



Preface
When normal cells become confluent, they get arrested: a phenomenon known as contact inhibition [1-7]. Certainly, this is the most physiologically relevant type of cell cycle arrest. In the organism, cells are predominantly contact-inhibited. Yet, contact inhibition is the least studied type of cell cycle arrest. Instead, scientific attention has been attracted to two types of arrest: (a) starvation-induced arrest and (b) Cyclin Dependent Kinase-inhibitor (CDKi)-induced arrest.
As a classic example of starvation-induced arrest, serum withdrawal causes reversible quiescence in normal cells. During serum-starvation, mitogen-activated pathways become silent [8]. Cells neither grow in size nor cycle. Re-addition of serum causes cell activation and proliferation.
As an example of CDKi-induced arrest, DNA damage and telomere shortening induce p53, which in turn induces p21 and p16, inhibiting CDKs. In other cases, stresses induce both p21 and p16 [8-23]. When serum growth factors and nutrients stimulate growth, then inhibition of CDKs leads to senescence [8]. All stresses that induce senescence inhibit CDKs in part by inducing CDKi such as p21, p16, p15. Oncogenic Ras and Raf activate MAPK and mTOR pathways and induce p21 and p16, causing senescence [9, 24-27].
Numerous studies have been aimed to pinpoint the difference between quiescence and senescence based on either the point of arrest, the nature of stresses or peculiarities of CDKi (p21 versus p16). Yet, despite all efforts, the distinction remained elusive.
In fact, the difference between quiescence and senescence lies outside the cell cycle [8, 28, 29]. A senescent program consists of two steps: cell cycle arrest and gerogenic conversion or geroconversion, for brevity [29]. It is geroconversion that distinguishes quiescence from senescence. Geroconversion is “futile cellular growth” driven by mTOR as well as related mitogen-activated and growth-promoting signaling pathways [29-31]. Rapamycin suppresses gero-conversion, maintaining quiescence instead [32-38]. Furthermore, any condition that directly or indirectly inhibits mTOR in turn suppresses geroconversion [39-49]. Two-step model of senescence is applicable to all forms of senescence: from replicative and stress-induced to physiological cellular aging in the organism [29]. Senescent cells are hyper-active, hyper-functional (for example, hyper-secretory phenotype or SASP) compensatory signal-resistant, secondary malfunctional and eventually atrophic [28, 36-38, 50-55]. Hyper-function and secondary malfunction lead to age-related diseases from cancer and atherosclerosis to diabetes and Alzheimer's disease [54, 56-73]. MTOR-driven gero-conversion activates stem cells, eventually leading to their exhaustion [34, 46, 74-82].
Rapamycin extends life span and prevents age-related diseases, including cancer in mice and humans [33, 57-73, 83-110].
The two-step model is applicable to contact inhibition. Given that contact inhibition is reversible, we predicted that mTOR is inhibited. In fact, we found that mTORC1 targets - S6K and S6 – are dephosphorylated in CI cells [41]. Furthermore, activation of mTOR (by depletion of TSC2) shifts reversible contact inhibition towards senescence [41]. Thus, it is deactivation of mTOR that suppresses geroconversion in contact inhibited cells. Deactivation of mTOR was associated with induction of p27. In cancer cells, there is no induction of p27 in high cell density. Accordingly, cancer cells do not get arrested in confluent cultures. There is a complex relationship between p27 and mTOR [111-113].
To cause arrest of cancer cells, we induced ectopic p21. Remarkably, p21-mediated arrest, which leads to senescence of HT-p21 cells in regular density, did not cause senescence in confluent cultures [41]. Why? It turned out that the mTOR pathway was inhibited in dense cultures of cancer cells. Yet, cancer cells do not induce p27 and do not undergo contact inhibition. mTOR is constitutively activated in cancer, [114-118]. And induction of p21 by itself does not inhibit mTOR. So why mTOR is deactivated not only in contact-inhibited but also in confluent cancer cells? The answer is that cancer cells with highly increased metabolism rapidly exhaust and acidify the medium, thus inhibiting mTOR by starvation-like mechanism [41]. In fact, change of the medium restored mTOR activity. Therefore, in normal cells with low metabolism, mTOR is deactivated by contact inhibition and the change of the medium only marginally affects mTOR. In cancer cells, mTOR is inhibited due to exhaustion of the medium. And some cell lines are somewhere in between.
Illustrations
In agreement with previous report, pS6 was barely detectable in contact inhibited cells (Fig. 1A). Inhibition of pS6 was associated with induction of p27. Treatment of contact-inhibited (CI) cells with etoposide did not affect either pS6 or p27. Change of the medium also did not affect pS6, as measured on second day after the change. Yet, the change of the medium transiently activated pS6 up to 6 hours (Fig. 1 B). This transient activation was not result in medium exhation, because CM by itseld did not inhibit pS6 in sparse culture [41]. Transient induction was in part due to hyper-sensitivity of CI-cells to slight signals.
