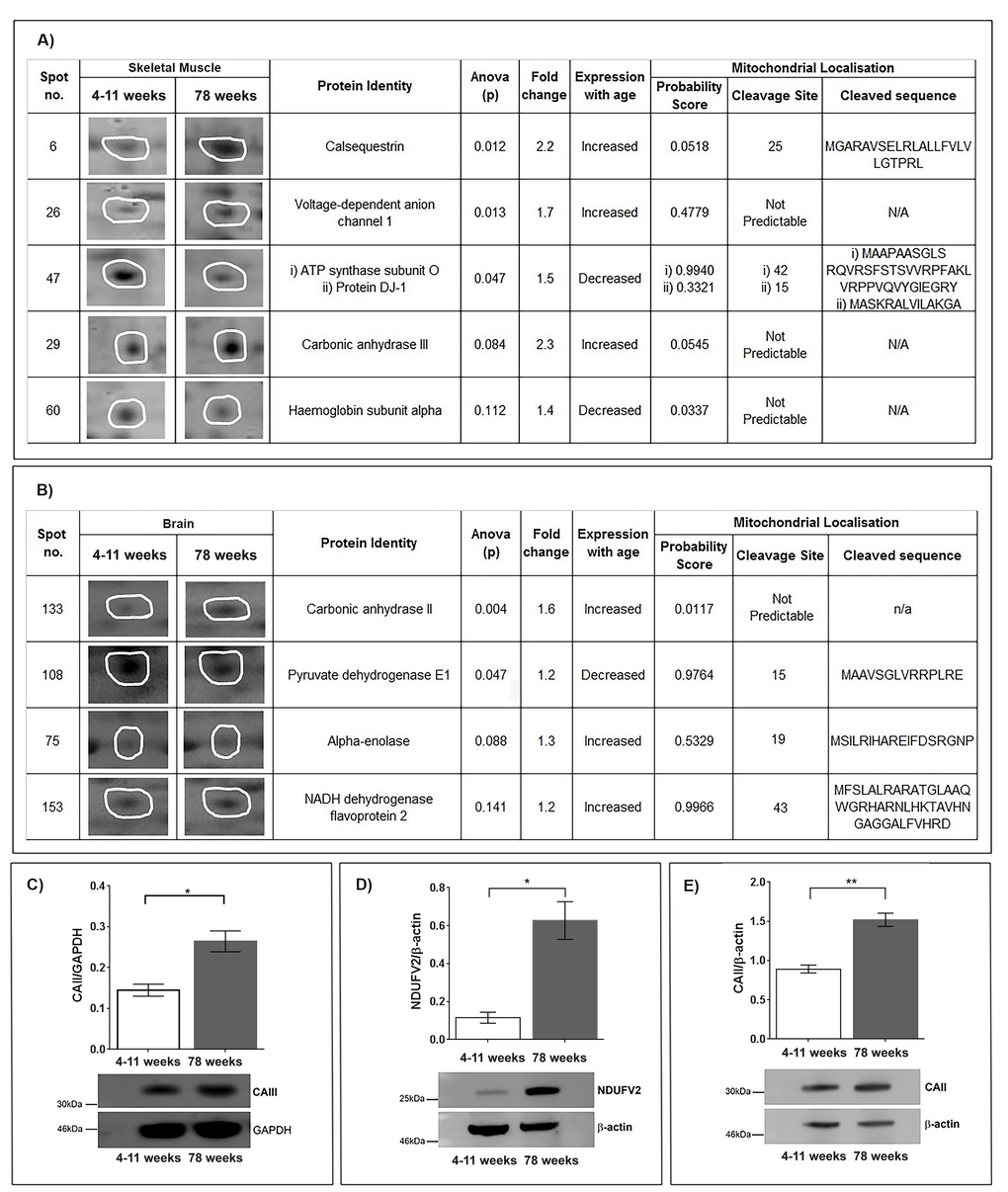
Figure 1. (A) Identification of protein changes with age in the skeletal muscle mitochondrial proteome. Five protein spots were selected after (SameSpots) analysis comparing murine skeletal muscle mitochondria aged 4-11 weeks (n=5) and 78 weeks (n=5). Representative protein spot images, statistical analyses (one-way ANOVA) and identities of the proteins (MASCOT) are shown. Mitochondrial localisation probability was calculated (Mitoprot) and is shown along with predicted cleavage sites and sequence. (B) Identification of proteins that change with age in the skeletal muscle mitochondrial proteome. Four protein spots were selected after (SameSpots) analysis comparing murine brain mitochondria aged 4-11 weeks (young) (n=3) and 78 weeks (old) (n=3). Representative protein spot images, statistical analyses (one-way ANOVA) and identities of the proteins (MASCOT) are shown. Mitochondrial localisation probability was calculated (Mitoprot) and is shown along with predicted cleavage sites and sequence. (C) Carbonic anhydrase III protein levels increase in aged skeletal muscle mitochondria. Carbonic anhydrase III, normalised to GAPDH, is significantly increased in 78 week old (n=4) skeletal muscle mitochondria compared with 4-11 week old (n=4) skeletal muscle mitochondria, p=0.0105. (D) NADH dehydrogenase flavoprotein 2 protein levels increase in aged brain mitochondria. NADH dehydrogenase flavoprotein 2, normalised to beta-actin, is significantly increased in 78 week old (n=4) brain mitochondria compared to 4-11 week old (n=4) brain mitochondria, p=0.0109. (E) Carbonic anhydrase II protein levels increase in aged brain mitochondria. Carbonic anhydrase II, normalised to beta-actin, is significantly increased in 78 week old (n=4) brain mitochondria compared to 4-11 week old (n=4) brain mitochondria p=0.0015. Columns display mean activity ± SEM. * = p<0.05 and **= p<0.03 two-tailed unpaired t-test with Welch’s correction.
