



Introduction
Alzheimer disease (AD) is a progressive and fatal neurodegenerative disease that is clinically characterized by dementia and neurobehavioral deterioration [1-4].
While the hallmark features of amyloid plaques, neurofibrillary tangles (NFTs), and neuronal loss are well established, the cause(s) of the disease remain elusive. Nonetheless, one mechanism that is gaining increased prominence is the ectopic re-entry of neurons into the cell cycle [5], which accumulate cyclins, CDKs, and other mitotic factors [6-22]. While neuronal cell cycle re-entry mediates AD-type changes [23] and is linked with cell death [24-27], a number of unanswered questions remain [28]. For example, it is still unclear whether the presence of various cell cycle markers represent a bona fide cell cycle or are they, instead, consequential to other pathological processes (e.g., apoptosis). Also, if representative of cell cycle, it is unclear why neurons do not progress and enter cytokinesis. One fitting hypothesis is that some cells undergo hypermitogenic cell cycle arrest, as an alternative to apoptosis, which would result in cell senescence and survival [29].
The minichromosome maintenance proteins are a eukaryotic family of six distinct protein subtypes (Mcm2-7) that are necessary for DNA replication initiation and progression in the cell cycle [30]. During the G1-phase of the cell cycle, the hexameric Mcm2-7 complex assembles at origins of replication on nuclear DNA [31]. Once in S-phase, the complex is phosphorylated by the Cdc7/Dbf4 kinase and the B-type CDKs, and acting as the DNA helicase initiates DNA replication at origins and allows progression of the replication forks [32-37]. The assembly of the Mcm complex is tightly regulated, can occur only in G1 when the activity of CDKs and Cdc7 is low, and is actively prevented once cells enter S-phase till exit of mitosis when the activity of these kinases is high [38], such that replication only occurs once per cell cycle. Expression of Mcm proteins is restricted to actively cycling cells and is a good proliferation marker [39]. While in budding yeast Mcm2-7 proteins shuttle in and out of the nucleus, human Mcms are generally detected in the nuclear compartment [40,41]. Phosphorylation can occur at multiple sites, however phosphorylation of Mcm2 in two adjacent sites Ser40 and Ser41, carried out in succession by CDKs and Cdc7, strictly correlates with cells undergoing or having terminated DNA synthesis [42]. As such, antisera specific for pSer40/41 Mcm2 phosphorylation provides an excellent marker for the detection of cells in a late stage of the cell cycle.
In this study, we compared Ser40/41 Mcm2 phosphorylation in AD and aged-matched control brain. In AD, phosphorylated Mcm2 localized to the cytoplasm of neurons, and strikingly with the characteristic NFT. These findings further support the notion that neurons in AD re-enter the cell cycle, pass through S-phase by activating the only two essential S-phase promoting kinases, and provide evidence for aberrant localization of an essential DNA replication protein.
Results
Phosphorylated Mcm2 protein at a CDK- and Cdc7- dependent site is localized to the cytoplasm of AD neurons and targets neurofibrillary tangles and amyloid plaques
The presence of pSer40/41 Mcm2 (pMcm2) protein was detected using the immunocytochemistry methods discussed in the corresponding section. All of the AD cases examined demonstrated significant accumulation of pMcm2 in NFTs, dystrophic neurites, and neuropil threads (Figure 1B). In most cases, glial nuclei were often stained, and in a small number of cases, some pyramidal cell nuclei within the CA3 region showed significant pMcm2 reactivity (Figure 1D, arrows). In similar areas in most control cases, no staining was seen (Figure 1A), in a small number of aged control cases, pyramidal neuron nuclei showed high pMcm2 protein levels (Figure 1C). In some of the aged controls, a small number of pathological structures (NFT, neuropil threads,etc) were labeled with the pMcm2 antisera (data not shown).
