



Introduction
Medial vascular calcification (VC) increases with advanced age [1]. This process is strongly accelerated by the uremic environment in chronic kidney disease (CKD) [2], which is considered a state of premature vascular aging [3]. VC increases vascular stiffness and elevates pulse pressure, resulting in impaired organ perfusion and increased left ventricular afterload [4]. Accordingly, the presence of calcifications in the arteries is associated with an increased mortality risk [5].
VC is the result of a complex process that ultimately culminates in deposition of calcium-phosphate in the vascular wall [6]. Under physiological conditions, extraosseous calcifications are actively prevented by calcification inhibitors, most importantly pyrophosphate, matrix GLA protein or fetuin-A [6]. In aged or diseased conditions, these mechanisms may be weakened and pro-calcific effects may be augmented in arterial tissue [2]. An especially important role in VC is attributed to vascular smooth muscle cells (VSMCs) [6]. These cells can take up pro-calcific functions and induce alteration of extracellular matrix, as well as release of pro-calcific vesicles and tissue-nonspecific alkaline phosphatase (ALPL), which cleaves the ubiquitous calcification inhibitor pyrophosphate [7]. The reprogramming of VSMCs towards a pro-calcific state is coordinated by various complex signaling pathways [8]. The mitogen-activated protein kinase (MAPK) p38 plays a prominent, but incompletely understood role in VC [9, 10]. P38 MAPK is activated by phosphorylation in response to various stimuli and inactivated by phosphatases such as dual-specificity phosphatase 1 (DUSP1, also known as MAPK phosphatase 1 or MKP1) [11]. P38 MAPK inhibition is sufficient to ameliorate VSMC calcification [12].
Phosphate has been recognized as a powerful stimulator of VSMC reprogramming and VC and may act, at least partly, through the formation of calciprotein particles [8]. Elevated phosphate conditions induce metabolic alterations with increased oxidative stress generation and activate inflammatory pathways to promote pro-calcific signaling in VSMCs [2, 13]. These alterations may involve VSMC senescence, inducing a proinflammatory senescence-associated secretory phenotype (SASP) [14]. Phosphate exposure is able to up-regulate senescence markers in VSMCs [15]. In turn, treatment of uremic rats with phosphate binders reduced expression of senescence markers in the vasculature [16]. Senolytic substances may eliminate senescent cells [17] and some have been shown to reduce VSMC calcification, such as quercetin [18], dasatinib [19], piperlongumine [20] or curcumin [21]. Thus, senolytics have been discussed as potential treatment options in uremic calcification, but their specificity and exact mechanisms might still be debatable.
One substance with senolytic potential is the flavonoid fisetin [22], which exhibits potent anti-inflammatory and antioxidant properties [23]. Fisetin increases lifespan in aged mice [24], attenuates renal fibrosis in cisplatin nephrotoxicity [25] and ameliorates atherosclerosis [26]. This study, therefore, investigated the vasculo-protective potential of fisetin in phosphate-induced VSMC calcification and the underlying mechanisms.
Results
To investigate a possible impact of fisetin on VSMC calcification, a first series of experiments was performed in cultured primary human aortic VSMCs during control or pro-calcific conditions with addition of calcium and the phosphate donor β-glycerophosphate, in the absence and presence of increasing concentrations of fisetin (0 – 20 μM). As illustrated in Figure 1, calcification medium significantly up-regulated BMP2 and ALPL mRNA expression in VSMCs, effects significantly suppressed in the presence of 1 μM fisetin concentration. Lower fisetin concentrations did not significantly affect calcification medium-induced BMP2 and ALPL mRNA expression (Supplementary Figure 1).
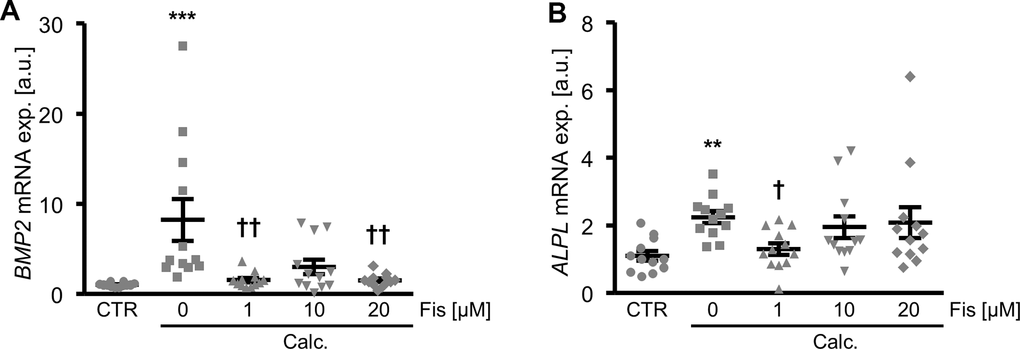
Figure 1. Dose-dependent effects of fisetin on calcific marker expression in VSMCs during pro-calcific conditions. Relative mRNA expression (n=12) of BMP2 (A) and ALPL (B) in HAoSMCs treated for 48h with control (CTR) or calcification medium (Calc.) without and with the indicated concentrations of fisetin (Fis, 0 - 20 μM). **(p<0.01), ***(p<0.001) significant vs. control group; †(p<0.05), ††(p<0.01) significant vs. Calc.-treated group.
The calcification medium-induced increase of pro-calcific markers CBFA1, SP7, BGLAP and SPP1 mRNA expression (Figure 2A–2D), ALP activity (Figure 2E) as well as CBFA1 nuclear localization (Figure 2F) were all suppressed by 1 μM fisetin supplementation. Co-treatment with fisetin reduced expression of markers associated with senescence of VSMCs promoted by calcification medium, as shown by the mRNA expression of CDKN1A and GLB1 and further indicated by senescence-associated (SA)-β-galactosidase staining (Supplementary Figure 2). Treatment with fisetin alone did not consistently modify pro-calcific and senescence markers expression at 1 μM concentration, but high concentrations of fisetin (20 μM) had adverse effects in VSMCs (Supplementary Figure 3). Moreover, pre-treatment of VSMCs with 1 μM fisetin did not significantly modify calcification medium-induced pro-calcific marker expression (Supplementary Figure 4). More importantly, as illustrated by Osteosense fluorescence imaging and determination of calcium content (Figure 3), co-treatment with 1 μM fisetin significantly reduced calcification of VSMCs exposed to calcification medium. Taken together, fisetin inhibited pro-calcific signaling and calcification of VSMCs induced by mineral stress during high phosphate and calcium exposure.
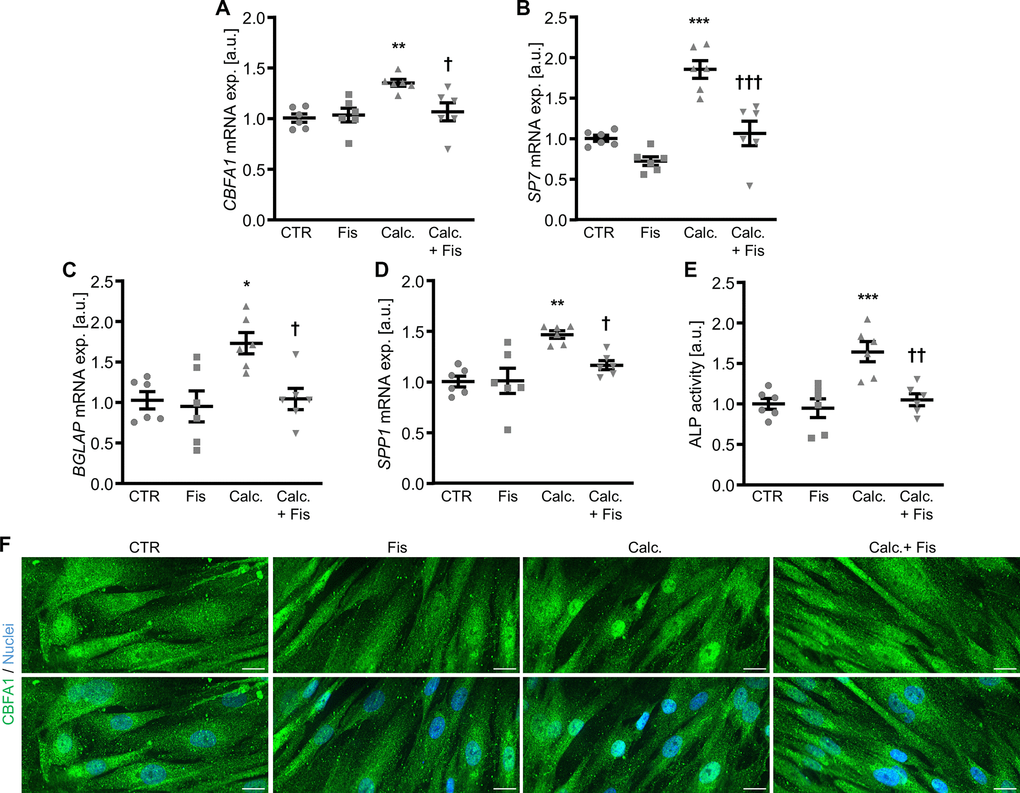
Figure 2. Effects of fisetin on pro-calcific signaling in VSMCs during pro-calcific conditions. Relative mRNA expression (n=6) of CBFA1 (A), SP7 (B), BGLAP (C) and SPP1 (D) in HAoSMCs treated for 48h with control (CTR) or calcification medium (Calc.) without and with 1 μM fisetin (Fis). (E) Normalized ALP activity (n=6) in HAoSMCs treated for 7d with control (CTR) or calcification medium (Calc.) without and with 1 μM fisetin (Fis). *(p<0.05), **(p<0.01), ***(p<0.001) significant vs. control group; †(p<0.05), ††(p<0.01), †††(p<0.001) significant vs. Calc.-treated group. (F) CBFA1 (green) and nuclei (blue) shown by confocal imaging in HAoSMCs treated for 48h with control (CTR) or calcification medium (Calc.) without and with 1 μM fisetin (Fis). Scale bar: 20 μm.
