



Introduction
In live cells, DNA is continuously being damaged by reactive oxygen species (ROS), the by-products of aerobic respiration in mitochondria [1-6]. Exogenous oxidants originating from environmental pollutants [7],phagocyte-oxidative burst [8-10], and even iatrogenic factors [11], additionally contribute to DNA damage. Such DNA damage involves oxidation of the constituent DNA bases, particularly of guanine by formation of 8-oxo-7,8-dihydro-2'-deoxyguanosine (oxo8dG), base ring fragmentation, modification of deoxyglucose, crosslinking of DNA and protein, and induction of DNA double strand breaks (DSBs) [12, 13]. Another important injurious effect of endogenous and exogenous oxidants is peroxidation of lipids in cell membranes [14].
The extent of ROS-induced DNA damage varies widely in different studies [1-6]. According to one rather conservative estimate, about 5,000 DNA single-strand lesions (SSLs) are generated per nucleus during a single cell cycle of approximately 24 h duration [6]. About 1% of those lesions become converted to DSBs, mostly during DNA replication. This leads to formation of ~50 “endogenous DSBs”, the most severe and potentially mutagenic lesions [6]. DSBs can be repaired by two mechanisms, recombinatorial repair or nonhomologous DNA-end joining (NHEJ). The template-assisted recombinatorial repair is essentially error-free but takes place only when cells have already replicated their DNA which can serve as a template, namely in late-S and G2 phase of the cell cycle. DNA repair in cells lacking a template such as in G1 and early S phase occurs via the NHEJ mechanism. The latter is error-prone and may result in deletion of some base pairs [15, 16]. When such change occurs at the site of an oncogene or tumor suppressor gene it may promote carcinogenesis [17, 18]. It can also lead to translocations and telomere fusion, hallmarks of tumor cells [19]. The progressive accumulation of DNA damage with each sequential cell cycle has been considered to be the primary cause of cell aging and senescence [20]. However, the notion that persistent stimulation of mTOR-driven pathways (rather than the ROS-induced DNA damage) is the major mechanism responsible for aging appears to have more merit [21-27]. Oxidative DNA damage, on the other hand, by contributing to replication stress may be a factor enhancing the TOR-driven aging or senescence process [28].
Strategies for preventing cancer or slowing down aging are often directed at protecting DNA from oxidative damage. Protective agents can be identified by their ability to reduce formation of “endogenous DSBs”. The direct detection of endogenous DSBs in individual cells has been difficult because the leading methodology, single cell electrophoresis (comet) assay [29], lacks the desired sensitivity. The TUNEL assay, developed to label DSBs in apoptotic cells, also lack sufficient sensitivity [30, 31]. While the assays of DNA damage measurement in bulk offer greater sensitivity, these approaches do not allow one to relate the damage to individual cells, reveal any heterogeneity within cell populations, or the relationship of DSBs to cell cycle phase or apoptosis.
Among the early and most sensitive reporters of DNA damage, and in particular formation of DSBs, is the activation of the Ataxia Telangiectasia mutated protein kinase (ATM) through its autophosphorylation on Ser1981 [32], and the phosphorylation of histone H2AX on Ser139; the phosphorylated H2AX is designated as γH2AX [33]. Immunocytochemical detection of these events offers high sensitivity in assessment of DSBs formation in individual cells [34-37]. These biosensors of DNA damage have been used in conjunction with flow- or image-cytometry to assess DNA damage in cells exposed to a variety of exogenous genotoxins (reviews, [31, 38]). In fact, the high sensitivity of these biomarkers makes it possible to use them to detect and measure the extent of constitutive DNA damage induced by the metabolically generated ROS in untreated cells [39-41]. Furthermore, these markers can be used to explore the effectiveness of factors protecting nuclear DNA from endogenous oxidants [42-45]. Thus, the anti-oxidants (N-acetyl-L-cysteine, ascorbate, Celecoxib), inhibitors of glycolysis and oxidative phosphorylation (2-deoxy-D-glucose and 5-bromo-pyruvate), hypoxia (3-5% O2), confluency, low serum concentration, were all shown to distinctly reduce the level of constitutive ATM activation and H2AX phosphorylation [40-45]. Conversely, the factors enhancing metabolic activity (aerobic glycolysis) such as cell mitogenic activation, glucose, or dichloroacetate amplified the level of constitutive expression of γH2AX and activated ATM [42-45]. Collectively, these observations provide strong evidence that the extent of the ongoing DNA damage imposed by endogenous oxidants as well as the effectiveness of factors that protect from (or enhance) the damage can be assessed by analysis of the level of constitutive DNA damage signaling.
In the present study we tested whether metformin, a drug widely prescribed to treat type 2 diabetes, has the ability to modulate the level of constitutive DNA damage signaling. Metformin is a specific activator of 5'AMP-activated protein kinase (AMPK), a phylo-genetically conserved serine/threonine kinase that plays a key role in cellular energy homeostasis (reviews, [46-52]). AMPK is the energy sensor (“fuel gauge”) monitoring and regulating cellular energy in response to metabolic needs and nutritional environmental variations. This kinase is activated by low cellular energy status (increased AMP/ATP ratio) and responds by: (i) activating ATP-producing catabolic pathways such as glycolysis and fatty acids oxidation and (ii) suppressing the energy (ATP)-consuming anabolic pathways such as lipogenesis, gluconeogenesis and protein synthesis. Another effect of AMPK activation is inhibition of mammalian target of rapamycin (mTOR), the downstream effector of growth factor signaling pathways [51]. AMPK affects these activities by phosphorylating proteins regulating these pathways (instant effect) as well as by modulating transcription of genes encoding proteins of these pathways (delayed effect) [53-55]. AMPK itself is activated by the upstream mediator liver kinase B1 (LKB1) [52]. Activation of AMPK by metformin was shown to reduce intracellular reactive oxygen species (ROS) levels via upregulation of expression of the antioxidant thioredoxin through the AMPK-FOXO3 pathway [55].
