



Introduction
Ischemic stroke is mainly induced by cerebral microcirculation obstruction or destruction resulting from internal or external factors-mediated cerebrovascular vascular blockage, thrombosis, or insufficient blood supply to the brain, which eventually contribute to brain parenchyma cell damage or death, brain dysfunction or damage from acute cerebrovascular disease. Ischemic stroke is difficult to treat due to such characteristics as high incidence and multiple sequelae [1]. It has been reported that destruction of the blood-brain barrier (BBB) after ischemic stroke is an important cause of death due to secondary brain injury, and effective repair of the BBB is one of the main means to treat ischemic stroke. The BBB is composed of brain microvascular endothelial cells (BMECs), an astrocyte terminus, pericytes, and a basement membrane [2], with endothelial cells being its main structure. Endothelial cells and tight junction (TJ) proteins together strictly control BBB permeability [3], which protects the brain from potential neurotoxic substances and facilitates the exchange of nutrients and waste products between the brain and blood to maintain the optimal extracellular environment for nerve function [4]. Damage of the BBB is an important physiopathological basis of ischemic stroke [5] and one of the important causes of death from ischemic stroke. Therefore, the repair of BBB damage may be a possible approach for the treatment of ischemic stroke. Studies have shown that the BBB junction complex is a complicated cellular system with active regulation, consisting TJs and adherent junctions (AJs) [6]. Tight junctions possess highly regulated dynamic structures, consisting of three membrane proteins (Claudin, Occludin, and junction adhesion molecules), cytoplasmic helper proteins (ZO-1, ZO-2, and ZO-3), and Cingulin [7, 8]. Reduced or altered tight junction protein expression in cells will affect cellular permeability and thus impair BBB permeability [9]. It is recently reported that vascular endothelial growth factor (VEGF)/VEGFR2 is involved in the regulation of tight junction protein expression [10]. Thus, VEGF/VEGFR2-mediated BBB permeability might be a promising target for treating ischemic stroke. Ischemic stroke demands an early diagnosis and treatment in a narrow time frame [11]. The standard medical treatment for acute ischemic stroke, such as with intravenous alteplase, is considered for use within 4.5 hours after the onset of symptoms [12, 13]. However, 14 to 27% of strokes occur at night when patients are sleeping, thus symptoms cannot be recognized on time [14]. Such patients are generally excluded from treatment with intravenous alteplase, and only some of them are candidates for mechanical thrombectomy. Therefore, the approaches for preventing and treating ischemic stroke are still being explored.
Cepharanthine (CEP) is an isoquinoline alkaloid and the main component of Tetrandrine tablets, mainly used for the clinical treatment of leukocyte elevation. Previous studies have shown that CEP represses the growth and metastasis of tumor cells, and reduces the resistance of tumor cells to various chemotherapy drugs [15]. Its anti-tumor mechanism is closely related to its regulation of tumor autophagy and apoptosis [16, 17]. Studies have claimed the anti-angiogenesis property of CEP by repressing the expression of VEGF [18]. However, the function of CEP in BBB permeability and ischemic stroke remains unclear. The present study proposes to assess the regulatory function of CEP on middle cerebral artery occlusion (MCAO) mice to explore the potential application of CEP in treating ischemic stroke.
Materials and Methods
Animal models and treatments
40 C57BL/6J male mice (20-22 g) were sectionalized into 4 groups: Vehicle, CEP, MCAO, and MCAO+CEP (10 mice/group). In the MCAO groups, a surgical filament was inserted into the middle cerebral artery of mice, followed by closing the sutures 2 h later. Then, a reperfusion was allowed for 24 h. In the CEP groups, 1.5 mg/kg CEP was administered into the peritoneal cavity of mice for 3 weeks prior to MCAO modeling [16, 18]. The animals were euthanized after the neurological scores were measured.
Neurological score
The neurological scoring was utilized to determine the neurological deficit in each animal. Four grades were established: grade 0: neurological deficit was not observed; grade 1: the contralateral forepaw cannot be extended when being grasped on the tail; grade 2: animals moved in circles toward the ipsilateral side; grade 3: animals fell to the contralateral side of brain damage; grade 4: decreased mobility with rare signs of consciousness [19].
Diffusion of sodium fluorescein assay
Animals were injected with 0.1 mL 10% NaFI, followed by being intraperitoneally injected with 4% chloral hydrate for anesthesia, 45 min later. The chest cavity was then opened and the cannula was intubated to the aorta through the left ventricle, followed by cutting the right atrium to perfuse the normal saline until the clear fluid flowed out of the right atrium. The brain was taken and 0.6 mL phosphate-buffered saline (PBS) homogenate was added into the brain tissue, followed by being centrifuged at 3000 r/min for 5 min and the supernatant was diluted with 20% Trichloroacetic acid (TCA) (1:10) and incubated at 4° C for 24 h. The samples were centrifuged at 10000 r/min for 15 min and the supernatant was collected and then diluted with the same volume of Tris-HCL buffer. Fluorescence intensity (490/514 nm) was measured and the BBB permeability was determined by the amount of NaFI per milligram of brain tissue protein.
Real-time polymerase chain reaction (PCR)
Trizol reagent was utilized to isolate RNA from brain tissues or cells, followed by being transformed into cDNA using the reverse transcription kit (Takara, Japan). Then the 2×SYBR Green PCR Master Mix kit (Lifeint, China) was utilized to conduct the PCR reaction, and the 2−ΔΔCt method was performed for the calculation of targeted genes [20]. The following primers were used in the study: ZO-1 Forward: 5’-CCCCTCTGTC CAGCTCTTC-3’; Reverse: 5’-CACCGGAGTGATGGTTTTCT-3’; VEGF-A Forward: 5’-TCTACCTCCACCATGCCAAGT-3’, Reverse: 5’-TGCGCTGATA GACATCCATGA-3’; VEGFR2 Forward: 5’-AATTATTGCAGGGGACAGAG-3’, Reverse: 5’-TTCTGGGTATCTTGCACAAAG-3’; β-Actin Forward: 5’-AGGTG ACAGCATTGCTTCTG-3’; Reverse: 5’-GCTGCCTCAACACCTCAAC-3’.
