



Introduction
Obesity and diabetes have reached epidemic proportions in all age groups, but are most pronounced among the elderly [1, 2]. Obesity is characterized by increased fat mass and dysfunction of adipose tissues. There are 2 types of adipose tissues: white adipose tissue (WAT) is a specialized lipid storage organ for storing excess calories; in contrast, brown adipose tissue (BAT) contains large amounts of mitochondria and uses lipids to generate heat [3, 4]. Upon cold-stimulus, the sympathetic nervous system (SNS) is stimulated, which releases norepinephrine (NE) into BAT to activate β3-adrenergic receptor (β3-AR). Subsequently, uncoupling protein 1 (UCP1) is recruited into mitochondria, which promotes lipolysis and heat production [3, 4]. Non-shivering thermogenesis in BAT was recently recognized to play a crucial role in energy balance in rodents and human neonates [3, 5-7]. Thermogenic activity of BAT is positively correlated with energy expenditure, and dysregulation of thermogenesis in BAT is linked to obesity in humans [6]. Most recently, studies have shown that enhanced thermogenesis also improves glucose homeostasis and insulin sensitivity in animals [8] and humans [9]. These results suggest that interventions to increase BAT mass and/or activity may be very attractive strategies for prevention/treatment of obesity and diabetes.
Aging is associated with severe thermogenic impairment; BAT declines 95% in mass and 75% in activity in old men, as compared to young men [10, 11]. Thermogenic impairment likely contributes to age-associated obesity [12], However, the factors underlying the dysfunction of BAT in aging are currently unknown. During aging, cumulative molecular damage leads to impairment and functional decline [13, 14]. Age-associated mitochondrial dysfunction is involved in pathogenesis of metabolic disorders and neuro-degenerative diseases [15-18]. Mitochondria are dynamic organelles responsible for cellular energy production in response to cell signals [19]. Mitochondrial dynamics are determined by fusion and fission processes; the balance between fusion and fission is essential for the maintenance of normal mitochondrial function. Fusion is a ‘joining event’ between two different mitochondria, mediated by mitofusins (Mfns) and optic atrophy gene 1 (OPA1); fission is a process dividing one mitochondrion into two mitochondria, mediated by dynamin-related protein 1 (Drp1) and fission 1 (Fis1) protein [20]. It was recently reported that expression of Mfn2 and Drp1 genes is reduced in the skeletal muscle of elderly humans [21], and smaller and fragmented mitochondria are more abundant in the muscle of obese and type 2 diabetic subjects [22]. Furthermore, reduced expression of fission-related protein Drp1 has been linked to decreased mitochondrial complex IV activity in HeLa cells [23]. Drp1 deletion has been shown to decrease mtDNA content [24], and cause brain developmental defects and severe neuro-degeneration [25].
Ghrelin is the only known orexigenic hormone to increase appetite and promote obesity [26-28]. We and others have reported that ghrelin's effects on GH release and appetite are mediated through its receptor, the Growth Hormone Secretagogue Receptor (GHS-R) [29-31]. Ghrelin is ubiquitously expressed, but the highest level of expression is detected in the stomach and intestine [32]. The expression of GHS-R is more restricted; the highest expression is detected in pituitary and brain, but lower levels of expression are detectable in some peripheral tissues, including WAT and BAT [31, 33, 34]. It has been reported that ghrelin stimulates lipid accumulation in WAT [35, 36], but suppresses norepinephrine (NE) release of BAT [37, 38]. Aging is associated with insulin resistance, and ghrelin is known to increase insulin resistance [39]. We have found that deletion of Ghsr gene enhances BAT thermogenesis in aged mice, resulting in a lean and insulin-sensitive phenotype [34].
Ghrelin (aka “active ghrelin”), and its related peptides des-acyl ghrelin (DAG) and obestatin, are all derived from the same preproghrelin gene [40, 41]. However, only ghrelin activates GHS-R; the effects of DAG and obestatin are mediated through other receptors [40]. We have shown that DAG has ghrelin-like effects on feeding, but it cannot activate GHS-R [42]. It has also been shown that DAG stimulates lipid accumulation in visceral fat [35], has adipogenic effects in bone marrow [43], and prevents diet-induced adipose inflammation and development of diabetes [44]. In contrast, obestatin is an anorexic hormone; its effect on appetite is opposite from ghrelin [41]. We previously reported that ghrelin-null nice (Ghrelin−/−), absent of ghrelin, DAG and obestatin, show no thermogenic phenotype [45]. On the other hand, Ghsr knockout mice (Ghsr−/−, where ghrelin's effect is blocked but the effects of DAG and obestatin remain intact) show enhanced thermogenesis [34]. The differential thermogenic phenotypes of Ghrelin−/− and Ghsr−/− mice raise the question of whether DAG and/or obestatin have opposing effects on thermogenesis as compared to ghrelin.
To better understand the role of ghrelin signaling in the regulation of age-associated decline of thermogenesis, we analyzed ghrelin and its related peptides in young, middle-aged and old mice, and characterized thermogenic signaling cascades, insulin activation, and mitochondrial biogenesis and dynamics in BAT of old Ghsr−/− mice. We also investigated whether GHS-R mediated thermogenesis is likely regulated centrally and/or peripherally.
Results
Obestatin increases UCP1 expression in HIB1B cells, but DAG has no effect
Our previous study showed that ghrelin inhibits adipogenesis and suppresses UCP1 expression in brown adipocyte HIB1B cells [34]. DAG and obestatin have been reported to have different physiological effects from ghrelin in several cell types and tissues [40, 41, 46, 47]. To assess the effects of DAG and obestatin on thermogenesis, we treated differentiated HIB1B cells with different concentrations of DAG or obestatin. Consistent with our previous report [34], 1 nM ghrelin inhibits UCP1 expression in differentiated HIB1B cells (Fig. 1A). In contrast, DAG had no effect on UCP1 expression even at high concentration (Fig. 1B), but obestatin increased UCP1 gene expression in a dose-dependent manner (Fig. 1C). Ghrelin and obestatin showed opposite effects on UCP1 expression in brown adipocytes (Fig. 1D), suggesting that ghrelin and obestatin may have opposing effects on thermogenesis.
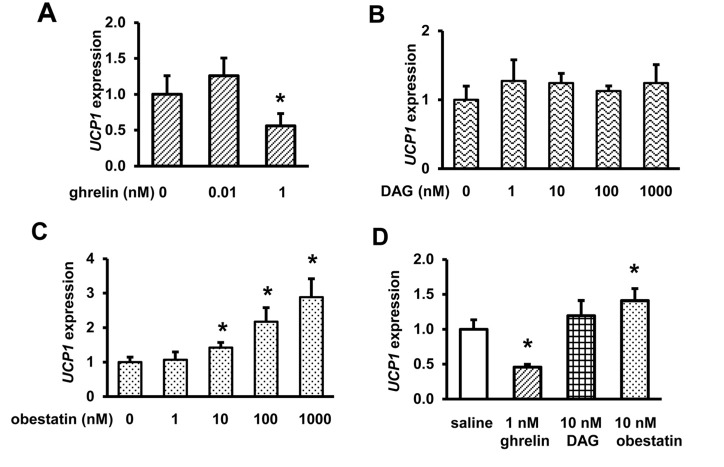
Figure 1. Ghrelin and obestatin exhibit differential effects on UCP1 expression in differentiated brown adipocyte HIB1B cells UCP1 expression in HIB1B cells treated with saline or different concentrations of ghrelin (A), des-acyl ghrelin, DAG (B), and obestatin (C). Summary of UCP1 expression in HIB1B cells treated with saline, 1 nM ghrelin, 10 nM DAG, or 10 nM obestatin (D). N = 9, and each assay was measured in triplicate. *p<0.05, Treatments vs. Controls.