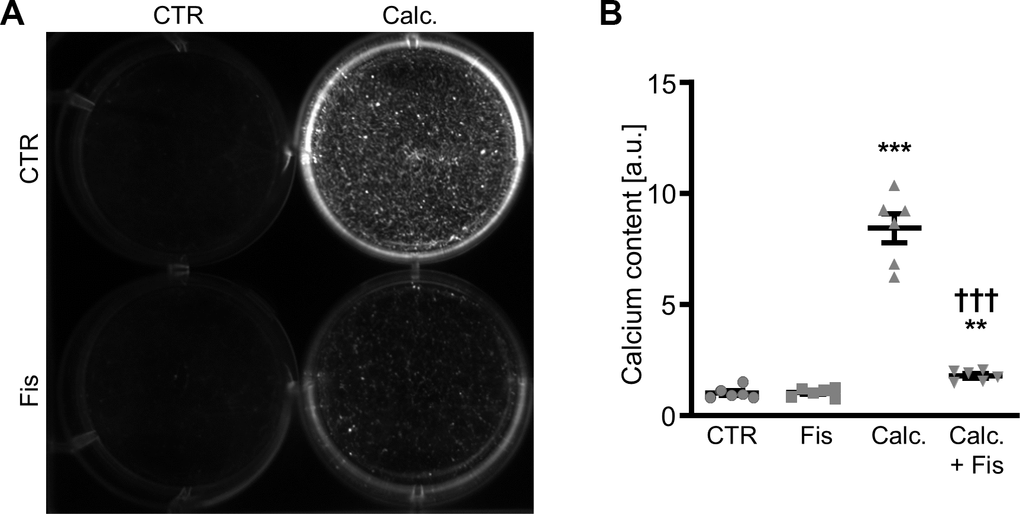
Figure 3. Effects of fisetin on calcification of VSMCs during pro-calcific conditions. Calcification detected by Osteosense fluorescence imaging (A) and normalized calcium content (n=6, B) in HAoSMCs treated for 11d with control (CTR) or calcification medium (Calc.) without and with 1 μM fisetin (Fis). Calcified areas: white pseudocolor. **(p<0.01), ***(p<0.001) significant vs. control group; †††(p<0.001) significant vs. Calc.-treated group.
To elucidate the underlying mechanisms of the anti-calcific effects of fisetin in VSMCs, its potential to interfere with p38 MAPK activation was explored. As illustrated in Figure 4, calcification medium induced phosphorylation of p38 MAPK in VSMCs, effects significantly blunted in the presence of fisetin. Subsequently, the role of dual-specificity phosphatase 1 (DUSP1), a negative regulator of p38 MAPK activity was investigated. As shown in Figure 5, fisetin increased total DUSP1 protein abundance in VSMCs as well as the phosphorylation of DUSP1 at Ser359, a direct phosphorylation site for p44/42 MAPK that could inhibit DUSP1 degradation via the ubiquitination pathway [27]. Moreover, fisetin increased the abundance of phosphorylated and total p44/42 MAPK protein in VSMCs (Supplementary Figure 5). Pharmacological inhibition of p44/42 MAPK with LY3214996 blunted fisetin-induced phosphorylation of DUSP1 at Ser359 (Supplementary Figure 6).
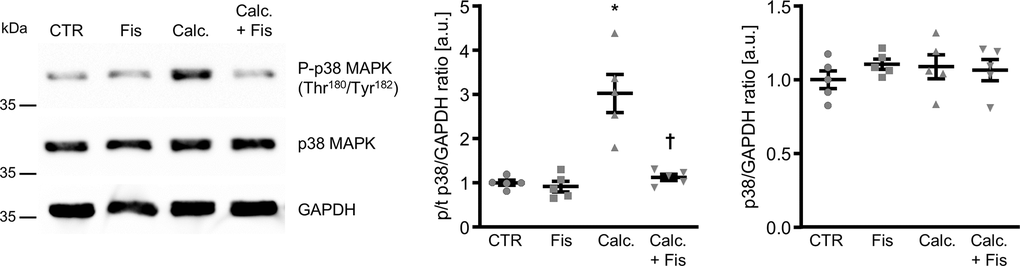
Figure 4. Effects of fisetin on p38 MAPK phosphorylation in VSMCs during pro-calcific conditions. Representative Western blots and normalized phospho-p38 and total p38 MAPK protein abundance (n=5) in HAoSMCs treated for 30 min with control (CTR) or calcification medium (Calc.) without and with 1 μM fisetin (Fis). *(p<0.05) significant vs. control group; †(p<0.05) significant vs. Calc.-treated group.
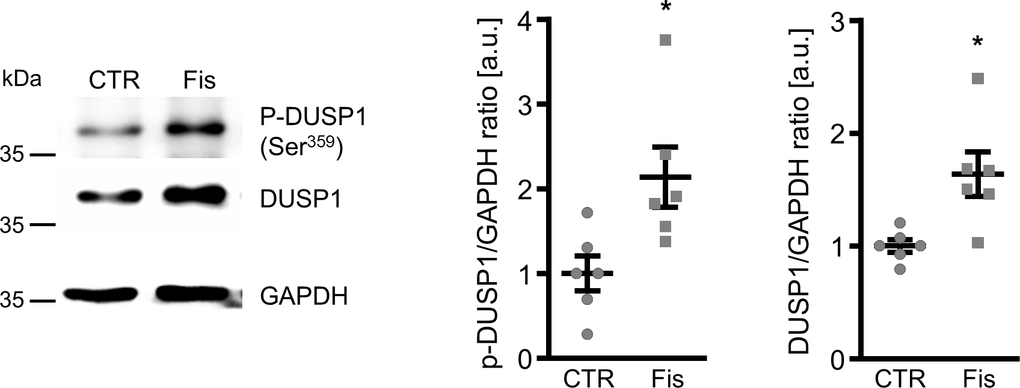
Figure 5. Effects of fisetin on dual-specificity phosphatase 1 regulation in VSMCs. Representative Western blots and normalized phospho-DUSP1 and total DUSP1 protein abundance (n=6) in HAoSMCs treated for 30 min with control (CTR) or 1 μM fisetin (Fis). *(p<0.05) significant vs. control group.
In further experiments, the endogenous expression in VSMCs was suppressed by silencing of the DUSP1 gene using small interfering RNA (siRNA), during control and pro-calcific conditions with and without fisetin supplementation. As a result, transfection with DUSP1 siRNA significantly reduced DUSP1 expression in VSMCs as compared to negative control siRNA-transfected cells (Figure 6A). Calcification medium increased DUSP1 mRNA expression in VSMCs (Figure 6A and Supplementary Figure 7), which was not significantly affected by fisetin (Figure 6A). Knockdown of DUSP1 alone was sufficient to significantly up-regulate mRNA expression of pro-calcific markers in VSMCs (Figure 6B–6E). Furthermore, DUSP1 silencing abolished the protective effects of fisetin during pro-calcific conditions. In accordance, silencing of DUSP1 aggravated VSMC calcification induced by calcification medium and virtually abrogated the anti-calcific properties of fisetin (Figure 7).
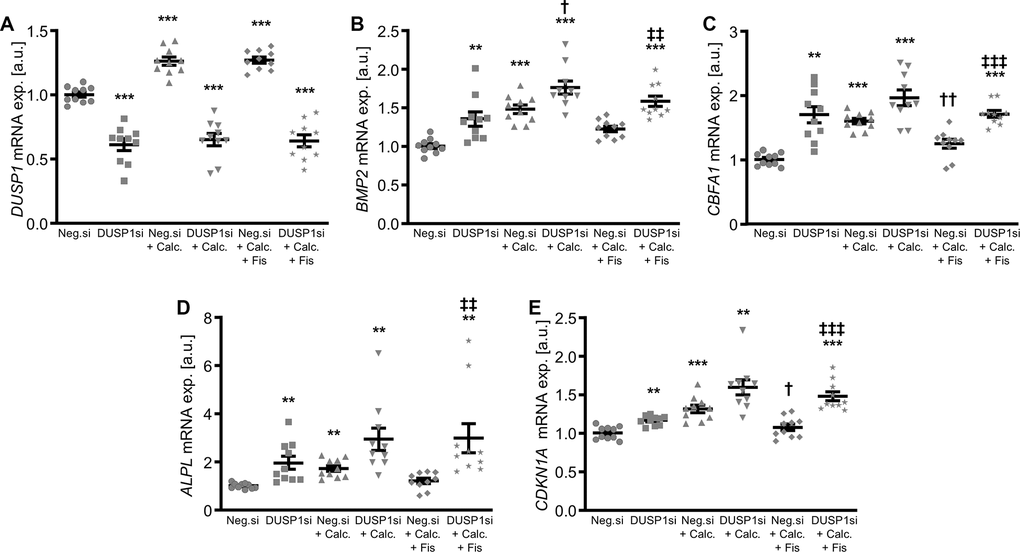
Figure 6. Effects of dual-specificity phosphatase 1 knockdown on anti-calcifying properties of fisetin in VSMCs. Relative mRNA expression (n=10) of DUSP1 (A), BMP2 (B), CBFA1 (C), ALPL (D) and CDKN1A (E) in HAoSMCs transfected for 72h with negative control (Neg.si) or DUSP1 (DUSP1si) siRNA and treated for 48h with control (CTR) or calcification medium (Calc.) without and with 1 μM fisetin (Fis). **(p<0.01), ***(p<0.001) significant vs. Neg.si group; †(p<0.05), ††(p<0.01) significant vs. Neg.si+Calc.-treated group; ‡‡(p<0.01), ‡‡‡(p<0.001) significant between Neg.si+Calc.+Fis- and DUSP1si+Calc.+Fis-treated groups.
