



Introduction
Aging is a process by which an individual's body changes in its own unique way. Aging proceeds through complex processes involving inherited genetics, the environment to which one has been exposed, and individual status/traits, such as epigenetics [1, 2]. An individual and their discrete tissues may encounter a variety of environmental stressors (chemicals, mutagens, ultraviolet and other radiation exposures) during the course of their life which may enhance biological damage (including oxidative stress), which in turn may cause mutagenesis [1, 3, 4], and contribute to age-associated diseases [5]. Studies of cellular aging have largely focused on telomere shortening. While telomere shortening is a dynamic genomic change, the impact of shortening is apparently minor, perhaps because the genome has built protective and compensatory mechanisms to shield ‘important’ genes from chromosome end erosion [6, 7]. Additionally, gene targeting studies show that telomerase-deficient mice do not age rapidly; in fact, overt phenotypes are not observed for several generations [8]. A recent report describes changes in the exome of a normal single cell and those changes are inherited as the cell propagates [9]. With age, such additive changes in coding regions may promote aging and age related diseases, and result in significant mosaicism. In population-scale studies, however, bulk measurement of an individual genome and the assumption that it completely describes the individual is the preferred approach over sequencing individual cells [10-12]. These studies characterize a variety of important diseases, especially cancer, and ethnic differences found within the population. Recently, three studies reporting exome sequencing for autism spectrum disorder (ASD) compared three very large cohorts of children to the reference genome, which was determined from adult sequences [13-15]. These studies, although characterizing a single disease lacked consensus, perhaps in-part as a consequence of an inappropriate (not age-matched) reference [16].
The inability of genomic variants to fully explain known or suspected inherited and spontaneous components of a wide variety of diseases may indicate that there may be additional undiscovered factors that complicate analysis. These factors include the number and rapidity with which one accumulates genomic variants, which if known could be compensated for, like ethnicity and sex. Genetic characterization of aging, therefore, may hold a key to questions regarding the importance of acquired somatic variants, variation in aging within a population, and their role in human diseases. Adding time and the accompanying mosaic changes as variables may enhance the accuracy and utility of population-scale analysis of human traits and disease. In an attempt to begin to address this gap, we hypothesized that the inherited genome is not static but rather dynamic with time with individual experiences punctuating genomes differently. To test this hypothesis we used exome sequencing of normal epithelial samples from three healthy individuals serially collected at different ages in their life.
Results and Discussion
Single nucleotide variant analysis
We targeted 201,071 exons (62.2 Mb target sequence) in epithelial samples serially collected from three individuals and sequenced them at high coverage (152x, average). Overall, exome enrichment efficiency was 99% (200,264 average target exons sequenced). This enabled 61.6 Mb of target sequence to be analyzed for aging induced exomic changes (SNVs, indels and microsatellites).
These individuals had between 11,100 to 11,500 nsSNVs (nonsynonymous single nucleotide variants) at any age time-point studied when compared to the human genome reference (Table S1). The 1000 Genome Project (1kGP) estimated that an individual typically differs from the reference human genome sequence at 10,000-11,000 non-synonymous sites [17]. In other independent studies, Dr. Venter and Dr. Watson's exome were reported to show 10,389 and 10,569 nsSNVs, respectively [18, 19]. These data indicate that the individual's genomic changes in our study are in close agreement with the range of previously reported nsSNVs across individuals (Figure 1), and that this is not a small effect. Using the human genome reference (hg19), we found an overall increase of 725 and 156 SNVs and indels at different ages for two individuals (1 and 2), respectively, and a decrease of 301 and 255 variants in individual-3 upon nine and fifteen years of aging (Table S1), respectively. List of all the variations called is presented in Table S2.
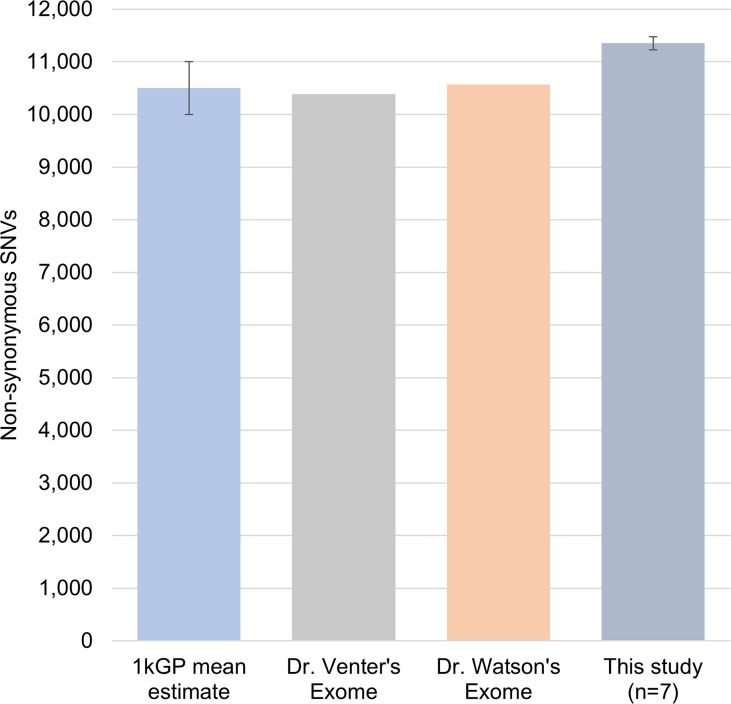
Figure 1. Comparison of number of non-synonymous SNVs Number of non-synonymous SNVs found in samples in our study were comparable with the 1kGP average 10500 ± 500 (n=882), Dr.Venter, and Dr. Watson's exomes. This indicates that the amount of variation identified in the individuals for our study is comparable to previously reported population scale studies.
