



Introduction
Hepatocellular carcinoma (HCC) is a common liver cancer with a substantial global death rate, ranking as the second most frequent cause of cancer death in men and sixth in women. In 2020, HCC was reported to have afflicted 906,000 new patients and caused 830,000 deaths [1]. HCC is highly malignant, rapidly metastasizes, is mostly detected late, and recurs > 70% of the time after five years of treatment [2]. For accurate HCC diagnosis and prognosis and improved survival time, the identification and validation of prognostic markers and therapeutic targets are urgently required.
N7-methylguanosine (m7G) is a conserved RNA modification of the seventh N of RNA guanine. It comprises a methyl group of eukaryotes, prokaryotes, and some archaea. This modification occurs not only in mRNA caps but also at some positions in mRNAs, tRNAs, and rRNAs [3, 4] and is essential for RNA metabolism, including nucleation [5], pre-mRNA splicing [6], transcription elongation [7], and mRNA translation [8]. The literature suggests that m7G modification is crucial for regulating biological processes and tumor disease development [9–11]. Aberrant m7G expression may affect the incidence and progression of cancer by modulating various tumor suppressors and oncogenes [12]. m7G methylation complexes comprise methyltransferase like1 (METTL1) and WD repeat domain 4 (WDR4). According to a study, m7G modification and its abovementioned catalyzing enzymes are elevated in HCC cases and linked with a substandard prognosis. Functionally, m7G modification promotes HCC progression and tumorigenesis [13]. Nevertheless, the mechanism of m7G RNA methylation in HCC is still not fully determined.
MicroRNAs (miRNAs) are small, non-coding, highly conserved RNAs. Although they cannot be translated into protein, they are essential for targeting messenger RNA (mRNA) in gene expression and are thus involved in many biological processes, such as tumorigenesis, progression, and responses to therapy [14, 15]. Individuals with HCC have shown aberrant miRNA levels in multiple studies. Because of their robust presence in body tissues and fluids and easy detectability [16], miRNA expression is not only used as a biomarker for diagnosis and metastasis but also predicts therapeutic response, recurrence, and overall HCC survival rate [17–20]. Research has proven that METTL1 can regulate microRNAs in an m7G-dependent pattern. Pandolfini et al. revealed that METTL1 stimulates the processing of let-7 miRNA via m7G methylation [21]. METTL1 sensitizes colon cancer cells resistant to cisplatin by modulating miR-149-3p/S100A4/p53 [22]. However, the direct functions of m7G and miRNAs in HCC and their associations with HCC, including the molecular markers for predicting HCC prognosis using m7G-miRNAs, are still insufficiently understood and require further investigation.
This investigation established a prognostic model based on m7G-linked differentially expressed miRNAs (DEmiRNAs) and validated its prognostic and clinical significance. Furthermore, to elucidate the molecular mechanisms dysregulated in HCC, biological pathways and the immune landscape were explored via relevant public databases.
Results
Identification of DEmiRNAs
Figure 1 shows the study workflow; 792 miRNAs that targeted WDR4 or METTL1 were identified in TargetScan (Supplementary Table 1). Of these, 63 m7G-related miRNAs (m7G-miRNAs) were identified as DEmiRNAs in 375 HCC and 50 healthy samples, with 40 defined as up-regulated and 23 as down-regulated according to the cutoff criteria (Figure 2A). The top 20 DEmiRNAs are shown in the heatmap in Figure 2B.
Figure 1. The workflow.
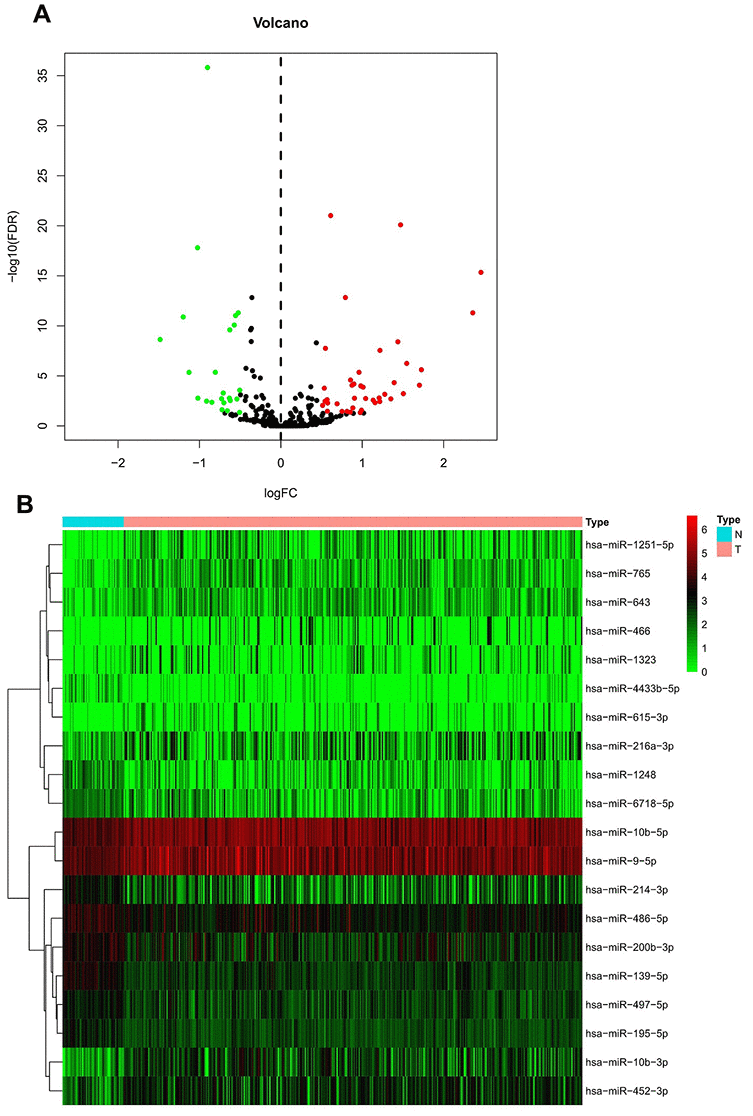
Figure 2. Differential expression of m7G-related miRNA in HCC and non-tumor samples. (A) Heatmap of top 20 DEmiRNAs. Normal and tumor samples are represented in N and T, respectively. (B) Volcano plot of all DEmiRNAs. Red and green dots represent up- and down-regulated m7G-miRNAs, respectively.