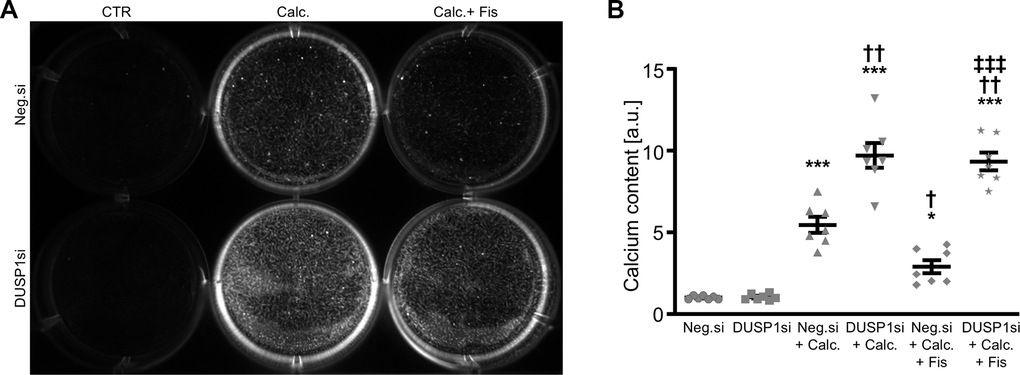
Figure 7. Effects of dual-specificity phosphatase 1 knockdown on the protective role of fisetin during VSMC calcification. Calcification detected by Osteosense fluorescence imaging (A) and normalized calcium content (n=7, B) in HAoSMCs transfected with negative control (Neg.si) or DUSP1 (DUSP1si) siRNA and treated for 11d with control (CTR) or calcification medium (Calc.) without and with 1 μM fisetin (Fis). Calcified areas: white pseudocolor. *(p<0.05), ***(p<0.001) significant vs. Neg.si group; †(p<0.05), ††(p<0.01) significant vs. Neg.si+Calc.-treated group; ‡‡‡(p<0.001) significant between Neg.si+Calc.+Fis- and DUSP1si+Calc.+Fis-treated groups.
Moreover, knockdown of DUSP1 was sufficient to increase phosphorylation of p38 MAPK in VSMCs (Figure 8A), but DUSP1 knockdown did not significantly affect SAPK/JNK or p44/42 MAPK phosphorylation (Supplementary Figure 8). Pharmacological inhibition of p38 MAPK with SB203580 significantly blunted the increased BMP2, CBFA1, ALPL and CDKN1A mRNA expression in DUSP1-silenced VSMCs (Figure 8C–8F). Treatment with the SB203580 inhibitor alone did not significantly modify DUSP1 or calcific marker mRNA expression in VSMCs (Supplementary Figure 9). Thus, knockdown of DUSP1 increased p38 MAPK-dependent pro-calcific signaling and aggravated calcification of VSMCs.
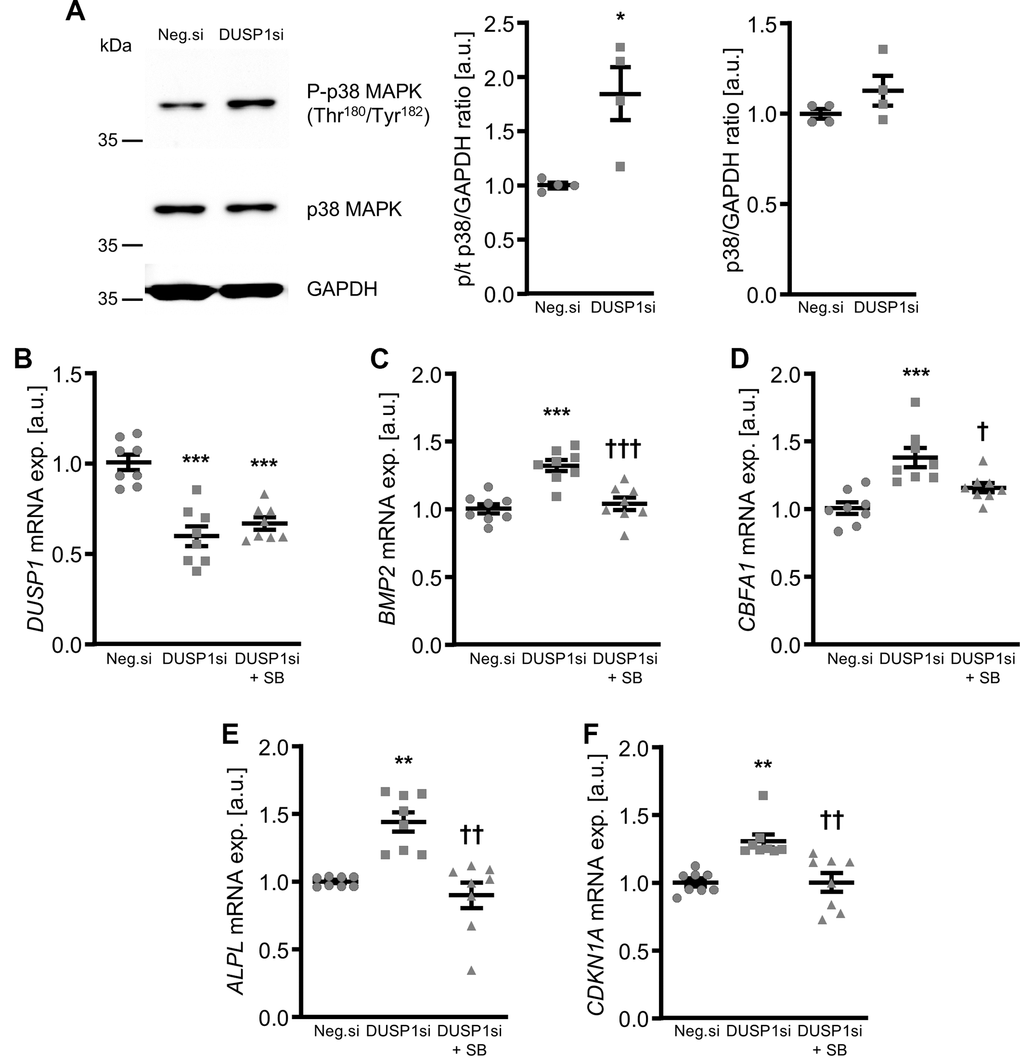
Figure 8. Role of p38 MAPK in dual-specificity phosphatase 1 knockdown-induced calcific marker expression in VSMCs. Representative Western blots and normalized phospho-p38 and total p38 MAPK protein abundance (n=4, A) in HAoSMCs transfected for 24h with negative control (Neg.si) or DUSP1 (DUSP1si) siRNA. Relative mRNA expression (n=8) of DUSP1 (B), BMP2 (C), CBFA1 (D), ALPL (E) and CDKN1A (F) in HAoSMCs transfected for 72h with negative control (Neg.si) or DUSP1 (DUSP1si) siRNA and treated without and with 10 μM p38 MAPK inhibitor SB203580 (SB). *(p<0.05), **(p<0.01), ***(p<0.001) significant vs. Neg.si group; †(p<0.05), ††(p<0.01), †††(p<0.001) significant vs. DUSP1si group.
Next experiments explored the effects of fisetin on pro-calcific marker expression in VSMCs during uremic conditions. Exposure of VSMCs to uremic serum from hemodialysis patients significantly increased BMP2, CBFA1, ALPL and CDKN1A mRNA expression as compared to VSMCs exposed to control serum from healthy volunteers (Figure 9). Treatment with fisetin significantly suppressed uremic serum-induced BMP2, CBFA1 and CDKN1A mRNA expression and tended to reduce ALPL mRNA expression, a difference, however, not reaching statistical significance (p=0.0551). Thus, fisetin treatment induced protective effects in VSMCs during uremic conditions.