Overall, we found that ~94%, ~ 62%, and ~93% of the variations with respect to the reference genome were present before and after various aging time intervals in individuals 1, 2 and 3, respectively. These suggest the persistent presence of these sites in an individual's specific genome sequence. Thus, although different from the reference genome, as somatic variants, they may not be informative. This sets this study apart from current approaches, which typically would have based the interpretation including all of these variations.
Important to this study is therefore not the number of variants with respect to a (arbitrary) reference genome, rather the differences observed between samples collected from the same individual at different times. To detect the overall frequency of age-specific genomic changes, we examined unique variants in each individual. To avoid false negative results due to inconsistencies in sequenced loci between samples, we report only on those loci completely covered by at least five reads in both samples under comparison. There were 6,005 variants (SNVs and indels) in individual-1 (after 13 years of aging), 45,505 in individual-2 (after 16 years of aging), 5,003 in individual-3 (after 9 years of aging) and 4,948 in individual-3 upon 15 years of aging (Table 1, Table S1).
Table 1.
Exome variations that differ upon aging in three individuals
| Individual- 1 | Individual- 2 | Individual- 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (13 years apart) | (16 years apart) | (9 years apart) | (15 years apart) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total SNVs and indel variations | 6,005 | 45,505 | 5,003 | 4,948 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Microsatellite variations | 173 | 801 | 151 | 164 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Synonymous SNVs | 604 | 8455 | 455 | 478 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nonsynonymous SNVs | 1,239 | 8,398 | 904 | 901 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Novel nsSNVs* | 849 | 776 | 594 | 576 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stop-gain SNVs** | 37 | 90 | 15 | 22 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stop-loss SNVs*** | 1 | 8 | 2 | 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Splicing† | 3 | 6 | 2 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Frameshift indels | 21 | 128 | 13 | 18 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nonframeshift indels | 19 | 165 | 32 | 27 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Functionally damaging‡ | 295 | 1263 | 207 | 175 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| COSMIC variants | 107 | 1609 | 100 | 104 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Novel nsSNVs are those not previously reported in the dbSNP 137. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stop-gain: A variant that leads to the creation of stop codon at the variant site compared to the reference. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stop-loss: A variant that leads to the elimination of stop codon at the variant site compared to the reference. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Splicing: A variant within 2-bp of a splicing junction. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Functionally damaging variants are those predicted by Polyphen v2. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Exomes of these individuals exhibited a variation rate of 9.6×10−7 to 8.4×10−6 bp−1 year−1 for nsSNVs (considering the 62.2Mb of target sequence), which have aged experiencing different stress and environmental exposures and because the genomic region was exclusively exomes may represent a selection pressure that is different from the whole genome (Figure 2). Compared to the reference human genome, individual-2 does not appear very different from other individuals in terms of the total number of genomic variations with respect to the reference (Table S1). However, he exhibits a very high nsSNVs variation rate (8.4×10−6 bp−1 year−1) when referenced to his own previous genomic state. This finding is of particular significance because it highlights the importance of genomic status relative to personal genomics, rather than observations made from a standard reference genome that are missing valuable details especially, individual specific genomic variation trends and behavior.
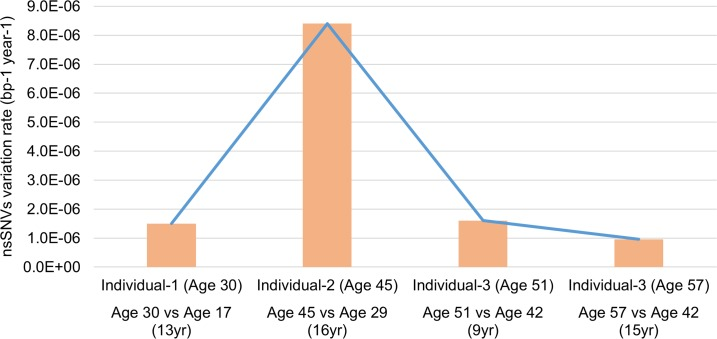
Figure 2. Comparison of nsSNVs variation rate at different ages in three individuals The variation rate varies dramatically between three individuals in our study. This indicates the individual specific genome dynamic of samples in our study.