The mitochondrial proteome is also distinctly different between young and old murine brain tissue
In brain mitochondrial fractions our top list defines pyruvate dehydrogenase E1, alpha enolase and NADH flavoprotein 2 as changed between the young (4-11 weeks) and old (78 weeks) brain mitochondrial proteome (Figure 1B). Pyruvate dehydrogenase is known to decline through the brain with age and enolase has recently found to be decreased on the CD4(+) T cell surface in a small study of older males [21]. However, this is the first time these have been shown to be changed in association with ageing of the mitochondrial organelle. NADH dehydrogenase flavoprotein 2 (NDUFV2) a complex 1 protein is confirmed to be increased in the old mitochondria (p<0.005), agreeing with the perceived increase in complex 1 enzymes reported in senescent muscle [11] (Figure 1D). Mutations and variation in NDUFV2 are associated with disorders of the brain and ageing [22–25]. Our finding could suggest that the variations and mutations have a subtle effect on NDUFV2, which is most detrimental when upregulated, perhaps for neuroprotection in middle-age.
Carbonic anhydrase II and III are significantly increased in mitochondria isolated from older mice
Interestingly we show that two isoforms of carbonic anhydrase (CAII and CAIII) increase in both 78 week old brain mitochondria and 78 week old skeletal muscle mitochondria, respectively. Carbonic anhydrases are zinc metalloenzymes that catalyse the reversible hydration of carbon dioxide to bicarbonate. Carbonic anhydrase II binds to Na+/H+ exchanger altering pH [26]. Carbonic anhydrases also catalyse the reversible hydration of CO2, HCO3- and H+ [27]. Therefore, the role of carbonic anhydrases in maintaining the pH environment of the cell and specifically the mitochondrion is important to delineate.
CAIII is the muscle-specific isoenzyme [28] whilst CAII is located in the cytosol and widely expressed in most tissues [29]. In our study carbonic anhydrase III is significantly increased in old skeletal muscle mitochondria compared with young muscle mitochondria p<0.005 (t-test) (Figure 1C). We also see that CAII is significantly increased in mitochondria isolated from old brain tissue p<0.05 (t-Test) (Figure 1E). Calculated probability scores suggest that CAII and CAIII are not predicted to cleave into mitochondrial targeted forms. It is possible that these proteins associate very tightly with the mitochondrion without necessarily entering the organellar space [30,31]. Carbonic anhydrase III is not essential for survival in mouse and has been known for some time to increase in muscle with ageing and contraction [28,32,35]. A role in mitochondrial function has not yet been postulated for it, though we clearly observe a significant increase in the quantity of this protein in aged-muscle mitochondrial fractions.
CAII, belongs to the group of these isoenzymes that are pharmacologically targeted by inhibitors (such as acetazolamide) to treat a variety of disorders including glaucoma, cancer osteoporosis, epilepsy, neuropsychiatric disorders and acute mountain sickness [36–39]. Methazolamide also a carbonic anhydrase inhibitor, has also been shown recently to prevent amyloid-beta induced mitochondrial dysfunction and is neuroprotective in mouse models of Alzheimer’s disease [40].
The esterase activity of carbonic anhydrase II is increased in the mitochondrial fraction of brain tissue from older animals
We tested whether an upregulation of CAII protein corresponded with an increase in enzymatic activity. We measured the esterase activity of carbonic anhydrase II by monitoring the hydrolysis of 4-nitrophenyl acetate to form 4-nitrophenol. Mitochondria from aged brain tissue exhibited a higher rate of change in absorbance in comparison to mitochondria from young brain tissue throughout the 5-minute assay (Figure 2A). The rate of change at 1, 2 and 4 minutes were significantly higher in mitochondria from the old brain tissue (t-test) p<0.05. Our data show that the expression of CAII increases with ageing and this can be measured by the activity of CAII which is greater per mitochondrial unit in old versus young brain mitochondrial fractions.