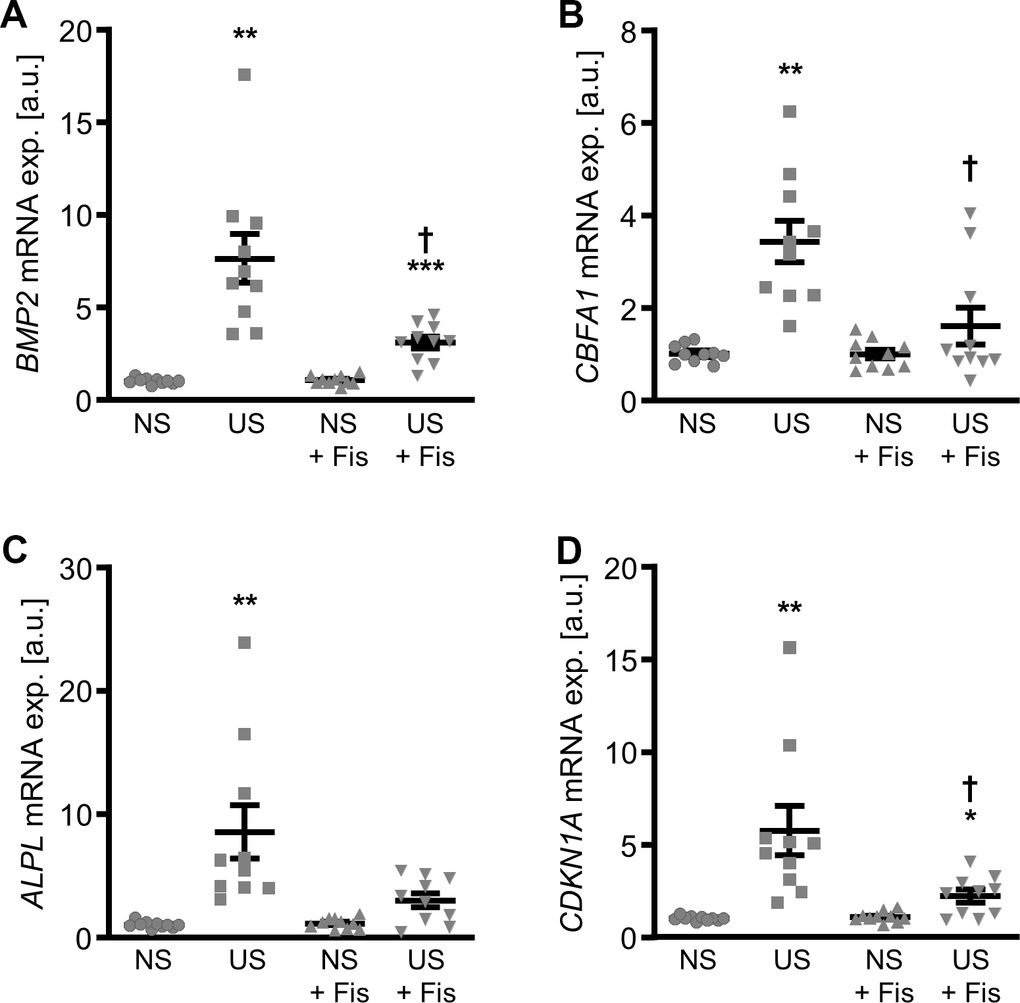
Figure 9. Effects of fisetin on calcific marker expression in VSMCs during uremic conditions. Relative mRNA expression (n=10) of BMP2 (A), CBFA1 (B), ALPL (C) and CDKN1A (D) in HAoSMCs treated for 24h with 15% normal serum (NS) or uremic serum (US) without and with 1 μM fisetin (Fis). *(p<0.05), **(p<0.01), ***(p<0.001) significant vs. NS group; †(p<0.05) significant vs. respective serum alone group.
Additional experiments explored the effects of fisetin following angiotensin II stimulation. Again, fisetin ameliorated the expression of pro-calcific markers in VSMCs during angiotensin II treatment (Supplementary Figure 10).
To confirm the anti-calcific properties of fisetin, additional experiments were performed ex vivo in mouse aortic explants cultured during control or high phosphate conditions without and with co-treatment with fisetin. As shown in Figure 10, fisetin reduced calcification as well as the increased mRNA expression of Bmp2, Cbfa1, Alpl and Cdkn1a induced by phosphate exposure in mouse aortic explants. In addition, fisetin significantly reduced the abundance of phosphorylated p38 Mapk and tended to reduce total p38 Mapk protein abundance (p=0.0798) in mouse aortic explants during high phosphate conditions (Supplementary Figure 11).
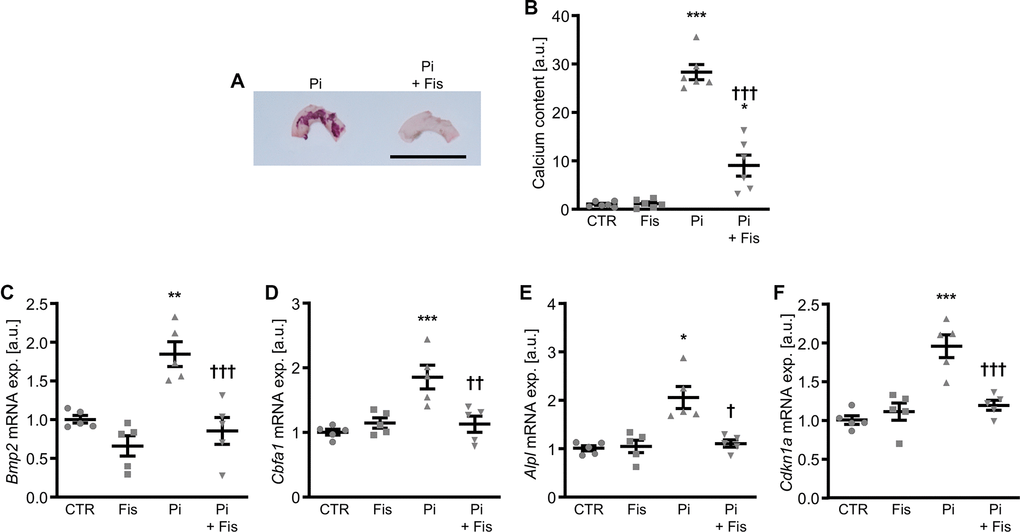
Figure 10. Effects of fisetin ex vivo in calcifying mouse aortic explants. Alizarin Red staining (A) of mouse aortic arches cultured for 7d in medium supplemented with 1.6 mM phosphate (Pi) without and with 1 μM fisetin (Fis). Calcification: red staining; scale bar: 5 mm. Normalized calcium content (n=6, B) in mouse aortic explants cultured for 7d in medium supplemented with control (CTR) or 1.6 mM phosphate (Pi) without and with 1 μM fisetin (Fis). Relative mRNA expression (n=5) of Bmp2 (C), Cbfa1 (D), Alpl (E) and Cdkn1a (F) in mouse aortic explants cultured for 7d in medium supplemented with control (CTR) or 1.6 mM phosphate (Pi) without and with 1 μM fisetin (Fis). *(p<0.05), **(p<0.01), ***(p<0.001) significant vs. control group; †(p<0.05), ††(p<0.01), †††(p<0.001) significant vs. Pi-treated group.
Further experiments investigated the effects of fisetin in vivo in the cholecalciferol-induced VC mouse model. As shown by alizarin Red staining and quantification of calcium content, cholecalciferol overload induced aortic calcification in mice, effects significantly reduced by additional fisetin treatment (Figure 11A, 11B). Furthermore, fisetin suppressed cholecalciferol-induced Bmp2, Cbfa1, Alpl and Cdkn1a mRNA expression in the aortic tissues (Figure 11C–11F). High-dosed cholecalciferol significantly increased serum calcium and altered cystatin C, phosphate and fetuin A concentrations, while fisetin ameliorated most effects of cholecalciferol treatment (Supplementary Table 1).
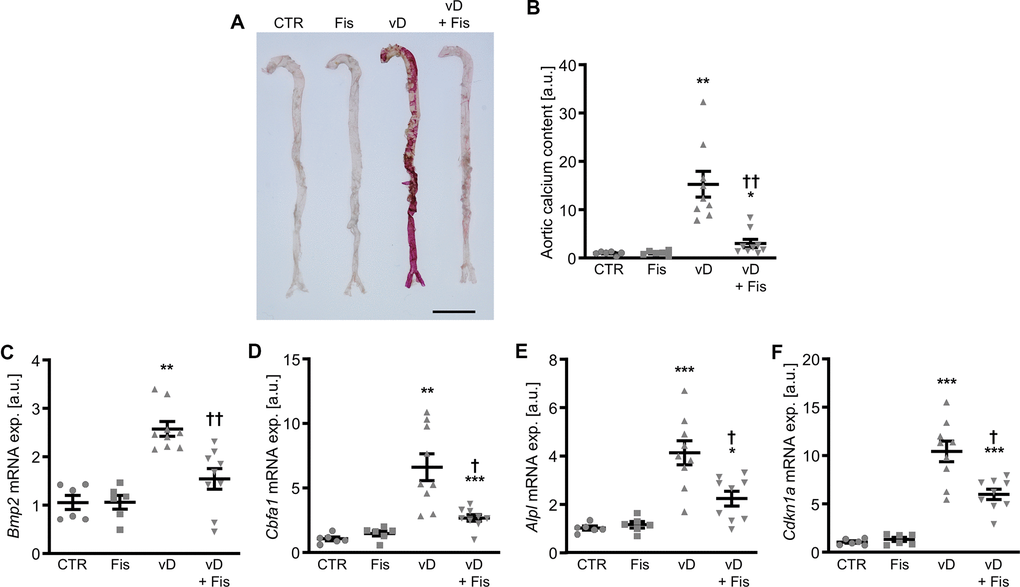
Figure 11. Effects of fisetin in vivo during cholecalciferol overload. Alizarin Red staining (A) of aortas from mice receiving vehicle (CTR) or high-dosed cholecalciferol (vD) without and with fisetin (Fis). Calcification: red staining; scale bar: 5 mm. Normalized calcium content (n=6-9, B) in aortic tissue from mice receiving vehicle (CTR) or high-dosed cholecalciferol (vD) without and with fisetin (Fis). Relative mRNA expression (n=6-9) of Bmp2 (C), Cbfa1 (D), Alpl (E) and Cdkn1a (F) in aortic tissue from mice receiving vehicle (CTR) or high-dosed cholecalciferol (vD) without and with fisetin (Fis). *(p<0.05), **(p<0.01), ***(p<0.001) significant vs. control group; †(p<0.05), ††(p<0.01) significant vs. vD-treated group.