There is a growing body of evidence that metformin may be considered a promising anti-aging candidate, applicable for life span extension, prevention and even treatment of cancer [22-27, 50, 56]. Given the above, it is of additional interest to know how metformin affects the level of constitutive DNA signaling in normal and tumor cells. Our present data show that in normal lymphocytes, as well as in cells of tumor lines the level of constitutive ATM activation and γH2AX expression was distinctly attenuated upon exposure to metformin. Also reduced was the level of intracellular ROS.
Results
The effect of metformin was tested on the level of constitutive expression of γH2AX and Ser1981-phoshorylated ATM in human lung adenocarcinoma A549 cells. The cells were grown attached on slides and the expression of these phospho-proteins was measured by laser scanning cytometry (LSC) [57]. The data provide clear evidence that expression of γH2AX in A549 cells growing in the presence of metformin for 48 h was reduced (Figure 1). The reduction was apparent at 1 mM, and was progressively more pronounced following exposure to 5 and 20 mM concentrations of metformin.
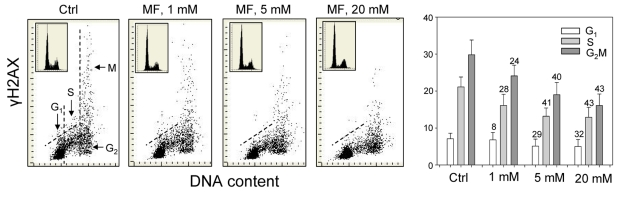
Figure 1. Effect of metformin (MF) on the level of constitutive γH2AX expression in A549 cells Exponentially growing A549 cells were left untreated (Ctrl) or treated with 1, 5 or 20 mM metformin for 48 h. Left panels present bivariate distributions of cellular DNA content versus intensity of γH2AX immunofluorescence (IF) detected with H2AX-Ser139 phospho-specific Ab in cells of these cultures; fluorescence of individual cells was measured by laser scanning cytometry (LSC) [76]. Based on differences in DNA content the cells were gated in G1, S and G2M phases of the cell cycle, as shown in the left panel, and the mean values of γH2AX IF for cells in each of these cell cycle phases by were obtained gating analysis. These mean values (+SD) are presented as the bar plots (right panel). The percent decrease in mean values of γH2AX expression of the metformin-treated cells with respect to the same phase of the cell cycle of the untreated cells is shown above the respective bars. The skewed dash line shows the upper level of γH2AX IF intensity for 97% of G1- and S- phase cells in Ctrl. The insets show cellular DNA content frequency histograms in the respective cultures.
Across all the three metformin concentrations, the degree of reduction in γH2AX expression was more distinct in G2M- and S- phase cells compared to cells in the G1-phase of the cycle. The DNA content frequency histograms did not show major changes in the cell cycle distribution following 48 h treatment with up to 10 mM metformin, while only a modest decrease in the proportion of S-phase cells was apparent following exposure to 20mM metformin (Figure 1, insets).
The effect of metformin on the level of constitutive expression of ATM phosphorylated on Ser1981 in A549 cells was strikingly similar to that of γH2AX (Figure 2). The degree of reduction of ATM-S1981P was metformin-concentration dependent. While the decline in ATM activation was seen in all phases of the cell cycle, the most pronounced reduction was evident in S-phase cells (Figure 2).
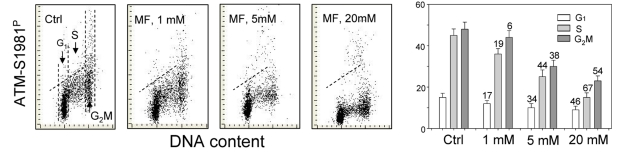
Figure 2. Effect of metformin (MF) on the level of constitutive ATM phosphorylation on Ser1981 in A549 cells Similar as in Figure 1, the cells were treated with 1, 5 or 20 mM MF for 48 h. Left panels present bivariate distributions of cellular DNA content vs intensity of ATM-S1981P IF. The mean values of ATM-S1981P for cells in G1, S, and G2M were obtained by gating analysis and are shown (+SD) as the bar plots (right panel). The skewed dash line shows the upper level of ATM-S198P IF intensity for 97% of G1- and S- phase cells in Ctrl.
In the next set of experiments we have tested the effect of metformin on human lymphoblastoid TK6 cells. These cells grow in suspension and their fluorescence, upon staining with phospho-specific Abs, was measured by flow cytometry [57]. The data show that, similar to A549, the expression of γH2AX was also reduced in TK6 cells exposed to metformin (Figure 3). The effect could be seen (7 - 10% decrease) even at a metformin concentration as low as 0.1 mM, and was more pronounced (up to 44% reduction) at higher concentrations. In TK6 cells the reduction in γH2AX was more pronounced in G1 and S phase than in G2M phase cells. The level of constitutively activated ATM was also decreased in TK6 cells growing in the presence of metformin (Figure 4).