The m7G-miRNA risk model’s generation and validation
Based on a univariate Cox regression analysis, seven DEmiRNAs (hsa-miR-195-5p, hsa-miR-9-5p, hsa-miR-504-3p, has-miR-892a, hsa-miR-6764-5p, hsa-miR-4652-3p, hsa-miR-152-5p) were selected for use in constructing a prognostic risk regression model (Figure 3A, 3B). A total of 366 HCC case records with survival time data were used to construct and verify this model. Risk scores were determined for all subjects, and the latter were divided into two groups according to the risk score median. The low-risk (LR) and high-risk (HR) score cohorts included 183 patients each (Figure 4A). It is clear from Figure 4B that the mortality rate in the HR group was significantly higher than that in the LR group, which indicates valid sample grouping. The grouping’s accuracy was also ensured using principal component analysis (PCA), the results of which show that the HCC cases in the LR and HR groups were clearly separated (Figure 4C). According to a Kaplan–Meier survival test, the HR HCC patients’ overall survival rate was poorer than that of the LR patients (Figure 4D). The risk model’s predictive ability was evaluated by time receiver operating characteristic (ROC) analysis; at 1, 3, and 5 years, the areas under the curve (AUCs) of OS were 0.706, 0.695, and 0.715 (Figure 4E). This result indicates that the risk model has moderate diagnostic strength.
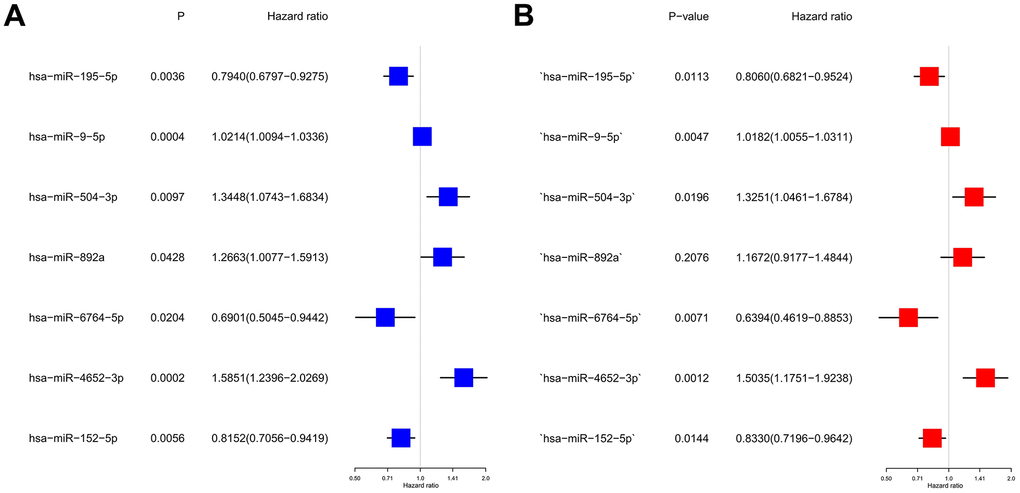
Figure 3. Construction of risk scores for m7G-related miRNAs. (A) Univariate Cox regression analysis was used to screen seven prognosis-related miRNAs. (B) Forest plot of multivariate analyses.
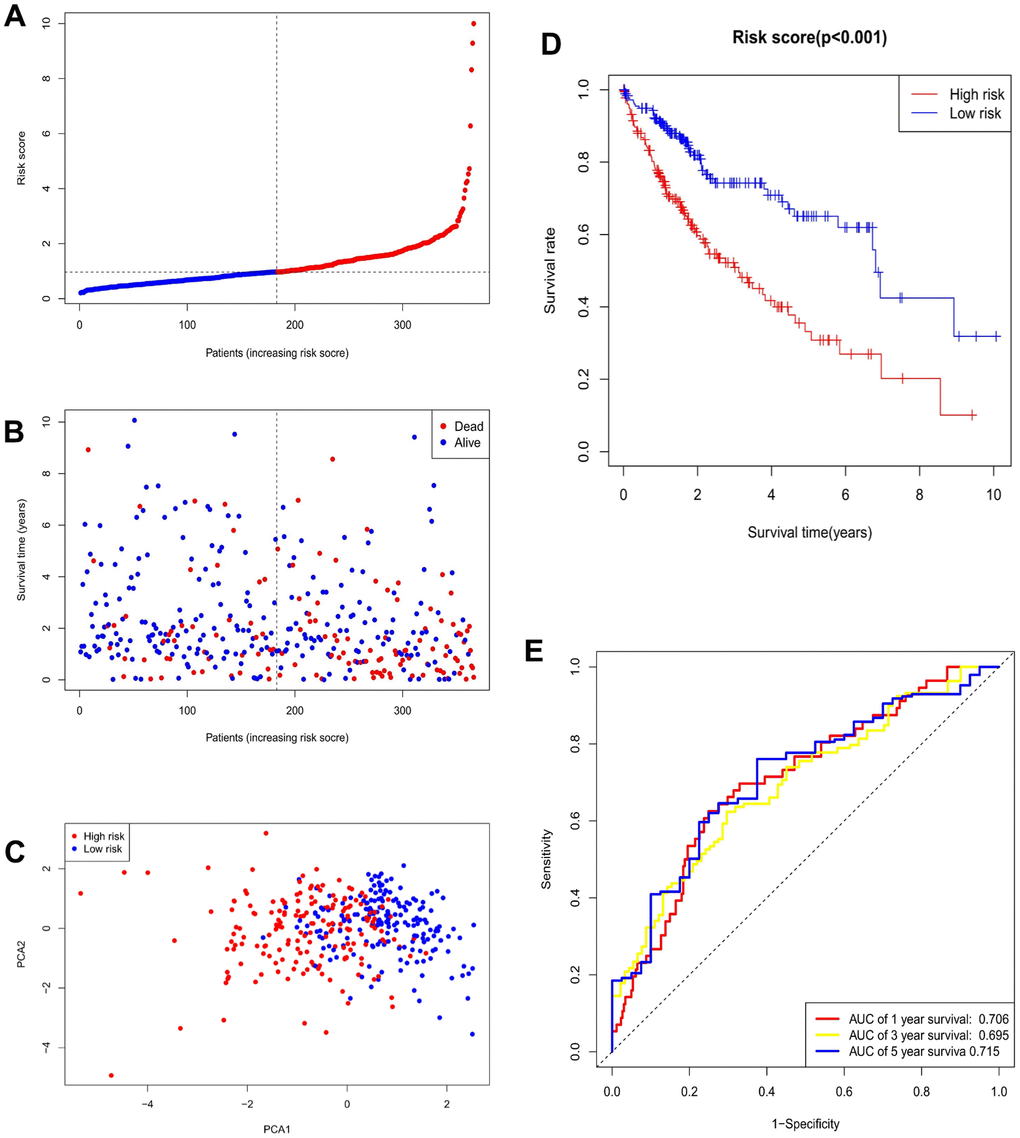
Figure 4. Validation of prognostic models for seven m7G-related miRNAs. (A) Risk score distribution. (B) Survival status. (C) PCA plot. (D) Survival curve for low- and high-risk groups. (E) ROC curve for 1-, 3-, and 5-year overall survival.