Discussion
This study discloses a powerful anti-calcific effect of fisetin in VSMCs during mineral stress, a condition typically observed in CKD [2]. Mechanistically, fisetin requires the phosphatase DUSP1 to inhibit p38 MAPK in order to mediate its protective effect on VSMC calcification (Figure 12).
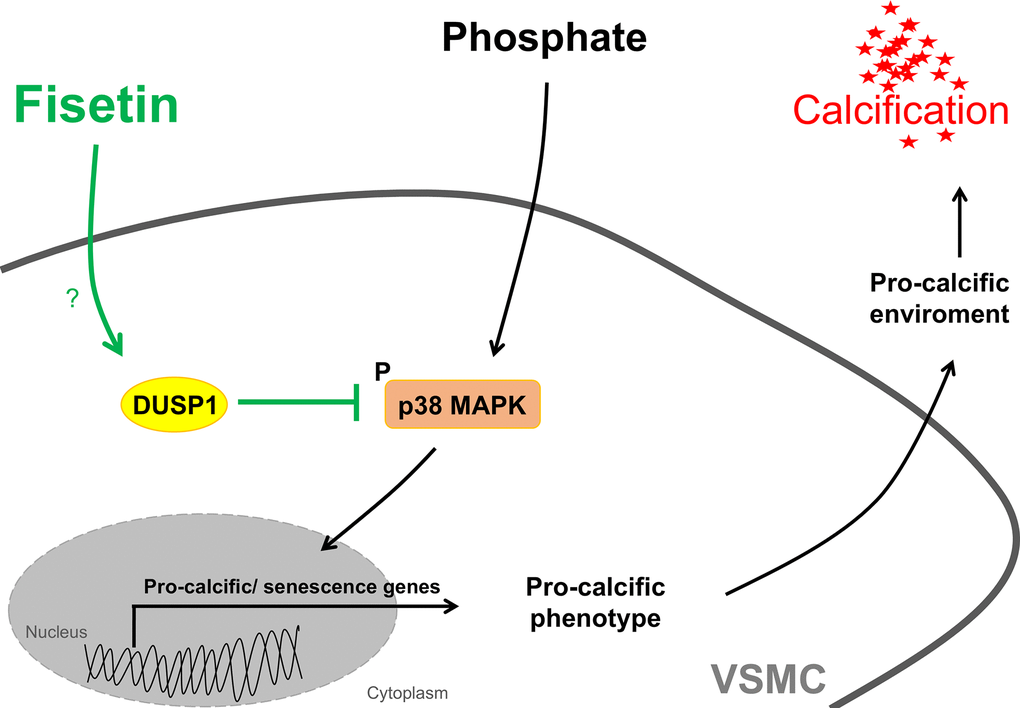
Figure 12. Schematic illustration of mechanisms involved in the protective effects of fisetin during VSMC calcification. Mineral stress with disturbed phosphate and calcium homeostasis may trigger activation of pro-calcific signaling including phosphorylation and activation of p38 MAPK in VSMCs, which leads to a pro-calcific environment causing vascular calcification. Fisetin, a natural flavonol, induces the dual-specificity phosphatase 1 (DUSP1). Fisetin thereby inactivates p38 MAPK signaling through DUSP1 and inhibits further pro-calcific signaling and calcification of VSMCs.
Fisetin has previously been associated with protective effects in the vasculature and prevents neointimal hyperplasia [28], atherosclerosis [26], glucose-induced inflammation [29] and endothelial dysfunction [30] in animal models or cell culture. Especially the ability of fisetin to decrease vascular senescence has been well established [30, 31]. Also in the kidney, fisetin is associated with attenuation of fibrosis [25, 32]. The current observations now extend the protective effects of fisetin on the calcifying vasculature in vitro, ex vivo and in vivo. Although fisetin treatment altered calcium-phosphate homeostasis in cholecalciferol-treated mice and this might affect the calcification response, a direct effect on the vasculature is supported by aortic explant calcification model and cell culture experiments. A potential translational relevance of these observations for the CKD environment is underscored by the protective effects of fisetin in VSMCs exposed to uremic serum.
Mechanistically, the effects of fisetin appear to be mediated by inhibition of p38 MAPK during pro-calcific conditions. P38 MAPK appears to have a central role in pro-calcific signaling of VSMCs [9, 10, 33]. P38 MAPK inhibition has been shown to ameliorate VSMC calcification after phosphate exposure, while ERK1/2 MAPK or SAPK/JNK inhibition showed no effect [12]. The downstream effects of p38 MAPK activation in VC apparently are mediated through a mechanism involving RUNX2/CBFA1 [10]. P38 MAPK also exerts a critical role in the intracellular signaling pathways of inflammatory mediators, which are able to augment VC.
Accordingly, the pro-calcific effects of CRP [33], IL18 [34], ceramide [9] or angiotensin II [35] involve p38 MAPK.
P38 kinases react to a variety of cellular stress conditions and control a diverse array of cellular functions [11]. Phosphatases such as DUSP1 are induced by p38 MAPK and limit or terminate p38 MAPK signaling [11]. The current observations identify DUSP1 as a novel regulator of VSMC calcification. DUSP1 is expressed in VSMCs and plays an important role in maintaining the contractile phenotype by regulating p38 MAPK [36]. Fisetin reduces p38 MAPK phosphorylation in calcifying VSMCs apparently through DUSP1. Similarly, fisetin inhibits ubiquitination and proteasomal degradation of DUSP1 in osteoclasts and thereby limits p38 MAPK signaling [37]. In T-cells, angiotensin II increases proteasome activity and degrades DUSP1 [38]. Angiotensin II and aldosterone attenuate DUSP1 expression in VSMCs [30]. In accordance, we observed a reduced pro-calcific effect of angiotensin II in VSMCs treated with fisetin. Mechanistically, fisetin might activate ERK1/2 MAPK [39]. ERK1/2 could phosphorylate DUSP1 on Ser359, leading to its stabilization [27], but other ERK1/2-dependent phosphorylation sites could actually promote DUSP1 proteasomal degradation [40]. Therefore, the regulatory network of DUSP1 activation and its effect on MAPK appear intricate and multi-factorial [41]. The exact mechanisms how fisetin regulates DUSP1 are currently unknown.
Although the gene silencing experiments indicate a major role for DUSP1 in the anti-calcific effects of fisetin, the current experiments cannot rule out involvement of other pathways or mechanisms. Also, ubiquitination of NRF2 is negatively regulated by fisetin [42]. NRF2 is a powerful modulator of VC [43]. Other pathways might therefore contribute to the effects of fisetin in phosphate-treated VSMCs. In addition, the role of cellular senescence in the current observations should be interpreted with caution. P38 MAPK is important for the development of a senescence-associated secretory phenotype (SASP) [44] and the increased expression of senescence markers in calcifying VSMCs are ameliorated by fisetin. However, the role of DUSP1 in senescence appears complex [45, 46] and the primary effect of fisetin in this model may be mediated through other mechanisms than directly through senolytic properties [22]. In neuronal cells, DUSP1 is polyubiquitylated and increasingly degraded in response to glutamate-induced oxidative stress [47]. Thus, oxidative stress might shift the balance of DUSP1 expression and degradation. Increased oxidative stress is also a hallmark of phosphate-induced VC [2]. In turn, fisetin has been attributed with substantial antioxidant properties [23], which could therefore be involved in its protective effect during VC and subsequently alter expression of senescence markers.
Fisetin treatment has already been used in animal models of CKD and renal fibrosis [25, 32]. In addition, first clinical studies are being conducted on the beneficial effects of fisetin [48]. While the reduced renal function and uremic environment in CKD patients warrants caution, our observations suggest a potential benefit of fisetin treatment on VC. However, the current observations are limited by the model systems used. The artificial cell culture environment does not allow extrapolation to the in vivo situation, especially in terms of effective concentrations of fisetin. The cholecalciferol model rapidly induces VC, but differs from the situation in CKD, where active vitamin D levels are typically reduced. Therefore, CKD-specific or sex-dependent effects cannot be interpreted and further translational studies are required to delineate whether the putative benefits outweigh the risks of fisetin treatment in CKD.
In conclusion, this study shows a novel role of fisetin as powerful protective agent during phosphate-induced VSMC calcification. Mechanistically, this effect identifies a critical role of DUSP1 in the pro-calcific p38 MAPK signaling of VSMCs during calcifying conditions.
Materials and Methods
Cell culture
Primary human aortic smooth muscle cells (HAoSMCs, Fisher Scientific and Cell Applications) were routinely cultured as described previously [49–51] and used in experiments up to passage 12. HAoSMCs were treated for the indicated times with 10 mM β-glycerophosphate and 1.5 mM CaCl2 (Sigma Aldrich) as calcification medium [52], 1 μM or the indicated concentrations of fisetin (stock in DMSO, HY-N0182, MedChemExpress), 10 μM of p38 MAPK inhibitor SB203580 (stock in DMSO, 13067, Cayman Chemical) [33] and 1 μM of p44/42 MAPK inhibitor LY3214996 (stock in DMSO, HY-101494, MedChemExpress) [53]. Where indicated, cells were pre-treated for 48 hours with 1 μM fisetin prior to treatments. After informed consent, serum was collected from dialysis patients (uremic serum, US) or control serum from apparently healthy individuals with absence of known CKD (normal serum, NS) and stored at -80° C. HAoSMCs were treated with 15% uremic serum or control serum [52]. Treatments with control (age 53.6 ± 1.7 years) or uremic (age 63.3 ± 3.0 years) serum was sex-matched (n =5 female / n=5 male). Where indicated, HAoSMCs were transfected with 10 nM DUSP1 (ID: s4363) or negative control (ID: 4390843) siRNA using siPORT amine transfection reagent (all from Fisher Scientific). Treatment with equal amounts of vehicle was used as control. For long-term treatments, fresh medium with agents were added every 2-3 days.