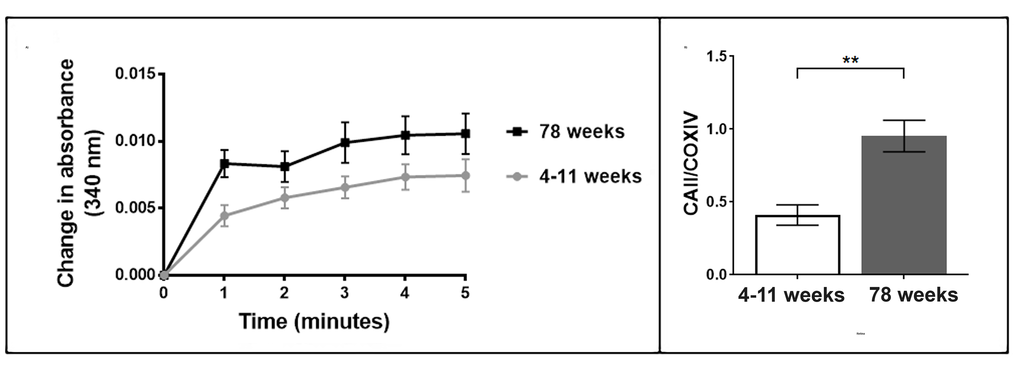
Figure 2. (A) Enzymatic activity of carbonic anhydrase II increases in aged brain mitochondria. The enzymatic activity of carbonic anhydrase II is higher in the 78 week old brain mitochondria compared to the 4-11 week brain mitochondria (three mitochondrial samples from individual animals in each age group and three technical replicates of each sample). Each mitochondrial sample contained 30 µg/µl of protein. (B) Carbonic anhydrase II protein levels increase in aged retina mitochondria. The protein levels of carbonic anhydrase II were measured in the murine retina mitochondria aged 4-11 weeks and 78 weeks. Carbonic anhydrase II significantly increases in retina mitochondria from older mice, p=0.028 (two-tailed unpaired t-test with Welch’s correction). Replicates were obtained from individual animals (young n=6, old n=6). Columns display mean activity ± SEM. **= p<0.03 two-tailed unpaired t-test with Welch’s correction.
Carbonic anhydrase II protein levels in retina mitochondria significantly increase in aged animals
Advanced age leads to an increased risk of developing neurodegenerative diseases. Glaucoma is a neurodegenerative disease that has been associated with oxidative stress and age related mitochondrial dysfunction [41,42]. We investigated the protein levels of CAII in retina mitochondria from aged mice (78 weeks). We observe a large increase in CAII in the retina mitochondria from aged mice in comparison to young mice (4-11 weeks), p<0.05 (Figure 2B). Our data indicate that CAII protein levels also increase in the retina with age.
Carbonic anhydrase II protein levels significantly increase in brain mitochondria from the neurodegenerative mouse model pcd5J
The Purkinje Cell Degeneration mouse, pcd, is an autosomal recessive mutant and a model of neurodegeneration. The pcd5J mouse model has a mutation in the Nna1 gene that encodes a protein that is localised in the mitochondrion. Initially the pcd5J mice are born with normal development of Purkinje cells but after 15 days rapid degeneration of the Purkinje cells occur, with over a 99% loss of Purkinje cells by around 3 weeks of age [13]. We used the pcd5J mouse model to investigate whether the changes in CAII levels are specific to the ageing process or are also a sign of neurodegeneration. Mitochondria were isolated from the cerebellum of pcd5J and aged matched wild type animals (10-13 weeks). CAII protein levels are significantly elevated in the pcd5J cerebellum mitochondria compared to wild type mitochondria, p<0.05 (Figure 3A). We propose that the increase in CAII in the brain with ageing is an early symptom of neurodegenerative decline.
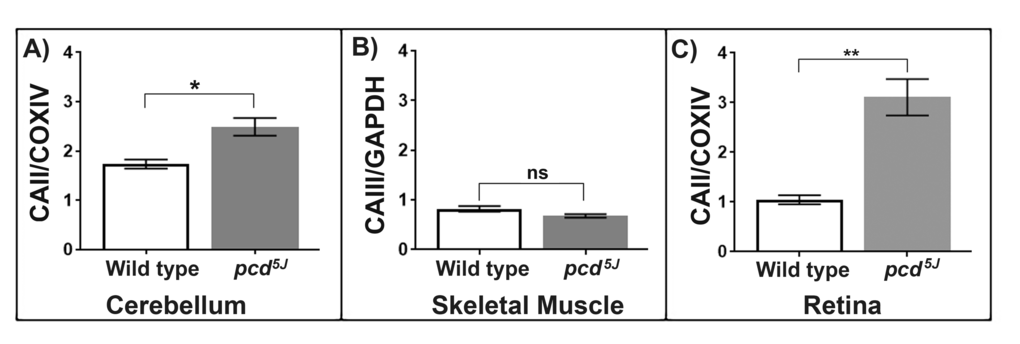
Figure 3. (A) Carbonic anhydrase II protein levels increase in the neurodegenerative mouse model, pcd5J cerebellum mitochondria. Carbonic anhydrase II, normalised to COXIV, is significantly increased in pcd5J cerebellum mitochondria compared to wild type animals aged (10-13 weeks old). Replicates were obtained from individual animals (wild type n=4, pcd5J n=4). (B) Carbonic anhydrase III protein levels are not significantly different between the pcd5J and wild type skeletal muscle mitochondria. CAIII protein levels, normalised to GAPDH, were compared between wild type (n=3) and pcd5J (n=3) mice aged 10-13 weeks. Replicates were obtained from individual animals. (C) Carbonic anhydrase II accumulates in the pcd5J retina mitochondria. Carbonic anhydrase II levels are significantly higher in pcd5J mice compared to wild type animals, p=0.0019. Replicates were obtained from individual animals (wild type n=6, pcd5J n=6), all animals were between 9-17 weeks old. Columns display mean activity ± SEM. * = p<0.05, **= p<0.03 two-tailed unpaired t-test with Welch’s correction.