Independent prognostic factors of the final model
The risk scores were combined with the clinical parameters to perform univariate and multivariate Cox regression analyses to elucidate the independent predictive potential of this risk model. Risk score and stage were indicated to be independent predictors (Figure 5A, 5B). A univariate Cox regression analysis (p < 0.001) of risk score [hazard ratio (HR) = 2.488 and 95% confidence interval (CI) = 1.681–3.682] and stage [HR = 2.463 and 95% CI = 1.693–3.583] as well as a multivariate Cox regression analysis (p < 0.001) of risk score [HR = 2.159 and 95% CI = 1.439–3.240] and stage [HR = 2.235 and 95% CI = 1.526–3.275] were performed. Furthermore, a nomogram was established for risk measurement and clinical variables (stage, sex, age) to predict HCC patient outcomes (Figure 5C). Based on the calibration curve, the nomogram’s predictive performance was good (Figure 5D).
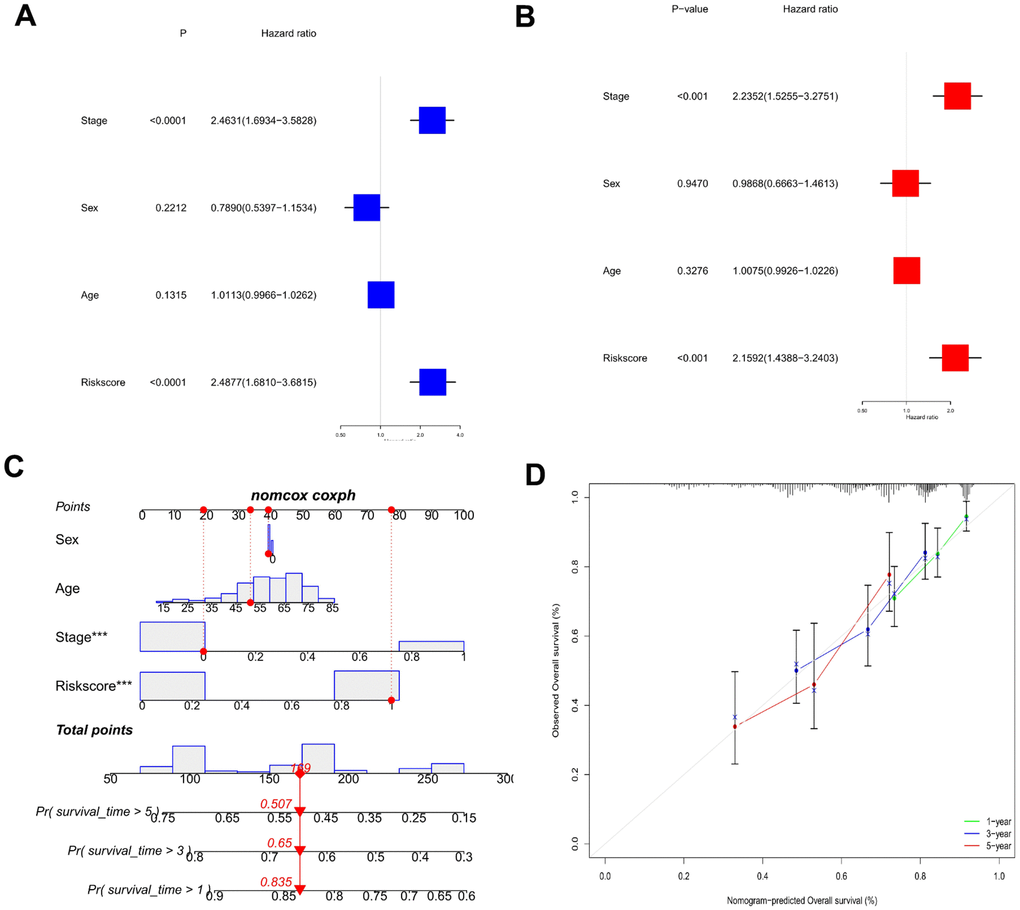
Figure 5. Establishment of a predictive nomogram. (A) Univariate Cox regression analysis of risk scores and clinical characteristics in HCC samples. (B) Multivariate Cox regression analysis of risk scores and clinical characteristics in HCC samples. (C) Nomogram used to predict the survival of the HCC patients. (D) Calibration curve for 1-, 3-, and 5-year overall survival.
Association of miRNA candidates with HCC patients’ survival
Survival analysis was performed to evaluate the prognostic value of the candidate miRNAs for HCC. Patients with high hsa-miR-195-5p and hsa-miR-152-5p expression had a notably higher survival rate than patients with reduced expression, while those with high hsa-miR-9-5p expression showed a markedly lower survival rate (Figure 6).