Animal experiments
Calcification was induced in female C57BL/6 mice [54] by daily injections with cholecalciferol of 500 IU/g on d1-d3. Mice were gavaged daily with control or 100 mg/kg fisetin (5% ETOH, 55% PEG 300, 40% Water). Blood was collected by retroorbital puncture on d6 and tissues were snap frozen or stained with Alizarin Red (0.0016% in 0.5% KOH) [55]. Serum concentrations of calcium and phosphate were determined by using a photometric method (FUJI Dri-Chem Nx700). Serum concentrations of Cystatin C (RD291009200R, BioVendor) and Fetuin A (MFTA00, R&D Systems) were determined by ELISA.
Ex vivo mouse aortic explants culture
C57BL/6 mice were sacrificed by cervical dislocation in isoflurane anaesthesia and aortic tissues were rapidly excised, cut into rings and cultured for 1 hour or 7 days in DMEM high glucose medium supplemented with 5% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin and 0.25 μg/ml Fungizone (all from Fisher Scientific). Aortic rings were treated with 1.6 mM sodium phosphate buffer (Sigma Aldrich) and 1 μM fisetin (stock in DMSO, HY-N0182, MedChemExpress). For long-term treatments, fresh medium with agents were added every 2 days. Tissues were snap frozen or stained with Alizarin Red (0.0016% in 0.5% KOH).
RNA isolation and RT-PCR
Total RNA was isolated by using Trizol Reagent and reverse transcription was performed by using oligo(dT)12-18 primers and SuperScript III Reverse Transcriptase (all from Fisher Scientific). RT-PCR was performed with iQ Sybr Green Supermix (Bio-Rad Laboratories) and the primers listed below (Fisher Scientific) [56–58]. Relative mRNA fold changes were calculated by the 2-ΔΔCt method using GAPDH as housekeeping gene.
Human primers:
ALPL fw: GGGACTGGTACTCAGACAACG; ALPL rev: GTAGGCGATGTCCTTACAGCC; BGLAP fw: CACTCCTCGCCCTATTGGC; BGLAP rev: CCCTCCTGCTTGGACACAAAG; BMP2 fw: TTCGGCCTGAAACAGAGACC; BMP2 rev: CCTGAGTGCCTGCGATACAG; CBFA1 fw: GCCTTCCACTCTCAGTAAGAAGA; CBFA1 rev: GCCTGGGGTCTGAAAAAGGG; CDKN1A fw: TGTCCGTCAGAACCCATGC; CDKN1A rev: AAAGTCGAAGTTCCATCGCTC; DUSP1 fw: AGTACCCCACTCTACGATCAGG; DUSP1 rev: GAAGCGTGATACGCACTGC; GAPDH fw: GAGTCAACGGATTTGGTCGT; GAPDH rev: GACAAGCTTCCCGTTCTCAG; GLB1 fw: TATACTGGCTGGCTAGATCACTG; GLB1 rev: GGCAAAATTGGTCCCACCTATAA; SP7 fw: CACAAAGAAGCCGTACTCTGT; SP7 rev: GGGGCTGGATAAGCATCCC; SPP1 fw: GAAGTTTCGCAGACCTGACAT; SPP1 rev: GTATGCACCATTCAACTCCTCG.
Mouse primers:
Alpl fw: TTGTGCCAGAGAAAGAGAGAGA; Alpl rev: GTTTCAGGGCATTTTTCAAGGT; Bmp2 fw: TCTTCCGGGAACAGATACAGG; Bmp2 rev: TGGTGTCCAATAGTCTGGTCA; Cbfa1 fw: AGAGTCAGATTACAGATCCCAGG; Cbfa1 rev: AGGAGGGGTAAGACTGGTCATA; Cdkn1a fw: CCTGGTGATGTCCGACCTG; Cdkn1a rev: CCATGAGCGCATCGCAATC; Gapdh fw: AGGTCGGTGTGAACGGATTTG; Gapdh rev: TGTAGACCATGTAGTTGAGGTCA.
Protein isolation and Western blotting
Total proteins were isolated by using ice-cold Pierce IP lysis buffer containing complete protease and phosphatase inhibitors cocktail (all from Fisher Scientific) and protein concentrations were determined by the Bradford assay (Bio-Rad Laboratories). Equal amounts of protein were boiled in Roti-Load1 Buffer (Carl Roth) at 100° C for 10 minutes and then separated on SDS-PAGE gels and transferred to PVDF membranes (Roche Applied Science). Membranes were incubated with primary antibodies: rabbit anti-phospho-p38 MAPK (Thr180/Tyr182) (1:1000, 9215, Cell Signaling), rabbit anti-p38 MAPK (1:1000, 9212, Cell Signaling), rabbit anti-phospho-SAPK/JNK (Thr183/Tyr185) (1:1000, 4668, Cell Signaling), rabbit anti-SAPK/JNK (1:1000, 9258, Cell Signaling), rabbit anti-phospho-p44/42 MAPK (Thr202/Tyr204) (1:1000, 4379, Cell Signaling), rabbit anti-p44/42 MAPK (1:1000, 4695, Cell Signaling), rabbit anti-phospho-DUSP1 (Ser359) (1:1000, 2857, Cell Signaling), rabbit anti-DUSP1 (1:1000, 35217, Cell Signaling) and rabbit anti-GAPDH (1:3000, 2118, Cell Signaling) at 4° C overnight and with secondary anti-rabbit HRP-conjugated antibody (1:1000, Cell Signaling) at room temperature for 1 hour. Membranes were stripped with Restore Plus Western blot stripping buffer (Fisher Scientific) at room temperature. Bands were detected with Clarity Western ECL substrate (Bio-Rad Laboratories) using the ChemiDoc MP imaging system (Bio-Rad Laboratories) and quantified using the ImageJ software. Data are shown as the ratio of phosphorylated to total protein to GAPDH, phosphorylated protein to GAPDH and of total protein to GAPDH, normalized to the control group [33, 59].
Immunofluorescence staining and confocal microscopy
Cells were fixed in 4% PFA/PBS for 15 minutes, permeabilized in 0.3% TritonX-100/PBS for 10 minutes and blocked with 5% goat serum in 0.1% TritonX-100/PBS for 1 hour at room temperature. Cells were incubated with primary rabbit anti-RUNX2 antibody (1:100 in 0.1% TritonX-100/PBS, 12556, Cell Signaling) [54] at 4° C overnight and then with goat anti-rabbit Alexa488-conjugated antibody (1:500 in 0.1% TritonX-100/PBS, Invitrogen) for 2 hours at room temperature. Nuclei were stained with 0.5 μg/ml DAPI (Carl Roth) for 5 minutes at room temperature and slides were mounted with Prolong Diamond antifade reagent (Invitrogen). Images were acquired on a Nikon Ti-2 microscope (x60 oil immersion, NA 1.42) equipped with a Clarity Laser Free Confocal Unit (Aurox).
Senescence-associated (SA)-β-galactosidase staining
HAoSMCs were stained for SA-β-Galactosidase by using the Senescence β-Galactosidase staining kit (9860, Cell Signaling).
ALP activity
ALP activity was determined in cell lysates by using a colorimetric kit (Abcam) and protein concentration was determined by the Bradford assay (Bio-Rad Laboratories). Data are shown normalized to total protein concentration and to the control group [49].
Calcification analysis
HAoSMCs were incubated with OsteoSense 680EX (1:250, NEV10020EX, Perkin Elmer) at 37° C overnight and images were acquired with the ChemiDoc MP imaging system (Bio-Rad Laboratories) [60, 61]. HAoSMCs and aortic tissues were decalcified in 0.6M HCl at 4° C and 37° C, respectively overnight and the calcium content in the supernatant was quantified by using the QuantiChrom Calcium assay kit (DICA-500, BioAssay Systems). Proteins were isolated by using 0.1M NaOH/0.1% SDS buffer and quantified by the Bradford assay (Bio-Rad Laboratories). Data are shown normalized to total protein concentration and to the control group.
Statistics
Data are shown as scatter dot plots and arithmetic means ± SEM and n represents the number of independent experiments performed. Normalized data are shown as arbitrary units (a.u.). Normality was determined by Shapiro-Wilk test. For two groups, statistical testing was performed using unpaired T-test, Mann-Whitney-U-test or one-sample T-test. For multiple group comparison, statistical testing was performed by using one-way ANOVA with Tukey test (homoscedastic data) or Games-Howell test (heteroscedastic data) and Kruskal-Wallis test with Steel-Dwass test (non-normal data). P<0.05 was considered statistically significant.
Author Contributions
IA and JV designed research and acquired funding; MR, SB, AS, MM, IJ and IA performed experiments; MR, SB, GB, IA and JV analyzed and interpreted data; IA and JV wrote the manuscript with comments and edits from all authors.
Acknowledgments
The authors gratefully acknowledge the technical assistance of A. Kurishev.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Statement and Consent
The blood collection was approved by the local ethics commission (approval number 1141/2019) and all volunteers provided informed consent. All animal experiments were approved by the local authorities (BMBWF Vienna, Austria), protocol number 2022-0.406.924.