Variable indel and microsatellite analysis
Indels that cause frameshifts are usually under negative selection pressure [18]. Indels are the second most abundant type of genetic variation, following single nucleotide substitutions and account for almost a quarter of the genetic variation implicated in disease [20]. As shown in Table S3, we identified a reduction in frame-shift indels and the ratio (frameshift/nonframeshift indels) in older individuals (individual-2 and 3) indicating the negative selection of frameshift indels in these individuals at later age. This is consistent with previous observations [18], and suggests the presence of sustained selection pressure throughout the lifespan of an individual.
Since regions with repetitive sequences exhibit a higher-than-background frequency of indel variation [21], we inspected repeat regions (microsatellites) independently. Repeat containing loci are considered to be highly vulnerable to genomic variation, and they represent as much as 3% of the genome; twice the size of the coding region [22, 23]. We identified 173 variable microsatellites within the 53,161 total microsatellite loci called in both samples from individual-1 (Table 2, S4). This translates to a variation rate of 2.5×10−4 locus−1 year−1 for microsatellite loci, significantly higher than what we measured for nsSNVs. Four of the microsatellite variants were found in exons of known genes (DIAPH1: AGG, RPL14: CTG, BCL6B: CAG and FLJ32682: CCTT), 5' UTR (2) and 3' UTR (27). Three of the four coding variable repeats were nonframeshift.
Table 2.
Microsatellite variants that differ between samples at different ages and their distributions
| Individual- 1 | Individual- 2 | Individual- 3 | ||
|---|---|---|---|---|
| (13 years apart) | (16 years apart) | (9 years apart) | (15 years apart) | |
| Total microsatellites called | 53,161 | 49,988 | 51,902 | 52,301 |
| Total microsatellites difference | 173 | 801 | 151 | 164 |
| % Global microsatellite index | 0.3% | 1.6% | 0.3% | 0.3% |
| Location of variable microsatellites | ||||
| Exonic | 4 | 26 | 1 | 2 |
| Intronic | 95 | 351 | 80 | 96 |
| 3' UTRs exon | 27 | 224 | 36 | 28 |
| 5' UTRs exon | 2 | 17 | 2 | 4 |
| 3' UTRs intron | 2 | 12 | 4 | 2 |
| 5' UTRs intron | 6 | 37 | 3 | 7 |
| Downstream | 6 | 36 | 4 | 4 |
| Upstream | 8 | 15 | 4 | 5 |
| Intergenic | 23 | 83 | 17 | 16 |
Individual-3 has a comparable number of microsatellite variations to individual-1. Both sets indicate 0.3% of the callable microsatellites vary upon aging. Individual-3 with 9 years of aging shows one acquired variation in an exon (BTN2A1: CCT). This mutation appears to have been acquired between age 42 to 51 with age 57 having the same genotype as age 51. Individual-3 acquired an additional variation in an exon (MEF2A, CAG). It is not clear if this mutation is present at 9 years of aging in this individual as we were not able to genotype this microsatellite locus in the 51 year old sample. The exonic variations found in both of these genes are in frame (Table S4).
Individual-2 has 801 microsatellite variations representing 1.6% of the callable microsatellites upon aging. This is a five times higher microsatellite variation rate compared to the other two individuals (0.3%) in this study, which cannot be attributed to experimental variations for the total number of microsatellite loci called were similar across all samples. The samples for this individual do span the largest amount of time, in total 16 years, though this is not much more than samples from individual-3 that span 15 years. In total there are 26 exonic microsatellite loci found to vary in individual-2 upon 16 years of aging. One of these microsatellites is the same as that found to vary in the samples from individual-3 (MEF2A, CAG). Additionally, one of the exonic microsatellites, which vary in the samples from individual-1, is also indicated to vary in individual-2 (BCL6B, CAG). All the 26 microsatellite loci found to vary in this sample set are in frame. Together, in all three individuals, we identified 33 exomic microsatellite variations of which 32 were nonframeshift. This supports the previous observation that tandem insertions or deletions of repeated motifs is a conservative event (minimizing the number of changed amino acids and the introduction/ elimination of stop codons) [24]. Together, this is the first observation demonstrating persistent effects of selection pressure on indels and repeats among cells in the life span of an individual.