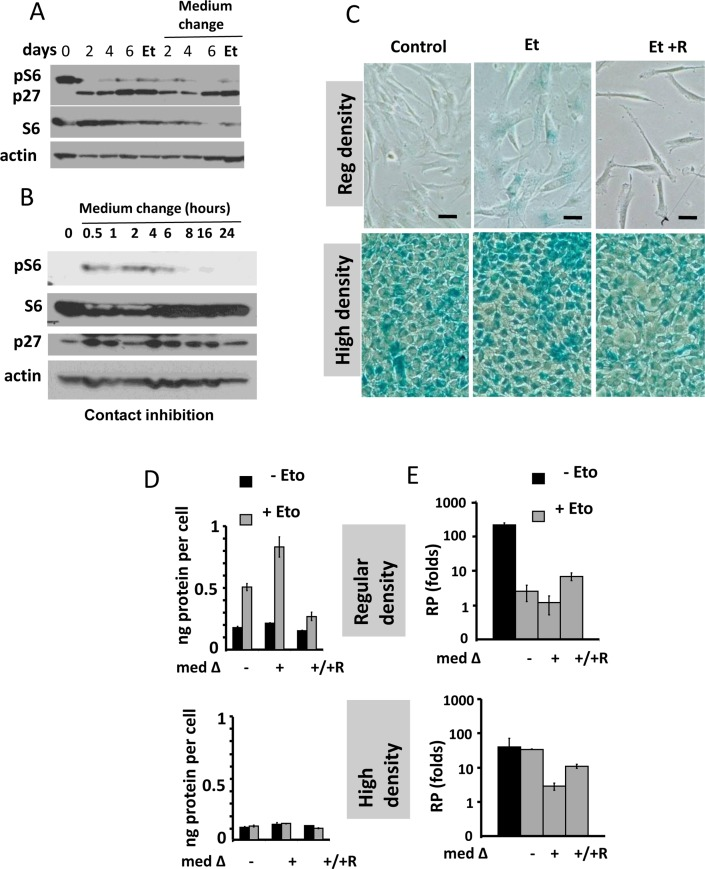
Figure 1. Contact inhibition suppresses etoposide-induced senescence in WI-38t cells (A-B) Immunoblot analysis [41]. (A) WI-38t cells [41] were plated at high density and lysed on the days indicated. When indicated “Medium change”, the medium was changed to fresh one every day. Et: cells were treated with 0.5 μg/ml etoposide on day 3 and lysed on day 6. p-S6(S240/244). (B) The effect of medium change on Contact Inhibited cells, measured in hours. (C) Beta-gal staining. WI38t cells were plated at regular or high density. After 3 days, 0.5 μg/ml etoposide (Et) and +/− 10 nM rapamycin (R) was added, if indicated. After 3 days, cells were stained for beta-Gal. Bar – 100 μm. (D-E) Cells were treated as described in panel C. Data are mean ± SD. (D) Cell size, protein per cell. (E) Reversibility potential or Replicative potential (RP). On day 6, cells were counted and re-plated at 1000/well in 12-well plates in fresh drug-free medium. Cells were counted after 9 days of growth. Fold increase in cell numbers were calculated. Mean ± SD.
We treated CI-culture and regular (exponentially growing) culture with etoposide for 3 days (Fig. 1C). In regular culture, WI-38 cells acquired a large flat morphology with beta-Gal staining. This senescent morphology was prevented by rapamycin (Fig. 1C). So etopiside caused mTOR-dependent senescence in regular culture conditions.
In CI-culture, cells were beta-Gal positive to start with. In fact, beta-Gal-staining is a marker of both contact inhibition, senescence and serum-starvation [119-130]. In contact-inhibition, beta-Gal staining, a marker of lysosomal overactivation, is mTOR-independent [41] and Figure 1 C. We next, employed two non-morphological tests to evaluate senescence: (a) cellular hypertrophy, measured as protein per cell and (b) the reversibility potential (RP) or the potential to restart proliferation and to regenerate cell culture after splitting in drug-free media. (Note: the reversibility also means that regenerated culture consists of cells identical to initial regular culture).
In regular culture, etoposide induced hypertrophy, which was prevented by rapamycin (Fig. 1D). In CI-culture, the cells were small and etoposide failed to cause any increase in cellular size. A small cell morphology is due to deactivated mTOR pathway in CI-culture. As expected, rapamycin did not decrease cell size further. Thus, etoposide caused mTOR-dependent hypertrophy only in regular but not in CI-culture.
The regenerative/reversibility potential can be tested after washing etoposide out. It is importantly that etoposide is easily washable [131].
In regular culture, etoposide dramatically eliminated the reversibility potential (RP), meaning that etoposide-treated cells did not proliferate after re-plating in low density in drug-free culture. This effect was in part mTOR dependent, because co-addition of rapamycin and etoposide caused a lesser loss of RP (Fig. 1E).
In CI-culture, etoposide did not cause loss of RP. Etoposide-pretreated cells resumed proliferation, similar to untreated cells. Noteworthy, when treatment with rapamycin was combined with a daily-change of the medium, which caused transient mTOR activation in CI-culture, the cells indeed lost some RP (Fig. 1 E, low panel). This loss was m-TOR-dependent, reversed by rapamycin (Fig. 1E). Therefore, we confirmed that, due to deactivation mTOR, etoposide did not cause senescence in CI-cultures. Also, etoposide did not cause hypertrophy in CI-cultures of RPE cells, while causing hypertrophy in regular density RPE cells (Fig. 2A). Etoposide-pretreated CI-cells retained RP, capable to proliferate and regenerate culture after splitting in low cell density (Fig. 2B).