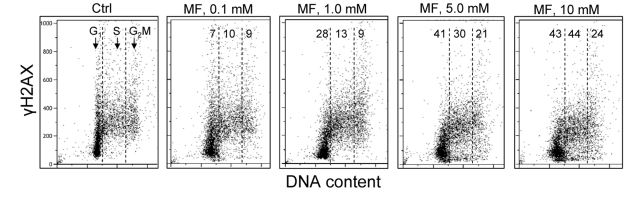
Figure 3. Effect of metformin on the level of constitutive expression of γH2AX in TK6 cells Exponentially growing TK6 cells were untreated (Ctrl) or were grown in the presence of 0.1, 1.0, 5.0 and 10 mM metformin (MF) for 48 h. The expression of γH2AX was detected with phospho-specific (Ser139-P) Ab and cell fluorescence was measured by flow cytometry. Based on differences in DNA content the cells were gated in G1, S and G2M phases of the cell cycle and the mean values of γH2AX IF for cells in each of these cell cycle phases were calculated. The numerical figures show the percent reduction in mean values of γH2AX IF of the metformin-treated cells with respect to the mean values of the untreated cells (Ctrl) in the respective phases of the cell cycle.
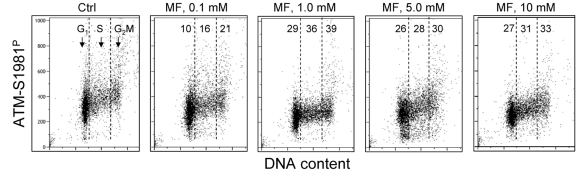
Figure 4. Effect of metformin on the level of constitutive expression of ATM-S1981P Exponentially growing TK6 cells were untreated (Ctrl) or were grown in the presence of 0.1, 1.0, 5.0 and 10 mM metformin (MF) for 48 h. The expression of ATM-S1981P was detected with phospho-specific Ab. As in Fig. 3, the cells were gated in G1, S and G2M phases of the cell cycle and the mean values of ATM-S1981P for cells in each of these cell cycle phases were estimated. The figures show the percent reduction in mean values of ATM-S1981P IF of the metformintreated cells with respect to the mean values of the untreated cells (Ctrl) in the respective phases of the cell cycle.
Figure 5 illustrates the effect of metformin on proliferating human lymphocytes. The peripheral blood lymphocytes were stimulated to proliferate by the polyvalent mitogen phytohemagglutinin for 48 h and subsequently were grown in the absence or presence of 5 mM metformin for 24 h. The data show that, as was the case with the tumor cell lines A549 and TK6, growth of lymphocytes in the presence of 5mM metformin distinctly reduced both the level of constitutive expression of γH2AX as well as of ATM-S1981P.
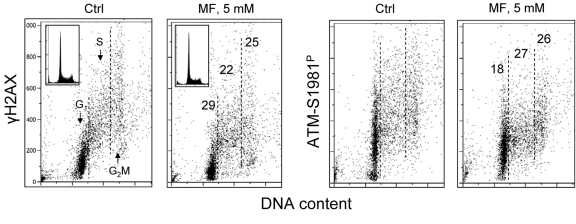
Figure 5. Effect of metformin on constitutive expression γH2AX and ATM-S1981P in normal human proliferating lymphocytes Peripheral blood lymphocytes were mitogenically stimulated by phytohemagglutinin for 48 h and then were grown in the absence (Ctrl) or presence of 5 mM metformin (MF) for additional 24 h. The expression of γH2AX and ATM-S1981P was detected with phospho-specific Abs and cell fluorescence we as measured by flow cytometry. The numerical figures show the percent reduction in expression of γH2AX and ATM-S1981P of cells treated with metformin with respect to Ctrl, in the respective phases of the cell cycle.
As mentioned in the Introduction, the decline in the level of constitutive expression of γH2AX and phosphorylation of ATM was observed in cells treated with agents that decrease the level of endogenous oxidants such as ROS scavengers or antioxidants [39-45, 58]. Therefore, we assessed the effect of metformin on the abundance of reactive oxidants in human leukemic TK6 cells in the same cultures in which we observed the decline in expression of γH2AX (Figure 3) and ATM-S1981P (Figure 4). As is quite evident from the data shown in Figure 6, the growth of TK6 cells for 48 h in the presence of metformin led to a decrease in the level of ROS that were detected by their ability to oxidize 2',7'-dihydro-dichlorofluorescein diacetate (H2DCF-DA); following oxidation by ROS the non-fluorescent substrate H2DCF-DA is converted to the highly fluorescent product DCF [59]. The effect was concentration dependent and the oxidation of H2DCF-DA was reduced by nearly two orders of magnitude at a 10 mM concentration of metformin compared to untreated cells (Figure 6).