Immunofluorescence labeling
The immunofluorescence labeling was performed as per a previous study [21]. The brain tissues were embedded in tissue-tek optimal cutting temperature (OCT) compound to be snap-frozen in dry ice and then stored in a -80° C freezer. 7-μm frozen sections were cut using a cryostat-microtome (Leica CM3050S, Leica Biosystems, Germany) at -20° C and were mounted on glass slides. The tissues were then fixed with 4 % paraformaldehyde solution for 10 mins, blocked with 5% BSA for half an hour, and then incubated with primary antibodies against ZO-1 (1:200, Cat# 2847, CST, USA), VEGF (1:200, Cat# sc-365578, Santa Cruz, USA) and VEGFR2 (1:500, Cat# 9698, CST, USA) at room temperature for 2 hours. Then the secondary antibodies were labeled with the corresponding secondary antibodies at room temperature for 1 h: Alexa Fluor 488 AffiniPure goat anti-mouse IgG, (1:800; Cat# 33206ES60, Yeasen, China), and Alexa Fluor plus 555 goat ant-rabbit IgG (H + L) (1:1000; Cat# ab150078, Abcam, USA) were used. Finally, the sections were mounted using the fluorescent mounting medium DAPI G-Fluoromount medium (Southern Biotech, USA), A fluorescence microscope (×40 objective, Nikon Eclipse Ni, Nikon, Japan) coupled with a digitizing camera was used to capture the pictures.
Human bEnd.3 brain microvascular endothelial cells and cell treatment
Cells were purchased from ATCC (USA) and cultured in a high-glucose Dulbecco’s modified Eagle medium (DMEM) medium containing 10% Fetal Bovine Serum (FBS), which were incubated at 37° C and 5% CO2. For the establishment of the H/R model, cells were placed in the anaerobic incubator, followed by introducing 5% CO2+95% N2 into the incubator. After incubating for 6 h, cells were placed in the incubator with normal conditions and incubated for 24 h.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cells were implanted in a 96-well plate (3×103/well) and incubated for 12 h, followed by adding 20 μL 0.5 mg/mL MTT solution. After 4h-incubation at 37° C, the supernatant was disregarded and 200 μL dimethyl sulfoxide (DMSO) was added for 15 mins, followed by measuring the optical density (OD) value at 570 nm using a microplate reader (Tecan, Switzerland).
Lactate dehydrogenase (LDH) release assay
After treatments, 20 μL supernatant was collected and mixed with 250 μL matrix buffer and 50 μL coenzyme I solution, followed by incubation for 15 min at 37° C. Then 250 μL 2, 4-dinitrophenylhydrazine was introduced for 15 min at 37° C, followed by introducing NaOH to terminate the reaction. Lastly, the OD value was achieved at 440 nm with a microplate reader (Tecan, Switzerland).
Fluorescein isothiocyanate (FITC)-dextran assay
Cells were planted on the luminal side of filters (Millipore, USA), followed by introducing FITC-dextran (AbMole, USA) into the upper compartment. Following incubation for 1 h, the microplate reader (Tecan, Switzerland) was used to detect the OD value of the solution in the lower chamber at 492/520 nm.
Trans-endothelial electrical resistance (TEER)
The in vitro endothelial permeability was evaluated using the TEER assay with the 1600R ECIS System (Applied Biophysics, Australia) according to the method described previously [22] (Rom et al. 2015). The data were expressed with an average of the resistance values (Ω·cm2) and the average percent change from baseline TEER (Mean ± SD).
Western blotting assay
Proteins were isolated and quantified using the Bicinchoninic acid (BCA) method and loaded to sodium dodecyl sulfate (SDS)-polyacrylamide gel (PAGE) for separation. Then, the proteins in the gel were transferred onto the polyvinylidene fluoride (PVDF) membrane, followed by being blocked using the 5% BSA reagent. Primary antibodies against ZO-1 (1:2000, Cat# 2847, CST, USA), VEGF (1:2000, Cat# sc-365578, Santa Cruz, USA), VEGFR2 (1:1500, Cat# 9698, CST, USA), and β-actin (1:8000, Cat# GTX100313, GeneTex, USA) were then added and incubated for 12 h at 4° C, followed by introducing the anti-rabbit IgG, horseradish peroxidase (HRP)-linked antibody (Cat#7074, CST, USA), anti-mouse IgG, and HRP-linked antibody (Cat# 7076, CST, USA). After 1.5 h incubation, an enhanced chemiluminescence (ECL) solution was added for exposure, followed by quantification of the protein expression using the Image J software [23].
Statistical analysis
Data were expressed as mean ± standard deviation (S.D.) and the comparison was analyzed using an analysis of variance (ANOVA) method by the software GraphPad Prism 8. The Chi-square test was used as a post-hoc test. P<0.05 was taken as a statistically significant difference.
Data availability statement
Data will be made available on reasonable request.
Results
CEP improved neurological dysfunction in a MCAO mice model
The molecular structure of CEP is shown in Figure 1A. 15 mg/kg CEP was injected into the peritoneal cavity of mice for 3 weeks before the MCAO modeling. The neurological score (Figure 1B) in the control and CEP group was 0, while it was elevated to 3.3 in the MCAO group, then declined to 1.9 by treatment with 15 mg/kg CEP, suggesting a protective property of CEP against neurological dysfunction in MCAO mice.