Carbonic anhydrase III protein levels are not significantly different between pcd5J and wild type skeletal muscle mitochondria
Skeletal muscle is unaffected by the loss of Nna1 function in pcd5J animals. The muscle specific isoform of carbonic anhydrase CAIII was found to be altered in the ageing skeletal muscle mitochondria. We wanted to test whether CAIII protein levels changed in the pcd5J mouse muscle to see whether the changes in CAII in neural tissues were indeed specific for ageing and neurodegeneration or whether the pcd5J mutant has a systemic alteration in carbonic anhydrases, even in unaffected tissues. We compared pcd5J skeletal muscle from the young mice that already showed signs of neurodegeneration in retina and cerebellum with age-matched wild type animals (10-13 weeks). We found that the quantity of CAIII is not significantly different in the pcd5J and wild type animals (Figure 3B). Since the pcd5J animals have functionally healthy skeletal muscle it is unsurprising that there is no significant difference between the quantity of CAIII in the wild type and mutant animals. However, this suggests very strongly that the alterations in carbonic anhydrases found in ageing and neurodegeneration are a harbinger of the dysfunction which ensues.
The neurodegenerative mouse model, pcd5J, also shows significantly increased carbonic anhydrase II protein levels in retinal mitochondria
Retinal degeneration is a feature of the pcd5J animals and so we used this mouse model to investigate whether CAII increases in mitochondria in a disease state. We show CAII protein levels significantly increase in retina from pcd5J mice compared to aged matched wild type mice (2-4 weeks old), p<0.05 (Figure 3C). Our data suggest that the accumulation of CAII in retina mitochondria occurs during the normal ageing process and that increasing levels of CAII is also a feature of retinal degeneration. The CAII inhibitor dorzolamide hydrochloride is commonly used in the treatment of glaucoma to improve ocular perfusion. Dorzolamide hydrochloride has also been suggested to act as antioxidant, exerting its effect through intact mitochondria [43]. We now suggest that the action of dorzolamide hydrochloride in glaucoma should be analysed for its likely effect on the raised CAII levels we find in ageing retinal mitochondria.
Increased carbonic anhydrase II reduces lifespan
The six alpha-carbonic anhydrase isoforms 1 to 6 are encoded for in the Caenorhabditis elegans genome (cah-1, cah-2, cah-3, cah-4, cah-5 and cah-6) [44]. Wormbase searches revealed that the murine (Mus musculus) CAII gene has similar homology to the C.elegans gene cah-3. Both cah-3 and cah-4 in C.elegans are orthologs of the murine carbonic anhydrase II gene [45]. Ensemble searches showed that the amino acid sequence for the murine carbonic anhydrase II protein has sequence homology with four carbonic anhydrase proteins in C.elegans, cah-3, cah-5, cah-1 and cah-2. We tested whether the increase in CAII is a protective mechanism or a sign of dysfunction by exploring the effect of CAII on C. elegans. C. elegans (strain CB5600) were treated with three different concentrations of CAII (1500 units, 150 units and 15 units). Animals treated with CAII have a significant reduction in lifespan p=0.0006 (Log-rank test). The animals show a dose dependant response to increased levels of CAII (Figure 4). The median lifespans were 6, 8 and 12 days for animals treated with 1500 units, 150 units, 15 units respectively. In the same experiments control animals had a median lifespan of 19 days. The absolute maximum lifespans were 13, 14 and 18 days for animals treated with 1500 units, 150 units and 15 units respectively and the control animals had an absolute maximum lifespan of 31 days. Animals treated with the highest concentration of CAII (1500 units) had a 58% reduction in lifespan compared to the control animals, whilst animals treated with 150 and 15 units showed a reduction in lifespan by 55% and 42% respectively compared to the controls. Based on these findings we suggest that carbonic anhydrase inhibitors could be targeting the effects of mitochondrial ageing in neurons by reducing carbonic anhydrase levels to physiologically more youthful levels. CAII is likely to be an important regulator of the ageing process.