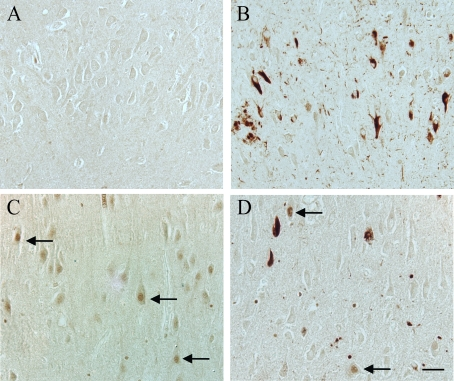
Figure 1. In an 87 year old AD case, hippocampal tissue sections demonstrate significant
localization of pMcm2 protein in NFT, dystrophic neurites, and neuropil
threads (B). In another AD case, in the CA3 region, in addition to
pathological structures, a few pyramidal neuron nuclei (arrows) have significant
pMcm2 accumulation (D). Most control cases, representative case age
61 years, demonstrate no neuronal staining for pMcm2 protein (A),
while a few older control cases demonstrate significant nuclear
immunolocalization in the pyramidal neurons (control case age 74 years, C)
Scale bar= 50 μm.
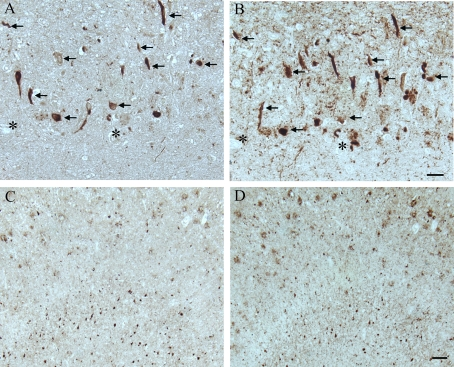
Figure 2. In another AD case, age 63, adjacent hippocampal
tissue sections demonstrate many of the AD-related pathological
structures (arrows) containing pMcm2 (A) are also positive
for hyper-phosphorylated tau (B) in the CA1 region.
Lower magnification of adjacent sections of the subiculum
shows the large number of NFT and plaques recognized by
pMcm2 (C) and AT8 (D). * denotes landmark vessel. Scale
bars= 50 μm (A,B), 100 μm (C,D).
All AD cases examined, both with formalin and methacarn fixation, contained many immunoreactive NFT throughout the hippocampus. Additionally, the binding of the anti-pMcm2 antibody to NFT within AD brains was striking and showed some co-localization with phosphorylated tau on adjacent sections of AD tissue In particular, many of the same NFT and senile plaques demonstrated co-localization of tau with pMcm2 in all AD cases (Figure 2). In Figure 3, the specificity of the antibody to pMcm2 protein was confirmed by absorbing antibodies to pMcm2 with phosphorylated and non-phosphorylated peptides. As expected, the phosphorylated peptide completely absorbed the antibody producing no visible staining on the section (Figure 3C) whereas the peptide lacking phosphorylation failed to absorb the antibody (Figure 3B) and produced staining similar to that of the unabsorbed sample (Figure 3A). Further confirmation of the specificity was obtained by treating some sections with alkaline phosphatase to remove phosphate groups. Figure 4 shows that nearly all of the reactivity of the pMcm2 antisera is abolished following dephosphorylation on adjacent sections with (Figure 4B) and without (Figure 4A) alkaline phosphatase pretreatment.