Circulating ghrelin is increased in aging, but DAG and obestatin are not changed
To assess whether ghrelin, DAG and obestatin regulate thermogenesis and insulin resistance during aging, we measured circulating ghrelin, DAG, obestatin and insulin concentrations in young, middle-aged and old wild-type (WT) mice, under either fed or fasting conditions. Under fed condition, ghrelin concentration in old mice was significantly higher than young and middle-aged mice (Fig. 2A). Interestingly, fasting only increased ghrelin in young and middle-aged mice, but not old mice (Fig. 2A), suggesting that ghrelin regulation in old mice is less responsive to fasting. The concentrations of DAG and obestatin didn't change during aging (Fig. 2B and 2C); while fasting did not affect DAG (Fig. 2B), fasting significantly decreased obestatin in middle-aged and old mice (Fig. 2C). Elevated ghrelin in aging increases lipolysis and exacerbates insulin resistance [39]. Consistently, our data showed that fed insulin levels were higher in middle-aged and old mice relative to young mice, and fasting reduced insulin (Fig. 2D). The data support that increased ghrelin in old mice promotes age-associated insulin resistance, which may further impair BAT function in aging.
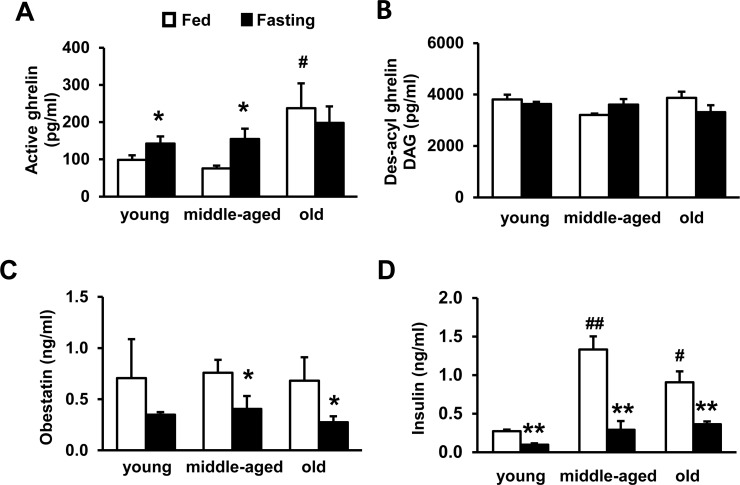
Figure 2. Plasma concentrations of active ghrelin, des-acylated ghrelin, and obestatin during aging Active ghrelin (A), des-acyl ghrelin, DAG (B), obestatin (C), and insulin (D) of young, middle-aged and old WT mice under fed or fasting conditions. Young (4-months), middle-aged (10-months) and old (22-months) mice were used. The mice were fasted overnight. N = 10-12. *p<0.05, fed vs. fasting. #p<0.05, ## P<0.001, young vs. middle-aged or old.
GHS-R ablation attenuates age-associated decline of thermogenesis and activates insulin signaling in BAT
Similar to our previous reports [34, 45], energy expenditure of WT mice decreased with age in both light and dark cycles, and GHS-R ablation increased energy expenditure in middle-aged and old mice (Fig. 3A). GHS-R ablation did not alter food intake or physical activity in any age group, indicating that GHS-R exerts its effects primarily by regulating energy expenditure. We studied GHS-R expression in BAT of young, middle-aged and old mice, and found that Ghsr expression in BAT increased during aging (Fig. 3B).
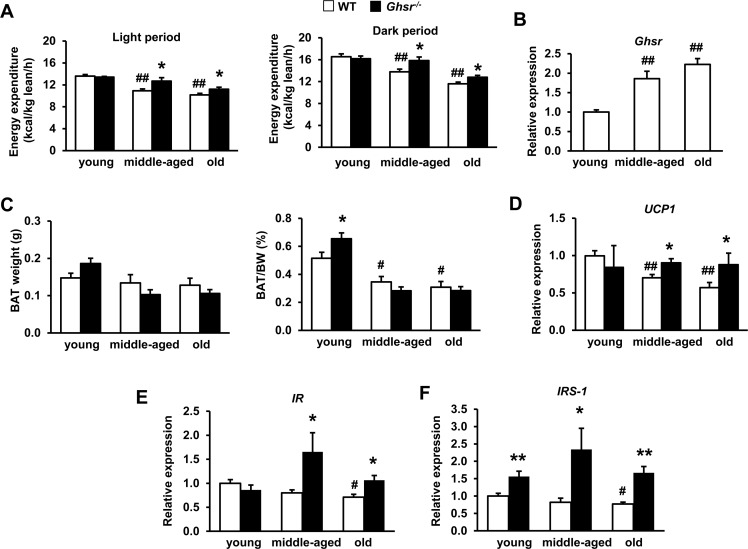
Figure 3. Ghrelin receptor ablation attenuates age-associated decline of thermogenesis and enhances insulin signaling in BAT (A) Energy expenditure of young, middle-aged and old WT and Ghsr−/− mice normalized by lean body mass. (B) Ghsr expression in BAT of young, middle-aged and old WT mice. (C) BAT mass and percentage of BAT weight over body weight in young, middle-aged and old WT and Ghsr−/− mice. (D) UCP1 expression in BAT of young, middle-aged and old WT and Ghsr−/− mice. IR (E) and IRS-1 (F) expression in BAT of young, middle-aged and old WT and Ghsr−/− mice. Young (4-months), middle-aged (10-months) and old (22-months) mice were used. N = 6-14. *p<0.05, **p<0.001, WT vs. Ghsr−/−; #p<0.05, ##p<0.001, old or middle-aged vs. young.
Higher circulating ghrelin and increased GHS-R expression in BAT of aged mice suggest enhanced ghrelin signaling in BAT during aging, which may promote age-associated decline of thermogenesis. To further define the role of GHS-R in thermogenesis of BAT, we compared BAT mass and UCP1 expression in BAT of young, middle-aged and old WT and Ghsr−/− mice. Although the absolute weight of BAT didn't change with age, BAT mass normalized to body weight decreased during aging in both WT and Ghsr−/− mice (Fig. 3C). There was no significant difference in the ratio of BAT/body weight between WT and Ghsr−/− mice in middle-aged and old mice, while the ratio of BAT/body weight in young Ghsr−/− mice was higher than that of young WT mice (Fig. 3C). While UCP1 expression in BAT of WT mice declined during aging, higher UCP1 expression was detected in BAT of GHS-R ablated aged (middle-aged and old) mice, maintaining expression levels similar to that of young mice (Fig. 3D). These results suggest that GHS-R ablation improves thermogenesis through regulation of BAT activity, but not BAT mass.
It was reported that BAT regulates glucose homeostasis and insulin sensitivity [8, 9]. It has been shown that insulin signaling in BAT is activated by cold stress [48]. Our previous studies showed that aged Ghsr−/− mice have improved insulin sensitivity [34]. Indeed, we detected lower expression of insulin receptor (IR) and insulin receptor substrate 1 (IRS-1) in BAT of old WT mice, but GHS-R ablation increased IR and IRS-1 expression in BAT (Fig. 3E and 3F). Together, these data suggest that GHS-R expression in BAT is increased during aging, and ablation of GHS-R attenuates age-associated decline of thermogenesis and insulin sensitivity in BAT.