Biological implications
Three unrelated and ‘healthy’, individuals demonstrate acquired genetic variants as a consequence of aging and the accumulation of variants occurs at different rates. Mutations were observed in up to 13,000 genes in these individuals including some common genes with consistently high numbers of mutations. To identify the most frequently mutated genes upon aging, we compared the lists of the mutated genes from each individual. This resulted in the identification of 20 genes that are consistently and highly mutated upon aging (Figure 3A). We identified MUC4 as the most frequently mutated gene in all three individuals acquiring >75 mutations. In-fact, four MUC genes (MUC4, MUC5B, MUC6, and MUC12) were among the top mutated genes in all three individuals (Figure 3A). These top 20 genes did not have known protein-protein interactions except for the MUC family genes (Figure 3B). This indicates a previously unknown association among these genes and aging. Previous studies have associated differential MUC4 expression with a number of cancers, including pancreatic, lung, breast, gall bladder, salivary gland, prostate and ovarian cancer, indicating that MUC4 may be a good candidate as a diagnostic and prognostic marker [25]. In one breast cancer study, silencing MUC4 led to reduced expression of HER2, although the molecular mechanism of this interaction is unknown [26]. Over-expression of HER2 occurs in 30% of breast cancers and has been used effectively as an adjuvant therapy drug target in these patients [27]. It is interesting to note here that MUC genes were not previously reported as the most frequently mutated genes in any disease or condition. In addition, the functional significance of the MUC, especially the MUC4 mutations upon aging is unclear at this time and warrants further investigation.
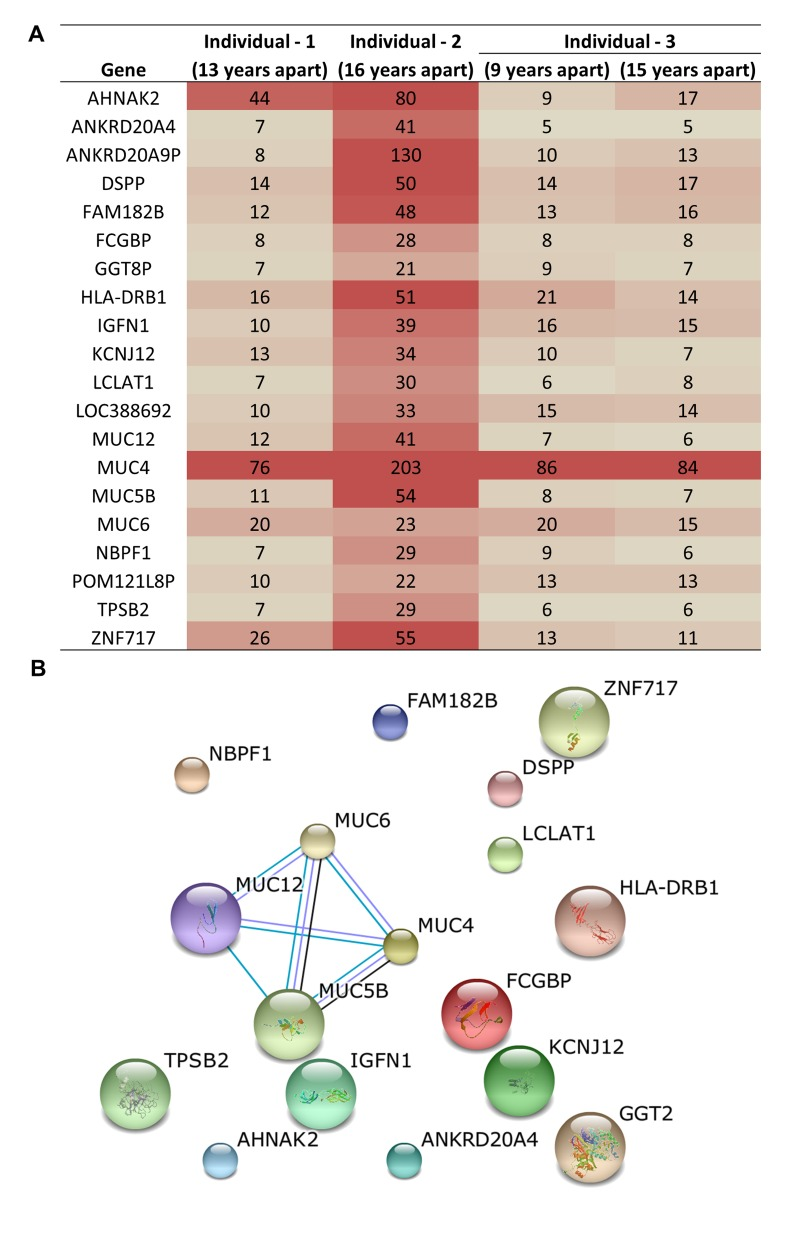
Figure 3. Common genes that acquired genetic variation upon aging and were among the top 100 most frequent variants (A) Table shows the number of mutations in each of the genes most frequently impacted in three individuals. (B) Protein-protein interaction analysis reveals that for the highly related MUC genes are significant targets for genetic variation in aged samples
To explore the biological significance of genomic variants in these individuals, we performed pathway enrichment analysis of the affected genes (all genes that showed variations upon aging (Individual-1= 3,138, Individual-2= 13,014, and Individual-3= 2,782 and 2,768 genes, at 9 and 15 years of aging respectively). The PANTHER classification system identified Wnt signaling (involving 5.5% of the total genes on average) and Gonadotropin-releasing hormone receptor (GnRHR) (involving 5% of the total genes on average) pathways with the highest gene associations. Interestingly, it was recently reported that the Wnt signaling pathway enhances the protection of telomeres, and is downregulated in aged skin [28, 29]. The GnRHR pathway is proposed to control central nervous physiology and pathophysiology modulating cognitive changes associated with aging and age-related neurodegenerative disorders [30]. These changes, if found within neurological cells, may indicate the possibility of gradual loss of cognitive function with aging may be due to age-associated genomic modifications.