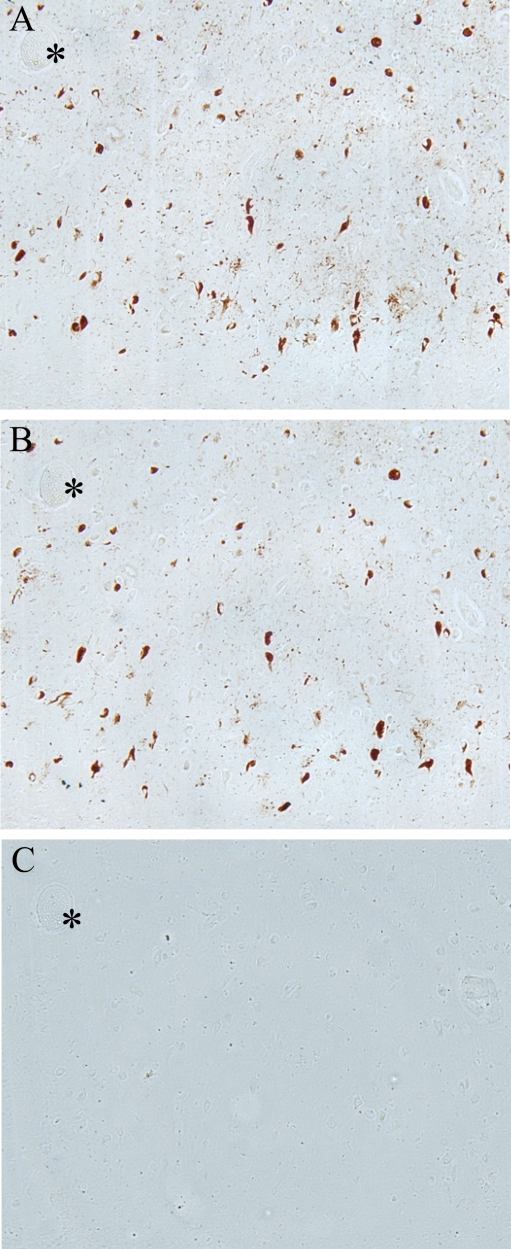
Figure 3. Adsorption
of pMcm2 antibody confirms specificity
to corresponding pMcm2 antigen. (A) AD hippocampal tissue stained
with pMcm2 antibody. (B) Adjacent section treated with pMcm2
antibody absorbed with non-phosphorylated Mcm peptide demonstrates similar
staining. (C) Adjacent section treated with pMcm2 antibody absorbed
with phosphorylated Mcm2 peptide demonstrates complete absorption. *
denotes landmark vessel.
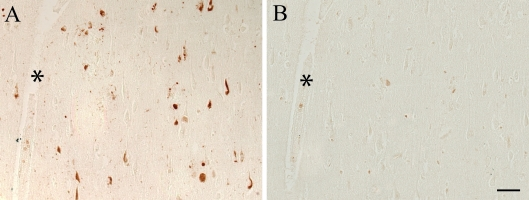
Figure 4. Pretreatment with alkaline phosphatase to remove phosphate
groups, results in elimination of pMcm2 reactivity (B) compared to an
untreated adjacent serial section of an AD case (A). * denotes
landmark vessel. Scale bar = 50 μm.
Discussion
In AD, multiple lines of evidence suggest that neurons vulnerable to degeneration emerge from the post-mitotic, quiescent state and are phenotypically suggestive of cells that are cycling, rather than being in the normal, terminally differentiated, non-dividing state [43]. Such cell cycle re-entry has not only been linked to cell death [44], but has also been implicated in the hallmark pathologies of the disease, namely tau phosphorylation and amyloid-β (Aβ) [23]. Nonetheless, despite the identification of a variety of cell cycle proteins in AD, there remains controversy over whether these are truly indicative of a bona fide reaction of the cell cycle or, instead, reflect the pleotrophic actions of these protein markers [28]. Indeed, proteins previously detected in AD such as Ki67, PCNA, cdc2, cdk4, BRCA1 and pRb [9,44-49], although noted regulators of the mitotic process, are also involved in neuronal processes unrelated to the cell cycle such as DNA repair [50], apoptosis [51], and oxidative stress [52]. Here, however, the detection of a key component of the DNA replication machinery Mcm2, phosphorylated in the Cdk and Cdc7 dependent site Ser40/41 in AD neuronal cytoplasm and NFT not only provides additional support for the cell cycle hypothesis of AD [10], but supports an authentic re-entrant phenotype associated with DNA replication [53]. Mcm2 is in fact not expressed in non-proliferating tissues, as shown in neurons in age-matched control brain, but it accumulates in G1 cells re-entering the cell cycle. Dual phosphorylation of Mcm2 at serine 40 and serine 41, then requires the activity of two kinases whose activity is upregulated in S-phase by the periodic expression of regulatory subunits, Cyclin and Dbf4 [54].
Very intriguingly, pMcm2 in AD neurons, unlike in most cancer cell lines [42], appears to accumulate mostly in the cytoplasm suggesting further degree of deregulation of the MCM complex in disease tissues that may explain the inability of neurons to progress through cytokinesis.