Ablation of GHS-R activates thermogenic signaling and improves mitochondrial dynamics of BAT
BAT thermogenic capacity is determined by the availability of fuel substrate in BAT and UCP1 activity in mitochondria [3]. Protein kinase A (PKA) activates hormone-sensitive lipase (HSL), which is a predominant lipase for catecholamine-stimulated lipolysis in brown adipocytes [49]. Our data revealed that ablation of GHS-R stimulates PKA and HSL activation in BAT, indicative of stimulated thermogenic signaling (Fig. 4A). cAMP response element-binding protein (Creb) is a known downstream target of PKA; phosphorylation of Creb is known to increase the expression of mitochondrial respiratory chain proteins and UCP1 expression [50, 51]. As expected, Creb phosphorylation is increased in BAT of Ghsr−/− mice (Fig. 4A). Consistently, our ex vivo lipolysis study showed that β-adrenergic agonist CL316243-treated brown adipocytes of Ghsr−/− mice exhibited higher basal and stimulated glycerol release when compared with those of WT mice (Fig. 4B). These data together demonstrate that GHS-R ablation activates thermogenic signaling cascade and promotes lipolysis in BAT.
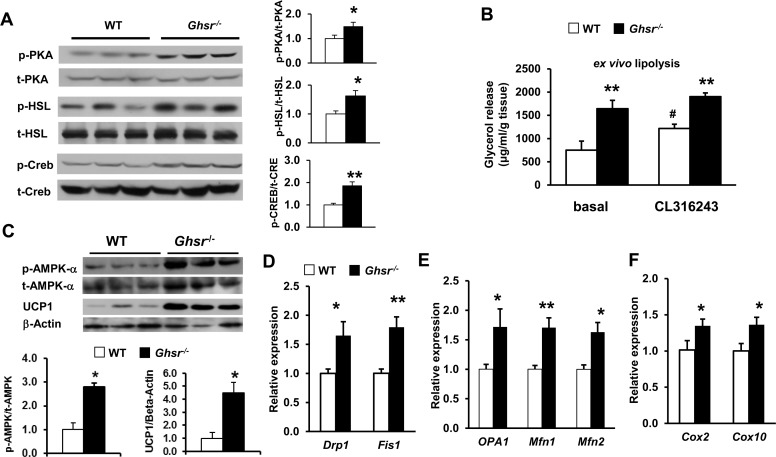
Figure 4. Ablation of GHS-R activates thermogenic signaling cascade and improves mitochondrial dynamics of BAT in aged mice The representative data presented here were from 22 month-old WT and Ghsr−/− mice. (A) Representative Western blots of key thermogenic regulators in BAT of aged WT and Ghsr−/− mice. (B) Ex vivo lipolysis of BAT of aged WT and Ghsr−/− mice, treated with or without 10 μM CL316243. (C) Representative Western blots show phosphorylated AMPK α (p-AMPK-α), total AMPK (t-AMPK-α) and UCP1 protein expression in BAT of old WT and Ghsr−/− mice after 4 hours of cold exposure. (D) Expression of mitochondrial fission genes in BAT of aged WT and Ghsr−/− mice. (E) Expression of mitochondrial fusion genes in BAT of aged WT and Ghsr−/− mice. (F) Expression of mitochondrial complex biogenesis markers in BAT of aged WT and Ghsr−/− mice. N = 6. *p<0.05, **p<0.001, WT vs. Ghsr−/−; #p<0.05, basal vs. CL316243 treatment.
The 5′ AMP-activated protein kinase (AMPK), a key nutrient and energy sensor, is the master regulator of cellular energy metabolism in many tissues, including BAT [52, 53]. Cold exposure increases AMPK activity [52], and aging is associated with decreased AMPK activity [54]. To test whether GHS-R ablation in BAT activates AMPK, we investigated AMPK activity by studying protein levels of phosphorylated AMPK and total AMPK. Enhanced phosphorylated AMPK was detected in BAT of Ghsr−/− mice, and was correlated with increased UCP1 (Fig. 4C). This result suggests that AMPK activity may facilitate GHS-R mediated thermogenic regulation.
Mitochondrial biogenesis and dynamics are critical for mitochondrial function [20], and mitochondrial dynamics have been suggested to play a critical role in the pathogenies of insulin resistance [55-57]. We thus studied expression of key mitochondrial dynamics genes in BAT of aged WT and Ghsr−/− mice. The expression of Drp1, Fis1, OPA1, Mfn1, and Mfn2 was significantly increased in aged Ghsr−/− mice (Fig. 4D and 4E). Consistently, the expression of subunits of mitochondrial respiratory chain complexes IV, cox2 and cox10 was increased in the BAT of Ghsr−/− mice, indicative of increased mitochondrial activity (Fig. 4F). These data suggest that mitochondrial dynamics may also play a role in GHS-R mediated thermogenic regulation.
GHS-R regulates BAT thermogenesis through both central and peripheral mechanisms
We have evidence that GHS-R regulates thermogenic function, and ablation of GHS-R attenuates the age-associated decline of thermogenesis [34]. However, since the data were obtained from global GHS-R knockout mice, we could not determine with certainty whether the effect of GHS-R on thermogenesis is mediated through central SNS and/or peripheral brown adipocytes. SNS activation increases catecholamine (norepinephrine, NE) release at SNS endings in BAT, which in turn activates β3-AR in brown adipocytes. To determine the role of SNS-induced catecholamine release in GHS-R mediated thermogenesis, we assessed NE in the circulation and in BAT of WT and Ghsr−/− mice. Since urinary cate-cholamines are less likely to be affected by handling stress [58], we measured NE concentration in urine of young, middle-aged and old WT and Ghsr−/− mice. As expected, urinary NE decreased with age in WT mice, while GHS-R ablated mice maintained NE at a level comparable to that of young mice (Fig. 5A).
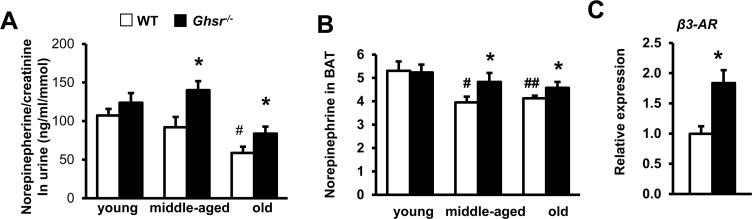
Figure 5. Ablation of GHS-R prevents age-associated decline of norepinephrine (NE) and enhances NE-responsiveness NE concentrations in the urine (A) and NE levels in BAT (B) of young, middle-aged and old WT and Ghsr−/− mice. Young (4-months), middle-aged (10-months) and old (22-months) mice were used. (C) β3-AR mRNA expression in BAT of 22 month-old WT and Ghsr−/− mice. N = 6-10. *p<0.05, WT vs. Ghsr−/−; #p<0.05, ##p<0.001, old or middle-aged vs. young.
Similarly, NE content in BAT of WT mice declined with age, while GHS-R ablation prevented NE decline in BAT of middle-age and old mice (Fig. 5B). Consistently, the expression of β3-AR in BAT was higher in old Ghsr−/− mice than in old WT mice (Fig. 5C). Increased SNS-induced NE release and β3-AR expression in BAT of old Ghsr−/− mice suggest that GHS-R ablation regulates BAT thermogenesis, at least in part, by activating central SNS-mediated thermogenic signaling pathway of SNS-NE-β3-AR.