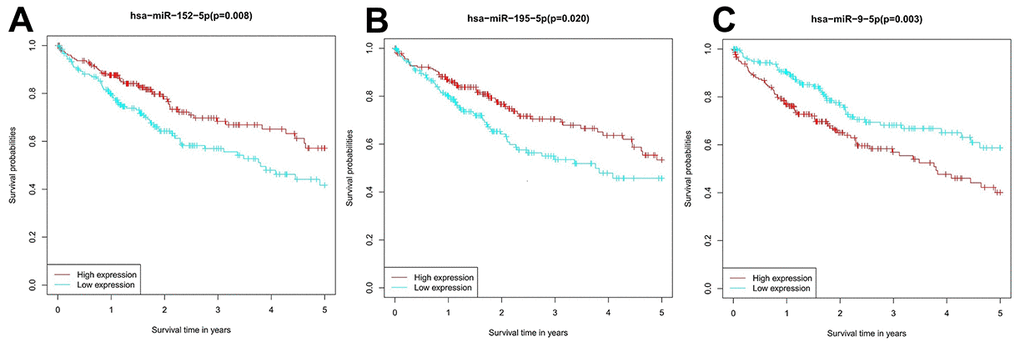
Figure 6. Survival analysis of the prognostic value of the miRNA candidates in HCC patients. (A) hsa-miR-195-5p, (B) hsa-miR-152-5p, and (C) hsa-miR-9-5p.
Enrichment analyses
A differential expression analysis identified 3,247 differentially expressed genes (DEGs) after comparing the LR and HR cohorts and 6,654 DEGs after comparing high immune score (HIS) and low immune score (LIS) cohorts. As the Venn diagram depicts, 2,378 overlapping DEGs were identified and considered co-differentially expressed genes (coDEGs) (Figure 7A). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses uncovered the possible functions and molecular mechanisms of these coDEGs. The KEGG analysis revealed the top three pathways, namely the cytokine–cytokine receptor interaction, PI3K-Akt signaling pathway, and neuroactive ligand–receptor interaction (Figure 7B). The GO analysis indicated that the biological processes (BP) of the coDEGs were primarily enriched in leukocyte-regulated immunity, ossification, and extracellular matrix organization. The cellular components (CC) include an extracellular matrix containing collagen, the plasma membrane’s external side, and the neuronal cell body. The molecular functions (MF) include receptor ligand, signaling receptor activator, and channel activity (Figure 7C).
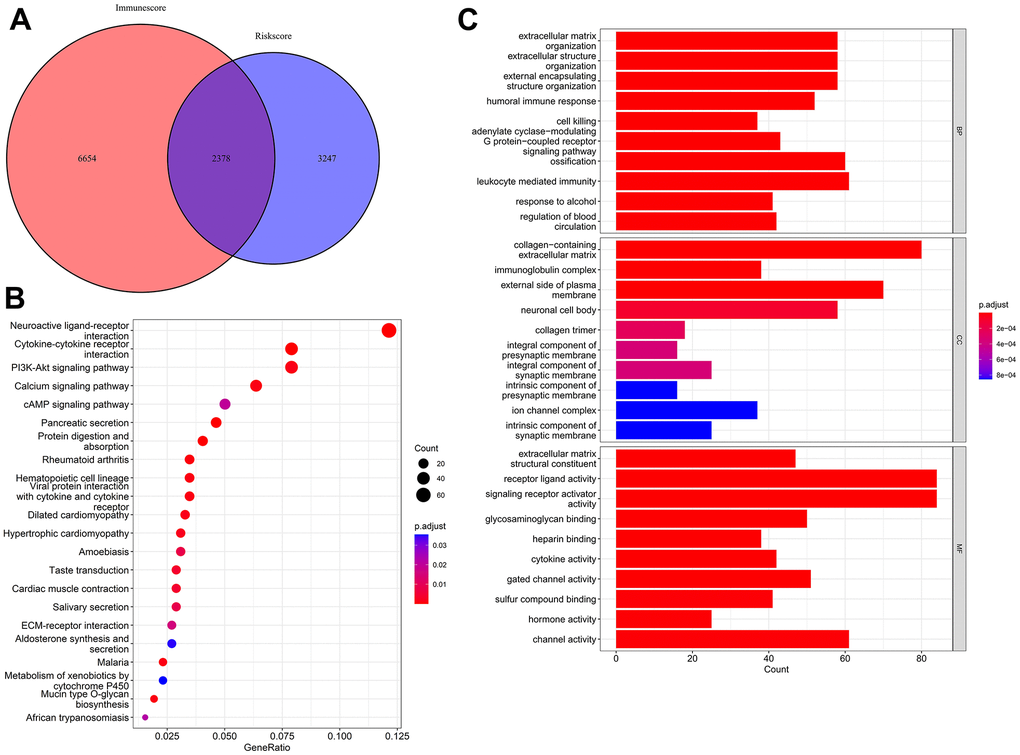
Figure 7. Identification and functional enrichment analysis of coDEGs. (A) A total of 2,378 coDEGs were obtained by finding the intersections. (B) KEGG enrichment analysis shown in dot plots. (C) GO enrichment analysis shown in bar plots.
Immune infiltration in the tumor microenvironment
A single sample gene set enrichment analysis (ssGSEA) revealed different degrees of 28 immune cell infiltration (ICI) and 13 immune functions in the HCC samples. In Figure 8A, a heatmap of immune infiltration based on the ssGSEA scores demonstrates the prominent expression of major histocompatibility complex class 1 (MHC class I), T helper cells, human leukocyte antigen, a type I interferon response, and parainflammation in the tumor immune microenvironment of HCC patients.