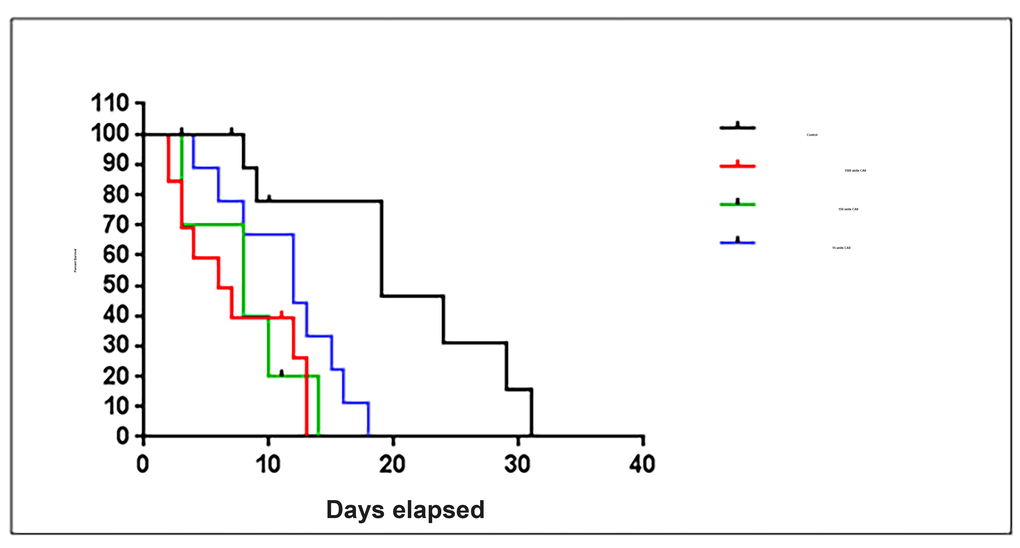
Figure 4. Exposure to carbonic anhydrase II treatment significantly shortens lifespan of C.elegans. C. elegans (strain CB5600) exposed to carbonic anhydrase II were recorded by Kaplan Meier survival plot (Log-rank test, p=0.0006). Three concentrations of carbonic anhydrase II 1500 units, 150 units and 15 units were used (10 animals in each condition).
Conclusions
We present a picture of complex proteomic profile changes in mitochondrial fractions with ageing. In particular, we observe accumulations of carbonic anhydrase isoenzymes with increased age. CAII protein levels were also found to increase in the cerebellum and retina mitochondria of the neurodegenerative disease mouse model, pcd5J. We propose that increased quantities of CAII play a detrimental role in the ageing process. Therapeutic use of carbonic anhydrase inhibitors are likely to be exerting an effect on mitochondrial populations and may be offering protection through maintenance or stabilisation of carbonic anhydrase to physiologically young levels.
Materials and Methods
Mitochondrial preparations
Brain, skeletal muscle and retinal tissue were dissected from young (4-11 week old) and old (78 weeks) C57BL/6J mice (Charles River). The Purkinje cell degeneration mice (pcd5J) and wild type animals (9-17 weeks old) were sourced from the University of Nottingham.
2D gel analysis
Mitochondrial samples, 4-11 weeks brain (n=5), 78 weeks brain (n=5), 4-11 weeks skeletal muscle (n=3) and 78 weeks skeletal muscle (n=3), were subject to iso-electric focussing using ZOOM IPG (Life Technologies) system and pH 3-10 (non-linear) ZOOM IPG strips following the manufacturers protocol. Gels were stained (SimplyBlue™ SafeStain, Life Technologies) and imaged (ImageQuant 300, GE Healthcare Life Sciences). Analysis was performed using SameSpots software (Totallab). Protein spots with a p value of less than 0.15 and a fold change greater than 1.2 were further analysed (one-way ANOVA). Proteins were identified from the gel pieces as described previously5.
Western blotting
Western blotting was carried out as described previously [5].
Antibody dilutions: Carbonic anhydrase II ab6621 (Abcam) 1:7000 dilution in 3% (w/v) BSA in TBS-T; NADH dehydrogenase flavoprotein 2 ARP57510-PO50 (Cambridge Bioscience) 1:5000 dilution in 3% (w/v) BSA in TBS-T; Beta-actin ab8227 (Abcam) 1:5000 dilution in 3% (w/v) BSA in TBS-T; Carbonic anhydrase III AP7633a (ABGENT) 1:2500 dilution in 3% (w/v) BSA in TBS-T, GAPDH G9545 (SIGMA) 1:5000 dilution in 3% (w/v) BSA in TBS-T and COXIV (ab16056) 1:5000 dilution in 3% (w/v) BSA in TBS-T. Brain mitochondrial samples were normalised to beta-actin level. The average of four samples for each condition (old and young) were plotted showing the mean +/- SEM. The muscle mitochondrial samples were normalised to GAPDH level. The average of the four samples for each condition (old and young) were plotted showing the mean +/- SEM. The retina mitochondrial samples were normalised to COXIV level. The average of the six samples for each condition (young and old) were plotted showing the mean +/- SEM. Statistical analyses (unpaired t-tests with Welch’s correction) were carried out in GraphPad Prism.
Carbonic anhydrase II enzyme assay
Esterase activity of carbonic anhydrase II was measured by monitoring the release of 4-nitrophenol at A348 nm in a Thermo Scientific Helios Epsilon spectrophotometer using standard methods [46]. Cuvettes contained 900 μl of 15 mM Tris Sulphate Buffer, pH 7.6 at 0°C (Sigma), 500 μl of 3 mM 4-nitrophenyl-acetate (Sigma) and 30 μg/μl of mitochondrial sample (3 mitochondrial preparations and 3 replicates for each condition, 4-11 weeks and 78 weeks). The rate of change in absorbance of the assay was plotted. Unpaired t-tests (GraphPad Prism) were carried out at each time point.