We subsequently investigated a sub-set of genes that contained damaging (high impact) variants as predicted by Polyphen, and performed disease ontology analysis to find patterns of disease relationship enrichment (Individual-1= 296, Individual-2= 1,267, and Individual-3= 208 and 175 genes, at 9 and 15 years of aging respectively). FunDO (Functional Disease Ontology Annotations) identified genes with a statistically significant (Bonferroni corrected p-value <0.05) disease association for disease ontology terms in these three individuals. Individual-2 showed the highest number of genes enriched for disease associations including genes for diabetes mellitus, cancer, HIV infection, and Rheumatoid arthritis, Individual-1 showed enrichment for diabetes mellitus, leukemia, and cancer (Table 3). Whereas, individual-3 showed the least disease enrichment, except, notably gene variants implicated in mental retardation (Table 3). This analysis revealed the individual specific patterns of disease susceptibility as a direct result of the age associated variants. No metadata are available for these individuals for any correlation with disease history, except that Individual-2 has a history of bone fractures. Osteoporosis is a feature of rheumatoid arthritis and can cause bone loss early in disease [31]. Therefore, because individual-2 is at risk for rheumatoid arthritis, we would have predicted/ expected this individual to be the susceptible to bone fracture. This is one example of possible impactful clinical significance that can emerge from this type of analysis.
Table 3.
Disease ontology analysis of genes with functionally damaging variants indicates increased risk of developing age-related diseases.
| Individual- 1 | Individual- 2 | Individual- 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (13 years apart) | (16 years apart) | (9 years apart) | (15 years apart) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diabetes mellitus (11) | Diabetes mellitus (31) | Long QT syndrome (2) | Adenovirus infection (3) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Leukemia (10) | Breast cancer (32) | Mental retardation (3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Kidney failure (5) | Prostate cancer (28) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Colon cancer (8) | Neoplasm metastasis (17) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cancer (13) | Cancer (40) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alzheimer's disease (6) | Rheumatoid arthritis (21) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melanoma (14) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HIV infection (12) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Colon cancer (19) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Leukemia (20) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chronic simple glaucoma (5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atherosclerosis (15) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cirrhosis (6) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A sub-set of genes, which contained predicted damaging variants (by Polyphen v2) were analyzed using Functional Disease Ontology Annotations (FunDO). The significance of each disease association is evaluated by Fisher's exact test, and diseases that showed Bonferroni corrected p-value <0.05 are considered as significant. Numbers in parenthesis indicates the number of genes associated with disease. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
There are additional examples of associations with disease via genes in which there were frequent variants. Recently, a melanoma sequencing study revealed frequent mutations in PREX2 (phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 2) as a consequence of ultraviolet exposure [11]. We found that, with age, individual-1 acquired a PREX2 mutation (Chr8:69143713, delT) in the 3' UTR and Individual-2 acquired five mutations (four are synonymous and one deletion). Individual-3 acquired a PREX2 mutation identical to Individual-1 upon nine years of aging, however that variation was not seen in the sample taken at 15 years. This suggests the possibility that the PREX2 mutation, while it may be acquired with ultraviolet exposure, is negatively selected in this individual probably to protect from the pathogenesis of skin cancer. Environmental exposure selects for genomic changes for better adaptation and to increase fitness, a process that usually spans several generations [32]. With the relatively longer and growing life spans of humans and the spectrum of stressors to which humans are exposed, it is very likely that individual cells and tissues may exhibit genomic changes shaped by local selective forces within the life span of an individual. This finding further suggests the influence of environmental exposures on the genome with aging and importance of serial measurements to monitor genome dynamics. Therefore, it will be of interest to establish measures to quantify individual genome dynamics with age and with tissue specificity, which may help detect the rate of functional decline, the vulnerability of an individual to age-related diseases, and potentially predict longevity.