The ectopic re-entry of neurons into the cell cycle likely plays an important role mediating other aspects of AD pathology. Specifically, the microtubule associated protein tau, in cases of AD, exists in a highly phosphorylated form and composes the NFTs that burden the diseased brain, and this increased phosphorylation of tau destabilizes microtubular dynamics and results in neuronal dysfunction [55,56]. Interestingly, while cells are mitotically active, the cell cycle regulator proteins CDKs initiate a similar phosphorylation of tau that precedes the appearance of the NFTs [8] and suggests a possible cause-effect relationship [23]. Similarlythe major protein component of senile plaques is a 4.2 kDa polypeptide termed Aβ, which is derived from a larger precursor (APP) encoded on chromosome 21. Attesting to the importance of this protein, mutations in the APP gene are linked to the inevitable onset of familial AD [57]. Given the probable role of mitotic re-entry in AD, it is notable that APP is upregulated secondary to mitogenic stimulation [58] and that APP metabolism is regulated by cell cycle-dependent changes [59]. Interestingly, Aβ itself is mitogenic in vitro [60,61] and therefore may play a direct role in the induction and/or propagation of cell cycle-mediated events in AD. Additionally, Aβ-mediated cell death, at least in vitro, is dependent on the presence of various cell cycle-related elements [62]. Most importantly, the ectopic re-entry of neurons into the cell cycle was recently shown to lead to cell death, gliosis, and cognitive deficits—all cardinal features of AD [24].
In conclusion, our results provide further support for the role of cell cycle re-entry in the initiation and progression and AD. As such, cell cycle inhibitors present potential therapies for the disease [63].
Methods
Tissue. Autopsy tissue samples were obtained using a protocol approved by the Institutional Review Board at University Hospitals of Cleveland. Hippocampal or cortical tissue samples were obtained post mortem from patients (n = 10, ages 63-91 years, mean = 81.8 years) with clinically and histopathologically confirmed AD, as well as from aged-matched controls (n = 8, ages 56-86 years, mean = 70.2 years) with similar post mortem intervals (AD: 2-31 h, mean = 14.5 h; controls: 5-27 h, mean = 15.6 h). All cases were categorized based on clinical and pathological criteria established by CERAD and NIA consensus panel [64]. From the clinical reports available to us, we found no obvious differences in agonal status or other potential confounders between the groups. Tissue was fixed in methacarn (methanol: chloroform: acetic acid; 6: 3: 1 v/v/v) at 4°C overnight or in routine formalin. Following fixation, tissue was dehydrated through ascending ethanol, embedded in paraffin, and 6-μm sections were cut.
Immunohistochemistry. Tissue sections were deparaffinized in xylene, hydrated through descending ethanol, and endogenous peroxidase activity was quenched by 30 minute incubation in 3% hydrogen peroxide in methanol. Non-specific binding sites were blocked with 30 minute incubation in 10% normal goat serum. Sections of both AD and control were immunostained with rabbit polyclonal antibody to Mcm2 phosphorylated at sites Ser40/41 (1:150) [42] or mouse monoclonal antibody to tau (AT8 1:1000) recognizing phosphorylated tau (Ser202/Thr205) (Pierce, Rockford, IL) to identify the location of neuronal pathological structures. Absorption experiments were performed to verify the binding of the Mcm2 Ser40/41 antibody to the appropriate phosphorylated peptide. The primary antibody was incubated in 0.2mg/ml peptide containing 0 or 2 phosphates for 16 hours at 4°C prior to immunostaining. All sections were immunostained using the peroxidase-antiperoxidase with 3-3'-diaminobenzidine as co-substrate as previously described [65].
Acknowledgments
Work in the authors' laboratories is supported by the National Institutes of Health (AG031364, AG030096, AG028679) and the Alzheimer's Association.
Conflicts of Interest
Dr. Smith is, or has in the past been, a paid consultant for, owns equity or stock options in and/or receives grant funding from Canopus BioPharma, Medivation, Neurotez, Neuropharm, Panacea Pharmaceuticals, and Voyager Pharmaceuticals. Dr. Perry is, or has in the past been, a paid consultant for and/or owns equity or stock options in Takeda Pharmaceuticals, Voyager Pharmaceuticals, Panacea Pharmaceuticals and Neurotez Pharmaceuticals.