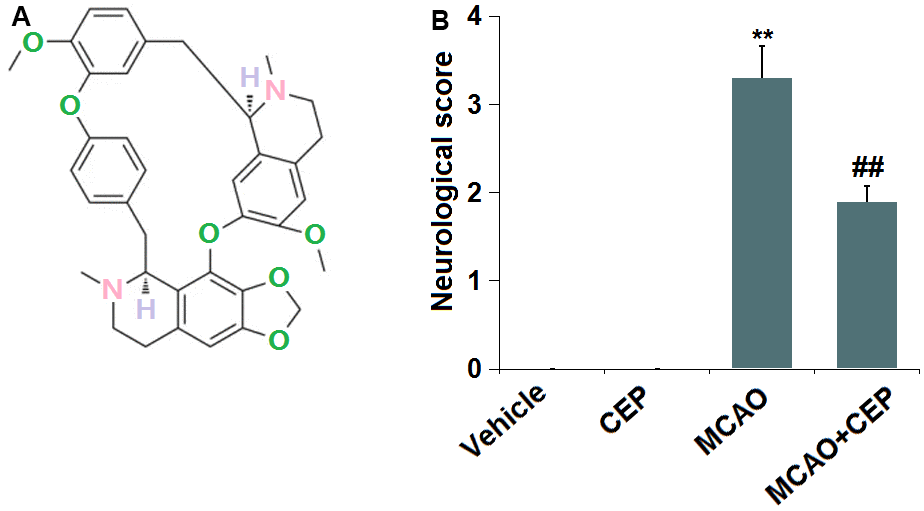
Figure 1. Cepharanthine (CEP) improved neurological dysfunction in a middle cerebral artery occlusion (MCAO) mice model. (A) Molecular structure of Cepharanthine; (B) Neurological score graph of the three experimental groups (**, P<0.01 vs. vehicle group; ##, P<0.01 vs. MCAO group).
CEP prevented the increase in BBB permeability in MCAO mice
Diffusion of sodium fluorescein assay was used to assess BBB permeability. We found that the BBB permeability (Figure 2) was minorly changed from 21.3 to 20.7 ng/mg protein in CEP-treated normal mice, but was greatly promoted to 46.5 ng/mg protein in MCAO mice. After treatment with 15 mg/kg CEP, the BBB permeability was reduced to 33.7 ng/mg protein, implying an inhibitory effect of CEP on the increased BBB permeability in MCAO mice.
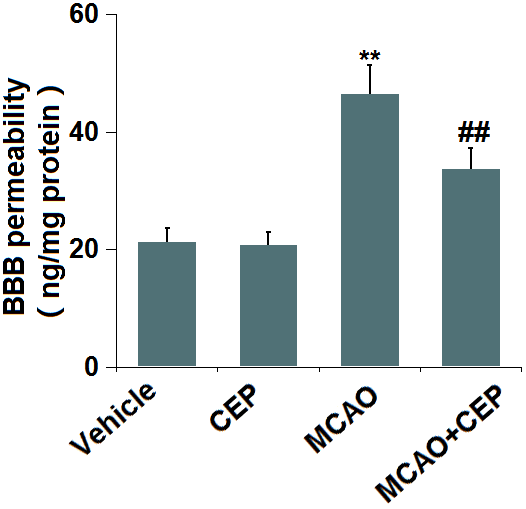
Figure 2. Cepharanthine (CEP) prevented the increase in blood-brain barrier (BBB) permeability in MCAO mice model. Blood-brain barrier permeability was measured by diffusion of sodium fluorescein assay (**, P<0.01 vs. vehicle group; ##, P<0.01 vs. MCAO group).
CEP restored the expression of ZO-1 in the cortex of MCAO mice
We further checked the impact of CEP on TJ protein level. ZO-1 was found (Figure 3A) significantly upregulated in CEP-treated normal mice, but downregulated in MCAO mice, which was greatly reversed by 15 mg/kg CEP, suggesting a protective effect of CEP on the downregulation of ZO-1 in MCAO mice.
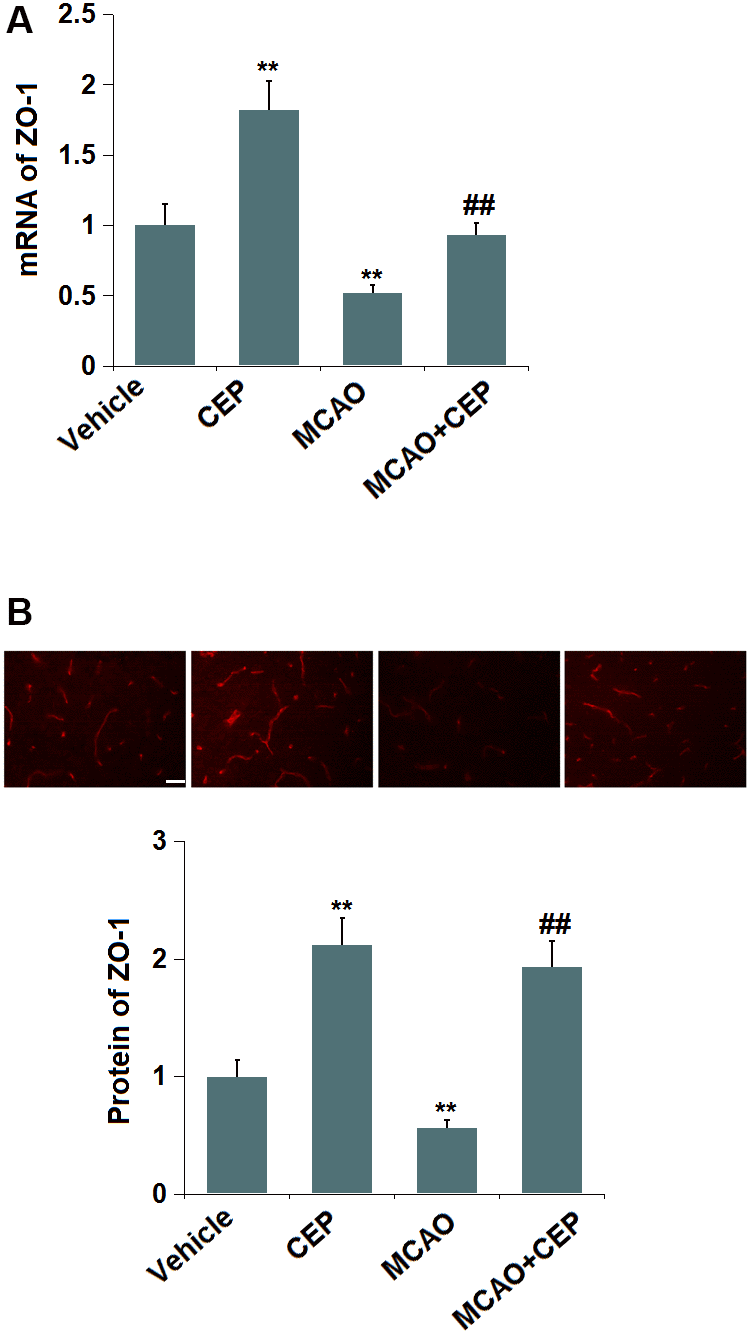
Figure 3. Cepharanthine (CEP) restored the expression of ZO-1 in the cortex of MCAO mice model. (A) mRNA of ZO-1; (B) Protein of ZO-1 as measured using immunostaining. Scale bars, 100 μm (**, P<0.01 vs. vehicle group; ##, P<0.01 vs. MCAO group).