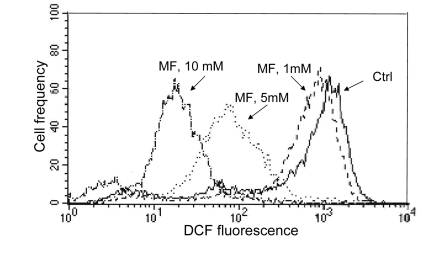
Figure 6. Effect of metformin on ability of TK6 cells to oxidize 2',7'-dihydro-dichlorofluorescein diacetate (H2DCF-DA) TK6 cells were untreated (Ctrl) or treated with 1, 5 or 10 mM metformin (MF) for 48 h. The cells were then incubated for 30 min with 10 μM H2DCF-DA and their fluorescence was measured by flow cytometry. While H2DCF-DA is not fluorescent, the product of its oxidation (DCF) by intracellular ROS shows strong green fluorescence. Note dramatic decline in fluorescence intensity of cells treated with 5 or 10 mM metformin.
Discussion
The present data demonstrate that exposure of either normal, mitogenically activated lymphocytes, or tumor cell lines (A549, TK6) to metformin leads to a decrease in the level of constitutive phosphorylation of H2AX on Ser139 and constitutive activation of ATM. The observed decrease was evident even at a concentration as low as 0.1 mM metformin (Figures 3 and 4). Pharmacokinetic data indicate that this concentration of metformin is of pharmacological relevance [60].Since the level of constitutive expression of γH2AX and ATM-S1981P to a large extent reports DNA damage signaling in response to DNA damage by endogenous oxidants generated during aerobic respiration [39-45, 58]. the present findings would be consistent with a notion that metformin exerts protective effect on nuclear DNA against oxidative damage. These findings are consistent with the observation that exposure of cells to metformin lowered the extent of reactive oxidants that were able to oxidize the H2DCF-DA substrate (Figure 5). They are also in accordance with numerous studies in which a decrease in the level of ROS in cells treated with metformin has been observed [55, 61-65]. It appears that the mechanisms activated by metformin for neutralizing ROS such as upregulation of the antioxidant thioredoxin [55], and/or suppression of NAD(P)H oxidase activity [61] may prevail over the ROS-generating inhibitory effect on mitochondrial respiratory complex I or catabolic processes activated by AMPK [66, 67].
It should be noted that DNA damage signaling such as reported by H2AX phosphorylation and ATM activation do not necessarily indicate the actual DNA damage that involves formation of DNA strand breaks [68]. While some breaks may be formed during replication of DNA sections containing the primary oxidative lesions (e.g. oxo8dG) the presence of such lesions by themselves can induce persistent replication stress. The persistent replication stress combined with activation of mTOR pathways is considered to be the main mechanism contributing to aging and senescence [22-27, 69, 70]. Induction of replication stress by arrest in the cell cycle e.g. by upregulation of the CKI p21, with no evidence of actual DNA damage, elevates the level of constitutive DNA damage signaling (“pseudo-DNA damage response”) whereas attenuation of this “pseudo-DNA damage response” can be achieved by reduction in mTOR-signaling [29]. Likewise, the cell senescence induced by the replication stress triggered by low doses (1 – 2 nM) of the DNA damaging agent mitoxantrone, that is also accompanied by elevated levels of DNA damage signaling, was shown to be attenuated by the caloric restriction-mimicking drug 2-deoxy-D-glucose [71]. All this evidence collectively indicates that the observed constitutive DNA damage signaling occurs as a response to persistent DNA replication stress. Thus, by reducing the level of DNA damage signaling, as we presently see, metformin appears to alleviate the extent of the persistent DNA replication stress. Since metformin inhibits mTOR pathways, the reduction of replication stress by metformin may not only be mediated by attenuation of the oxidative stress through reduction of ROS, but also may be mediated by its direct inhibitory effect on mTOR [50-53].
Our observation that cells exposure to metformin reduces expression of γH2AX and ATM-S1981P remains in contrast to recent data by Vazquez-Martin et al., that show the opposite, namely an activation of ATM and phosphorylation of H2AX in cells treated with metformin [72]. This report prompted us to repeat our experiments numerous times, using a variety of positive and negative controls. Yet in each experiment we observed that treatment of proliferating lymphocytes, TK6 or A549 cells led to a decline in expression of γH2AX and ATM-S1981P. We have also tested the A431 epidermoid carcinoma cells used by these authors [72]. The data show that treatment of A431with metformin decreased the level of H2AX and ATM phosphorylation (Supplemental data, Figure 1). To exclude the possibility of bias resulting from different methodologies we also assessed the effect of metformin on expression of γH2AX and ATM-S1981P in TK6 cells using immonoblotting, the methodology used by the authors [72]. The results obtained by immunobloting (Figure 7) confirm all our immunocytochemical data (Figs. 1-5) by showing a distinct reduction of γH2AX and ATM-S1981P in cells treated with metformin. In fact, the reduction in expression of ATM-S1981P was nearly 45% related to the control. We have also observed that constitutive H2AX phosphorylation and ATM activation in quiescent A549 cells, maintained for 5 days at high cells density (>106 cells/ml) with no medium change also was reduced by treatment with metformin (Supplemental data Figure 2). The effect of metformin, thus, was unrelated as to whether the cells were in exponential- or stationary- phase of growth. Our data also concur with the findings of Nilsson et al., who did not detect any induction of γH2AX in U2OS or HT1080 cells treated with 40 mM metformin [73]. Actually, careful inspection of their data provides some evidence of a decline in expression of γH2AX upon treatment with metformin [73]. At present we see no explanation for the apparent discrepancy of our results (and the data of Nilsson et al., [73]) versus the data presented by Vazquez-Martin al., [72].