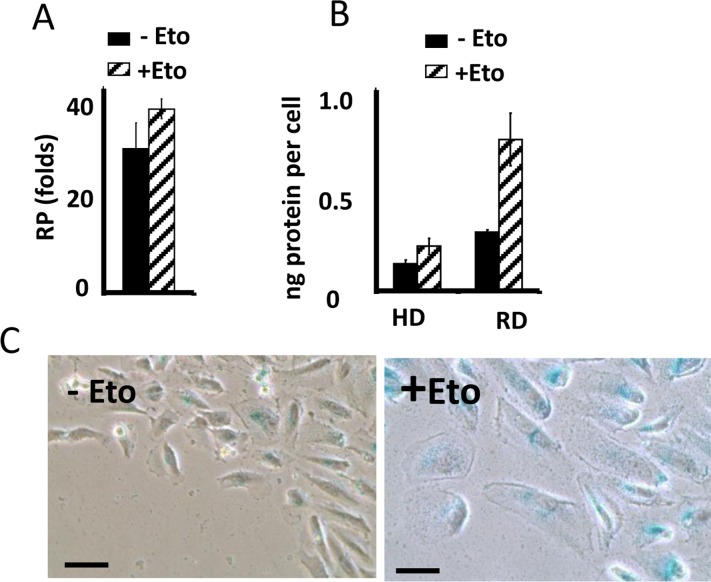
Figure 2. Suppression of etoposide-induced senescence in RPE cells (A) Reversibility potential (RP) in RPE cells [41] treated with 0.5 μg/ml etoposide (+Eto) in high cell density. After 2 day-treatment with etoposide (total 6 days in culture), cells were counted and re-plated in fresh medium at 1000/well in 24-well plates. After 7 days, cells were re-counted. Fold increase in cell numbers mean ± SD. (B) Protein per cell in regular (RD) versus high cell density (HD). RPE cells were plated at regular or high density. If indicated, cells were treated with 0.5 μg/ml etoposide (+Eto). After 2 days, cells were counted and lysed, protein amount was determined and protein (ng) amounts per cell was calculated. Data are mean ± SD. (C) Wounding. RPE cells were plated at high density. 0.5 μg/ml etoposide (+Eto) was added, if indicated “+”. Wounds were made in cell monolayer without changing the medium. After 3 days, cells were stained for beta-Gal.
Importantly, senescence could be induced in CI-cultures by wounding in the presence of rapamycin. At the edge of wounds, mTOR is reactivated [41]. When CI-monolayer was wounded, cells acquired senescent morphology in the presence of the same medium, containing etoposide (Fig. 2C). We emphasize that failure of etoposide to induce senescence in CI-cultures cannot be explained by cell cycle arrest in CI-cultures. It was extensively studied and shown that etoposide caused DNA damage in G1 phase of the cell cycle [132-134]. Furthermore its toxicity is high in non-cycling cells [132-134].
Previously we showed that geroconversion is suppressed in p21-arrested cancer cells in very high density [41]. This gerosuppression was associated with deactivated mTOR in exhausted media, [41]. Here we confirmed this observation and extended it to p16-induced arrest (Fig. 3). This indicated that high density suppresses geroconversion regardless of whether p21 or p16 caused arrest.
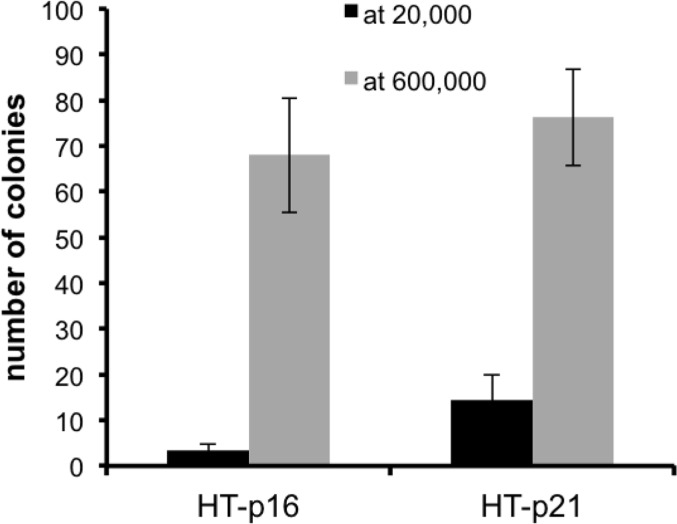
Figure 3. Effect of high density (HD) on p21- and p16-induced senescence HT-p21 and HT-p16 cells (see [37]) were plated at 20,000 (low density) and 600,000 (high density) cells in 6 well plates and then treated with IPTG to induce p21 and p16. After 3 days, cells were trypsinized, counted and 1000 cells were re-plated in IPTG-free medium in 6 well plates. Colonies were stained and counted after 8 days.