References
- 1. Smith MA Alzheimer disease. Int Rev Neurobiol. 1998; 42: 1 -54. [PubMed] .
- 2. Folstein MF and Bylsma FW. Terry RD, Katzman R, Bick KL, Sisodia SS. Noncognitive symptoms of Alzheimer disease Alzheimer disease. Philadelphia Lippincott Williams & Wilkins 1999; 25 -37. .
- 3. Morris JC Terry RD, Katzman R, Bick KL, Sisodia SS. Clinical presentation and course of Alzheimer disease Alzheimer disease. Philadelphia Lippincott Williams & Wilkins 1999; 11 -24. .
- 4. West MJ , Kawas CH , Stewart WF , Rudow GL and Troncoso JC. Hippocampal neurons in pre-clinical Alzheimer's disease. Neurobiol Aging. 2004; 25: 1205 -1212. [PubMed] .
- 5. Zhu X , Lee HG , Perry G and Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007; 1772: 494 -502. [PubMed] .
- 6. Zhu X , Raina AK and Smith MA. Cell cycle events in neurons. Proliferation or death. Am J Pathol. 1999; 155: 327 -329. [PubMed] .
- 7. Vincent I , Jicha G , Rosado M and Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997; 17: 3588 -3598. [PubMed] .
- 8. Vincent I , Zheng JH , Dickson DW , Kress Y and Davies P. Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging. 1998; 19: 287 -296. [PubMed] .
- 9. McShea A , Harris PL , Webster KR , Wahl AF and Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997; 150: 1933 -1939. [PubMed] .
- 10. McShea A , Wahl AF and Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med Hypotheses. 1999; 52: 525 -527. [PubMed] .
- 11. McShea A , Zelasko DA , Gerst JL and Smith MA. Signal transduction abnormalities in Alzheimer's disease: evidence of a pathogenic stimuli. Brain Res. 1999; 815: 237 -242. [PubMed] .
- 12. Vincent I , Rosado M and Davies P. Mitotic mechanisms in Alzheimer's disease. J Cell Biol. 1996; 132: 413 -425. [PubMed] .
- 13. Arendt T , Rodel L , Gartner U and Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. Neuroreport. 1996; 7: 3047 -3049. [PubMed] .
- 14. Smith TW and Lippa CF. Ki-67 immunoreactivity in Alzheimer's disease and other neurodegenerative disorders. J Neuropathol Exp Neurol. 1995; 54: 297 -303. [PubMed] .
- 15. Arendt T , Holzer M , Grossmann A , Zedlick D and Bruckner MK. Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen-activated protein kinase in Alzheimer's disease. Neuroscience. 1995; 68: 5 -18. [PubMed] .
- 16. Nagy Z , Esiri MM , Cato AM and Smith AD. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl). 1997; 94: 6 -15. [PubMed] .
- 17. Nagy Z , Esiri MM and Smith AD. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol (Berl). 1997; 93: 294 -300. [PubMed] .
- 18. Arendt T , Holzer M and Gartner U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer's disease. J Neural Transm. 1998; 105: 949 -960. [PubMed] .
- 19. Zhu X , Raina AK , Boux H , Simmons ZL , Takeda A and Smith MA. Activation of oncogenic pathways in degenerating neurons in Alzheimer disease. Int J Dev Neurosci. 2000; 18: 433 -437. [PubMed] .
- 20. Zhu X , Rottkamp CA , Boux H , Takeda A , Perry G and Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000; 59: 880 -888. [PubMed] .
- 21. Zhu X , Rottkamp CA , Raina AK , Brewer GJ , Ghanbari HA , Boux H and Smith MA. Neuronal CDK7 in hippocampus is related to aging and Alzheimer disease. Neurobiol Aging. 2000; 21: 807 -813. [PubMed] .
- 22. Zhu X , Raina AK , Perry G and Smith MA. Alzheimer's disease: the two-hit hypothesis. Lancet Neurol. 2004; 3: 219 -226. [PubMed] .
- 23. McShea A , Lee HG , Petersen RB , Casadesus G , Vincent I , Linford NJ , Funk JO , Shapiro RA and Smith MA. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta. 2007; 1772: 467 -472. [PubMed] .