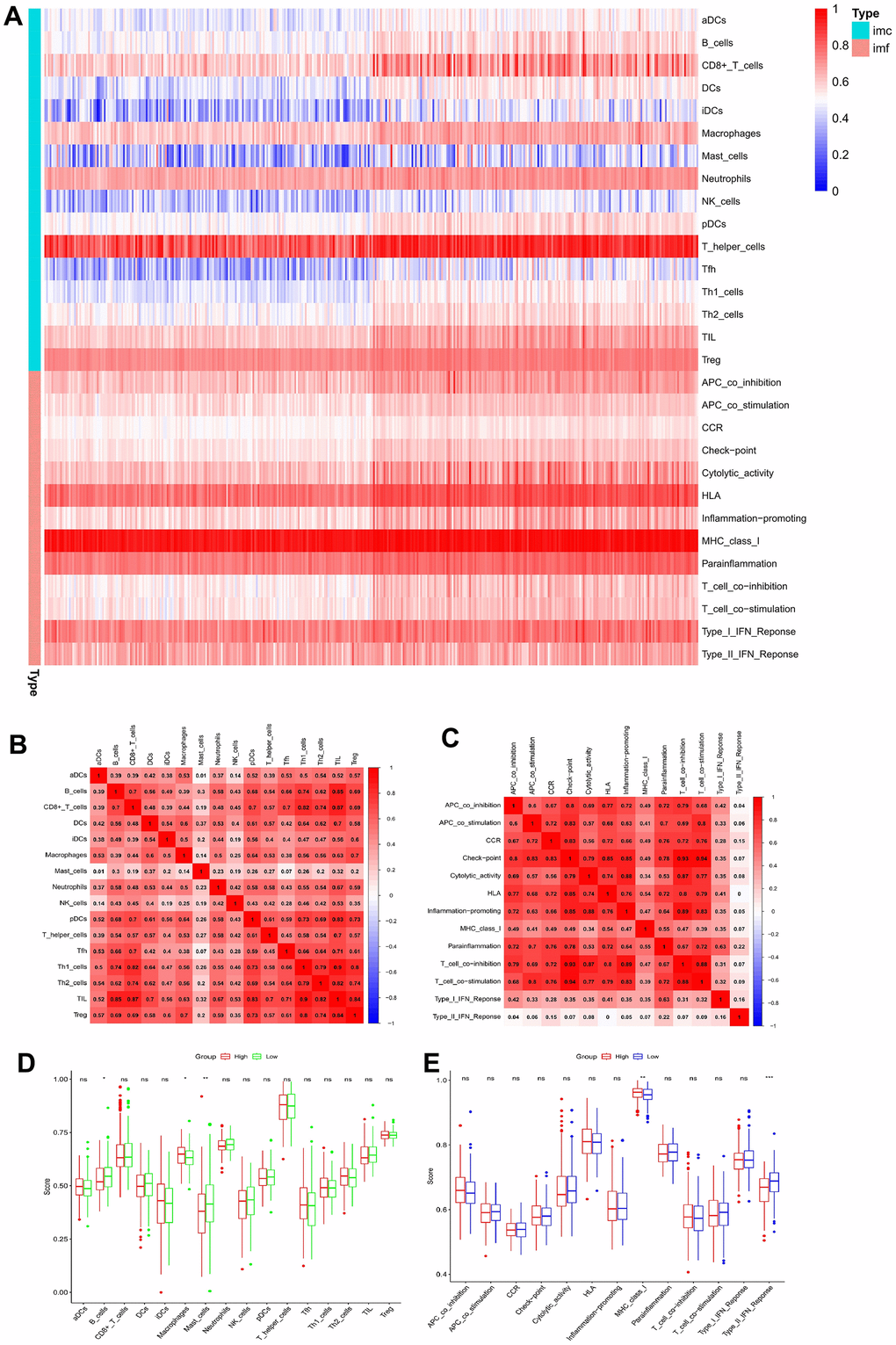
Figure 8. Correlation analysis between ssGSEA scores and immune cells or immune functions. (A) Heatmap of immune infiltration based on ssGSEA. Immune cells and immune functions are represented by imc and imf, respectively. (B) Correlation analysis of various immune cells. (C) Correlation analysis of various immune functions. (D) Box plots for comparing the ssGSEA scores of various immune cells in the high-risk and low-risk groups. (E) Box plots for comparing the ssGSEA scores of various immune functions in the high-risk and low-risk groups (*p < 0.05, **p < 0.01, and ***p < 0.001; ns: no significance).
Spearman correlation was used to assess the associations between various immune cells and immune functions in the immune microenvironment of HCC cases. The closer the correlation coefficient is to 1, the higher the correlation of immune cells or immunity functions. For ICI, Th1 cells and tumor-infiltrating lymphocytes showed the highest correlation, with an r-value of 0.90 (Figure 8B). Among the various immunity functions, the correlation between immune checkpoint and T-cell co-stimulation was the highest (r = 0.94), followed by that between immune checkpoint and T-cell co-inhibition (r = 0.93) (Figure 8C).
Furthermore, the ssGSEA scores of immune cells and functions in the HCC cohort were compared, indicating that the proportions of B and mast cells were substantially reduced and that those of macrophages were higher in the HR cohort than in the LR cohort (Figure 8D). In the immune functions, only type II interferon indicated a greater response in an HR cohort (Figure 8E). Finally, the association between the coDEGs’ expression and 16 different immune cell subtypes or 13 immune functions revealed that most coDEG expressions were positively linked with the degree of ICI and immune functions (Figure 9).

Figure 9. Correlation analysis of coDEG expression with immune cell infiltration and functions.
Discussion
Currently, various miRNA signatures can accurately predict disease prognoses, as when a signature comprising 11 miRNAs was used to forecast an endometrial cancer prognosis [23]. A necroptotic miRNA-based signature can predict colon cancer patients’ overall survival rate [24]. Furthermore, m7G-miRNA-based signatures have been reported in tumor research [25]. For instance, Hong et al. constructed a risk model including seven m7G-miRNAs in kidney renal clear cell carcinoma that could be used to predict patient prognosis and personalize immunotherapy [26]. Xu et al. developed an m7G-miRNA signature to predict the overall survival rate and immune landscape of triple-negative breast cancer [27]. However, the systematic analysis of m7G-miRNA signatures to predict HCC survival has not been undertaken. Therefore, this investigation established an m7G-miRNA-based signature to predict HCC sufferers’ survival. Additionally, the variations in the risk models in terms of ICI and immune function were determined.