To determine NE sensitivity of BAT, we studied NE-induced thermogenesis in old WT and Ghsr−/− mice. O2 consumption after NE injection was significantly higher in old Ghsr−/− compared with old WT mice (Fig. 6A), suggesting that BAT of old Ghsr−/− mice is more sensitive to NE stimulation. To assess whether GHS-R has direct effect in brown adipocytes, we further knocked down GHS-R in brown adipocytes (HIB1B) using shGHS-R. Transfection with shGHS-R resulted in 70% knockdown of GHS-R compared with scrambled (shScr) control (Fig. 6B). The expression of thermogenic marker UCP1 and adipogenic marker Peroxisome Proliferator-Activated Receptor γ (PPARγ) was significantly increased in isoproterenol-stimulated GHS-R knockdown HIB1B cells (Fig. 6B). These data suggest that GHS-R also directly inhibits thermogenic activity in brown adipocytes.
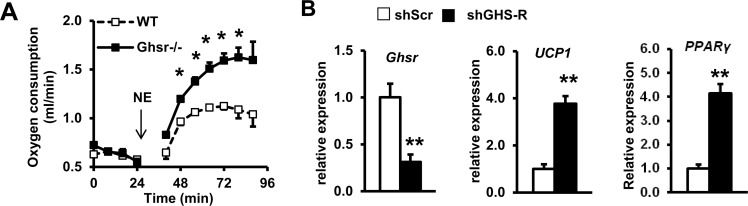
Figure 6. GHS-R directly regulates thermogenic activation in brown adipocytes (A) Oxygen consumption of old WT and Ghsr−/− mice after 1.5 mg/Kg subcutaneous injection of NE. N = 10-12. *p<0.05, WT vs. Ghsr−/−. (B) Expression of Ghsr, UCP1 and PPARγ in shGHS-R knockdown HIB1B cells. shScr represents scramble shRNA; shGHS-R represents shRNA specific for GHS-R. N = 9. **p<0.001, shScr vs. shGHS-R.
Discussion
We previously showed that ghrelin decreases UCP1 expression in brown adipocyte HIB1B cells through GHS-R [34]. Current data suggest that obestatin increases UCP1 expression in HIB1B cells, while DAG has no effect. Thus, ghrelin and obestatin exert opposing effects on thermogenesis. Our data are in agreement with published reports showing that ghrelin decreases UCP1 expression in BAT [59], while obestatin attenuates the hypothermia of Ghrelin−/− mice [60]. We observed improved thermogenesis in Ghsr−/− mice, but not in Ghrelin−/− mice [34, 45]. The absence of a thermogenic phenotype in Ghrelin−/− mice may be explained by the fact that both ghrelin and obestatin are absent in Ghrelin−/− mice; the opposing thermogenic effects of ghrelin and obestatin are eliminated, thus showing no thermogenic phenotype. On the other hand, Ghsr−/− mice express both ghrelin and obestatin. Since GHS-R is exclusively activated by ghrelin, only ghrelin signaling is blocked in Ghsr−/− mice, while obestatin signaling is intact. In Ghsr−/− mice, the stimulatory effect of obestatin on thermogenesis is unopposed, which may explain the increased thermogenesis observed in Ghsr−/− mice. Thus, the differential thermogenic phenotypes of Ghrelin−/− and Ghsr−/− mice are likely due to the opposing effects of ghrelin and obestatin on thermogenesis.
Fasting is associated with decreased energy expenditure, and UCP1 expression in brown fat decreases during fasting [61, 62]. Fasting is associated with increased ghrelin and decreased obestatin in the circulation [26, 41]. The changes of ghrelin and obestatin during fasting may contribute to the decrease of thermogenesis and energy expenditure. After meals, postprandial ghrelin and obestatin decrease, the ratio of ghrelin and obestatin may be involved in the regulation of diet-induced thermogenesis. Aging is associated with severely diminished thermogenic function [10, 11]. Here we found that circulating ghrelin is increased during aging. We previously reported that low levels of GHS-R expression are detectable in BAT [34]. Current data reveal GHS-R expression in BAT is increased during aging. Higher concentration of ghrelin has been shown to be associated with lower resting metabolic rate and postprandial thermogenesis in humans [63]. Ghrelin promotes fat deposition in white fat and suppresses sympathetic nerve activity in brown fat [64, 65]. Our new data show that thermogenic suppressor ghrelin increases during aging; conversely, thermogenic stimulator obestatin remains steady during aging, which may contribute to the age-associated decline of thermogenesis. Collectively, our data suggest that ghrelin signaling contributes to the age-associated thermogenic dysfunction, which in turn promotes obesity and insulin resistance in aging.
Our data further demonstrate that GHS-R regulates thermogenic signaling cascades in BAT. GHS-R ablation activates thermogenic signaling pathway PKA-Creb-UCP1, and lipolytic pathway PKA-HSL-UCP1. It has been reported that insulin resistance in BAT is associated with thermogenic defect in obese and diabetic rats [48, 66]. Age-associated mito-chondrial dysfunction is a well-known cause for insulin resistance [15-17]. It has been shown that acute cold exposure improves glucose clearance and insulin sensitivity of BAT by activating the PKA-HSL-Lipolysis pathway [48]. We observed decreased expression of IR and IRS-1 in BAT of old Ghsr−/− mice, suggesting that GHS-R ablation improves insulin sensitivity of BAT, which then further promotes BAT thermogenic activation.
AMPK is the master regulator of cellular energy metabolism in BAT [52, 53]. It is well documented that activation of AMPK enhances PGC1α-dependent transcription, which senses energy availability and stimulates mitochondrial DNA and protein synthesis [53, 67]. Ghrelin has been shown to have differential effects on AMPK activity: stimulatory in hypothalamus and heart, but inhibitory in liver and WAT [68, 69]. Our previous study showed that GHS-R ablation increases mitochondrial content [34]. Our current data reveal that higher levels of phosphorylated AMPK in BAT of Ghsr−/− mice are correlated with increased UCP1. This suggests that GHS-R ablation may activate AMPK to increase mitochondrial biosynthesis of BAT, thus enhancing thermogenesis.
Mitochondrial dynamics play a crucial role in mitochondrial function [20]. Abnormal mitochondrial structure has been observed i n obese and type 2 diabetic patients [22]. GHS-R deletion increased the expression of both fusion- and fission-related genes in BAT of aged Ghsr−/− mice, indicative of an active state of mitochondrial dynamics and improved mitochondrial homeostasis. Improved mitochondrial homeostasis will enhance the sensitivity of mitochondria to FFA to further promote mitochondrial uncoupling. The data suggest that suppressing ghrelin signaling preserves youthful mitochondrial dynamics in BAT during aging. It has been shown that obesity induces Mfn2 deficiency which results in reduced mitochondrial activity, and Drp1 deletion leads to reduction of mtDNA content [24, 70]. We found that the expression of Mfn2 and Drp1 was increased in BAT of aged Ghsr−/− mice, consistent with the increased thermogenic activity and mtDNA content detected in BAT of aged Ghsr−/− mice [34]. In addition, the subunits of mitochondrial respiratory chain complexes IV, such as mtDNA-encoded cox2 and nuclear DNA-encoded cox10, were also increased in Ghsr−/− mice, lending further support to the increased mitochondrial biogenesis. Taken together, we conclude that GHS-R ablation activates thermogenic signaling cascades and promotes lipolysis in BAT; GHS-R ablation enhances mitochondrial function of BAT by increasing mitochondrial biogenesis and stimulating mitochondrial dynamics.