Previous hypotheses on molecular damage and aging suggested that, 1) molecular damage limits life span not because of cellular decline but because of decline in cellular robustness, 2) although, the accumulation of molecular damage is observed, it is not the driving force for aging, and 3) mutations are not random, they are selected to activate/ inactivate particular pathways [33]. In this study, we observed that the genome dynamic, influenced by aging itself or environmental factors, could influence pathogenesis, and might initiate organ or system level damage that are frequently observed upon aging and in age related diseases in humans. Environmental exposures are known to influence the genome in a way that may increase the fitness or induce deleterious mutations [32, 34]. Therefore, age, genome dynamic and environmental exposures are interlinked and together could result in sustained selection and/or accumulation of variations and/or non-mutational hyper-activation of signaling pathways involved in aging. This could reduce overall robustness at a cellular level and cause degeneration of cells, tissues, and organ systems. Although, it is arguable to define aging as a consequence of molecular damage or vice versa at this time, both are positively correlated.
Taken together, the exome sequencing of individual's genome upon aging indicates that implementation of personalized genomics health strategies will require more thorough and potentially continuous analysis of individual's genomes to optimize outcomes. These data demonstrate that the exome of an individual is dynamic and constantly experiences environmental and evolutionary pressures and over time enriches for deleterious variants. This finding indicates that the accumulation of somatic variants and possibly the rate of accumulation will contribute to how an individual ages, and prompting age-related diseases. It challenges our existing approach in population-scale sequencing studies and establishes “age” as an important variable that must be accounted for in the analysis and interpretation of any given human genome. These observations are supportive of new paradigm, “Multiple genomes per individual”.
Methods
Sample details
DNA from primary skin fibroblasts was obtained from the Aging Cell Repository, NIA at the Coriell Institute (Camden, NJ). These samples were serially collected from three Caucasian male individuals at different time point in their life: Individual-1 (age 17- AG06234 and age 30-AG13153), Individual-2 (age 29-AG05415 and age 45-AG13353) and Individual-3 (age 42-AG05416, age 51-AG11364 and age 57-AG13145) (Total numbers of samples were seven).
Library construction and Exome enrichment
DNA-seq libraries were constructed using Illumina's TruSeq DNA Sample Preparation Kit-Set A/B (P/N FC-121-2001/2002). Briefly, 1.5 μg DNA was fragmented using Covaris M220 to 400bp. A gel-free method recommended in the protocol was used to prepare the library. The ends were repaired and a ‘A’ base added to the 3', which prepares the DNA fragments for ligation to the adapters that have a single ‘T’ base overhang at their 3' end. The adapters enable PCR amplification and hybridization to the flow cell. The library generated was validated using Agilent 2100 Bioanalyzer and quantitated using Quant-iT dsDNA HS Kit (Invitrogen; Carlsbad, CA). Exome enrichment was performed using TruSeq Exome Enrichment Kit (FC-121-1024; Illumina). Samples were pooled at concentrations of 500 ng each and enriched following the manufacturer's standard protocol. Enriched samples were quantitated based on Quant-iT dsDNA HS Kit (Invitrogen) and qPCR.
Cluster Generation and HiSeq Sequencing using RapidRun Mode
Libraries were clustered onto a flow cell using TruSeq® Rapid PE Cluster Kit – HS (PE-402-4001), and sequenced 2X for150 cycles using TruSeq® Rapid SBS Kits – HS (FC-402-4001) on HiSeq 2500®. Reads that passed the Illumina chastity filter were kept. Reads passed the chastity filter if they had, within the first 25 cycles, no more than one cycle of a chastity below 0.6 (Chastity = Highest intensity/(Highest intensity + Next highest intensity)). An average of 112.2 million high quality 150bp reads (passed Chastity filter) were generated from exome-enriched samples equivalent to 16.8 billion DNA bases per exome. We opted for longer (400bp) DNA fragments for library preparation and longer read length (150bp) for sequencing to enhance the quality and results, especially within repeat regions.
Variant discovery and quality control
The exome enrichment kit targeted 201,071 exons equivalent to 62.2 Mb target sequence in these epithelial samples serially collected. The read sequences were aligned to hg19 with BWA [35] resulting 110.6 million average reads mapped to hg19 per sample. Exome enrichment efficiency was 99%, as measured by the number of target exons sequenced (average 200,264) out of the total target exons (201,071) per sample. 62.8 million reads mapped on target providing 152x average depth of coverage. Reads were locally realigned around Indels, and raw variants (Single nucleotide variants and Indels) were called using GATK Unified Genotyper [36, 37].
We filtered variants with minimum read depth of ≥5x and mapping quality >30 for high confidence true positive variant calls. In addition, next-generation sequencing has overall very high true positive rate for identified SNVs [38]. In our test using Sanger sequencing, we validated 5 out of 6 loci for true positives. To minimize false negatives, we used a custom Perl script. The script verified loci of age specific variants for the presence of supporting sequence reads (≥5) in both comparison samples. Further, this study complies with the proposed standardization criteria for NGS studies as all samples were uniformly sequenced with identical protocols, sequencing instruments, and the data analysis criteria [16]. Together, these criteria ensure that variant identification is of very high confidence minimizing, false positive and false negative variants as well other technical variability.