Low basal activation of mTOR in vivo
In the organism, cells are predominantly contact inhibited. Our in vitro data predict that mTOR activity should be low in the organism compared with cells in vitro. We compared levels of p-S6 (a marker of mTOR activity) in the organs (the heart and the liver) with the levels in mouse embrionyc fibroblasts (MEF). Use of the same species (mouse) ensures that there is no species-dependent differences in detection of p-S6 by antibody. Due to extra-cell matrix in vivo, we loaded a lesser amount of MEF protein (5 microg) than tissue-extracted protein (30 microM). What was important is a ratio between pS6 and S6 and between p-Akt and Akt (Fig. 4). In the livers and the hearts, pS6/S6 ratios were lower (approximately 4-100 fold) compared to MEF.
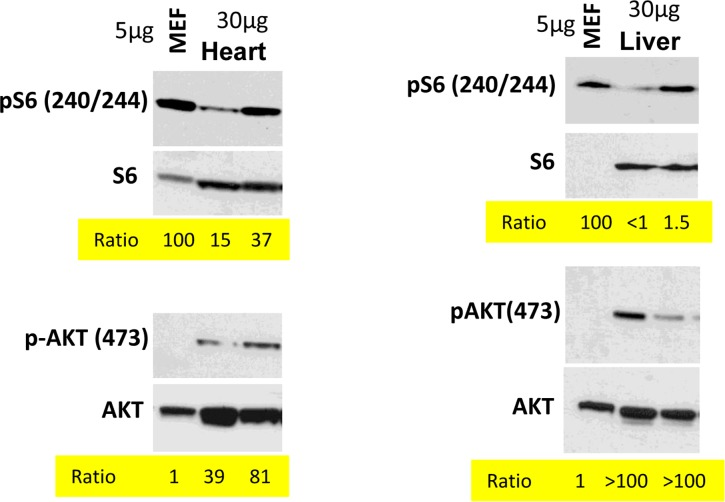
Figure 4. Comparison of p-S6(S240/244) and pAKT(S473) levels in murine heart and liver vs cultured MEFs Immunoblot analysis. 5 μg protein MEF lysate and 30 μg protein mice tissue were separated on the same gel and blotted for pS6/S6 and pAKT(S473)/AKT. Signal intensities were quantified using ImageJ program and normalized levels of p-S6 and p-AKT in mice organs were estimated. Ratio in MEFs is 100 and 1 (indicated as numbers). Methods are described previously and corresponding tissues samples blots published [103, 138-140].
In contrast, the ratio p-Akt/Akt was much higher in the tissues than in MEF in culture (Fig. 4).
Physiological and clinical applications
Contact inhibition suppresses geroconversion. In the organism, most cells are contact inhibited. Even proliferating cellular pools exist in a relatively high cell density, albeit they may occupy special niches. Also, near-anoxia suppresses mTOR, thus exerting gerosuppression during cell cycle arrest [40, 130, 135, 136]. Low glucose and amino-acid levels in the organism (compared with cell culture in vitro culture conditions [137]) are also gerosuppressive. Therefore, normal cells senesce slowly in the organism. In vivo, physiological geroconversion may take decades, culminating in age-related diseases.
The gerosuppression model shed light on the treatment with DNA damaging agents. Despite DNA damage, if the organism survives, it does not become old. One of explanations is that contact inhibition is gero-suppressive. This further supports the notion that accumulation of DNA damage is not a cause of aging, which instead is driven by the mTOR pathway.
Also, the model is applicable to tumors. In tumors, necrotic regions coincide with exhaustion of the medium. Thus in large tumors (and any detectable tumors are already large), geroconversion is suppressed. This may explain lack of senescence by conventional drugs, which easily cause senescence in cell culture. Also, solitary cancer cells, trapped among contact-inhibited normal cells, such as epithelial cells are resistant to therapy-induced senescence [41]. This is a subject of our ongoing investigation.
Conflicts of Interest
The authors of this manuscript declare no conflict of interests.
References
- 1. Abercrombie M. Contact inhibition and malignancy. Nature. 1979; 281: 259 -262. [PubMed] .
- 2. Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994; 8: 9 -22. [PubMed] .
- 3. Dietrich C, Wallenfang K, Oesch F, Wieser R. Differences in the mechanisms of growth control in contact-inhibited and serum-deprived human fibroblasts. Oncogene. 1997; 15: 2743 -2747. [PubMed] .
- 4. Levenberg S, Yarden A, Kam Z, Geiger B. p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene. 1999; 18: 869 -876. [PubMed] .
- 5. Faust D, Dolado I, Cuadrado A, Oesch F, Weiss C, Nebreda AR, Dietrich C. p38alpha MAPK is required for contact inhibition. Oncogene. 2005; 24: 7941 -7945. [PubMed] .
- 6. Slisz M, Rothenberger E, Hutter D. Attenuation of p38 MAPK activity upon contact inhibition in fibroblasts. Mol Cell Biochem. 2008; 308: 65 -73. [PubMed] .
- 7. Seluanov A, Hine C, Azpurua J, Feigenson M, Bozzella M, Mao Z, Catania KC, Gorbunova V. Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc Natl Acad Sci U S A. 2009; 106: 19352 -19357. [PubMed] .