Last, we explore the potential site(s) of action of GHS-R mediated thermogenesis. The phenotype we observed in Ghsr−/− mice could result from GHS-R mediated effects in central and/or peripheral sites. Thyroid hormones are important regulators of thermogenesis [71]. However, serum T3 and T4 concentrations were comparable in WT and Ghsr−/− mice [34], indicating that the elevated thermogenesis observed in old Ghsr−/− mice is not due to changes in circulating thyroid hormones. Sympathetic activity is known to play a dominant role in thermogenic regulation [3, 4]. Aging is associated with decreased stress responsiveness, because of reduced circulating NE and impaired hypothalamic-pituitary-adrenal axis [72]. In the current study, we found that GHS-R ablation prevents age-associated decrease of NE in the circulation, suggesting that GHS-R mediated thermogenesis is likely regulated centrally via SNS-induced NE release. On the other hand, our data also showed that GHS-R ablation increases NE-induced O2 consumption, indicative that BAT of Ghsr−/− mice has increased sensitivity to NE stimulation; this suggests that GHS-R ablation may directly affect the thermogenic response of BAT. Indeed, we found that knockdown of GHS-R in HIB1B cells up-regulates the thermogenic mediator UCP1, suggesting that ghrelin signaling regulates thermogenesis directly in BAT. Together, our data suggest that ghrelin signaling regulates BAT ther-mogenesis via both central and peripheral mechanisms.
In summary, our studies show that ghrelin and GHS-R are important thermogenic regulators, and increased ghrelin signaling in BAT during aging contributes to the age-associated thermogenic impairment. GHS-R mediated thermogenesis in BAT is attributable to thermogenic signaling activation, insulin sensitivity, mitochondrial biogenesis and dynamics of BAT (Fig. 7). GHS-R likely regulates thermogenesis via both central mechanisms involving SNS-induced NE release and direct effect in BAT. Thus, GHS-R antagonists may serve as a unique class of drugs that can prevent/treat age-associated obesity and insulin resistance by enhancing thermogenesis.
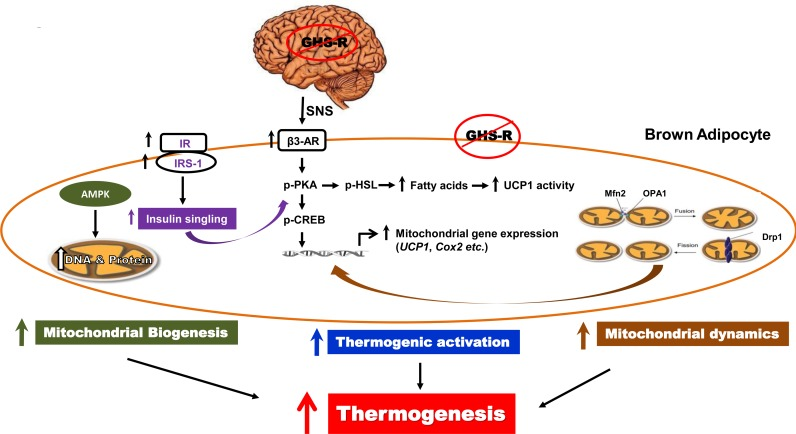
Figure 7. Schematic diagram of GHS-R mediated thermogenic regulation in brown adipocytes Our data suggest that ghrelin signaling may regulate thermogenesis in BAT via the following 4 independent and interconnected signaling pathways: 1) Ablation of GHS-R stimulates SNS-mediated NE release, which in turn induces β3-AR expression, subsequently activating thermogenic signaling cascades in BAT. This involves activation of thermogenic signaling pathway PKA-CREB-UCP1 and lipolytic pathway PKA-HSL-UCP1. 2) Ablation of GHS-R enhances insulin signaling in BAT, which improves insulin sensitivity of BAT and activates key thermogenic regulator PKA. 3) Ablation of GHS-R enhances AMPK activity in BAT, which increases DNA and protein synthesis of mitochondria, thus increasing mitochondrial biogenesis. 4) Ablation of GHS-R augments mitochondrial dynamics, enhancing both mitochondrial fission and fussion; this restores mitochondrial architecture and improves mitochondrial homeostasis. Improved mitochondrial homeostasis enhances the sensitivity of mitochondria to FFA to further promote mitochondrial uncoupling. Collectively, GHS-R ablation increases thermogenesis in BAT by activating thermogenic signaling, sensitizing insulin signaling, increasing mitochondrial biogenesis, and enhancing mitochondrial dynamics.
Methods
Animals
Ghsr−/− mice were generated and genotyped as described previously [29]. All mice used in the experiments are male mice, which have been backcrossed 13 generations onto C57BL/6J background. Wild-type (WT) and homozygous knockout mice (Ghsr−/−) were housed and bred in a pathogen-free facility at Baylor College of Medicine. Animals were housed under controlled temperature and lighting cycle (75±1 °F; 12h light-dark cycle) with free access to regular chow and water. All experiments were approved by the Animal Care Research Committee of Baylor College of Medicine. To determine data-relevant age cohorts, we tested young mice at 3-4 months-of-age, middle-aged mice at 10-12 months-of-age, and old mice at 18-24 months-of-age. “Aged” is defined as middle-aged or old mice.
Real-time RT-PCR
Total cellular RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA), following the manufacturer's instructions. In order to remove potential genomic DNA contamination, RNA was treated with DNAse and run on gels to validate the purity and quality. cDNA was synthesized from 1 μg RNA using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Real-time RT-PCR was performed on an ABI 7900 using the SYBR Green PCR Master Mix, the Taqman gene expression Master Mix (Applied Biosystems, Carlsbad, CA), or Bio-Rad using iQ SYBR GREEN supermix (Bio-Rad, Hercules, CA), according to the protocols provided by the manufacturers. 18s RNA and β-actin were used as internal controls. All primer and probe information is available upon request.
Plasma analyses
For active ghrelin and des-acyl ghrelin (DAG): about 200 μl blood was collected in EDTA-coated capillary tubes with or without 18 hours fasting. After spin down, plasma was transferred to an ice-cold tube with 5 μl 1N HCl. Then, 1 μl fresh PMSF (10 mg/ml in methanol, Sigma, St. Louis, MO) was added and gently mixed. 25 μl plasma was measured with either RIA assay kits (EMD Millipore, Billerica, MA) or ELISA kits (Mitsubishi Kagaku Iatron, Tokyo, Japan). For obestatin: about 200 μl blood was collected in EDTA-coated capillary tubes. The blood sample was then transferred to tubes and mixed with 20 μl aprotinin (6 TIU/ml, Phoenix, Milpitas, CA). After spin down, plasma was transferred to a new ice-chilled tube. 50 μl of plasma was measured with an EIA kit (Phoenix, Milpitas, CA). For insulin: about 200 μl blood was collected in EDTA-coated capillary tubes. The blood sample was then transferred to tubes. After spin down, plasma was transferred to a new ice-chilled tube. 50 μl of plasma was measured with a RIA kit (Linco Research, St. Charles, MI), following manufacture's instruction.
Metabolic characterizations
Metabolic parameters were obtained using an Oxymax open-circuit indirect calorimetry system (Columbus Instruments, Columbus, OH), as previously described [34, 73]. Briefly, mice were individually caged in chambers and given free access to regular chow and water for one week prior to tests. The first 24 hours in calorimetry chambers was considered the acclimation phase, and data were analyzed only for the following 48 hours. Oxygen consumption (VO2) and carbon dioxide production (VCO2) by each animal were measured. Energy expenditure (EE, or heat generation) was calculated from VO2 and VCO2 gas exchange data as follows: EE = (3.815+1.232×VCO2/VO2) × VO2. Energy expenditure was then normalized to lean body mass.