Microsatellite variation and quality control
After aligning the reads to hg19 with BWA version 5.10, we applied our microsatellite genotyping software, requiring a minimum of 15 reads completely covering a loci in order to call a genotype for each sample at these challenging loci that require more stringency [39, 40]. This software was recently updated to accept hg19 alignments by converting the prior microsatellite coordinate using the UCSC Genome Lift-Over tool [41]. The software was also updated to speed up the sub-functions allowing us to run an exome-sequenced sample in under 3 hours on a single core of an Intel Xeon 5500/5600 processor. We performed tests between our original hg18 software and the new, faster hg19 version to determine if there are any microsatellites calls that differed. We identified 530 microsatellites for which different genotypes were obtained. These microsatellites were removed from our analysis set leaving at most 856,104 callable microsatellite loci per sample. We were able to call on average 51,769 microsatellite loci from the exome-enriched samples with annotations. We used the locations of Refseq genes obtained from the UCSC Table Browser to determine the relative position of the called microsatellites with respect to genes [42]. We have previously validated that our microsatellite-based genotyping method has 96.5% accuracy [40].
Gene annotation, disease enrichment, and functional impact analysis
All identified variants (SNVs and indels) were annotated using ANNOVAR package using Reference Gene [43]. Splice site variants were identified as occurring within two base pairs of any intron/exon boundary. Variants that created a stop codon at the variant site were considered as stop-gain variants. Variants that eliminated stop codon at the variant site were considered as stop-loss variants. All identified variants were annotated for a variety of characteristics and analyzed. Single Nucleotide Polymorphism database (dbSNP 137) was used to check novel variants. Polyphen 2.0 was used to predict the functional impact of non-synonymous variants [44]. The Catalogue of Somatic Mutations in Cancer (COSMIC) database v64 was used to identify somatic cancer variants. Ingenuity Pathway Analysis (IPA) was used for canonical pathway enrichment analysis. The Functional Disease Ontology (FunDO) used for disease enrichment analysis [45].
Supplementary Materials
Acknowledgments
This work was supported by the Medical Informatics and Systems Division director's annual fund at Virginia Bioinformatics Institute. The high-performance computing infrastructure on which microsatellite analysis was conducted was supported by a grant from the National Science Foundation (OCI-1124123). We thank Heather Lewenczuk for technical help. The Genomics Research Laboratory (GRL), Virginia Bioinformatics Institute performed exome sequencing.
Conflicts of Interest
HRG is a founder of Genomeon, LLC, which may license these findings. Genomeon, LLC did not fund or influence this research.
References
- 1. Martin GM, Austad S, Johnson TE. Genetic analysis of ageing: role of oxidative damage and environmental stresses. Nature genetics. 1996; 13: 25 -34. [PubMed] .
- 2. Boyd-Kirkup JD, Green CD, Wu G, Wang D, Han JD. Epigenomics and the regulation of aging. Epigenomics. 2013; 205 -227. [PubMed] .
- 3. Sharp NP and Agrawal AF. Evidence for elevated mutation rates in low-quality genotypes. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109: 6142 -6146. [PubMed] .
- 4. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003; 17: 1195 -1214. [PubMed] .
- 5. Poduri A, Evrony GD, Cai X, Walsh CA, Somatic mutation. Somatic mutation, genomic variation, and neurological disease. Science. 2013; 341: 1237758 [PubMed] .
- 6. Christensen K, Johnson T, Vaupel JW. The quest for genetic determinants of human longevity: challenges and insights. Nature reviews Genetics. 2006; 7: 436 -448. .
- 7. Zhang F, Kato BS, Gardner JP, Kimura M, Spector T, Ahmadi KR. Lack of association between leukocyte telomere length and genetic variants in two ageing-related candidate genes. Mechanisms of ageing and development. 2007; 128: 415 -422. [PubMed] .
- 8. Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho R, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997; 91: 25 -34. [PubMed] .
- 9. Zong C, Lu S, Chapman A, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012; 338: 1622 -1626. [PubMed] .
- 10. Genomes Project C. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth G, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491: 56 -65. [PubMed] .
- 11. Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, Zhang H, Zeid R, Ren X, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012; 485: 502 -506. [PubMed] .
- 12. Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, McKenna A, Carter SL, Cibulskis K, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nature genetics. 2013; 45: 478 -486. [PubMed] .
- 13. Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012; 485: 237 -241. [PubMed] .
- 14. Neale BM, Kou Y, Liu L, Ma' ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012; 485: 242 -245. [PubMed] .
- 15. O' Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012; 485: 246 -250. [PubMed] .
- 16. Bavarva JH, McMahon W, Bavarva MJ, Karunasena E, Garner HR. Standardizing next-generation sequencing experiments and analysis methods. Clinical chemistry. 2012; 58: 1720 -1722. [PubMed] .
- 17. Genomes Project C. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles M, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010; 467: 1061 -1073. [PubMed] .