- 8. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358 -362. [PubMed] .
- 9. Serrano M, Lim AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK1A. Cell. 1997; 88: 593 -602. [PubMed] .
- 10. McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998; 8: 351 -354. [PubMed] .
- 11. Serrano M and Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol. 2001; 13: 748 -753. [PubMed] .
- 12. Chen QM, Prowse KR, Tu VC, Purdom S, Linskens MH. Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp Cell Res. 2001; 265: 294 -303. [PubMed] .
- 13. Campisi J, Kim SH, Lim CS, Rubio M. Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol. 2001; 36: 1619 -1637. [PubMed] .
- 14. Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJ, Van Lohuizen M, Band V, Campisi J, Dimri GP. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003; 23: 389 -401. [PubMed] .
- 15. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5: 741 -747. [PubMed] .
- 16. Itahana K, Campisi J, Dimri GP. Mechanisms of cellular senescence in human and mouse cells. Biogerontology. 2004; 5: 1 -10. [PubMed] .
- 17. Satyanarayana A, Greenberg RA, Schaetzlein S, Buer J, Masutomi K, Hahn WC, Zimmermann S, Martens U, Manns MP, Rudolph KL. Mitogen stimulation cooperates with telomere shortening to activate DNA damage responses and senescence signaling. Mol Cell Biol. 2004; 24: 5459 -5474. [PubMed] .
- 18. Jacobs JJ and de Lange T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle. 2005; 4: 1364 -1368. [PubMed] .
- 19. Jeyapalan JC and Sedivy JM. How to measure RNA expression in rare senescent cells expressing any specific protein such as p16Ink4a. Aging (Albany NY). 2013; 5: 120 -129. [PubMed] .
- 20. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014; 28: 99 -114. [PubMed] .
- 21. Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M, Gorgoulis VG. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY). 2013; 5: 37 -50. [PubMed] .
- 22. Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013; 152: 340 -351. [PubMed] .
- 23. Leontieva OV and Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013; 12: 3063 -3069. [PubMed] .
- 24. Zhu JY, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998; 12: 2997 -3007. [PubMed] .
- 25. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998; 12: 3008 -3019. [PubMed] .
- 26. Benanti JA and Galloway DA. The normal response to RAS: senescence or transformation? Cell Cycle. 2004; 3: 715 -717. [PubMed] .
- 27. Malumbres M, De Castro IP, Hernandez MI, Jimenez M, Corral T, Pellicer A. Cellular response to oncogenic Ras involves induction of the Cdk4 and Cdk6 inhibitor p15(INK4b). Mol Cell Biol. 2000; 20: 2915 -2925. [PubMed] .
- 28. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4: 159 -165. [PubMed] .
- 29. Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle. 2014; in press .
- 30. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 -3361. [PubMed] .
- 31. Cho S and Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells. 2012; 33: 597 -604. [PubMed] .
- 32. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8: 1888 -1895. [PubMed] .
- 33. Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009; 2: ra75 [PubMed] .
- 34. Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, Liu L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014; 97: 20 -29. [PubMed] .
- 35. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R, Pestell RG, Blagosklonny MV, Lisanti MP. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012; 181: 278 -293. [PubMed] .
- 36. Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012; 11: 2402 -2407. [PubMed] .
- 37. Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012; 11: 4642 -4649. [PubMed] .
- 38. Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Deth Diff. 2013; 20: 1241 -1249. .
- 39. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 -9664. [PubMed] .
- 40. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012; 109: 13314 -13318. [PubMed] .
- 41. Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A. 2014; 111: 8832 -8837. [PubMed] .
- 42. Zhao H, Halicka HD, Li J, Darzynkiewicz Z. Berberine suppresses gero-conversion from cell cycle arrest to senescence. Aging (Albany NY). 2013; 5: 623 -636. [PubMed] .
- 43. Dulic V. Senescence regulation by mTOR. Methods Mol Biol. 2013; 965: 15 -35. [PubMed] .
- 44. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, Gutkind JS. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012; 11: 401 -414. [PubMed] .
- 45. Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage-signaling. Aging (Albany NY). 2012; 4: 952 -965. [PubMed] .
- 46. Menendez JA, Vellon L, Oliveras-Ferraros C, Cufi S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle. 2011; 10: 3658 -3677. [PubMed] .
- 47. CoppŽ JP, Patil CK, Rodier F, Sun Y, Mu-oz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008; 6: 2853 -2868. [PubMed] .
- 48. Serrano M. Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle. 2012; 11: 2231 -2232. [PubMed] .
- 49. Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol. 2013; 14: R32 [PubMed] .
- 50. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11: 973 -979. [PubMed] .
- 51. Hubackova S, Krejcikova K, Bartek J, Hodny Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging (Albany NY). 2012; 4: 932 -951. [PubMed] .
- 52. Ohanna M, Cheli Y, Bonet C, Bonazzi VF, Allegra M, Giuliano S, Bille K, Bahadoran P, Giacchero D, Lacour JP, Boyle GM, Hayward NF, Bertolotto C, Ballotti R. Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget. 2013; 4: 2212 -2224. [PubMed] .