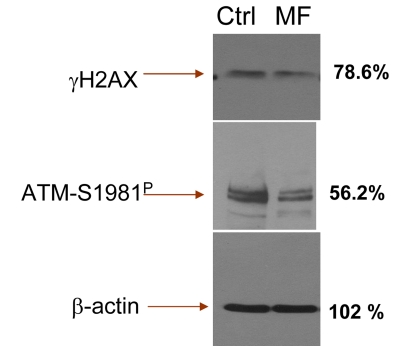
Figure 7. Detection of γH2AX and ATM-S1981P in TK6 cells untreated (Ctrl) and treated with 5 mM metformin (MF) for 48 h, by immunoblotting The figures on right side of the blot represent the percent intensity of the scanned protein bands of the metformin-treated cells (UN-SCAN-IT gel 6.1) as that of the intensity of the respective protein bands of the untreated (Ctrl) cells.
As mentioned, cell aging and senescence appear to be driven by persistent mTOR activation in conjunction with DNA replication stress; the latter can be induced by ROS as well as by inhibition of cell cycle progression, such as activation of CKIs. DNA replication stress and mTOR activation are being reported by the elevated level of constitutive DNA damage signaling (“pseudo-DNA damage response”) [29, 71]. As shown in the present study, the effectiveness of potential anti-aging factors such as metformin may be tested by monitoring their effect on constitutive DNA damage signaling. This approach offers novel means to assess the anti-aging or aging-promoting properties of different factors suspected of such activities. Assessment of DNA damage signaling may serve to detect both genotoxicity [38, 74] as well as genome-protective mechanisms related to attenuation of DNA replication stress.
Materials and Methods
Cells, cell treatment
Human lung carcinoma A549 cells, epidermoid carcinoma A431 and lymphoblastoid TK6 cells were obtained from American Type Culture Collection (ATCC CCL-185, Manassas,VA). Human peripheral blood lymphocytes were obtained by venipuncture from healthy volunteers and isolated by density gradient centrifugation. A549 cells were cultured in Ham's F12K, TK6 and lymphocytes were cultured in RPMI 1640 and A431cells in Dulbecco modified Eagle medium, with 2 mM L-glutamine adjusted to contain 1.5 g/L sodium bicarbonate supplemented with 10% fetal bovine serum (GIBCO/Invitrogen, Carlsbad, CA). Adherent A549 and A431 cells were grown in dual-chambered slides (Nunc Lab-Tek II), seeded with 105 cells/ml suspended in 2 ml medium per chamber. TK6 cells and lymphocytes were grown in suspension; lymphocyte cultures were treated with the polyvalent mitogen phytohemaglutinin (Sigma /Aldrich; St Louis, MO) as described [75]. During treatment with metformin (1,1-dimethylbiguanide; Calbiochem, La Jolla, CA) the cells were in exponential phase of growth unless indicated otherwise. After exposure to metformin at various concentrations and for specified periods of time (as shown in figure legends) the cells were rinsed with phosphate buffered salt solution (PBS) and fixed in 1% methanol-free formaldehyde (Polysciences, Warrington, PA) for 15 min on ice The cells were then transferred to 70% ethanol and stored at −20 °C for up to 3 days until staining.
Detection of H2AX phosphorylation and ATM activation
The cells were washed twice in PBS and with 0.1% Triton X-100 (Sigma) in PBS for 15 min and with a 1% (w/v) solution of bovine serum albumin (BSA; Sigma) in PBS for 30 min to suppress nonspecific antibody (Ab) binding. The cells were then incubated in 1% BSA containing a 1:300 dilution of phospho-specific (Ser139) γH2AX mAb (Biolegend, San Diego, CA or with a 1:100 dilution of phospho-specific (Ser1981) ATM mAb (Millipore, Tamecula, CA). The secondary Ab was tagged with AlexaFluor 488 fluorochrome (Invitrogen/Molecular Probes, used at 1:200 dilution). Cellular DNA was counterstained with 2.8 μg/ml 4,6-diamidino-2-phenylindole (DAPI; Sigma). Each experiment was performed with an IgG control in which cells were labeled only with the secondary AlexaFluor 488 Ab, without primary Ab incubation to estimate the extent of nonspecific adherence of the secondary Ab to the cells. The fixation, rinsing and labeling of A549 or A431 cell was carried out on slides, and lymphocytes and TK6 cells in suspension. Other details have been previously described [38-40].
Analysis of cellular fluorescence
A549 and A431 cells: Cellular immunofluorescence representing the binding of the respective phospho-specific Abs as well as the blue emission of DAPI stained DNA was measured with an LSC (iCys; CompuCyte, Westwood, MA) utilizing standard filter settings; fluorescence was excited with 488-nm argon, helium-neon (633 nm) and violet (405 nm) lasers [76]. The intensities of maximal pixel and integrated fluorescence were measured and recorded for each cell. At least 3,000 cells were measured per sample. Gating analysis was carried out as described in Figure legends. TK6 cells and lymphocytes: Cellular fluorescence was measured by using a MoFlo XDP (Beckman-Coulter, Brea, CA) high speed flow cytometer/sorter. DAPI fluorescence was excited with the UV laser (355-nm) and AlexaFluor 488 with the argon ion (488-nm) laser.