- 24. Lee HG , Casadesus G , Nunomura A , Zhu X , Castellani RJ , Richardson SL , Perry G , Felsher DW , Petersen RB and Smith MA. The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am J Pathol. 2009; 174: 891 -897. [PubMed] .
- 25. Hamdane M and Buee L. The complex p25/Cdk5 kinase in neurofibrillary degeneration and neuronal death: The missing link to cell cycle. Biotechnol J. 2007; 2: 967 -977. [PubMed] .
- 26. Wung JK , Perry G , Kowalski A , Harris PL , Bishop GM , Trivedi MA , Johnson SC , Smith MA , Denhardt DT and Atwood CS. Increased expression of the remodeling- and tumorigenic-associated factor osteopontin in pyramidal neurons of the Alzheimer's disease brain. Curr Alzheimer Res. 2007; 4: 67 -72. [PubMed] .
- 27. Neve RL and McPhie DL. Dysfunction of amyloid precursor protein signaling in neurons leads to DNA synthesis and apoptosis. Biochim Biophys Acta. 2007; 1772: 430 -437. [PubMed] .
- 28. Bowser R and Smith MA. Cell cycle proteins in Alzheimer's disease: plenty of wheels but no cycle. J Alzheimers Dis. 2002; 4: 249 -254. [PubMed] .
- 29. Blagosklonny MV Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358 -362. [PubMed] .
- 30. Bailis JM and Forsburg SL. MCM proteins: DNA damage, mutagenesis and repair. Curr Opin Genet Dev. 2004; 14: 17 -21. [PubMed] .
- 31. Bell SP and Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002; 71: 333 -374. [PubMed] .
- 32. Aparicio T , Ibarra A and Mendez J. Cdc45-MCM-GINS, a new power player for DNA replication. Cell Div. 2006; 1: 18 [PubMed] .
- 33. Kudoh A , Daikoku T , Ishimi Y , Kawaguchi Y , Shirata N , Iwahori S , Isomura H and Tsurumi T. Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J Virol. 2006; 80: 10064 -10072. [PubMed] .
- 34. You Z and Masai H. DNA binding and helicase actions of mouse MCM4/6/7 helicase. Nucleic Acids Res. 2005; 33: 3033 -3047. [PubMed] .
- 35. Masai H , You Z and Arai K. Control of DNA replication: regulation and activation of eukaryotic replicative helicase, MCM. IUBMB Life. 2005; 57: 323 -335. [PubMed] .
- 36. Lee JK and Hurwitz J. Processive DNA helicase activity of the minichromosome maintenance proteins 4, 6, and 7 complex requires forked DNA structures. Proc Natl Acad Sci U S A. 2001; 98: 54 -59. [PubMed] .
- 37. Kaplan DL , Davey MJ and O'Donnell M. Mcm4,6,7 uses a "pump in ring" mechanism to unwind DNA by steric exclusion and actively translocate along a duplex. J Biol Chem. 2003; 278: 49171 -49182. [PubMed] .
- 38. Blow JJ and Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol. 2005; 6: 476 -486. [PubMed] .
- 39. Gonzalez MA , Tachibana KE , Laskey RA and Coleman N. Control of DNA replication and its potential clinical exploitation. Nat Rev Cancer. 2005; 5: 135 -141. [PubMed] .
- 40. Krude T , Musahl C , Laskey RA and Knippers R. Human replication proteins hCdc21, hCdc46 and P1Mcm3 bind chromatin uniformly before S-phase and are displaced locally during DNA replication. J Cell Sci. 1996; 109 ( Pt 2): 309 -318. [PubMed] .
- 41. Rialland M , Sola F and Santocanale C. Essential role of human CDT1 in DNA replication and chromatin licensing. J Cell Sci. 2002; 115: 1435 -1440. [PubMed] .
- 42. Montagnoli A , Valsasina B , Brotherton D , Troiani S , Rainoldi S , Tenca P , Molinari A and Santocanale C. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J Biol Chem. 2006; 281: 10281 -10290. [PubMed] .
- 43. Obrenovich ME , Raina AK , Ogawa O , Atwood CS and Smith MA. Copani A and Nicoletti F. Alzheimer disease - a new beginning, or a final exit? The Cell Cycle and Neuronal Cell Death. Georgestown, Texas Landes Bioscience 2005; 79 -93. .