For norepinephrine-induced thermogenesis, the animals were anesthetized with pentobarbital (90 mg/kg, i.p.), and indirect calorimetry was performed for 30 minutes to obtain basal values as earlier described [74]. The individual mouse was then briefly removed from the calorimetry chambers, injected with norepinephrine (1.5 mg norepinephrine/kg, subcutaneously), and then returned to the metabolic chamber, and oxygen consumption was then measured for another 60 minutes.
Urinary norepinephrine assay
The mice urinary NE concentration was determined as described previously [58, 73]. Briefly, mice were grabbed quickly to induce urination. About 200 μl urine sample was collected into a tube with 2 μl 6N HCl, to determine NE level using ELISA assay (IBL Inc., Minneapolis, MN). Meanwhile, about 10 μl urine sample was transferred to another tube for creatinine determination using ELISA assay (Quidel Corporation, San Diego, CA). Creatinine-normalized norepinephrine levels are shown.
BAT norepinephrine assay
The BAT NE concentration of the mice was determined as previously described [75]. Briefly, BATs were homogenized by sonication in homogenization buffer (1 N HCl, 0.25 M EDTA, 1 mM Na2S2O5). Cell debris was then pelleted by centrifugation at 13,000 r.p.m. for 15 min at 4 °C. The cleared homogenates were collected and stored at −80 °C before quantification. Fifty μl tissue lysate was used for measurement of NE using ELISA assay (Rocky Mountain Diagnostics, Colorado Springs, CO) following the manufacturer's protocol. All samples were normalized to total tissue protein content.
Western blot analyses
Brown adipose tissues were lysed in RIPA buffer with Complete Protease Inhibitor Cocktail (Roche Inc., Indianapolis, IN). Protein concentration was determined with BCA protein assay kit (Pierce, Rockford, IL). Twenty microgram of protein of each sample was separated by SDS-PAGE, and electro-transferred to nitrocellulose membrane for immunoblot analyses. The following antibodies were used: anti-p-PKA (Tyr197) (Cell Signaling, Danvers, MA, 4781S, 1:1000), anti-t-PKA (Cell Signaling, Danvers, MA, 4782, 1:1000), anti-p-HSL (Ser563) (Cell Signaling, Danvers, MA, 4139S, 1:1000), anti-t-HSL (Cell Signaling, Danvers, MA, 4107, 1:1000), anti-p-Creb (Ser133) (Cell Signaling, Danvers, MA, 9191S, 1:1000), anti-t-Creb (Cell Signaling, Danvers, MA, 9197, 1:1000), p-AMPKα (T172) (Cell Signaling, Danvers, MA, 2535L, 1:1000), anti t-AMPK (Cell Signaling, Danvers, MA, 2603S, 1:1000), anti-UCP1 (ABCAM, Cambridge, MA Ab10983, 1:10000), anti-β-actin (Cell Signaling, Danvers, MA, 4967S, 1:1000), HRP-conjugated anti-mouse (GE Healthcare UK Limited, 1:10,000), and anti-rabbit (GE Healthcare UK Limited, 1:10,000). The SuperSignal West Pico Chemiluminescent kit (Pierce, Rockford, IL) was used as substrates.
Ex vivo lipolysis assay
The lipolysis activity of BAT was measured, using ex vivo lipolysis assay as described [76]. Briefly, interscapular BAT was dissected and separated into two pieces. Each piece was put into culture medium {DMEM with 0.5% fatty acid free BSA (Sigma, St. Louis, MO)} and minced into tiny pieces with scissors. The tissues were incubated at 37°C with 10 μM CL316243 (Sigma, St. Louis, MO) as stimulated condition, or DMSO as basal condition. 2 hours later, medium was collected and heated at 85°C for 10 minutes. After spin down, clear supernatant was transferred to a new tube, and 10 μl was used to measure free glycerol content using Free Glycerol Reagent (Sigma, St. Louis, MO). Lipolysis activity was represented by glycerol concentrations, normalized by weight of the tissue.
HIB1B cell culture
HIB1B pre-adipocytes were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% bovine calf serum. At confluence, HIB1B cells were induced to differentiate for 3 days in DMEM with 10% Cosmic Calf Serum (Hyclone, Logan, UT), supplemented with 5 μg/ml insulin (Sigma, St. Louis, MO), 0.5 mM isobutylmethylxanthine (Sigma, St. Louis, MO), 1 μM dexamethasone (Sigma, St. Louis, MO), and 1 nM triiodothyronine (Sigma, St. Louis, MO). The cells were then fed every 2 days with 10% Cosmic Calf Serum in DMEM, containing only insulin and triiodothyronine, at the concentrations mentioned above. On day 6 of differentiation, cells were treated with different concentrations of ghrelin, DAG, obestatin or saline for 24h. Cells were stimulated with 1 μM isoproterenol (Sigma, St. Louis, MO) for 6h prior to harvest.
Generation of GHS-R knockdown HIB1B cell line
The predesigned GHS-R shRNA (TG506242) and scrambled shRNA (GCACTACCAGAGCTAACTCAGATAGTACT) in pGFP-V-RS vector were purchased from Origene (Rockville, MD). The shRNAs were transfected into HIB1B cells by Lipofectamine® 2000™ (Invitrogen, Carlsbad, CA), according to the manufacturer's instruction. Briefly, 1 million HIB1B cells were seeded in a 10 cm dish for 12 hours. 2 hours before transfection, medium was changed. Five μg shRNA plasmids were mixed with Lipofectamine® 2000™ reagent and kept at room temperature for 5 minutes. Then, shRNA mixture was added to the dishes. 12 hours later, shRNA mixture was removed, and fresh medium was added. Then 24 hours later, 1 μg/ml puromycin was added to the medium to select positive knockdown cells. After 8 days of selection, all positive cells were pooled together as a stable cell line.
Statistics
Two-factor ANOVA was used to evaluate the significance of interaction between genotype and age, and post hoc test was used to follow up the significant differences between ages and genotypes. Data are represented as mean ± SEM, and statistical significance is set to a minimum of p<0.05.
Author Contributions
LL designed and performed the experiments and wrote the manuscript. JHL, OYNB and XM performed experiments and edited the manuscript. XZ and DSH advised and critiqued the manuscript. YS designed the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Indirect calorimetry was performed in the Mouse Metabolic Research Unit at the Children's Nutrition Research Center, Baylor College of Medicine. The authors acknowledge the expert assistance of Mr. Firoz Vohra and the MMRU Core Director Dr. Marta Fiorotto. We also thank Mr. Michael R. Honig at Houston's Community Public Radio Station KPFT for his excellent editorial assistance. This study was supported by USDA/CRIS grant ARS 6250-51000-059 (YS), American Heart Association (AHA) innovative grant 12IRG9230004 (YS) and Grant-in-Aid 14GRNT18990019 (YS), NIH RDK080306 (DSH), VA BX002006 (DSH), NIH training grant 1T32HD071839 (PI: Morey W. Haymond), and partly supported by the NIH-Diabetes and Endocrinology Research Center of Baylor College of Medicine (P30DK079638). This study was also supported by Macao Science and Technology Development Fund 042/2013/A2 (LL) and the Research Fund of University of Macau MRG015/LLG/2014/ICMS (LL).