Protein immonoblotting
Nitrocellulose membrane was blocked with 5% w/v nonfat dry milk in TBST (20 mM TrisHCl, pH 7.4, 150 mM NaCl, 0.05% Tween 20) for 1h at room temperature. The blot was then incubated with the primary antibody either phospho-specific (Ser139) γH2AX mAb (Biolegend) or a phospho-specific (Ser1981) ATM mAb (Millipore) at 1:500 dilution overnight at 4 °C. After three washes in TBST, the blot was incubated with HRP-conjugated goat anti-mouse IgG (Pierce, Rockford, IL) for 1h at room temperature and washed with TBST three times. SuperSignal West Pico chemiluminescence substrate (Pierce) was used for signal production.
Supplementary Materials
Effect of metformin (MF) on the level of constitutive expression of γH2AX and ATM-S1981P in A431 cells Exponentially growing A431 cells were left untreated (Ctrl) or treated with 5 mM metformin for 48 h. γH2AX and ATM-S1981 immunofluorescence (IF) was detected with the phospho-specific Abs and cells fluorescence was measured by laser scanning cytometry.75 Based on differences in DNA content the cells were gated in G1, S and G2M phases of the cell cycle and the mean values of γH2AX and ATM-S1981P IF for cells in each of these cell cycle phases by were obtained gating analysis. The percent reduction of these mean values of the metformin-treated related to the untreated (Ctrl) cells is shown in the respective panels (the means of the three separate bands per each protein). The insets show DNA content frequency histograms in the untreated and metformin-treated cultures.
Effect of metformin (MF) on the level of expression of γH2AX in TK6 cells in stationary cultures TK6 cells were maintained at high cell density (>106 cells/ml) with no medium change for 5 days, then cells were left untreated (Ctrl) or treated with 5 mM metformin for 24 h (MF). The percent decline in mean values of γH2AX IF of cells in G1, S, and G2M phases of the cycle in the metformin-treated culture is shown in the MF panel.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Barzilai A and Yamamoto K. DNA damage responses to oxidative stress. DNA repair (Amst). 2004; 3: 1109 -1115. [PubMed] .
- 2. Nohl H. Generation of superoxide radicals as byproducts of cellular respiration. Ann Biol Clin. 1994; 52: 199 -204. .
- 3. Moller P and Loft S. Interventions with antioxidants and nutrients in relation to oxidative DNA damage and repair. Mutat Res. 2004; 551: 79 -89. [PubMed] .
- 4. Beckman KB and Ames BN. Oxidative decay of DNA. J Biol Chem. 1997; 272: 13300 -13305. .
- 5. Dianov GL and Parsons JL. Co-ordination of DNA single strand break repair. DNA repair (Amst). 2007; 6: 454 -460. [PubMed] .
- 6. Vilenchik MM and Knudson AG. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA. 2003; 100: 12871 -12876. [PubMed] .
- 7. Taioli E, Sram RJ, Garte BM, Kalina I, Popov TA, Farmer PB. Effects of polycyclic aromatic hydrocarbons (PAHs) in environmental pollution on exogenous and oxidative DNA damage (EXPAH project): description of the population under study. Mutat Res. 2007; 620: 1 -6. [PubMed] .
- 8. Tanaka T, Halicka HD, Traganos F, Darzynkiewicz Z. Phosphorylation of histone H2AX on Ser 139 and activation of ATM during oxidative burst in phorbol ester-treated human leukocytes. Cell Cycle. 2006; 5: 2671 -2675. [PubMed] .
- 9. Shacter E, Beecham EJ, Covey JM, Kohn KW, Potter M. Activated neutrophils induce prolonged DNA damage in neighboring cells. Carcinogenesis. 1988; 9: 2297 -2304. [PubMed] .
- 10. Chong YC, Heppner GH, Paul LA, Fulton AM. Macrophage-mediated induction of DNA strand breaks in target tumor cells. Cancer Res. 1989; 49: 6652 -6657. [PubMed] .
- 11. Demirbag R, Yilmaz R, Kocyigit A, Guzel S. Effect of coronary angiography on oxidative DNA damage observed in circulating lymphocytes. Angiology. 2007; 58: 141 -147. [PubMed] .
- 12. Altman SA, Zastawny TH, Randers-Eichorn L, Caciuttolo MA, Akman SA, Disdaroglu M, Rao G. Formation of DNA-protein cross-links in cultured mammalian cells upon treatment with iron ions. Free Radic Biol Med. 1995; 19: 897 -902. [PubMed] .
- 13. Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, Sauvaigo S. Hydroxyl radicals and DNA base damage. Mutat Res. 1999; 424: 9 -21. [PubMed] .
- 14. Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002; 219: 181 -182. .
- 15. Pastwa E and Blasiak J. Non-homologous DNA end joining. Acta Biochim Pol. 2003; 50: 891 -908. [PubMed] .
- 16. Jeggo PA and Lobrich M. Artemis links ATM to double strand end rejoining. Cell Cycle. 2005; 4: 359 -362. [PubMed] .
- 17. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res. 2011; Jan 7 (Epub) .
- 18. Gorbunova V and Seluanov A. Making ends meet in old age: DSB repair and aging. Mech Ageing Dev. 2005; 126: 621 -628. [PubMed] .
- 19. Espejel S, Franco S, Rodriguez-Perales S, Bouffler SD, Cigudosa JC, Blasco MA. Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J. 2002; 21: 2207 -2219. [PubMed] .