Carbonic anhydrase II lifespan study
CB5600 C. elegans strain is superficially wild type and expresses GFP in nuclei and mitochondria of body-wall muscles. Animals were maintained on solid NGM agar plates seeded with the Escherichia coli strain OP50 using standard methods and aged-synchronized [47,48]. Animals were exposed to three concentrations of synthetic carbonic anhydrase II (C2522 SIGMA) 1500 units, 150 units and 15 units. Control plates had 20 μl of dH20 spotted on to the surface whilst treated plates had 20 μl of carbonic anhydrase II spotted on. Each plate contained 10 L1 larvae (n=40). Animals were scored every day and those not moving that did not respond to stimulation with a needle were recorded as dead. The experiment was maintained at 20°C. Kaplan-Meier survival curve and statistical analysis (log-rank, Mantel-Cox, test) was performed using GraphPad Prism.
Ethical approval
Animals were bred and housed in accordance with strict Home Office stipulated conditions. The overall programme of work (in respect to the original UK Home Office Project Licence application) is reviewed by the Animal Welfare and Ethical Review Body at the University of Nottingham and then scrutinised by the UK Home Office Inspectorate before approval by the Secretary of State. Individual study protocols link to the overarching Home Office Project Licence and are made available to the Named Animal Care and Welfare Officer, the Named Veterinary Surgeon (both are members of the AWERB), the animal care staff and the research group. The Project Licence Number for the breeding and maintenance of this genetically altered line of mice is PPL 40/3576. The mice are typically group housed and maintained within solid floor cages containing bedding and nesting material with additional environmental enrichment including chew blocks and hiding tubes. Cages are Individually Ventilated Cage Units within a barrier SPF unit to maintain bio-security. Animals are checked daily by a competent and trained animal technician. Any animal giving cause for concern such as subdued behaviour, staring coat, loss of weight or loss of condition will be humanely killed using a Home Office approved Schedule 1 method of killing.
Supplementary Materials
Acknowledgements
This work on the biology of the mitochondrion is dedicated to Gottfried Schatz.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was funded by the BBSRC BB/J014508/1 and the University of Nottingham.
References
- 1. Apostolo J, Holland C, O’Connell MD, Feeney J, Tabares-Seisdedos R, Tadros G, Campos E, Santos N, Robertson DA, Marcucci M, Varela-Nieto I, Crespo-Facorro B, Vieta E, et al. Mild cognitive decline. A position statement of the Cognitive Decline Group of the European Innovation Partnership for Active and Healthy Ageing (EIPAHA). Maturitas. 2016; 83:83–93. https://doi.org/10.1016/j.maturitas.2015.10.008 [PubMed]
- 2. Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, Ganie SA. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed Pharmacother. 2015; 74:101–10. https://doi.org/10.1016/j.biopha.2015.07.025 [PubMed]
- 3. Lagouge M, Larsson N-G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med. 2013; 273:529–43. https://doi.org/10.1111/joim.12055 [PubMed]
- 4. Nóbrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Viña J, Serrano M. G6PD protects from oxidative damage and improves healthspan in mice. Nat Commun. 2016; 7:10894. https://doi.org/10.1038/ncomms10894 [PubMed]
- 5. Shephard F, Greville-Heygate O, Marsh O, Anderson S, Chakrabarti L. A mitochondrial location for haemoglobins-Dynamic distribution in ageing and Parkinson’s disease. Mitochondrion. 2013. https://doi.org/10.1016/j.mito.2013.12.001 [PubMed]
- 6. Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005; 58:495–505. https://doi.org/10.1002/ana.20624 [PubMed]
- 7. Chakrabarti L, Zahra R, Jackson SM, Kazemi-Esfarjani P, Sopher BL, Mason AG, Toneff T, Ryu S, Shaffer S, Kansy JW, Eng J, Merrihew G, MacCoss MJ, et al. Mitochondrial dysfunction in NnaD mutant flies and Purkinje cell degeneration mice reveals a role for Nna proteins in neuronal bioenergetics. Neuron. 2010; 66:835–47. https://doi.org/10.1016/j.neuron.2010.05.024 [PubMed]
- 8. Licker V, Kövari E, Hochstrasser DF, Burkhard PR. Proteomics in human Parkinson’s disease research. J Proteomics. 2009; 73:10–29. https://doi.org/10.1016/j.jprot.2009.07.007 [PubMed]
- 9. Stauch KL, Purnell PR, Villeneuve LM, Fox HS. Proteomic Analysis and Functional Characterization of Mouse Brain Mitochondria during Aging Reveals Alterations in Energy Metabolism. Proteomics. 2014. https://doi.org/10.1002/pmic.201400277
- 10. Villa RF, Gorini A, Ferrari F, Hoyer S. Energy metabolism of cerebral mitochondria during aging, ischemia and post-ischemic recovery assessed by functional proteomics of enzymes. Neurochem Int. 2013; 63:765–81. https://doi.org/10.1016/j.neuint.2013.10.004 [PubMed]
- 11. Staunton L, O’Connell K, Ohlendieck K. Proteomic Profiling of Mitochondrial Enzymes during Skeletal Muscle Aging. J Aging Res. 2011; 2011:908035. https://doi.org/10.4061/2011/908035 [PubMed]
- 12. Flurkey K, Currer JM. H. D. in (ed. American College Laboratory Animal Medicine) 637–672 (Elsevier, 2007).