- 53. Berman AE, Leontieva OV, Natarajan V, McCubrey JA, Demidenko ZN, Nikiforov MA. Recent progress in genetics of aging, senescence and longevity: focusing on cancer-related genes. Oncotarget. 2012; 3: 1522 -1532. [PubMed] .
- 54. Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY). 2012; 4: 861 -877. [PubMed] .
- 55. Vizioli MG, Santos J, Pilotti S, Mazzoni M, Anania MC, Miranda C, Pagliardini S, Pierotti MA, Gil J, Greco A. Oncogenic RAS-induced senescence in human primary thyrocytes: molecular effectors and inflammatory secretome involved. Oncotarget. 2014; 5: 8270 -8283. [PubMed] .
- 56. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging (Albany NY). 2009; 1: 281 -288. [PubMed] .
- 57. Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012; 181: 1142 -1146. [PubMed] .
- 58. Blagosklonny MV. Once again on rapamycin-induced insulin resistance and longevity: despite of or owing to. Aging (Albany NY). 2012; 4: 350 -358. [PubMed] .
- 59. Blagosklonny MV. How to save Medicare: the anti-aging remedy. Aging (Albany NY). 2012; 4: 547 -552. [PubMed] .
- 60. Blagosklonny MV. Rapalogs in cancer prevention: Anti-aging or anticancer? Cancer Biol Ther. 2012; 13: 1349 -1354. [PubMed] .
- 61. Blagosklonny MV. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis. 2013; 4: e964 [PubMed] .
- 62. Blagosklonny MV. MTOR-driven quasi-programmed aging as a disposable soma theory: blind watchmaker vs. intelligent designer. Cell Cycle. 2013; 12: 1842 -1847. [PubMed] .
- 63. Blagosklonny MV. M(o)TOR of aging: MTOR as a universal molecular hypothalamus. Aging (Albany NY). 2013; 5: 490 -494. [PubMed] .
- 64. Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging Cell. 2012; 11: 675 -682. [PubMed] .
- 65. Gems DH and de la Guardia YI. Alternative Perspectives on Aging in C. elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2013; 19: 321 -329. [PubMed] .
- 66. Cai D and Liu T. Inflammatory cause of metabolic syndrome via brain stress and NF-kappaB. Aging (Albany NY). 2012; 4: 98 -115. [PubMed] .
- 67. Kennedy BK and Pennypacker JK. Drugs that modulate aging: the promising yet difficult path ahead. Transl Res. 2014; 163: 456 -465. [PubMed] .
- 68. Kaeberlein M. mTOR Inhibition: From Aging to Autism and Beyond. Scientifica (Cairo). 2013; 2013: 849186 [PubMed] .
- 69. Kaeberlein M. Longevity and aging. F1000Prime Rep. 2013; 5: 5 [PubMed] .
- 70. Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013; 342: 1524 -1528. [PubMed] .
- 71. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013; 493: 338 -345. [PubMed] .
- 72. Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013; 23: 53 -62. [PubMed] .
- 73. Kolosova NG, Muraleva NA, Zhdankina AA, Stefanova NA, Fursova AZ, Blagosklonny MV. Prevention of age-related macular degeneration-like retinopathy by rapamycin in rats. Am J Pathol. 2012; 181: 472 -477. [PubMed] .
- 74. Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008; 11: 801 -808. [PubMed] .
- 75. Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009; 5: 279 -289. [PubMed] .
- 76. Gan B and DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009; 8: 1003 -1006. [PubMed] .
- 77. Hinojosa CA, Mgbemena V, Van Roekel S, Austad SN, Miller RA, Bose S, Orihuela CJ. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol. 2012; 47: 958 -965. [PubMed] .
- 78. Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, Perdiguero E, Munoz-Canoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014; 506: 316 -321. [PubMed] .
- 79. Iglesias-Bartolome R and Gutkind SJ. Exploiting the mTOR paradox for disease prevention. Oncotarget. 2012; 3: 1061 -1063. [PubMed] .
- 80. Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging (Albany NY). 2010; 2: 265 -273. [PubMed] .
- 81. Zhang XM, Li L, Xu JJ, Wang N, Liu WJ, Lin XH, Fu YC, Luo LL. Rapamycin preserves the follicle pool reserve and prolongs the ovarian lifespan of female rats via modulating mTOR activation and sirtuin expression. Gene. 2013; 523: 82 -87. [PubMed] .
- 82. Adhikari D, Zheng W, Shen Y, Gorre N, Hamalainen T, Cooney AJ, Huhtaniemi I, Lan ZJ, Liu K. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum Mol Genet. 2010; 19: 397 -410. [PubMed] .
- 83. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandezr E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogenous mice. Nature. 2009; 460: 392 -396. [PubMed] .
- 84. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011; 66: 191 -201. [PubMed] .
- 85. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092 -2097. [PubMed] .
- 86. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10: 4230 -4236. [PubMed] .
- 87. Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012; 4: 144ra103 .
- 88. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53−/− mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4: 715 -722. [PubMed] .
- 89. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/− mice. Aging (Albany NY). 2012; 4: 709 -714. [PubMed] .