CEP reduced the expression of VEGF-A and VEGFR2 in the cortex of MCAO mice
The VEGF-A/VEGFR2 axis is reported to regulate the expression of TJ proteins in the BBB [10, 24]. We found that the expression levels of VEGF-A and VEGFR2 (Figure 4A, 4B) were repressed in CEP-treated normal mice and increased in MCAO mice. After the administration of 15 mg/kg CEP to MCAO mice, the expression levels of VEGF-A and VEGFR2 were significantly declined, indicating its inhibitory effect on the VEGF-A/VEGFR2 axis in MCAO mice.
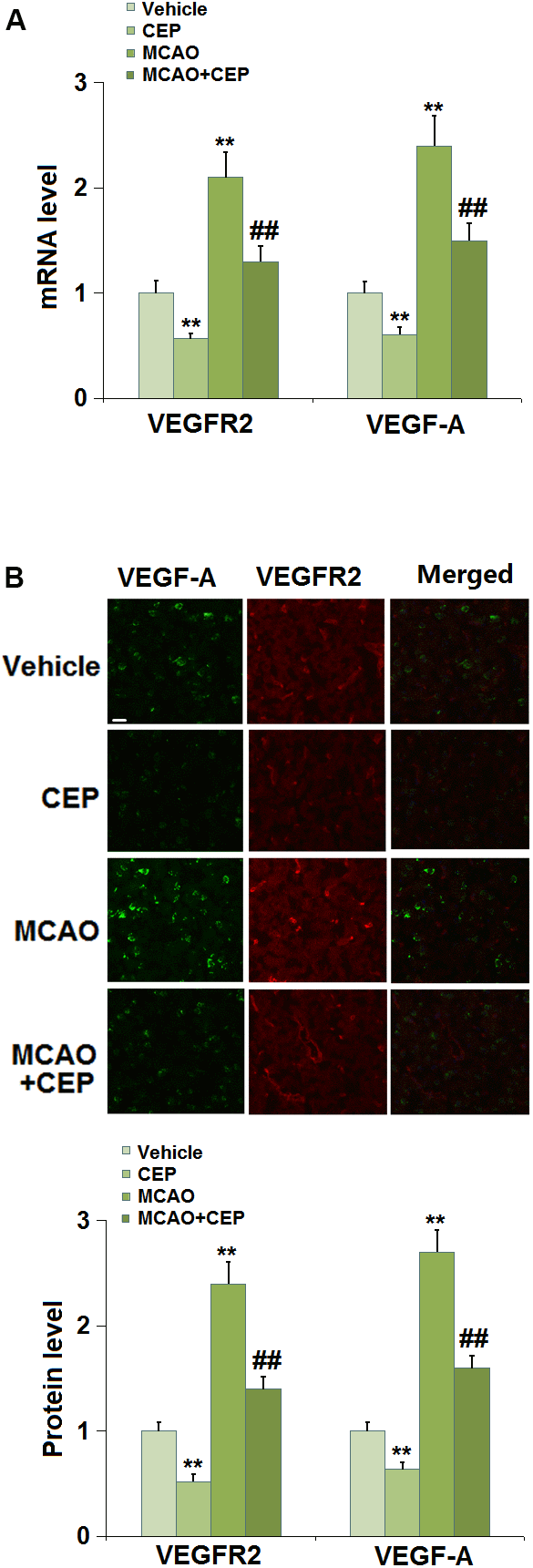
Figure 4. Cepharanthine (CEP) reduced the expression of VEGF-A and VEGFR2 in the cortex of MCAO mice model. (A) mRNA levels of VEGF-A and VEGFR2; (B) Protein levels of VEGF-A and VEGFR2 as measured using immunostaining. Scale bars, 100 μm (**, P<0.01 vs. vehicle group; ##, P<0.01 vs. MCAO group).
CEP ameliorated cytotoxicity of bEnd.3 cells against H/R
To explore the potential mechanism of the protective effects of CEP on stroke, we established an in vitro H/R (6 h/24 h) model, followed by treatment with 1.5 and 3 μM CEP. We found that the cell viability (Figure 5A) in H/R-treated cells declined from 100% to 62%, which was elevated to 81% and 93% by 1.5 and 3 μM CEP, respectively. Furthermore, the LDH release in the control, H/R, 1.5 CEP, and 3 μM CEP groups was 5%, 28.7%, 18.6%, and 12.5%, respectively. These data imply that the cytotoxicity in H/R-treated bEnd.3 cells was ameliorated by CEP.
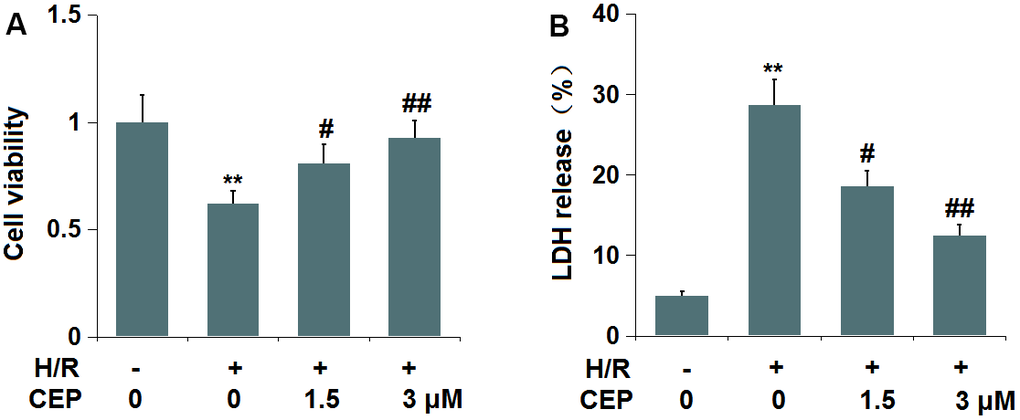
Figure 5. Cepharanthine (CEP) ameliorated cytotoxicity of human bEnd.3 brain microvascular endothelial cells against hypoxia/reperfusion (H/R). Cells were exposed to hypoxia/reperfusion condition (6 h/24 h) with or without CEP (1.5, 3 μM). (A) Cell viability measured by MTT assay; (B) LDH release (**, P<0.01 vs. vehicle group; #, ##, P<0.05, 0.01 vs. H/R group).