Funding
This work was funded by the Austrian Science Fund (FWF) (10.55776/P34724) and the Faculty of Medicine of the Johannes Kepler University Linz through its internal funding programme Impetus Nr. I-04-23. Supported by Johannes Kepler Open Access Publishing Fund and the federal state Upper Austria.
References
- 1. Elliott RJ, McGrath LT. Calcification of the human thoracic aorta during aging. Calcif Tissue Int. 1994; 54:268–73. https://doi.org/10.1007/BF00295949 [PubMed]
- 2. Voelkl J, Egli-Spichtig D, Alesutan I, Wagner CA. Inflammation: a putative link between phosphate metabolism and cardiovascular disease. Clin Sci (Lond). 2021; 135:201–27. https://doi.org/10.1042/CS20190895 [PubMed]
- 3. Tanriover C, Copur S, Mutlu A, Peltek IB, Galassi A, Ciceri P, Cozzolino M, Kanbay M. Early aging and premature vascular aging in chronic kidney disease. Clin Kidney J. 2023; 16:1751–65. https://doi.org/10.1093/ckj/sfad076 [PubMed]
- 4. Lanzer P, Hannan FM, Lanzer JD, Janzen J, Raggi P, Furniss D, Schuchardt M, Thakker R, Fok PW, Saez-Rodriguez J, Millan A, Sato Y, Ferraresi R, et al. Medial Arterial Calcification: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021; 78:1145–65. https://doi.org/10.1016/j.jacc.2021.06.049 [PubMed]
- 5. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009; 5:185–97. https://doi.org/10.2147/vhrm.s4822 [PubMed]
- 6. Voelkl J, Cejka D, Alesutan I. An overview of the mechanisms in vascular calcification during chronic kidney disease. Curr Opin Nephrol Hypertens. 2019; 28:289–96. https://doi.org/10.1097/MNH.0000000000000507 [PubMed]
- 7. Villa-Bellosta R. Vascular Calcification: A Passive Process That Requires Active Inhibition. Biology (Basel). 2024; 13:111. https://doi.org/10.3390/biology13020111 [PubMed]
- 8. Voelkl J, Lang F, Eckardt KU, Amann K, Kuro-O M, Pasch A, Pieske B, Alesutan I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. 2019; 76:2077–91. https://doi.org/10.1007/s00018-019-03054-z [PubMed]
- 9. Liao L, Zhou Q, Song Y, Wu W, Yu H, Wang S, Chen Y, Ye M, Lu L. Ceramide mediates Ox-LDL-induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PLoS One. 2013; 8:e82379. https://doi.org/10.1371/journal.pone.0082379 [PubMed]
- 10. Yang Y, Sun Y, Chen J, Bradley WE, Dell'Italia LJ, Wu H, Chen Y. AKT-independent activation of p38 MAP kinase promotes vascular calcification. Redox Biol. 2018; 16:97–103. https://doi.org/10.1016/j.redox.2018.02.009 [PubMed]
- 11. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol. 2021; 22:346–66. https://doi.org/10.1038/s41580-020-00322-w [PubMed]
- 12. Kang JH, Toita R, Asai D, Yamaoka T, Murata M. Reduction of inorganic phosphate-induced human smooth muscle cells calcification by inhibition of protein kinase A and p38 mitogen-activated protein kinase. Heart Vessels. 2014; 29:718–22. https://doi.org/10.1007/s00380-013-0427-x [PubMed]
- 13. Alesutan I, Moritz F, Haider T, Shouxuan S, Gollmann-Tepeköylü C, Holfeld J, Pieske B, Lang F, Eckardt KU, Heinzmann SS, Voelkl J. Impact of β-glycerophosphate on the bioenergetic profile of vascular smooth muscle cells. J Mol Med (Berl). 2020; 98:985–97. https://doi.org/10.1007/s00109-020-01925-8 [PubMed]
- 14. Sanchis P, Ho CY, Liu Y, Beltran LE, Ahmad S, Jacob AP, Furmanik M, Laycock J, Long DA, Shroff R, Shanahan CM. Arterial "inflammaging" drives vascular calcification in children on dialysis. Kidney Int. 2019; 95:958–72. https://doi.org/10.1016/j.kint.2018.12.014 [PubMed]
- 15. Zhang M, Li T, Tu Z, Zhang Y, Wang X, Zang D, Xu D, Feng Y, He F, Ni M, Wang D, Zhou H. Both high glucose and phosphate overload promote senescence-associated calcification of vascular muscle cells. Int Urol Nephrol. 2022; 54:2719–31. https://doi.org/10.1007/s11255-022-03195-4 [PubMed]
- 16. Yamada S, Tatsumoto N, Tokumoto M, Noguchi H, Ooboshi H, Kitazono T, Tsuruya K. Phosphate binders prevent phosphate-induced cellular senescence of vascular smooth muscle cells and vascular calcification in a modified, adenine-based uremic rat model. Calcif Tissue Int. 2015; 96:347–58. https://doi.org/10.1007/s00223-014-9929-5 [PubMed]
- 17. Power H, Valtchev P, Dehghani F, Schindeler A. Strategies for senolytic drug discovery. Aging Cell. 2023; 22:e13948. https://doi.org/10.1111/acel.13948 [PubMed]
- 18. Ceccherini E, Gisone I, Persiani E, Ippolito C, Falleni A, Cecchettini A, Vozzi F. Novel in vitro evidence on the beneficial effect of quercetin treatment in vascular calcification. Front Pharmacol. 2024; 15:1330374. https://doi.org/10.3389/fphar.2024.1330374 [PubMed]
- 19. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016; 15:973–7. https://doi.org/10.1111/acel.12458 [PubMed]
- 20. Shi W, Lu J, Li J, Qiu M, Lu Y, Gu J, Kong X, Sun W. Piperlongumine Attenuates High Calcium/Phosphate-Induced Arterial Calcification by Preserving P53/PTEN Signaling. Front Cardiovasc Med. 2021; 7:625215. https://doi.org/10.3389/fcvm.2020.625215 [PubMed]
- 21. Chen C, Li Y, Lu H, Liu K, Jiang W, Zhang Z, Qin X. Curcumin attenuates vascular calcification via the exosomal miR-92b-3p/KLF4 axis. Exp Biol Med (Maywood). 2022; 247:1420–32. https://doi.org/10.1177/15353702221095456 [PubMed]
- 22. Elsallabi O, Patruno A, Pesce M, Cataldi A, Carradori S, Gallorini M. Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection. Molecules. 2022; 27:738. https://doi.org/10.3390/molecules27030738 [PubMed]
- 23. Grynkiewicz G, Demchuk OM. New Perspectives for Fisetin. Front Chem. 2019; 7:697. https://doi.org/10.3389/fchem.2019.00697 [PubMed]
- 24. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018; 36:18–28. https://doi.org/10.1016/j.ebiom.2018.09.015 [PubMed]
- 25. Li S, Livingston MJ, Ma Z, Hu X, Wen L, Ding HF, Zhou D, Dong Z. Tubular cell senescence promotes maladaptive kidney repair and chronic kidney disease after cisplatin nephrotoxicity. JCI Insight. 2023; 8:e166643. https://doi.org/10.1172/jci.insight.166643 [PubMed]
- 26. Yan L, Jia Q, Cao H, Chen C, Xing S, Huang Y, Shen D. Fisetin ameliorates atherosclerosis by regulating PCSK9 and LOX-1 in apoE-/- mice. Exp Ther Med. 2021; 21:25. https://doi.org/10.3892/etm.2020.9457 [PubMed]
- 27. Brondello JM, Pouysségur J, McKenzie FR. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. 1999; 286:2514–7. https://doi.org/10.1126/science.286.5449.2514 [PubMed]
- 28. Pei F, Pei H, Su C, Du L, Wang J, Xie F, Yin Q, Gao Z. Fisetin Alleviates Neointimal Hyperplasia via PPAR γ/PON2 Antioxidative Pathway in SHR Rat Artery Injury Model. Oxid Med Cell Longev. 2021; 2021:6625517. https://doi.org/10.1155/2021/6625517 [PubMed]
- 29. Kwak S, Ku SK, Bae JS. Fisetin inhibits high-glucose-induced vascular inflammation in vitro and in vivo. Inflamm Res. 2014; 63:779–87. https://doi.org/10.1007/s00011-014-0750-4 [PubMed]
- 30. Mahoney SA, Venkatasubramanian R, Darrah MA, Ludwig KR, VanDongen NS, Greenberg NT, Longtine AG, Hutton DA, Brunt VE, Campisi J, Melov S, Seals DR, Rossman MJ, Clayton ZS. Intermittent supplementation with fisetin improves arterial function in old mice by decreasing cellular senescence. Aging Cell. 2024; 23:e14060. https://doi.org/10.1111/acel.14060 [PubMed]
- 31. Kim SG, Sung JY, Kang YJ, Choi HC. PPARγ activation by fisetin mitigates vascular smooth muscle cell senescence via the mTORC2-FoxO3a-autophagy signaling pathway. Biochem Pharmacol. 2023; 218:115892. https://doi.org/10.1016/j.bcp.2023.115892 [PubMed]
- 32. Ijima S, Saito Y, Nagaoka K, Yamamoto S, Sato T, Miura N, Iwamoto T, Miyajima M, Chikenji TS. Fisetin reduces the senescent tubular epithelial cell burden and also inhibits proliferative fibroblasts in murine lupus nephritis. Front Immunol. 2022; 13:960601. https://doi.org/10.3389/fimmu.2022.960601 [PubMed]