- 90. Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging (Albany NY). 2012; 4: 660 -661. [PubMed] .
- 91. Danilov A, Shaposhnikov M, Plyusnina E, Kogan V, Fedichev P, Moskalev A. Selective anticancer agents suppress aging in Drosophila. Oncotarget. 2013; 4: 1507 -1526. [PubMed] .
- 92. Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, Strong R, Bokov A, Javors MA, Ikeno Y, Hubbard G, Hasty P, Sharp ZD. Rapamycin extends life span of Rb1+/− mice by inhibiting neuroendocrine tumors. Aging (Albany NY). 2013; 5: 100 -110. [PubMed] .
- 93. Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, Diaz V, Sloane L, Maslin K, Treaster S, Rendon S, van Remmen H, Ward W, Javors M, Richardson A, Austad SN, et al. Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci. 2014; 69: 119 -130. [PubMed] .
- 94. Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, Hans W, Hettich MM, Holtmeier R, Holter SM, Moreth K, Prehn C, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013; 123: 3272 -3291. [PubMed] .
- 95. Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, Javors MA, Li X, Nadon NL, Nelson JF, Pletcher S, Salmon AB, Sharp ZD, Van Roekel S, Winkleman L, Strong R. Rapamycin-Mediated Lifespan Increase in Mice is Dose and Sex-Dependent and Appears Metabolically Distinct from Dietary Restriction. Aging Cell. 2014; 13: 468 -477. [PubMed] .
- 96. Ye L, Widlund AL, Sims CA, Lamming DW, Guan Y, Davis JG, Sabatini DM, Harrison DE, Vang O, Baur JA. Rapamycin doses sufficient to extend lifespan do not compromise muscle mitochondrial content or endurance. Aging (Albany NY). 2013; 5: 539 -550. [PubMed] .
- 97. Fok WC, Chen Y, Bokov A, Zhang Y, Salmon AB, Diaz V, Javors M, Wood WH 3rd, Becker KG, Perez VI, Richardson A. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS One. 2013; 9: e83988 [PubMed] .
- 98. Hasty P, Livi CB, Dodds SG, Jones D, Strong R, Javors M, Fischer KE, Sloane L, Murthy K, Hubbard G, Sun L, Hurez V, Curiel TJ, Sharp ZD. eRapa Restores a Normal Life Span in a FAP Mouse Model. Cancer Prev Res (Phila). 2014; 7: 169 -178. [PubMed] .
- 99. Fang Y and Bartke A. Prolonged rapamycin treatment led to beneficial metabolic switch. Aging (Albany NY). 2013; 5: 328 -329. [PubMed] .
- 100. Luo LL, Xu JJ, Fu YC. Rapamycin prolongs female reproductive lifespan. Cell Cycle. 2013; 12: 3353 -3354. [PubMed] .
- 101. Spong A and Bartke A. Rapamycin slows aging in mice. Cell Cycle. 2012; 11: 845 [PubMed] .
- 102. Selman C and Partridge L. A double whammy for aging? Rapamycin extends lifespan and inhibits cancer in inbred female mice. Cell Cycle. 2012; 11: 17 -18. [PubMed] .
- 103. Leontieva OV, Paszkiewicz GM, Blagosklonny MV. Weekly administration of rapamycin improves survival and biomarkers in obese male mice on high-fat diet. Aging Cell. 2014; 13: 616 -622. [PubMed] .
- 104. Khapre RV, Kondratova AA, Patel S, Dubrovsky Y, Wrobel M, Antoch MP, Kondratov RV. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 2014; 6: 48 -57. [PubMed] .
- 105. Kondratov RV and Kondratova AA. Rapamycin in preventive (very low) doses. Aging (Albany NY). 2014; 6: 158 -159. [PubMed] .
- 106. Popovich IG, Anisimov VN, Zabezhinski MA, Semenchenko AV, Tyndyk ML, Yurova MN, Blagosklonny MV. Lifespan extension and cancer prevention in HER-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol Ther. 2014; 15: 586 -592. [PubMed] .
- 107. Mabuchi S, Altomare DA, Connolly DC, Klein-Szanto A, Litwin S, Hoelzle MK, Hensley HH, Hamilton TC, Testa JR. RAD001 (Everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res. 2007; 67: 2408 -2413. [PubMed] .
- 108. Levine AJ, Harris CR, Puzio-Kuter AM. The interfaces between signal transduction pathways: IGF-1/mTor, p53 and the Parkinson Disease pathway. Oncotarget. 2012; 3: 1301 -1307. [PubMed] .
- 109. Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol. 2005; 56: 47 -60. [PubMed] .
- 110. Campistol JM, Eris J, Oberbauer R, Friend P, Hutchison B, Morales JM, Claesson K, Stallone G, Russ G, Rostaing L, Kreis H, Burke JT, Brault Y, Scarola JA, Neylan JF. Sirolimus Therapy after Early Cyclosporine Withdrawal Reduces the Risk for Cancer in Adult Renal Transplantation. J Am Soc Nephrol. 2006; 17: 581 -589. [PubMed] .
- 111. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996; 85: 707 -720. [PubMed] .