CEP attenuated H/R-induced aggravation of endothelial permeability in bEnd.3 cells
We further evaluated the impact of CEP on in vitro endothelial permeability. The fluorescence intensity of FITC (Figure 6A) was extremely increased in the H/R group, then reduced by 1.5 and 3 μM CEP. Moreover, the TEER value (Figure 6B) in H/R- treated cells was decreased from 113.5 Ωcm2 to 71.8 Ωcm2, which was increased to 92.3 Ωcm2 and 108.9 Ωcm2 by 1.5 and 3 μM CEP, respectively. These data imply the increased endothelial permeability in H/R-challenged bEnd.3 cells was attenuated by CEP.
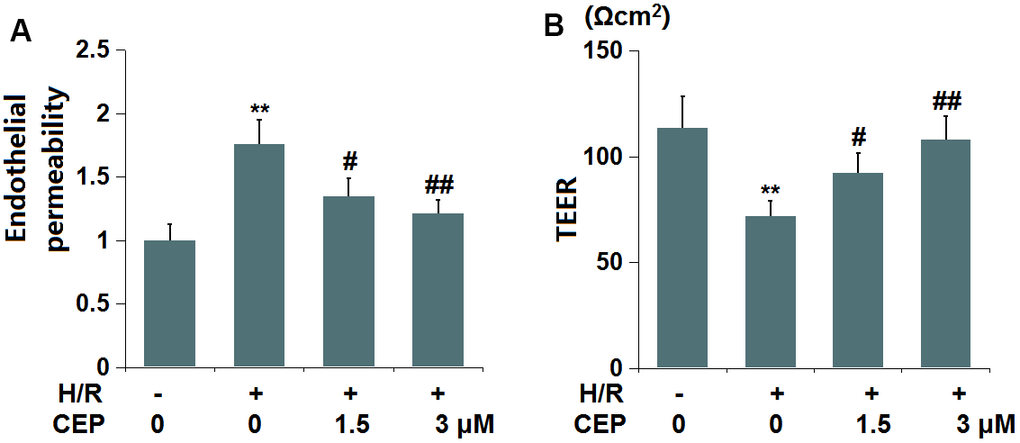
Figure 6. Cepharanthine (CEP) attenuated hypoxia/reperfusion-induced aggravation of endothelial permeability in brain bEND.3 endothelial cells. (A) Endothelial permeability was measured using FITC-dextran; (B) The trans-endothelial electrical resistance (TEER) (**, P<0.01 vs. vehicle group; #, ##, P<0.05, 0.01 vs. H/R group).
CEP restored the expression of ZO-1 in bEnd.3 cells
In accordance with the in vivo assay, the expression level of ZO-1 was investigated. We found that ZO-1 (Figure 7) was significantly downregulated in H/R-treated cells, which was greatly reversed by 1.5 and 3 μM CEP, suggesting that the level of ZO-1 in H/R-treated bEnd.3 cells was restored by CEP.
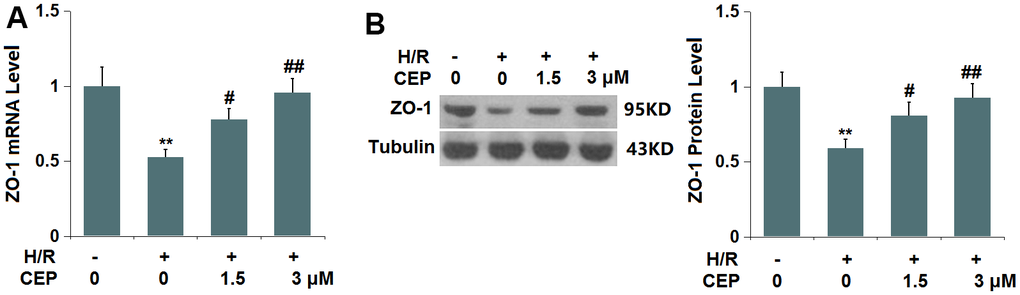
Figure 7. Cepharanthine (CEP) restored the expression of ZO-1 in brain bEND.3 endothelial cells. (A) mRNA of ZO-1; (B) Protein of ZO-1 as measured by western blot analysis (**, P<0.01 vs. vehicle group; #, ##, P<0.05, 0.01 vs. H/R group).
CEP repressed the level of VEGF-A and VEGFR2 in bEnd.3 cells
We further evaluated the state of the VEGF-A/VEGFR2 axis in bEnd.3 cells. The expression levels of VEGF-A and VEGFR2 (Figure 8A, 8B) were found significantly elevated in H/R- challenged cells, then greatly repressed by 1.5 and 3 μM CEP, suggesting an inhibitory effect of CEP on the VEGF-A/VEGFR2 axis in H/R-challenged bEnd.3 cells.