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Ahima RS. Connecting obesity, aging and diabetes. Nat Med. 2009; 15: 996 -997. [PubMed] .
- 2. Amati F, Dube JJ, Coen PM, Stefanovic-Racic M, Toledo FG, Goodpaster BH. Physical inactivity and obesity underlie the insulin resistance of aging. Diabetes Care. 2009; 32: 1547 -1549. [PubMed] .
- 3. Cannon B and Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004; 84: 277 -359. [PubMed] .
- 4. Kozak LP, Koza RA, Anunciado-Koza R. Brown fat thermogenesis and body weight regulation in mice: relevance to humans. Int J Obes (Lond). 2010; 34: Suppl 1 S23 -27. [PubMed] .
- 5. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009; 360: 1509 -1517. [PubMed] .
- 6. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009; 360: 1500 -1508. [PubMed] .
- 7. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009; 360: 1518 -1525. [PubMed] .
- 8. Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng YH, Goodyear LJ. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. The Journal of clinical investigation. 2013; 123: 215 -223. [PubMed] .
- 9. Chondronikola M, Volpi E, Borsheim E, Porter C, Annamalai P, Enerback S, Lidell ME, Saraf MK, Labbe SM, Hurren NM, Yfanti C, Chao T, Andersen CR, et al. Brown Adipose Tissue Improves Whole Body Glucose Homeostasis and Insulin Sensitivity in Humans. Diabetes. 2014; 63: 4089 -4099. [PubMed] .
- 10. Mattson MP. Perspective: Does brown fat protect against diseases of aging? Ageing Res Rev. 2010; 9: 69 -76. [PubMed] .
- 11. Pfannenberg C, Werner MK, Ripkens S, Stef I, Deckert A, Schmadl M, Reimold M, Haring HU, Claussen CD, Stefan N. Impact of age on the relationships of brown adipose tissue with sex and adiposity in humans. Diabetes. 2010; 59: 1789 -1793. [PubMed] .
- 12. Sellayah D and Sikder D. Orexin restores aging-related brown adipose tissue dysfunction in male mice. Endocrinology. 2014; 155: 485 -501. [PubMed] .
- 13. Chomyn A and Attardi G. MtDNA mutations in aging and apoptosis. Biochemical and biophysical research communications. 2003; 304: 519 -529. [PubMed] .
- 14. Detmer SA and Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nature reviews Molecular cell biology. 2007; 8: 870 -879. .
- 15. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005; 120: 483 -495. [PubMed] .
- 16. Trifunovic A and Larsson NG. Mitochondrial dysfunction as a cause of ageing. Journal of internal medicine. 2008; 263: 167 -178. [PubMed] .
- 17. Wallace DC. Mitochondrial DNA mutations in disease and aging. Environmental and molecular mutagenesis. 2010; 51: 440 -450. [PubMed] .
- 18. Su B, Wang X, Zheng L, Perry G, Smith MA, Zhu X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochimica et biophysica acta. 2010; 1802: 135 -142. [PubMed] .
- 19. Liesa M and Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell metabolism. 2013; 17: 491 -506. [PubMed] .
- 20. Westermann B. Mitochondrial fusion and fission in cell life and death. Nature reviews Molecular cell biology. 2010; 11: 872 -884. .
- 21. Crane JD, Devries MC, Safdar A, Hamadeh MJ, Tarnopolsky MA. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. The journals of gerontology Series, A, Biological sciences and medical sciences. 2010; 65: 119 -128. .
- 22. Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. The Journal of biological chemistry. 2003; 278: 17190 -17197. [PubMed] .
- 23. Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. Journal of cell science. 2007; 120: 838 -848. [PubMed] .
- 24. Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, Barja F, Martinou JC. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PloS one. 2008; 3: e3257 [PubMed] .
- 25. Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. The EMBO journal. 2006; 25: 2966 -2977. [PubMed] .
- 26. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999; 402: 656 -660. [PubMed] .
- 27. Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000; 407: 908 -913. [PubMed] .
- 28. Shimbara T, Mondal MS, Kawagoe T, Toshinai K, Koda S, Yamaguchi H, Date Y, Nakazato M. Central administration of ghrelin preferentially enhances fat ingestion. Neuroscience letters. 2004; 369: 75 -79. [PubMed] .
- 29. Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101: 4679 -4684. [PubMed] .
- 30. Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008; 454: 846 -851. [PubMed] .
- 31. Davies JS, Kotokorpi P, Eccles SR, Barnes SK, Tokarczuk PF, Allen SK, Whitworth HS, Guschina IA, Evans BA, Mode A, Zigman JM, Wells T. Ghrelin induces abdominal obesity via GHS-R-dependent lipid retention. Mol Endocrinol. 2009; 23: 914 -924. [PubMed] .
- 32. Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, Bhattacharya S, Carpenter R, Grossman AB, Korbonits M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. The Journal of clinical endocrinology and metabolism. 2002; 87: 2988 [PubMed] .
- 33. Sun Y, Garcia JM, Smith RG. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology. 2007; 148: 1323 -1329. [PubMed] .
- 34. Lin L, Saha PK, Ma X, Henshaw IO, Shao L, Chang BH, Buras ED, Tong Q, Chan L, McGuinness OP, Sun Y. Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell. 2011; 10: 996 -1010. [PubMed] .
- 35. Rodriguez A, Gomez-Ambrosi J, Catalan V, Gil MJ, Becerril S, Sainz N, Silva C, Salvador J, Colina I, Fruhbeck G. Acylated and desacyl ghrelin stimulate lipid accumulation in human visceral adipocytes. Int J Obes (Lond). 2009; 33: 541 -552. [PubMed] .
- 36. Miegueu P, St Pierre D, Broglio F, Cianfne K. Effect of desacyl ghrelin, obestatin and related peptides on triglyceride storage, metabolism and GHSR signaling in 3T3-L1 adipocytes. J Cell Biochem. 2011; 112: 704 -714. [PubMed] .
- 37. Mano-Otagiri A, Iwasaki-Sekino A, Nemoto T, Ohata H, Shuto Y, Nakabayashi H, Sugihara H, Oikawa S, Shibasaki T. Genetic suppression of ghrelin receptors activates brown adipocyte function and decreases fat storage in rats. Regulatory peptides. 2010; 160: 81 -90. [PubMed] .
- 38. Mano-Otagiri A, Ohata H, Iwasaki-Sekino A, Nemoto T, Shibasaki T. Ghrelin suppresses noradrenaline release in the brown adipose tissue of rats. The Journal of endocrinology. 2009; 201: 341 -349. [PubMed] .
- 39. Vestergaard ET, Gormsen LC, Jessen N, Lund S, Hansen TK, Moller N, Jorgensen JO. Ghrelin infusion in humans induces acute insulin resistance and lipolysis independent of growth hormone signaling. Diabetes. 2008; 57: 3205 -3210. [PubMed] .
- 40. Hassouna R, Zizzari P, Tolle V. The ghrelin/obestatin balance in the physiological and pathological control of growth hormone secretion, body composition and food intake. Journal of neuroendocrinology. 2010; 22: 793 -804. [PubMed] .
- 41. Zhang JV, Ren PG, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin's effects on food intake. Science. 2005; 310: 996 -999. [PubMed] .
- 42. Toshinai K, Yamaguchi H, Sun Y, Smith RG, Yamanaka A, Sakurai T, Date Y, Mondal MS, Shimbara T, Kawagoe T, Murakami N, Miyazato M, Kangawa K, et al. Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology. 2006; 147: 2306 -2314. [PubMed] .