- 13. Chakrabarti L, Neal JT, Miles M, Martinez RA, Smith AC, Sopher BL, La Spada AR. The Purkinje cell degeneration 5J mutation is a single amino acid insertion that destabilizes Nna1 protein. Mamm Genome. 2006; 17:103–10. https://doi.org/10.1007/s00335-005-0096-x [PubMed]
- 14. Chakrabarti L, Eng J, Martinez RA, Jackson S, Huang J, Possin DE, Sopher BL, La Spada AR. The zinc-binding domain of Nna1 is required to prevent retinal photoreceptor loss and cerebellar ataxia in Purkinje cell degeneration (pcd) mice. Vision Res. 2008; 48:1999–2005. https://doi.org/10.1016/j.visres.2008.05.026 [PubMed]
- 15. Chakrabarti L, Eng J, Ivanov N, Garden GA, La Spada AR. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain. 2009; 2:24. https://doi.org/10.1186/1756-6606-2-24 [PubMed]
- 16. Paolini C, Quarta M, Wei-LaPierre L, Michelucci A, Nori A, Reggiani C, Dirksen RT, Protasi F. Oxidative stress, mitochondrial damage, and cores in muscle from calsequestrin-1 knockout mice. Skelet Muscle. 2015; 5:10. https://doi.org/10.1186/s13395-015-0035-9 [PubMed]
- 17. Gueugneau M, Coudy-Gandilhon C, Gourbeyre O, Chambon C, Combaret L, Polge C, Taillandier D, Attaix D, Friguet B, Maier AB, Butler-Browne G, Béchet D. Proteomics of muscle chronological ageing in post-menopausal women. BMC Genomics. 2014; 15:1165. https://doi.org/10.1186/1471-2164-15-1165 [PubMed]
- 18. Guo A, Cala SE, Song L-S. Calsequestrin accumulation in rough endoplasmic reticulum promotes perinuclear Ca2+ release. J Biol Chem. 2012; 287:16670–80. https://doi.org/10.1074/jbc.M112.340927 [PubMed]
- 19. Groebe K, Klemm-Manns M, Schwall GP, Hübenthal H, Unterluggauer H, Jansen-Dürr P, Tanguay RM, Morrow G, Schrattenholz A. Age-dependent posttranslational modifications of voltage-dependent anion channel 1. Exp Gerontol. 2010; 45:632–37. https://doi.org/10.1016/j.exger.2010.02.006 [PubMed]
- 20. Su J, Ekman C, Oskolkov N, Lahti L, Ström K, Brazma A, Groop L, Rung J, Hansson O. A novel atlas of gene expression in human skeletal muscle reveals molecular changes associated with aging. Skelet Muscle. 2015; 5:35. https://doi.org/10.1186/s13395-015-0059-1 [PubMed]
- 21. Bennett SJ, Augustyniak EM, Dunston CR, Brown RA, Shantsila E, Lip GY, Torrao RD, Pararasa C, Remtulla AH, Ladouce R, Friguet B, Griffiths HR. CD4(+) T cell surface alpha enolase is lower in older adults. Mech Ageing Dev. 2015; 152:56–62. https://doi.org/10.1016/j.mad.2015.09.005 [PubMed]
- 22. Swerdlow RH, Weaver B, Grawey A, Wenger C, Freed E, Worrall BB. Complex I polymorphisms, bigenomic heterogeneity, and family history in Virginians with Parkinson’s disease. J Neurol Sci. 2006; 247:224–30. https://doi.org/10.1016/j.jns.2006.05.053 [PubMed]
- 23. Bénit P, Beugnot R, Chretien D, Giurgea I, De Lonlay-Debeney P, Issartel JP, Corral-Debrinski M, Kerscher S, Rustin P, Rötig A, Munnich A. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat. 2003; 21:582–86. https://doi.org/10.1002/humu.10225 [PubMed]
- 24. Nishioka K, Vilariño-Güell C, Cobb SA, Kachergus JM, Ross OA, Hentati E, Hentati F, Farrer MJ. Genetic variation of the mitochondrial complex I subunit NDUFV2 and Parkinson’s disease. Parkinsonism Relat Disord. 2010; 16:686–87. https://doi.org/10.1016/j.parkreldis.2010.09.007 [PubMed]
- 25. Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R, Koller D, Nurnberger JI, Corvin A, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012; 17:887–905. https://doi.org/10.1038/mp.2012.37 [PubMed]
- 26. Li X, Alvarez B, Casey JR, Reithmeier RA, Fliegel L. Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J Biol Chem. 2002; 277:36085–91. https://doi.org/10.1074/jbc.M111952200 [PubMed]
- 27. Nishimori I, et al. Carbonic Anhydrase Inhibitors. The Mitochondrial Isozyme VB as a New Target for Sulfonamide and Sulfamate Inhibitors. J Med Chem. 2005; 24:7860-66.