- 20. Karanjawala ZE and Lieber MR. DNA damage and aging. Mech Ageing Dev. 2004; 125: 405 -416. [PubMed] .
- 21. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 22. Blagosklonny MV and Campisi J. Cancer and aging: more puzzles, more promises? Cell Cycle. 2008; 7: 2615 -2618. [PubMed] .
- 23. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8: 1888 -1895. [PubMed] .
- 24. Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: Preliminary proposal. Cell Cycle. 2010; 9: 4788 -4794. [PubMed] .
- 25. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010; 9: 3151 -6. [PubMed] .
- 26. Blagosklonny MV. Why human lifespan is rapidly increasing: solving “longevity riddle” with “revealed-slow-aging” hypothesis. Aging. 2010; 2: 177 -182. [PubMed] .
- 27. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging. 2009; 1: 281 -288. [PubMed] .
- 28. Pospelova TV, Demidenko ZN, Bukreeva EI, Gudkov VA, Blagosklonny MV. Cell Pseudo-DNA damage response in senescent cells. Cycle. 2009; 8: 4112 -4118. .
- 29. Olive PL, Durand RE, Banath JP, Johnston PJ. Analysis of DNA damage in individual cells. Methods Cell Biol. 2001; 64: 235 -249. [PubMed] .
- 30. Gorczyca W, Gong J, Darzynkiewicz Z. Detection of DNA strand breaks in individual apoptotic cells by the in situ terminal deoxynucleotidyl transferase and nick translation assays. Cancer Res. 1993; 53: 1945 -1951. [PubMed] .
- 31. Huang X, Halicka HD, Traganos F, Tanaka T, Kurose A, Darzynkiewicz Z. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005; 38: 223 -243. [PubMed] .
- 32. Kitagawa R and Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol. 2005; 70: 99 -109. [PubMed] .
- 33. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273: 5858 -5868. [PubMed] .
- 34. Bartkova J, Bakkenist CJ, Rajpert-De Meyts E, Skakkebaek NE, Sehested M, Lukas J, Kastan MB, Bartek J. ATM activation in normal human tissues and testicular cancer. Cell Cycle. 2005; 4: 838 -845. [PubMed] .
- 35. Sedelnikova OA, Rogakou EP, Panuytin IG, Bonner W. Quantitive detection of 125IUdr-induced DNA double-strand breaks with γ-H2AX antibody. Ratiation Res. 2002; 158: 486 -492. .
- 36. Huang X, Traganos F, Darzynkiewicz Z. DNA damage induced by DNA topoisomerase I- and topoisomerase II- inhibitors detected by histone H2AX phosphorylation in relation to the cell cycle phase and apoptosis. Cell Cycle. 2003; 2: 614 -619. [PubMed] .
- 37. Banath JP and Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003; 63: 4347 -4350. [PubMed] .
- 38. Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, Dai W, Darzynkiewicz Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A. 2007; 71A: 648 -661. [PubMed] .
- 39. Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006; 5: 1940 -1945. [PubMed] .
- 40. Zhao H, Tanaka T, Halicka HD, Traganos F, Zarebski M, Dobrucki J, Darzynkiewicz Z. Cytometric assessment of DNA damage by exogenous and endogenous oxidants reports the aging-related processes. Cytometry A. 2007; 71A: 905 -914. [PubMed] .
- 41. Tanaka T, Kajstura M, Halicka HD, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation are strongly amplified during mitogenic stimulation of lymphocytes. Cell Prolif. 2007; 40: 1 -13. [PubMed] .
- 42. Tanaka T, Kurose A, Halicka HD, Traganos F, Darzynkiewicz Z. 2-Deoxy-D-glucose reduces the level of constitutive activation of ATM and phosphorylation of histone H2AX. Cell Cycle. 2006; 5: 878 -882. [PubMed] .
- 43. Zhao H, Tanaka T, Mitlitski V, Heeter J, Balazs EA, Darzynkiewicz Z. Protective effect of hyaluronate on oxidative DNA damage in WI-38 and A549 cells. Int J Oncol. 2008; 32: 1159 -1169. [PubMed] .
- 44. Halicka HD, Darzynkiewicz Z, Teodori L. Attenuation of constitutive ATM activation and H2AX phosphorylation in human leukemic TK6 cells by their exposure to static magnetic field. Cell Cycle. 2009; 8: 3236 -3238. .
- 45. Halicka HD, Ita M, Tanaka T, Kurose A, Darzynkiewicz Z. The biscoclaurine alkaloid cepharanthine protects DNA in TK6 lymphoblastoid cells from constitutive oxidative damage. Pharmacol Rep. 2008; 60: 93 -100. [PubMed] .
- 46. Towler MC and Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007; 100: 328 -341. [PubMed] .
- 47. Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans. 2011; 39: 1 -13. [PubMed] .
- 48. Viollet B and Andreelli F. AMP-activated protein kinase and metabolic control. Handb Exp Pharmacol. 2011; 203: 303 -330. [PubMed] .
- 49. Carling D, Mayer FV, Sanders MJ, Gamblin SJ. AMP-activated protein kinase: nature's energy sensor. Nat Chem Biol. 2011; 7: 512 -518. [PubMed] .
- 50. Anisimov VN. Metformin for aging and cancer prevention. Aging. 2010; 2: 760 -74. [PubMed] .