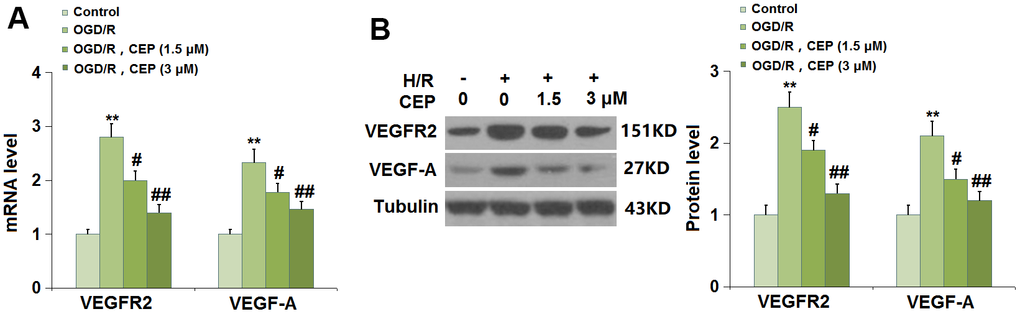
Figure 8. Cepharanthine (CEP) reduced the expression of VEGF-A and VEGFR2 in brain bEND.3 endothelial cells. (A) mRNA of VEGF-A and VEGFR2; (B) Protein of VEGF-A and VEGFR2 (**, P<0.01 vs. vehicle group; #, ##, P<0.05, 0.01 vs. H/R group).
VEGF-A abolished the protective effects of CEP on ZO-1 level and endothelial permeability
To examine whether the function of CEP was associated with the VEGF-A/VEGFR2 axis, cells were treated with H/R, with or without CEP (3 μM) or VEGF-A (10 ng/ml). We found that the downregulation of ZO-1 in H/R-challenged cells was greatly reversed by CEP, an effect that was abolished by the co-introduction of VEGF-A (Figure 9A). Furthermore, the enhanced fluorescence intensity of FITC in H/R-treated cells was reduced by CEP, and later elevated by the co-incubation with VEGF-A (Figure 9B). Moreover, the TEER values in the control, H/R, CEP, and CEP+ VEGF-A groups were 112.6, 71.8, 108.9, and 82.5 Ωcm2 (Figure 9C), respectively. These results imply that the protective function of CEP on ZO-1 level and endothelial permeability was abrogated by VEGF-A.
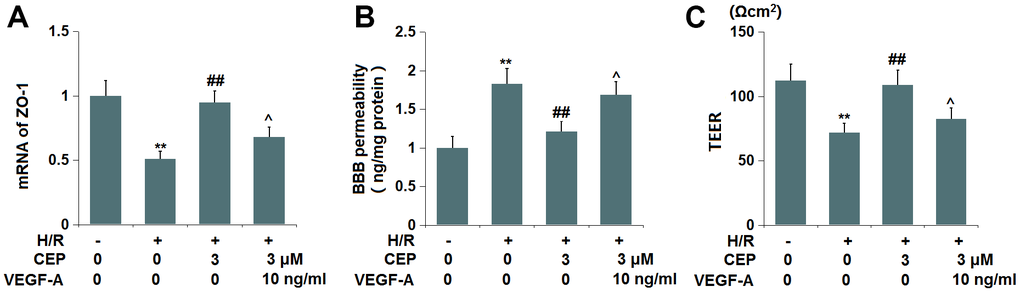
Figure 9. VEGF-A abolished the protective effects of CEP on the expression of ZO-1 and endothelial permeability. Cells were exposed to OGD/R in the presence or absence of CEP (3 μM) or VEGF-A (10 ng/ml). (A) mRNA of ZO-1; (B) Endothelial permeability was measured using FITC-dextran; (C) The trans-endothelial electrical resistance (TEER) (**, P<0.01 vs. vehicle group; ##, P<0.01 vs. H/R group group; ^, P<0.05 vs. H/R+CEP group).
Discussion
In the pathological process of ischemic stroke, disruption of the BBB is particularly significant. In the case of ischemic stroke complicated by diseases such as hypertension and hyperglycemia, destruction of the BBB is further aggravated after stroke due to changes in vascular anatomy and function of the central nervous system, accompanied by aggravation of nerve injury in stroke. The process of brain damage is sped up, and the risk of bleeding increased by damage to the BBB. Furthermore, cerebral edema results from BBB damage, which is the most serious disabling and fatal complication of ischemic stroke. Cerebral edema caused by ischemic stroke includes cytotoxic edema and vasogenic edema. Cerebral edema becomes progressively worse after ischemia, with cytotoxic edema occurring several minutes after an ischemic attack, and vasogenic edema occurring relatively later, which is particularly closely related to BBB damage [25, 26]. In patients with ischemic stroke, a large amount of blood-derived fluid flows into the brain parenchyma after BBB destruction, resulting in progressively increased water content in brain tissue [27]. We constructed a MCAO mice model with increased neurological scores and BBB permeability, consistent with the description by Du [28]. These pathological changes were significantly alleviated by CEP, suggesting that the neuroprotective effect of CEP on MCAO mice might be mediated by its impact on BBB permeability. Furthermore, increased BBB permeability was observed in H/R-challenged bEnd.3 cells, in accordance with the observation reported by Yang [29]. After incubation with CEP, the BBB permeability was greatly alleviated, which further evidenced the protective effect of CEP in MCAO mice.
ZO-1 is a TJ-related protein, the level change of which shows a critical impact on the structure and function of TJs. In the early stage of cerebral ischemia, the ZO-1 protein level is significantly reduced, accompanied by destroyed TJs and increased BBB permeability [30]. Changes in ZO-1 protein level are correlated with the degree of BBB damage to a certain extent, which can be used as a marker of BBB damage [31]. In the present study, ZO-1 was found downregulated in both MCAO mice and H/R- challenged bEnd.3 cells, also reported by Zhang [32]. The changes in ZO-1 protein level in both MCAO mice and H/R-challenged bEnd.3 cells were reversed by CEP, suggesting that CEP might exert its protective function by upregulating ZO-1.
VEGF is one of the main regulatory factors that increase microvascular permeability. Several studies have confirmed that the increased BBB permeability and cerebral edema can be induced by the interaction of VEGF [33, 34]. In central nervous immune diseases, hypoxia, and cerebral ischemia, the expression of VEGF in astrocytes is upregulated and induces an increase in BBB permeability [35, 36]. Furthermore, VEGF is reported to induce BBB dysfunction and increase its permeability by inducing tight junction protein degradation, occludin transport, and phosphorylation of Ser490 in occludin [37, 38]. We found that the activated VEGF-A/VEGFR2 axis in both MCAO mice and H/R-challenged bEnd.3 cells was significantly suppressed by CEP. Furthermore, the protective function of CEP on ZO-1 and endothelial permeability was extremely abrogated by VEGF-A, implying that CEP might exert its protective effect on stroke by regulating the VEGF-A/VEGFR2 axis. In future work, the mechanism will be further investigated by co-administering VEGF-A and CEP to MCAO mice.