- 28. Staunton L, Zweyer M, Swandulla D, Ohlendieck K. Mass spectrometry-based proteomic analysis of middle-aged vs. aged vastus lateralis reveals increased levels of carbonic anhydrase isoform 3 in senescent human skeletal muscle. Int J Mol Med. 2012; 30:723–33. [PubMed]
- 29. Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem. 1995; 64:375–401. https://doi.org/10.1146/annurev.bi.64.070195.002111 [PubMed]
- 30. Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008; 7:168–81. https://doi.org/10.1038/nrd2467 [PubMed]
- 31. Schroeder MA, Ali MA, Hulikova A, Supuran CT, Clarke K, Vaughan-Jones RD, Tyler DJ, Swietach P. Extramitochondrial domain rich in carbonic anhydrase activity improves myocardial energetics. Proc Natl Acad Sci USA. 2013; 110:E958–67. https://doi.org/10.1073/pnas.1213471110 [PubMed]
- 32. Côté CH, Ambrosio F, Perreault G. Metabolic and contractile influence of carbonic anhydrase III in skeletal muscle is age dependent. Am J Physiol. 1999; 276:R559–65. [PubMed]
- 33. Vasilaki A, Simpson D, McArdle F, McLean L, Beynon RJ, Van Remmen H, Richardson AG, McArdle A, Faulkner JA, Jackson MJ. Formation of 3-nitrotyrosines in carbonic anhydrase III is a sensitive marker of oxidative stress in skeletal muscle. Proteomics Clin Appl. 2007; 1:362–72. https://doi.org/10.1002/prca.200600702 [PubMed]
- 34. Kimura Y, Kurabe N, Ikegami K, Tsutsumi K, Konishi Y, Kaplan OI, Kunitomo H, Iino Y, Blacque OE, Setou M. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J Biol Chem. 2010; 285:22936–41. https://doi.org/10.1074/jbc.C110.128280 [PubMed]
- 35. Kim G, Lee TH, Wetzel P, Geers C, Robinson MA, Myers TG, Owens JW, Wehr NB, Eckhaus MW, Gros G, Wynshaw-Boris A, Levine RL. Carbonic anhydrase III is not required in the mouse for normal growth, development, and life span. Mol Cell Biol. 2004; 24:9942–47. https://doi.org/10.1128/MCB.24.22.9942-9947.2004 [PubMed]
- 36. Carradori S, Mollica A, De Monte C, Ganese A, Supuran CT. Nitric oxide donors and selective carbonic anhydrase inhibitors: a dual pharmacological approach for the treatment of glaucoma, cancer and osteoporosis. Molecules. 2015; 20:5667–79. https://doi.org/10.3390/molecules20045667 [PubMed]
- 37. Swenson ER. Carbonic anhydrase inhibitors and high altitude illnesses. Subcell Biochem. 2014; 75:361–86. https://doi.org/10.1007/978-94-007-7359-2_18 [PubMed]
- 38. Thiry A, Dogné J-M, Supuran CT, Masereel B. Anticonvulsant sulfonamides/sulfamates/sulfamides with carbonic anhydrase inhibitory activity: drug design and mechanism of action. Curr Pharm Des. 2008; 14:661–71. https://doi.org/10.2174/138161208783877956 [PubMed]
- 39. Farooq MU, Moore PW, Bhatt A, Aburashed R, Kassab MY. Therapeutic role of zonisamide in neuropsychiatric disorders. Mini Rev Med Chem. 2008; 8:968–75. https://doi.org/10.2174/138955708785740643 [PubMed]
- 40. Fossati S, Giannoni P, Solesio ME, Cocklin SL, Cabrera E, Ghiso J, Rostagno A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol Dis. 2016; 86:29–40. https://doi.org/10.1016/j.nbd.2015.11.006 [PubMed]
- 41. Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol. 2013; 13:12–15. https://doi.org/10.1016/j.coph.2012.09.008 [PubMed]
- 42. Cao L, Wang L, Cull G, Zhou A. Alterations in molecular pathways in the retina of early experimental glaucoma eyes. Int J Physiol Pathophysiol Pharmacol. 2015; 7:44–53. [PubMed]
- 43. Saccà SC, La Maestra S, Micale RT, Larghero P, Travaini G, Baluce B, Izzotti A. Ability of dorzolamide hydrochloride and timolol maleate to target mitochondria in glaucoma therapy. Arch Ophthalmol. 2011; 129:48–55. https://doi.org/10.1001/archophthalmol.2010.324 [PubMed]
- 44. Sherman TA, Rongali SC, Matthews TA, Pfeiffer J, Nehrke K. Identification of a nuclear carbonic anhydrase in Caenorhabditis elegans. Biochim. Biophys. Acta - Mol.. Cell Res. 2012; 1823:808–17.
- 45. Shaye DD, Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 2011; 6:e20085. https://doi.org/10.1371/journal.pone.0020085 [PubMed]
- 46. Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem. 1967; 242:4221–29. [PubMed]
- 47. Stiernagle T. Maintenance of C. elegans. WormBook. 2006; •••:1–11. https://doi.org/10.1895/wormbook.1.101.1 [PubMed]
- 48. Zdinak LA, Greenberg IB, Szewczyk NJ, Barmada SJ, Cardamone-Rayner M, Hartman JJ, Jacobson LA. Transgene-coded chimeric proteins as reporters of intracellular proteolysis: starvation-induced catabolism of a lacZ fusion protein in muscle cells of Caenorhabditis elegans. J Cell Biochem. 1997; 67:143–53. https://doi.org/10.1002/(SICI)1097-4644(19971001)67:1<143::AID-JCB15>3.0.CO;2-I [PubMed]