- 33. Henze LA, Luong TT, Boehme B, Masyout J, Schneider MP, Brachs S, Lang F, Pieske B, Pasch A, Eckardt KU, Voelkl J, Alesutan I. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging (Albany NY). 2019; 11:5445–62. https://doi.org/10.18632/aging.102130 [PubMed]
- 34. Zhang Y, Zhang K, Zhang Y, Zhou L, Huang H, Wang J. IL-18 Mediates Vascular Calcification Induced by High-Fat Diet in Rats With Chronic Renal Failure. Front Cardiovasc Med. 2021; 8:724233. https://doi.org/10.3389/fcvm.2021.724233 [PubMed]
- 35. Jeong J, Cho S, Seo M, Lee BS, Jang Y, Lim S, Park S. Soluble RAGE attenuates Ang II-induced arterial calcification via inhibiting AT1R-HMGB1-RAGE axis. Atherosclerosis. 2022; 346:53–62. https://doi.org/10.1016/j.atherosclerosis.2022.02.022 [PubMed]
- 36. Liu JX, Huang T, Xie D, Yu Q. Bves maintains vascular smooth muscle cell contractile phenotype and protects against transplant vasculopathy via Dusp1-dependent p38MAPK and ERK1/2 signaling. Atherosclerosis. 2022; 357:20–32. https://doi.org/10.1016/j.atherosclerosis.2022.08.010 [PubMed]
- 37. Léotoing L, Wauquier F, Guicheux J, Miot-Noirault E, Wittrant Y, Coxam V. The polyphenol fisetin protects bone by repressing NF-κB and MKP-1-dependent signaling pathways in osteoclasts. PLoS One. 2013; 8:e68388. https://doi.org/10.1371/journal.pone.0068388 [PubMed]
- 38. Qin XY, Zhang YL, Chi YF, Yan B, Zeng XJ, Li HH, Liu Y. Angiotensin II Regulates Th1 T Cell Differentiation Through Angiotensin II Type 1 Receptor-PKA-Mediated Activation of Proteasome. Cell Physiol Biochem. 2018; 45:1366–76. https://doi.org/10.1159/000487562 [PubMed]
- 39. Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, Maher P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer's disease transgenic mice. Aging Cell. 2014; 13:379–90. https://doi.org/10.1111/acel.12185 [PubMed]
- 40. Chen HF, Chuang HC, Tan TH. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int J Mol Sci. 2019; 20:2668. https://doi.org/10.3390/ijms20112668 [PubMed]
- 41. Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007; 26:3203–13. https://doi.org/10.1038/sj.onc.1210412 [PubMed]
- 42. Zhang H, Zheng W, Feng X, Yang F, Qin H, Wu S, Hou DX, Chen J. Nrf2-ARE Signaling Acts as Master Pathway for the Cellular Antioxidant Activity of Fisetin. Molecules. 2019; 24:708. https://doi.org/10.3390/molecules24040708 [PubMed]
- 43. Laget J, Hobson S, Muyor K, Duranton F, Cortijo I, Bartochowski P, Jover B, Lajoix AD, Söderberg M, Ebert T, Stenvinkel P, Argilés À, Kublickiene K, Gayrard N. Implications of Senescent Cell Burden and NRF2 Pathway in Uremic Calcification: A Translational Study. Cells. 2023; 12:643. https://doi.org/10.3390/cells12040643 [PubMed]
- 44. Odawara T, Yamauchi S, Ichijo H. Apoptosis signal-regulating kinase 1 promotes inflammation in senescence and aging. Commun Biol. 2024; 7:691. https://doi.org/10.1038/s42003-024-06386-0 [PubMed]
- 45. Lee JY, Yu KR, Kim HS, Kang I, Kim JJ, Lee BC, Choi SW, Shin JH, Seo Y, Kang KS. BMI1 inhibits senescence and enhances the immunomodulatory properties of human mesenchymal stem cells via the direct suppression of MKP-1/DUSP1. Aging (Albany NY). 2016; 8:1670–89. https://doi.org/10.18632/aging.101000 [PubMed]
- 46. Cheng CM, Liu F, Li JY, Song QY. DUSP1 promotes senescence of retinoblastoma cell line SO-Rb5 cells by activating AKT signaling pathway. Eur Rev Med Pharmacol Sci. 2018; 22:7628–32. https://doi.org/10.26355/eurrev_201811_16377 [PubMed]
- 47. Choi BH, Hur EM, Lee JH, Jun DJ, Kim KT. Protein kinase Cdelta-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. J Cell Sci. 2006; 119:1329–40. https://doi.org/10.1242/jcs.02837 [PubMed]
- 48. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, Kirkland JL. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019. https://doi.org/10.1016/j.ebiom.2018.12.052 [PubMed]
- 49. Alesutan I, Henze LA, Boehme B, Luong TT, Zickler D, Pieske B, Eckardt KU, Pasch A, Voelkl J. Periostin Augments Vascular Smooth Muscle Cell Calcification via β-Catenin Signaling. Biomolecules. 2022; 12:1157. https://doi.org/10.3390/biom12081157 [PubMed]
- 50. Alesutan I, Luong TTD, Schelski N, Masyout J, Hille S, Schneider MP, Graham D, Zickler D, Verheyen N, Estepa M, Pasch A, Maerz W, Tomaschitz A, et al. Circulating uromodulin inhibits vascular calcification by interfering with pro-inflammatory cytokine signalling. Cardiovasc Res. 2021; 117:930–41. https://doi.org/10.1093/cvr/cvaa081 [PubMed]
- 51. Voelkl J, Alesutan I, Leibrock CB, Quintanilla-Martinez L, Kuhn V, Feger M, Mia S, Ahmed MS, Rosenblatt KP, Kuro-O M, Lang F. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J Clin Invest. 2013; 123:812–22. https://doi.org/10.1172/JCI64093 [PubMed]
- 52. Voelkl J, Tuffaha R, Luong TTD, Zickler D, Masyout J, Feger M, Verheyen N, Blaschke F, Kuro-O M, Tomaschitz A, Pilz S, Pasch A, Eckardt KU, et al. Zinc Inhibits Phosphate-Induced Vascular Calcification through TNFAIP3-Mediated Suppression of NF-κB. J Am Soc Nephrol. 2018; 29:1636–48. https://doi.org/10.1681/ASN.2017050492 [PubMed]
- 53. Ren B, Kang J, Wang Y, Meng X, Huang Y, Bai Y, Feng Z. Transcranial direct current stimulation promotes angiogenesis and improves neurological function via the OXA-TF-AKT/ERK signaling pathway in traumatic brain injury. Aging (Albany NY). 2024; 16:6566–87. https://doi.org/10.18632/aging.205724 [PubMed]
- 54. Alesutan I, Razazian M, Luong TT, Estepa M, Pitigala L, Henze LA, Obereigner J, Mitter G, Zickler D, Schuchardt M, Deisl C, Makridakis M, Gollmann-Tepeköylü C, et al. Augmentative effects of leukemia inhibitory factor reveal a critical role for TYK2 signaling in vascular calcification. Kidney Int. 2024; 106:611–24. https://doi.org/10.1016/j.kint.2024.07.011 [PubMed]
- 55. Razazian M, Bahiraii S, Jannat I, Tiffner A, Beilhack G, Levkau B, Voelkl J, Alesutan I. Sphingosine kinase 1 inhibition aggravates vascular smooth muscle cell calcification. Pflugers Arch. 2025. [Epub ahead of print]. https://doi.org/10.1007/s00424-025-03068-6 [PubMed]
- 56. Voelkl J, Luong TT, Tuffaha R, Musculus K, Auer T, Lian X, Daniel C, Zickler D, Boehme B, Sacherer M, Metzler B, Kuhl D, Gollasch M, et al. SGK1 induces vascular smooth muscle cell calcification through NF-κB signaling. J Clin Invest. 2018; 128:3024–40. https://doi.org/10.1172/JCI96477 [PubMed]
- 57. Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003; 31:e154. https://doi.org/10.1093/nar/gng154 [PubMed]
- 58. Leibrock CB, Alesutan I, Voelkl J, Pakladok T, Michael D, Schleicher E, Kamyabi-Moghaddam Z, Quintanilla-Martinez L, Kuro-o M, Lang F. NH4Cl Treatment Prevents Tissue Calcification in Klotho Deficiency. J Am Soc Nephrol. 2015; 26:2423–33. https://doi.org/10.1681/ASN.2014030230 [PubMed]
- 59. Luong TT, Tuffaha R, Schuchardt M, Moser B, Schelski N, Boehme B, Gollmann-Tepeköylü C, Schramm C, Holfeld J, Pieske B, Gulbins E, Tölle M, van der Giet M, et al. Acid sphingomyelinase promotes SGK1-dependent vascular calcification. Clin Sci (Lond). 2021; 135:515–34. https://doi.org/10.1042/CS20201122 [PubMed]
- 60. Henze LA, Estepa M, Pieske B, Lang F, Eckardt KU, Alesutan I, Voelkl J. Zinc Ameliorates the Osteogenic Effects of High Glucose in Vascular Smooth Muscle Cells. Cells. 2021; 10:3083. https://doi.org/10.3390/cells10113083 [PubMed]
- 61. Moser B, Poetsch F, Estepa M, Luong TT, Pieske B, Lang F, Alesutan I, Voelkl J. Increased β-adrenergic stimulation augments vascular smooth muscle cell calcification via PKA/CREB signalling. Pflugers Arch. 2021; 473:1899–910. https://doi.org/10.1007/s00424-021-02621-3 [PubMed]