Collectively, our data reveal that CEP maintained integrity of the BBB in stroke by mediating VEGF/VEGFR2/ZO-1 signaling.
Author Contributions
Guarantor of integrity of the entire study: Yunfang Yang; Study concepts: Yunfang Yang; Study design: Jiang Huang; Definition of intellectual content: Sijin Yang; Literature research: Changjiang Li; Clinical studies: Yunfang Yang; Experimental studies: Jiang Huang; Data acquisition: Hongmei Tang; Data analysis: Zhuo Zhang; Statistical analysis: Xue Bai; Manuscript preparation: Jiang Huang; Manuscript editing: Yunfang Yang; Manuscript review: Jiang Huang; All the authors have read and approved the final submission of this study.
Conflicts of Interest
All authors declared that there are no financial interests that are directly or indirectly related to the work submitted for publication.
Ethical Statement
The animal experiments comply with the ARRIVE guidelines and were performed under a protocol approved by the Institutional Animal Care and Use Committee of The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University (SMU-EA-20190033).
Funding
This work was funded by the following: Special Project for Traditional Chinese Medicine Research of Sichuan Provincial Administration of Traditional Chinese Medicine (2023MS183), Major Science and Technology Special Project of Sichuan Provincial Department of Science and Technology (2022YFS0613), Joint Project of Southwest Medical University and Affiliated Hospital of Traditional Chinese Medicine of Southwest Medical University (2020XYLH-025).
References
- 1. George PM, Steinberg GK. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron. 2015; 87:297–309. https://doi.org/10.1016/j.neuron.2015.05.041 [PubMed]
- 2. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010; 37:13–25. https://doi.org/10.1016/j.nbd.2009.07.030 [PubMed]
- 3. Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011; 55:467–76. https://doi.org/10.1387/ijdb.103224sl [PubMed]
- 4. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004; 16:1–13. https://doi.org/10.1016/j.nbd.2003.12.016 [PubMed]
- 5. Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999; 842:92–100. https://doi.org/10.1016/s0006-8993(99)01843-0 [PubMed]
- 6. Go M, Kojima T, Takano K, Murata M, Ichimiya S, Tsubota H, Himi T, Sawada N. Expression and function of tight junctions in the crypt epithelium of human palatine tonsils. J Histochem Cytochem. 2004; 52:1627–38. https://doi.org/10.1369/jhc.4A6339.2004 [PubMed]
- 7. Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012; 32:3044–57. https://doi.org/10.1523/JNEUROSCI.6409-11.2012 [PubMed]
- 8. Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 2002; 38:323–37. https://doi.org/10.1016/s1537-1891(02)00200-8 [PubMed]
- 9. Thal SC, Luh C, Schaible EV, Timaru-Kast R, Hedrich J, Luhmann HJ, Engelhard K, Zehendner CM. Volatile anesthetics influence blood-brain barrier integrity by modulation of tight junction protein expression in traumatic brain injury. PLoS One. 2012; 7:e50752. https://doi.org/10.1371/journal.pone.0050752 [PubMed]
- 10. Bergman L, Acurio J, Leon J, Gatu E, Friis T, Nelander M, Wikström J, Larsson A, Lara E, Aguayo C, Torres-Vergara P, Wikström AK, Escudero C. Preeclampsia and Increased Permeability Over the Blood-Brain Barrier: A Role of Vascular Endothelial Growth Receptor 2. Am J Hypertens. 2021; 34:73–81. https://doi.org/10.1093/ajh/hpaa142 [PubMed]
- 11. Morotti A, Poli L, Costa P. Acute Stroke. Semin Neurol. 2019; 39:61–72. https://doi.org/10.1055/s-0038-1676992 [PubMed]
- 12. Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, Biller J, Brown M, Demaerschalk BM, Hoh B, Jauch EC, Kidwell CS, Leslie-Mazwi TM, et al, and American Heart Association Stroke Council. 2018 Guidelines for the Early Management of Patients With Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2018; 49:e46–110. https://doi.org/10.1161/STR.0000000000000158 [PubMed]
- 13. Lees KR, Emberson J, Blackwell L, Bluhmki E, Davis SM, Donnan GA, Grotta JC, Kaste M, von Kummer R, Lansberg MG, Lindley RI, Lyden P, Murray GD, et al, and Stroke Thrombolysis Trialists’ Collaborators Group. Effects of Alteplase for Acute Stroke on the Distribution of Functional Outcomes: A Pooled Analysis of 9 Trials. Stroke. 2016; 47:2373–9. https://doi.org/10.1161/STROKEAHA.116.013644 [PubMed]
- 14. Mackey J, Kleindorfer D, Sucharew H, Moomaw CJ, Kissela BM, Alwell K, Flaherty ML, Woo D, Khatri P, Adeoye O, Ferioli S, Khoury JC, Hornung R, Broderick JP. Population-based study of wake-up strokes. Neurology. 2011; 76:1662–7. https://doi.org/10.1212/WNL.0b013e318219fb30 [PubMed]
- 15. Rogosnitzky M, Danks R. Therapeutic potential of the biscoclaurine alkaloid, cepharanthine, for a range of clinical conditions. Pharmacol Rep. 2011; 63:337–47. https://doi.org/10.1016/s1734-1140(11)70500-x [PubMed]
- 16. Gao S, Li X, Ding X, Qi W, Yang Q. Cepharanthine Induces Autophagy, Apoptosis and Cell Cycle Arrest in Breast Cancer Cells. Cell Physiol Biochem. 2017; 41:1633–48. https://doi.org/10.1159/000471234 [PubMed]