The TargetScan database was used to search for m7G-miRNAs, and seven miRNAs were identified for use in constructing a novel prognostic signature for HCC: hsa-miR-195-5p, hsa-miR-9-5p, hsa-miR-504-3p, has-miR-892a, hsa-miR-6764-5p, hsa-miR-4652-3p, and hsa-miR-152-5p. The literature suggests that these miRNAs are notably linked with cancer pathogenesis. miR-195–5p expression is frequently downregulated in numerous tumors, including osteosarcoma [28], prostate cancer [29], and HCC [30]. miR-195–5p acts as a tumor suppressor by targeting various oncogenes, such as Fos-like antigen-1, basic fibroblast growth factor, programmed death-ligand 1, axin2, and myc binding protein [31]. Accordingly, miR-195–5p reportedly involves various tumorigenesis and developmental processes, such as migration, apoptosis, invasion, proliferation, and chemoresistance [32–34]. Therefore, investigating miR-195–5p might reveal diagnostic indices and therapeutic cancer targets [35]. miR-9-5p was aberrant expressed in various tumor tissues and developed different functions by targeting different mRNAs. For instance, miR-9-5p functions as an oncomiR, acting against curcumin and paclitaxels’ cytotoxic synergistic effects by inhibiting BRCA1 in ovarian cancer cases [36]. However, it has also acted as a tumor suppressor by inhibiting tumorigenesis and chemosensitivity by targeting neuropilin-1 in gastric cancer cases [37]. It was reported that miR-504-3p suppresses tumors’ malignant phenotypes by reducing interferon-induced transmembrane protein 1 or methylenetetrahydrofolate dehydrogenase 2 expression [38, 39]. In gastric cancer cases, it was found that miR-892a overexpression predicts a substandard prognosis and clinical pathological characteristic [40]. lncRNA CAR10 regulates the expression of gap junction protein beta 2 via miR-892a, thus promoting the migration and invasion capacity of non-small-cell lung cancer cells [41]. Dysregulation of miR-6764-5p was identified in pituitary adenoma cases [42]. Spen family transcriptional repressors activate PI3K/AKT signaling to modulate the c-JUN/miR-4652-3p/HIPK2 axis, thereby activating epithelial-mesenchymal transition signaling and promoting nasopharyngeal carcinoma metastasis [43]. In malignant meningioma cases, miR-4652-3p has been associated with LINC00702-regulated Wnt/β-catenin signaling and attenuated the cell proliferation and migration caused by this pathway [44]. miR-152-5p inhibits malignant progression and tumorigenesis by targeting FBXL7, potentiating gliomas’ temozolomide sensitivity [45]. Furthermore, miR-152-5p has been reported to suppress tumors from gastric cancer by targeting PIK3CA [46]. miR-152-5p overexpression enhances the expression of apoptosis-related factors and forkhead transcription factor O by activating the JNK pathway and inhibiting malignant liver cancer phenotypes [47].
In this study, risk-score-based LR and HR cohorts were used to establish a signature that could predict HCC patients’ OS with high accuracy, as evidenced by survival analysis. Furthermore, univariate and multivariate Cox regression analyses revealed that risk score was markedly linked with HCC OS. According to the PCA, the model differentiated the LR and HR cohorts efficiently. This suggests the reliability of the seven m7G-miRNAs mentioned above in predicting the OS of HCC patients.
HCC is induced by a chronic inflammatory state at its initiation and development. To understand the importance of the infiltration of various immune cells and alterations in immune function linked with HCC in the LR and HR cohorts, ssGSEA was applied to evaluate the differential immune scores. It has been suggested that tumor-infiltrating macrophages of the alternatively activated macrophage (M2) phenotype have pro-inflammatory and tumor-promoting effects by suppressing anti-tumor immune response [48]. Increased infiltration of M2 yielded a poorer prognosis [49]. Consistent with this study, an association between high macrophage infiltration and substandard HCC progression was observed. In addition, increased type II IFN response was observed in the LR cohort. These are essential components of immune response against infections and cancers, stimulating pro-inflammatory responses crucial for immune activation and inducing immune-repressing feedback circuits to inhibit cancer growth [50]. This indicates that immunotherapy is more effective in HR than in LR cohorts. Thus, an m7G-miRNA-based signature may provide potential clues for developing HCC immunotherapy. Furthermore, 2,378 coDEGs were identified for GO and KEGG enrichment analyses based on the signatures’ risk and immune scores. The coDEGs were primarily enriched in humoral immune response, cell killing, leukocyte-mediated immunity, and cytokine–cytokine receptor interaction. The association of coDEG expression with ICI and immune function revealed a correlation that was more positive than negative. All these results suggest that m7G-miRNA may affect HCC prognosis via immune mechanisms.
Limitations
The limitations of this investigation were as follows: 1) The conclusions drawn are based on integrative bioinformatics and experimental verification. Further study of the HCC cohort is required to validate these conclusions. 2) Functional experiments illustrating how these m7G-miRNAs affect HCC are missing. 3) The m7G-miRNA signature’s accuracy in HCC prognosis and immune regulation is crucial for clinical studies, and clinical findings are required to further validate this prognostic model.
Conclusions
An m7G-miRNA prognostic model with a substantial capacity to predict OS in HCC was established. Risk score’s influence on ICI and immune function was elucidated. Moreover, a functional analysis of coDEGs and their relationships with ICI and immune function was provided. As far as we know, this is the first m7G-miRNA-based prognostic model for HCC and could, therefore, help us discover novel therapeutic HCC targets.
Materials and Methods
Data collection
The clinical and RNA-seq data of 375 HCC and 50 healthy tissues were acquired from The Cancer Genome Atlas (TCGA) database in May 2023. The subsequent analyses were performed using the m7G methylation complexes METTL1 and WDR4.
Predictive nomogram construction and principal component analysis
The nomogram and calibration curves predicted 1-, 3-, and 5-year survival rates. PCA was carried out using the “scatterplot3D” R package to elucidate the number of individuals with HR and LR scores.