- 112. Roque M, Reis ED, Cordon-Cardo C, Taubman MB, Fallon JT, Fuster V, Badimon JJ. Effect of p27 deficiency and rapamycin on intimal hyperplasia: in vivo and in vitro studies using a p27 knockout mouse model. Lab Invest. 2001; 81: 895 -903. [PubMed] .
- 113. Diersch S, Wenzel P, Szameitat M, Eser P, Paul MC, Seidler B, Eser S, Messer M, Reichert M, Pagel P, Esposito I, Schmid RM, Saur D, Schneider G. Efemp1 and p27(Kip1) modulate responsiveness of pancreatic cancer cells towards a dual PI3K/mTOR inhibitor in preclinical models. Oncotarget. 2013; 4: 277 -288. [PubMed] .
- 114. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY). 2011; 3: 1130 -1141. [PubMed] .
- 115. Francipane MG and Lagasse E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget. 2013; 4: 1948 -1962. [PubMed] .
- 116. Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013; 23: 53 -62. [PubMed] .
- 117. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Basecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012; 3: 954 -987. [PubMed] .
- 118. Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007; 13: 252 -259. [PubMed] .
- 119. Knecht E, Hernandez-Yago J, Grisolia S. Regulation of lysosomal autophagy in transformed and non-transformed mouse fibroblasts under several growth conditions. Exp Cell Res. 1984; 154: 224 -232. [PubMed] .
- 120. Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res. 2000; 257: 162 -171. [PubMed] .
- 121. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000; 113: 3613 -3622. [PubMed] .
- 122. Yang NC and Hu ML. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp Gerontol. 2005; 40: 813 -819. [PubMed] .
- 123. Gary RK and Kindell SM. Quantitative assay of senescence-associated beta-galactosidase activity in mammalian cell extracts. Anal Biochem. 2005; 343: 329 -334. [PubMed] .
- 124. Coates PJ. Markers of senescence? J Pathol. 2002; 196: 371 -373. [PubMed] .
- 125. Krishna DR, Sperker B, Fritz P, Klotz U. Does pH 6 beta-galactosidase activity indicate cell senescence? Mech Ageing Dev. 1999; 109: 113 -123. [PubMed] .
- 126. Yegorov YE, Akimov SS, Hass R, Zelenin AV, Prudovsky IA. Endogenous beta-galactosidase activity in continuously nonproliferating cells. Exp Cell Res. 1998; 243: 207 -211. [PubMed] .
- 127. Papadopoulos T and Pfeifer U. Protein turnover and cellular autophagy in growing and growth-inhibited 3T3 cells. Exp Cell Res. 1987; 171: 110 -121. [PubMed] .
- 128. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006; 5: 187 -195. [PubMed] .
- 129. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995; 92: 9363 -9367. [PubMed] .
- 130. Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013; 9: 260 -262. [PubMed] .
- 131. Soubeyrand S, Pope L, Hache RJ. Topoisomerase IIalpha-dependent induction of a persistent DNA damage response in response to transient etoposide exposure. Mol Oncol. 2010; 4: 38 -51. [PubMed] .
- 132. Wang L, Roy SK, Eastmond DA. Differential cell cycle-specificity for chromosomal damage induced by merbarone and etoposide in V79 cells. Mutat Res. 2007; 616: 70 -82. [PubMed] .
- 133. Tanaka T, Halicka HD, Traganos F, Seiter K, Darzynkiewicz Z. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: relation to cell cycle phase. Cell Cycle. 2007; 6: 371 -376. [PubMed] .
- 134. Robison JG, Dixon K, Bissler JJ. Cell cycle-and proteasome-dependent formation of etoposide-induced replication protein A (RPA) or Mre11/Rad50/Nbs1 (MRN) complex repair foci. Cell Cycle. 2007; 6: 2399 -2407. [PubMed] .
- 135. Leontieva OV and Blagosklonny MV. Hypoxia and gerosuppression: the mTOR saga continues. Cell Cycle. 2012; 11: 3926 -3931. [PubMed] .
- 136. Demaria M and Campisi J. Matters of life and breath: A role for hypoxia in determining cell state. Aging (Albany NY). 2012; 4: 523 -524. [PubMed] .
- 137. Leontieva OV, Demidenko ZN, Blagosklonny MV. Rapamycin reverses insulin resistance (IR) in high-glucose medium without causing IR in normoglycemic medium. Cell Death Dis. 2014; 5: e1214 [PubMed] .
- 138. Leontieva OV, Geraldine M, Paszkiewicz GM, Blagosklonny MV. Mechanistic or mammalian target of rapamycin (mTOR) may determine robustness in young male mice at the cost of accelerated aging. Aging (Albany NY). 2012; 4: 899 -916. [PubMed] .
- 139. Leontieva OV, Paszkiewicz GM, Blagosklonny MV. Fasting levels of hepatic p-S6 are increased in old mice. Cell Cycle. 2014; 13: 2656 -2659. [PubMed] .
- 140. Leontieva OV, Paszkiewicz GM, Blagosklonny MV. Comparison of rapamycin schedules in mice on high-fat diet. Cell Cycle. 2014; 13: 3350 -3356. [PubMed] .