- 43. Thompson NM, Gill DA, Davies R, Loveridge N, Houston PA, Robinson IC, Wells T. Ghrelin and des-octanoyl ghrelin promote adipogenesis directly in vivo by a mechanism independent of the type 1a growth hormone secretagogue receptor. Endocrinology. 2004; 145: 234 -242. [PubMed] .
- 44. Delhanty PJ, Huisman M, Baldeon-Rojas LY, van den Berge I, Grefhorst A, Abribat T, Leenen PJ, Themmen AP, van der Lely AJ. Des-acyl ghrelin analogs prevent high-fat-diet-induced dysregulation of glucose homeostasis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013; 27: 1690 -1700. [PubMed] .
- 45. Ma X, Lin L, Qin G, Lu X, Fiorotto M, Dixit VD, Sun Y. Ablations of ghrelin and ghrelin receptor exhibit differential metabolic phenotypes and thermogenic capacity during aging. PloS one. 2011; 6: e16391 [PubMed] .
- 46. Gauna C, Meyler FM, Janssen JA, Delhanty PJ, Abribat T, van Koetsveld P, Hofland LJ, Broglio F, Ghigo E AJ. Administration of acylated ghrelin reduces insulin sensitivity, whereas the combination of acylated plus unacylated ghrelin strongly improves insulin sensitivity. The Journal of clinical endocrinology and metabolism. 2004; 89: 5035 -5042. [PubMed] .
- 47. Chen CY, Asakawa A, Fujimiya M, Lee SD, Inui A. Ghrelin gene products and the regulation of food intake and gut motility. Pharmacol Rev. 2009; 61: 430 -481. [PubMed] .
- 48. Wang X and Wahl R. Responses of the insulin signaling pathways in the brown adipose tissue of rats following cold exposure. PloS one. 2014; 9: e99772 [PubMed] .
- 49. Holm C, Osterlund T, Laurell H, Contreras JA. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Annual review of nutrition. 2000; 20: 365 -393. .
- 50. Chandrasekaran K and Jayaraman J. Effect of cyclic AMP on the biogenesis of cytochrome oxidase in yeast. FEBS letters. 1978; 87: 52 -54. [PubMed] .
- 51. De Rasmo D, Signorile A, Roca E, Papa S. cAMP response element-binding protein (CREB) is imported into mitochondria and promotes protein synthesis. The FEBS journal. 2009; 276: 4325 -4333. [PubMed] .
- 52. Mulligan JD, Gonzalez AA, Stewart AM, Carey HV, Saupe KW. Upregulation of AMPK during cold exposure occurs via distinct mechanisms in brown and white adipose tissue of the mouse. The Journal of physiology. 2007; 580: 677 -684. [PubMed] .
- 53. Canto C and Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current opinion in lipidology. 2009; 20: 98 -105. [PubMed] .
- 54. Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell metabolism. 2007; 5: 151 -156. [PubMed] .
- 55. Zorzano A, Liesa M, Palacin M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Archives of physiology and biochemistry. 2009; 115: 1 -12. [PubMed] .
- 56. Zorzano A, Liesa M, Palacin M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. The international journal of biochemistry & cell biology. 2009; 41: 1846 -1854. [PubMed] .
- 57. Liu R, Jin P, LiqunYu, Wang Y, Han L, Shi T, Li X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PloS one. 2014; 9: e92810 [PubMed] .
- 58. Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PloS one. 2010; 5: e10995 [PubMed] .
- 59. Tsubone T, Masaki T, Katsuragi I, Tanaka K, Kakuma T, Yoshimatsu H. Ghrelin regulates adiposity in white adipose tissue and UCP1 mRNA expression in brown adipose tissue in mice. Regulatory peptides. 2005; 130: 97 -103. [PubMed] .
- 60. Szentirmai E, Kapas L, Sun Y, Smith RG, Krueger JM. The preproghrelin gene is required for the normal integration of thermoregulation and sleep in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106: 14069 -14074. [PubMed] .
- 61. Landsberg L, Saville ME, Young JB. Sympathoadrenal system and regulation of thermogenesis. The American journal of physiology. 1984; 247: E181 -189. [PubMed] .
- 62. Champigny O and Ricquier D. Effects of fasting and refeeding on the level of uncoupling protein mRNA in rat brown adipose tissue: evidence for diet-induced and cold-induced responses. The Journal of nutrition. 1990; 120: 1730 -1736. [PubMed] .
- 63. St-Pierre DH, Karelis AD, Cianflone K, Conus F, Mignault D, Rabasa-Lhoret R, St-Onge M, Tremblay-Lebeau A, Poehlman ET. Relationship between ghrelin and energy expenditure in healthy young women. The Journal of clinical endocrinology and metabolism. 2004; 89: 5993 -5997. [PubMed] .
- 64. Theander-Carrillo C, Wiedmer P, Cettour-Rose P, Nogueiras R, Perez-Tilve D, Pfluger P, Castaneda TR, Muzzin P, Schurmann A, Szanto I, Tschop MH, Rohner-Jeanrenaud F. Ghrelin action in the brain controls adipocyte metabolism. The Journal of clinical investigation. 2006; 116: 1983 -1993. [PubMed] .
- 65. Yasuda T, Masaki T, Kakuma T, Yoshimatsu H. Centrally administered ghrelin suppresses sympathetic nerve activity in brown adipose tissue of rats. Neuroscience letters. 2003; 349: 75 -78. [PubMed] .
- 66. Marette A, Deshaies Y, Collet AJ, Tulp O, Bukowiecki LJ. Major thermogenic defect associated with insulin resistance in brown adipose tissue of obese diabetic SHR/N-cp rats. The American journal of physiology. 1991; 261: E204 -213. [PubMed] .
- 67. Reznick RM and Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. The Journal of physiology. 2006; 574: 33 -39. [PubMed] .
- 68. Kola B, Boscaro M, Rutter GA, Grossman AB, Korbonits M. Expanding role of AMPK in endocrinology. Trends in endocrinology and metabolism: TEM. 2006; 17: 205 -215. [PubMed] .
- 69. Kola B and Korbonits M. Shedding light on the intricate puzzle of ghrelin's effects on appetite regulation. The Journal of endocrinology. 2009; 202: 191 -198. [PubMed] .
- 70. Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Human molecular genetics. 2005; 14: 1405 -1415. [PubMed] .
- 71. Wolthers T, Groftne T, Moller N, Christiansen JS, Orskov H, Weeke J, Jorgensen JO. Calorigenic effects of growth hormone: the role of thyroid hormones. The Journal of clinical endocrinology and metabolism. 1996; 81: 1416 -1419. [PubMed] .
- 72. Cizza G, Gold PW, Chrousos GP. Aging is associated in the 344/N Fischer rat with decreased stress responsivity of central and peripheral catecholaminergic systems and impairment of the hypothalamic-pituitary-adrenal axis. Annals of the New York Academy of Sciences. 1995; 771: 491 -511. [PubMed] .
- 73. Lin L and Sun Y. Thermogenic characterization of ghrelin receptor null mice. Methods Enzymol. 2012; 514: 355 -370. [PubMed] .
- 74. Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell metabolism. 2009; 9: 203 -209. [PubMed] .
- 75. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011; 480: 104 -108. [PubMed] .
- 76. Qatanani M, Szwergold NR, Greaves DR, Ahima RS, Lazar MA. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. The Journal of clinical investigation. 2009; 119: 531 -539. [PubMed] .