Survival analysis of prognosis-relevant miRNAs
A survival analysis was conducted to explore the correlation between the expression of prognosis-relevant miRNAs and prognoses in HCC patients. HCC patients were categorized as high- and low-expression patients according to the median expression level of each miRNA. Patients whose miRNA expression level was above the median expression level of this gene were defined as high-expression patients; otherwise, they were low-expression patients. A Kaplan–Meier survival curve was drawn using the “survival” package in R, and the OS figures were compared using a log rank test.
Screening of differentially expressed genes
The HCC cohort’s immune scores were obtained via the “estimation of stromal and immune cells in malignant tumor tissues using expression data” method. On the basis of the immune score median, HCC individuals were categorized into HIS and LIS cohorts. The DEGs of different subgroups (HR vs. LR score and HIS vs. LIS) were screened using the “edgeR” package, and the intersecting points were acquired as coDEGs. The cutoff values were based on a |log (FC) | ≥ 0.5 and FDR < 0.05.
Enrichment analyses
To elucidate the primary biological processes and coDEG pathways, GO and KEGG enrichment assays were conducted using the “ClusterProfiler” package.
Immune infiltrate analysis
With the help of the “GSVA” package, ssGSEA scores were used to quantify ICI and immune function in HCC. Furthermore, the correlation between different ICI statuses and immune functions was assessed using Spearman analysis. ICI statuses and immune functions in the LR and HR cohorts were compared via the “ggpubr” package. Additionally, the association of coDEGs with ICI statuses and immune functions was evaluated.
Statistical measurements
The data were measured statistically using RStudio (version 4.0.4) and its packages (limma, ggplot2, survminer, timeROC, GSVA, etc.). Cox univariate and multivariate analyses were used to construct a prognosis-prediction model and evaluate the independent prognostic value of the clinical characteristics of OS. Kaplan–Meier analysis was used to compare OS in the various subgroups. The prognostic ability of the predictive models was evaluated based on the area under the ROC curve. Student’s t-test was used to compare the two groups’ differences. Spearman’s correlation was used to calculate the association between different ICI statuses and immune functions. A two-tailed P < 0.05 was considered to indicate statistical significance.
Data availability statement
The datasets presented in this study can be found in online repositories.
Supplementary Materials
Author Contributions
Xiaoxia Lao and Ping Du conceived and designed this study. Qiaomei Deng, Jilu Zhou and Zhihu Huang collected data. Yingpei Zhou, Qianqian Wei and Liping Ma analyzed the interpreted data, Liping Ma and Qingwei Ma drafted and revised the article. All authors read and approve the final manuscript.
Acknowledgments
We gratefully acknowledge Scribendi for proofreading this manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This research was supported by the Research Team Incubation Project of Minzu Hospital of Guangxi Zhuang Autonomous Region [grant number MKFY202104].
References
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71:209–49. https://doi.org/10.3322/caac.21660 [PubMed]
- 2. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021; 7:6. https://doi.org/10.1038/s41572-020-00240-3 [PubMed]
- 3. Furuichi Y. Discovery of m(7)G-cap in eukaryotic mRNAs. Proc Jpn Acad Ser B Phys Biol Sci. 2015; 91:394–409. https://doi.org/10.2183/pjab.91.394 [PubMed]
- 4. Zhang LS, Liu C, Ma H, Dai Q, Sun HL, Luo G, Zhang Z, Zhang L, Hu L, Dong X, He C. Transcriptome-wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian mRNA. Mol Cell. 2019; 74:1304–16.e8. https://doi.org/10.1016/j.molcel.2019.03.036 [PubMed]
- 5. Lewis JD, Izaurralde E. The role of the cap structure in RNA processing and nuclear export. Eur J Biochem. 1997; 247:461–9. https://doi.org/10.1111/j.1432-1033.1997.00461.x [PubMed]
- 6. Konarska MM, Padgett RA, Sharp PA. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell. 1984; 38:731–6. https://doi.org/10.1016/0092-8674(84)90268-x [PubMed]
- 7. Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA, McCleery JA, Yates JR 3rd, Hartzog GA. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol. 2003; 23:1368–78. https://doi.org/10.1128/MCB.23.4.1368-1378.2003 [PubMed]
- 8. Malbec L, Zhang T, Chen YS, Zhang Y, Sun BF, Shi BY, Zhao YL, Yang Y, Yang YG. Dynamic methylome of internal mRNA N7-methylguanosine and its regulatory role in translation. Cell Res. 2019; 29:927–41. https://doi.org/10.1038/s41422-019-0230-z [PubMed]
- 9. Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW, Gregory RI. Mettl1/Wdr4-Mediated m7G tRNA Methylome Is Required for Normal mRNA Translation and Embryonic Stem Cell Self-Renewal and Differentiation. Mol Cell. 2018; 71:244–55.e5. https://doi.org/10.1016/j.molcel.2018.06.001 [PubMed]
- 10. Ma J, Han H, Huang Y, Yang C, Zheng S, Cai T, Bi J, Huang X, Liu R, Huang L, Luo Y, Li W, Lin S. METTL1/WDR4-mediated m7G tRNA modifications and m7G codon usage promote mRNA translation and lung cancer progression. Mol Ther. 2021; 29:3422–35. https://doi.org/10.1016/j.ymthe.2021.08.005 [PubMed]