- 51. Xie Y, Wang Y, Yu L, Hu Q, Ji L, Zhang Y, Liao Q. Metformin promotes progesterone receptor expression via inhibition of mammalian target or rapamycin (mTOR) in endometrial cancer cells. J Ster Biochem Mol Biol. 2010; Dec 17 Epub .
- 52. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005; 310: 1642 -1646. [PubMed] .
- 53. Jalving M, Gietema JA, Lefrandt JD, De Jong S, Reyners AKL, Gans ROB, de Vries EGE. Metformin: taking away the candy for cancer? Eur J Cancer. 2010; 46: 2369 -2380. [PubMed] .
- 54. Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem. 2011; 286: 1 -11. [PubMed] .
- 55. Hou X, Song J, Li X-N, Zhang L, Wang XL, Chen L, Shen YH. Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochem Biophys Res Commun. 2010; 396: 199 -205. [PubMed] .
- 56. Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, Zabezhinski MA, Anikin IV, Karkach AS, Romanyukha AA. Metformin extents life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors. Cell Cycle. 2010; 9: 188 -197. [PubMed] .
- 57. Darzynkiewicz Z, Traganos F, Zhao H, Halicka HD, Skommer J, Wlodkowic D. Analysis of individual molecular events of DNA damage response by flow and image-assisted cytometry. Methods Cell Biol. 2011; 103: 115 -148. [PubMed] .
- 58. Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol. 2006; 29: 495 -501. [PubMed] .
- 59. Rothe G and Klouche M. Phagocyte functions. Methods Cell Biol. 2004; 75: 679 -708. [PubMed] .
- 60. Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong JR, Greenfield JR, Greenup LC, Kirkpatrick CM, Ray JE, Timmins P, Williams KM. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011; 50: 81 -98. [PubMed] .
- 61. Piwkowska A, Rogacka D, Jankowski M, Dominiczak MH, Stepinski JK, Angielski S. Metformin induces suppression of NAD(P)H oxidase activity in podocytes. Biochem Biophys Res Commun. 2010; 393: 268 -273. [PubMed] .
- 62. Ouslimani N., Peynet J., Bonnefont-Rousselot D., Therond P., Legrand A., Beaudeux J. L.. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism. 2005; 54: 829 -834. [PubMed] .
- 63. Kane DA, Andersen EJ, Price JW III, Woodlief TL, Lin C-T, Bikman BT, Cortright RN, Neufer PD. Metformin selectively attenuates mitochondrial H2O2 emission without affecting respiratory capacity in skeletal muscle of obese rats. Free Radical Biol Med. 2010; 49: 1082 -1087. [PubMed] .
- 64. Piro S, Rabuazzo AM, Renis M, Purello F. Effects of metformin on oxidative stress, adenine nucleotides balance and glucose-induced insulin release impaired by chronic FFA exposure in rat pancreatic islets. J Endocrinol Invest. 2011; Jul 12 (Epub) .
- 65. Bellin C, de Wiza DH, Wiernsperger NF, Rosen P. Generation of reactive oxygen species by endothelial and smooth muscle cells: Influence of hyperglycemia and metformin. Horm Metab Res. 2006; 38: 732 -739. [PubMed] .
- 66. Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Hohl H, Waldhansl W, Furnassin C. Thiazolidinediones, like metformin, inhibit respiratory complex I. A common mechanism contributing to their antidiabetic action. Diabetes. 2004; 53: 1052 -1059. [PubMed] .
- 67. Drose S, Hanley PJ, Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and II. Biochim Biophys Acta. 2009; 1790: 558 -565. [PubMed] .
- 68. Cleaver JA, Feeney L, Revet I. Phosphorylated H2Ax is not an unambiguous marker of DNA double strand breaks. Cell Cycle. 2011; 10: 3223 -3224. [PubMed] .
- 69. Burhans WC and Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007; 33: 7545 -7556. [PubMed] .
- 70. Bartkova J, Hamerlik P, Stockhausen MT, Ehrman J, Hlobikova A, Laursen H, Kalita O, Kolar Z, Paulsen HS, Broholm H, Lukas J, Bartek J. Replication stress and oxidative damage contributes to aberrant constitutive activation of DNA damage signaling in human gliomas. Oncogene. 2010; 29: 5095 -5102. [PubMed] .
- 71. Zhao H, Halicka HD, Jorgensen E, Traganos F, Darzynkiewicz Z. New biomarkers probing the depth of cell senescence assessed by laser scanning cytometry. Cytometry A. 2010; 77A: 999 -1007. [PubMed] .
- 72. Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Martin-Castillo B, Menendez Ja. Metformin activates an Ataxia Telangiectasia Mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011; 10: 1499 -1501. [PubMed] .
- 73. Nilsson S, Huelsenbeck J, Fritz G. Mevalonate pathway inhibitors affect anticancer drug-induced cell death and DNA damage response of human sarcoma cells. Cancer Lett. 2011; 304: 60 -69. [PubMed] .
- 74. Smart DJ, Ahmedi KP, Harvey JS, Lynch AM. Genotoxicity screening via the γH2AX by flow assay. Mutat Res. 2011; 71: 21 -31. .
- 75. Kajstura M, Halicka HD, Pryjma J, Darzynkiewicz Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytometry A. 2007; 71A: 125 -131. [PubMed] .
- 76. Pozarowski P, Holden E, Darzynkiewicz Z. Laser scanning cytometry: Principles and applications. Meth Molec Biol. 2006; 319: 165 -192. .