- 17. Seubwai W, Vaeteewoottacharn K, Hiyoshi M, Suzu S, Puapairoj A, Wongkham C, Okada S, Wongkham S. Cepharanthine exerts antitumor activity on cholangiocarcinoma by inhibiting NF-kappaB. Cancer Sci. 2010; 101:1590–5. https://doi.org/10.1111/j.1349-7006.2010.01572.x [PubMed]
- 18. Harada K, Ferdous T, Itashiki Y, Takii M, Mano T, Mori Y, Ueyama Y. Cepharanthine inhibits angiogenesis and tumorigenicity of human oral squamous cell carcinoma cells by suppressing expression of vascular endothelial growth factor and interleukin-8. Int J Oncol. 2009; 35:1025–35. https://doi.org/10.3892/ijo_00000417 [PubMed]
- 19. Chen B, Grothe C, Schaller K. Validation of a new neurological score (FOUR Score) in the assessment of neurosurgical patients with severely impaired consciousness. Acta Neurochir (Wien). 2013; 155:2133–9. https://doi.org/10.1007/s00701-013-1854-2 [PubMed]
- 20. Yu N, Wang Z, Chen Y, Yang J, Lu X, Guo Y, Chen Z, Xu Z. The ameliorative effect of bloodletting puncture at hand twelve Jing-well points on cerebral edema induced by permanent middle cerebral ischemia via protecting the tight junctions of the blood-brain barrier. BMC Complement Altern Med. 2017; 17:470. https://doi.org/10.1186/s12906-017-1979-6 [PubMed]
- 21. Guitart M, Lloreta J, Mañas-Garcia L, Barreiro E. Muscle regeneration potential and satellite cell activation profile during recovery following hindlimb immobilization in mice. J Cell Physiol. 2018; 233:4360–72. https://doi.org/10.1002/jcp.26282 [PubMed]
- 22. Rom S, Dykstra H, Zuluaga-Ramirez V, Reichenbach NL, Persidsky Y. miR-98 and let-7g* protect the blood-brain barrier under neuroinflammatory conditions. J Cereb Blood Flow Metab. 2015; 35:1957–65. https://doi.org/10.1038/jcbfm.2015.154 [PubMed]
- 23. Shin MK, Vázquez-Rosa E, Koh Y, Dhar M, Chaubey K, Cintrón-Pérez CJ, Barker S, Miller E, Franke K, Noterman MF, Seth D, Allen RS, Motz CT, et al. Reducing acetylated tau is neuroprotective in brain injury. Cell. 2021; 184:2715–32.e23. https://doi.org/10.1016/j.cell.2021.03.032 [PubMed]
- 24. Heinolainen K, Karaman S, D’Amico G, Tammela T, Sormunen R, Eklund L, Alitalo K, Zarkada G. VEGFR3 Modulates Vascular Permeability by Controlling VEGF/VEGFR2 Signaling. Circ Res. 2017; 120:1414–25. https://doi.org/10.1161/CIRCRESAHA.116.310477 [PubMed]
- 25. Dharmasaroja PA. Fluid Intake Related to Brain Edema in Acute Middle Cerebral Artery Infarction. Transl Stroke Res. 2016; 7:49–53. https://doi.org/10.1007/s12975-015-0439-1 [PubMed]
- 26. Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016; 36:513–38. https://doi.org/10.1177/0271678X15617172 [PubMed]
- 27. Rosenberg GA. Ischemic brain edema. Prog Cardiovasc Dis. 1999; 42:209–16. https://doi.org/10.1016/s0033-0620(99)70003-4 [PubMed]
- 28. Du J, Yin G, Hu Y, Shi S, Jiang J, Song X, Zhang Z, Wei Z, Tang C, Lyu H. Coicis semen protects against focal cerebral ischemia-reperfusion injury by inhibiting oxidative stress and promoting angiogenesis via the TGFβ/ALK1/Smad1/5 signaling pathway. Aging (Albany NY). 2020; 13:877–93. https://doi.org/10.18632/aging.202194 [PubMed]
- 29. Yang B, Li Y, Ma Y, Zhang X, Yang L, Shen X, Zhang J, Jing L. Selenium attenuates ischemia/reperfusion injury-induced damage to the blood-brain barrier in hyperglycemia through PI3K/AKT/mTOR pathway-mediated autophagy inhibition. Int J Mol Med. 2021; 48:178. https://doi.org/10.3892/ijmm.2021.5011 [PubMed]
- 30. Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008; 32:200–19. https://doi.org/10.1016/j.nbd.2008.08.005 [PubMed]
- 31. Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002; 282:H1485–94. https://doi.org/10.1152/ajpheart.00645.2001 [PubMed]
- 32. Zhang Y, Hu Y, Li M, Wang J, Guo G, Li F, Yu B, Kou J. The Traditional Chinese Medicine Compound, GRS, Alleviates Blood-Brain Barrier Dysfunction. Drug Des Devel Ther. 2020; 14:933–47. https://doi.org/10.2147/DDDT.S229302 [PubMed]
- 33. Bauer AT, Bürgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010; 30:837–48. https://doi.org/10.1038/jcbfm.2009.248 [PubMed]
- 34. Liu L, Fujimoto M, Kawakita F, Ichikawa N, Suzuki H. Vascular Endothelial Growth Factor in Brain Edema Formation After Subarachnoid Hemorrhage. Acta Neurochir Suppl. 2016; 121:173–7. https://doi.org/10.1007/978-3-319-18497-5_31 [PubMed]
- 35. Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, Ferrara N, Sofroniew MV, John GR. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012; 122:2454–68. https://doi.org/10.1172/JCI60842 [PubMed]
- 36. Chapouly C, Tadesse Argaw A, Horng S, Castro K, Zhang J, Asp L, Loo H, Laitman BM, Mariani JN, Straus Farber R, Zaslavsky E, Nudelman G, Raine CS, John GR. Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain. 2015; 138:1548–67. https://doi.org/10.1093/brain/awv077 [PubMed]
- 37. Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol. 2002; 39:225–37. https://doi.org/10.1016/s1537-1891(03)00011-9 [PubMed]
- 38. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999; 399:597–601. https://doi.org/10.1038/21218 [PubMed]