- 44. Smith MZ , Nagy Z and Esiri MM. Cell cycle-related protein expression in vascular dementia and Alzheimer's disease. Neurosci Lett. 1999; 271: 45 -48. [PubMed] .
- 45. Ueberham U , Hessel A and Arendt T. Cyclin C expression is involved in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2003; 24: 427 -435. [PubMed] .
- 46. Nagy Z Cell cycle regulatory failure in neurones: causes and consequences. Neurobiol Aging. 2000; 21: 761 -769. [PubMed] .
- 47. Ding XL , Husseman J , Tomashevski A , Nochlin D , Jin LW and Vincent I. The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer's disease. Am J Pathol. 2000; 157: 1983 -1990. [PubMed] .
- 48. Hoozemans JJ , Bruckner MK , Rozemuller AJ , Veerhuis R , Eikelenboom P and Arendt T. Cyclin D1 and cyclin E are co-localized with cyclo-oxygenase 2 (COX-2) in pyramidal neurons in Alzheimer disease temporal cortex. J Neuropathol Exp Neurol. 2002; 61: 678 -688. [PubMed] .
- 49. Yang Y , Mufson EJ and Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003; 23: 2557 -2563. [PubMed] .
- 50. Schwartz EI , Smilenov LB , Price MA , Osredkar T , Baker RA , Ghosh S , Shi FD , Vollmer TL , Lencinas A , Stearns DM , Gorospe M and Kruman II. Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle. 2007; 6: 318 -329. [PubMed] .
- 51. Zhang Y , Qu D , Morris EJ , O'Hare MJ , Callaghan SM , Slack RS , Geller HM and Park DS. The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin. J Neurosci. 2006; 26: 8819 -8828. [PubMed] .
- 52. Clement A , Henrion-Caude A , Besnard V and Corroyer S. Role of cyclins in epithelial response to oxidants. Am J Respir Crit Care Med. 2001; 164: S81 -84. [PubMed] .
- 53. Zhu X , Siedlak SL , Wang Y , Perry G , Castellani RJ , Cohen ML and Smith MA. Neuronal binucleation in Alzheimer disease hippocampus. Neuropathol Appl Neurobiol. 2008; 34: 457 -465. [PubMed] .
- 54. Jiang W , McDonald D , Hope TJ and Hunter T. Mammalian Cdc7-Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J. 1999; 18: 5703 -5713. [PubMed] .
- 55. Lindwall G and Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984; 259: 5301 -5305. [PubMed] .
- 56. Alonso AC , Grundke-Iqbal I and Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996; 2: 783 -787. [PubMed] .
- 57. Hardy J Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 2006; 9: 151 -153. [PubMed] .
- 58. Ledoux S , Rebai N , Dagenais A , Shaw IT , Nalbantoglu J , Sekaly RP and Cashman NR. Amyloid precursor protein in peripheral mononuclear cells is up-regulated with cell activation. J Immunol. 1993; 150: 5566 -5575. [PubMed] .
- 59. Suzuki T , Oishi M , Marshak DR , Czernik AJ , Nairn AC and Greengard P. Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J. 1994; 13: 1114 -1122. [PubMed] .
- 60. McDonald DR , Bamberger ME , Combs CK and Landreth GE. beta-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998; 18: 4451 -4460. [PubMed] .
- 61. Pyo H , Jou I , Jung S , Hong S and Joe EH. Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. Neuroreport. 1998; 9: 871 -874. [PubMed] .
- 62. Giovanni A , Wirtz-Brugger F , Keramaris E , Slack R and Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem. 1999; 274: 19011 -19016. [PubMed] .
- 63. Woods J , Snape M and Smith MA. The cell cycle hypothesis of Alzheimer's disease: Suggestions for drug development. Biochim Biophys Acta. 2007; 1772: 503 -508. [PubMed] .
- 64. Mirra SS , Heyman A , McKeel D , Sumi SM , Crain BJ , Brownlee LM , Vogel FS , Hughes JP , van Belle G and Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991; 41: 479 -486. [PubMed] .
- 65. Sternberger LA New York Wiley Immunocytochemistry. 1986; .