- 18. Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, Axelrod N, Busam DA, Strausberg R, Venter JC. Genetic variation in an individual human exome. PLoS genetics. 2008; 4: e1000160 [PubMed] .
- 19. Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008; 452: 872 -876. [PubMed] .
- 20. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Human mutation. 2003; 21: 577 -581. [PubMed] .
- 21. Nishizawa M and Nishizawa K. A DNA sequence evolution analysis generalized by simulation and the markov chain monte carlo method implicates strand slippage in a majority of insertions and deletions. Journal of molecular evolution. 2002; 55: 706 -717. [PubMed] .
- 22. Ellegren H. Microsatellites: simple sequences with complex evolution. Nature reviews Genetics. 2004; 5: 435 -445. .
- 23. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409: 860 -921. [PubMed] .
- 24. de la Chaux N, Messer P, Arndt PF. DNA indels in coding regions reveal selective constraints on protein evolution in the human lineage. BMC evolutionary biology. 2007; 7: 191 [PubMed] .
- 25. Singh AP, Chaturvedi P, Batra SK. Emerging roles of MUC4 in cancer: a novel target for diagnosis and therapy. Cancer research. 2007; 67: 433 -436. [PubMed] .
- 26. Singh AP, Moniaux N, Chauhan SC, Meza J, Batra SK. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer research. 2004; 64: 622 -630. [PubMed] .
- 27. Carlsson J, Nordgren H, Sjostrom J, Wester K, Villman K, Bengtsson NO, Ostenstad B, Lundqvist H, Blomqvist C. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. British journal of cancer. 2004; 90: 2344 -2348. [PubMed] .
- 28. Makrantonaki E, Brink TC, Zampeli V, Elewa RM, Mlody B, Hossini AM, Hermes B, Krause U, Knolle J, Abdallah M, Adjaye J, Zouboulis CC. Identification of biomarkers of human skin ageing in both genders. Wnt signalling - a label of skin ageing? PloS one. 2012; 7: e50393 [PubMed] .
- 29. Diala I, Wagner N, Magdinier F, Shkreli M, Sirakov M, Bauwens S, Schluth-Bolard C, Simonet T, Renault VM, Ye J, Djerbi A, Pineau P, Choi J, et al. Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO reports. 2013; 14: 356 -363. [PubMed] .
- 30. Wang L, Chadwick W, Park SS, Zhou Y, Silver N, Martin B, Maudsley S. Gonadotropin-releasing hormone receptor system: modulatory role in aging and neurodegeneration. CNS & neurological disorders drug targets. 2010; 9: 651 -660. [PubMed] .
- 31. Gough AK, Lilley J, Eyre S, Holder R, Emery P. Generalised bone loss in patients with early rheumatoid arthritis. Lancet. 1994; 344: 23 -27. [PubMed] .
- 32. Wirgin I, Roy NK, Loftus M, Chambers RC, Franks D, Hahn ME. Mechanistic basis of resistance to PCBs in Atlantic tomcod from the Hudson River. Science. 2011; 331: 1322 -1325. [PubMed] .
- 33. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging. 2011; 3: 1130 -1141. [PubMed] .
- 34. Bavarva JH, Tae H, McIver L, Garner HR. Nicotine and oxidative stress induced exomic variations are concordant and overrepresented in cancer-associated genes. Oncotarget. 2014; .
- 35. Li H and Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26: 589 -595. [PubMed] .
- 36. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011; 43: 491 -498. [PubMed] .
- 37. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010; 20: 1297 -1303. [PubMed] .
- 38. Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, Ju J, Mei J, Shi Y, He M, Wang G, Liang J, Wang Z, et al. Detection of Clinically Relevant Genetic Variants in Autism Spectrum Disorder by Whole-Genome Sequencing. American journal of human genetics. 2013; 93: 249 -263. [PubMed] .
- 39. McIver LJ, Fondon JW 3rd, Skinner MA, Garner HR. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics. 2011; 97: 193 -199. [PubMed] .
- 40. McIver LJ, McCormick JF, Martin A, Fondon JW 3rd, Garner HR. Population-scale analysis of human microsatellites reveals novel sources of exonic variation. Gene. 2013; 516: 328 -334. [PubMed] .
- 41. Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, Hillman-Jackson J, Kuhn RM, Pedersen JS, et al. The UCSC Genome Browser Database: update 2006. Nucleic acids research. 2006; 34: Database issue D590 -598. [PubMed] .
- 42. Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic acids research. 2004; 32: Database issue D493 -496. [PubMed] .
- 43. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38: e164 [PubMed] .
- 44. Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Human mutation. 2011; 32: 894 -899. [PubMed] .
- 45. Osborne JD, Flatow J, Holko M, Lin SM, Kibbe WA, Zhu LJ, Danila MI, Feng G, Chisholm RL. Annotating the human genome with Disease Ontology. BMC genomics. 2009; 10: Suppl 1 S6 [PubMed] .