- 11. Shaheen R, Abdel-Salam GM, Guy MP, Alomar R, Abdel-Hamid MS, Afifi HH, Ismail SI, Emam BA, Phizicky EM, Alkuraya FS. Mutation in WDR4 impairs tRNA m(7)G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 2015; 16:210. https://doi.org/10.1186/s13059-015-0779-x [PubMed]
- 12. Orellana EA, Liu Q, Yankova E, Pirouz M, De Braekeleer E, Zhang W, Lim J, Aspris D, Sendinc E, Garyfallos DA, Gu M, Ali R, Gutierrez A, et al. METTL1-mediated m7G modification of Arg-TCT tRNA drives oncogenic transformation. Mol Cell. 2021; 81:3323–38.e14. https://doi.org/10.1016/j.molcel.2021.06.031 [PubMed]
- 13. Chen Z, Zhu W, Zhu S, Sun K, Liao J, Liu H, Dai Z, Han H, Ren X, Yang Q, Zheng S, Peng B, Peng S, et al. METTL1 promotes hepatocarcinogenesis via m7G tRNA modification-dependent translation control. Clin Transl Med. 2021; 11:e661. https://doi.org/10.1002/ctm2.661 [PubMed]
- 14. Elrebehy MA, Al-Saeed S, Gamal S, El-Sayed A, Ahmed AA, Waheed O, Ismail A, El-Mahdy HA, Sallam AM, Doghish AS. miRNAs as cornerstones in colorectal cancer pathogenesis and resistance to therapy: A spotlight on signaling pathways interplay - A review. Int J Biol Macromol. 2022; 214:583–600. https://doi.org/10.1016/j.ijbiomac.2022.06.134 [PubMed]
- 15. Hashemi M, Arani HZ, Orouei S, Rostamnejad E, Ghorbani A, Khaledabadi M, Kakavand A, Tavakolpournegari A, Saebfar H, Heidari H, Salimimoghadam S, Taheriazam A, Entezari M, Khan H. Crosstalk of miRNAs with signaling networks in bladder cancer progression: Therapeutic, diagnostic and prognostic functions. Pharmacol Res. 2022; 185:106475. https://doi.org/10.1016/j.phrs.2022.106475 [PubMed]
- 16. Jung M, Schaefer A, Steiner I, Kempkensteffen C, Stephan C, Erbersdobler A, Jung K. Robust microRNA stability in degraded RNA preparations from human tissue and cell samples. Clin Chem. 2010; 56:998–1006. https://doi.org/10.1373/clinchem.2009.141580 [PubMed]
- 17. Cheng Z, Gong L, Cai Q. LncRNA00978 contributes to growth and metastasis of hepatocellular carcinoma cells via mediating microRNA-125b-5p/SOX12 pathway. Bioengineered. 2022; 13:11228–39. https://doi.org/10.1080/21655979.2022.2063648 [PubMed]
- 18. de la Cruz-Ojeda P, Schmid T, Boix L, Moreno M, Sapena V, Praena-Fernández JM, Castell FJ, Falcón-Pérez JM, Reig M, Brüne B, Gómez-Bravo MA, Giráldez Á, Bruix J, et al. miR-200c-3p, miR-222-5p, and miR-512-3p Constitute a Biomarker Signature of Sorafenib Effectiveness in Advanced Hepatocellular Carcinoma. Cells. 2022; 11:2673. https://doi.org/10.3390/cells11172673 [PubMed]
- 19. Yousuf T, Dar SB, Bangri SA, Choh NA, Rasool Z, Shah A, Rather RA, Rah B, Bhat GR, Ali S, Afroze D. Diagnostic implication of a circulating serum-based three-microRNA signature in hepatocellular carcinoma. Front Genet. 2022; 13:929787. https://doi.org/10.3389/fgene.2022.929787 [PubMed]
- 20. Zhang X, Zhang D, Bu X, Zhang X, Cui L. Identification of a novel miRNA-based recurrence and prognosis prediction biomarker for hepatocellular carcinoma. BMC Bioinformatics. 2022; 23:479. https://doi.org/10.1186/s12859-022-05040-y [PubMed]
- 21. Pandolfini L, Barbieri I, Bannister AJ, Hendrick A, Andrews B, Webster N, Murat P, Mach P, Brandi R, Robson SC, Migliori V, Alendar A, d’Onofrio M, et al. METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol Cell. 2019; 74:1278–90.e9. https://doi.org/10.1016/j.molcel.2019.03.040 [PubMed]
- 22. Liu Y, Yang C, Zhao Y, Chi Q, Wang Z, Sun B. Overexpressed methyltransferase-like 1 (METTL1) increased chemosensitivity of colon cancer cells to cisplatin by regulating miR-149-3p/S100A4/p53 axis. Aging (Albany NY). 2019; 11:12328–44. https://doi.org/10.18632/aging.102575 [PubMed]
- 23. Lu J, Liang J, Xu M, Wu Z, Cheng W, Wu J. Identification of an eleven-miRNA signature to predict the prognosis of endometrial cancer. Bioengineered. 2021; 12:4201–16. https://doi.org/10.1080/21655979.2021.1952051 [PubMed]
- 24. Huang Y, Zou Y, Xiong Q, Zhang C, Sayagués JM, Shelat VG, Wang X. Development of a novel necroptosis-associated miRNA risk signature to evaluate the prognosis of colon cancer patients. Ann Transl Med. 2021; 9:1800. https://doi.org/10.21037/atm-21-6576 [PubMed]
- 25. Jiang S, Xiao M, Shi Y, Wang Y, Xu Z, Wang K. Identification of m7G-Related miRNA Signatures Associated with Prognosis, Oxidative Stress, and Immune Landscape in Lung Adenocarcinoma. Biomedicines. 2023; 11:1569. https://doi.org/10.3390/biomedicines11061569 [PubMed]
- 26. Hong P, Du H, Tong M, Cao Q, Hu D, Ma J, Jin Y, Li Z, Huang W, Tong G. A Novel M7G-Related MicroRNAs Risk Signature Predicts the Prognosis and Tumor Microenvironment of Kidney Renal Clear Cell Carcinoma. Front Genet. 2022; 13:922358. https://doi.org/10.3389/fgene.2022.922358 [PubMed]
- 27. Xu J, Cen X, Yao Y, Zhao S, Li W, Zhang W, Qiu M. Identification of Six N7-Methylguanosine-Related miRNA Signatures to Predict the Overall Survival and Immune Landscape of Triple-Negative Breast Cancer through In Silico Analysis. J Oncol. 2022; 2022:2735251. https://doi.org/10.1155/2022/2735251 [PubMed]
- 28. Yang B, Li L, Tong G, Zeng Z, Tan J, Su Z, Liu Z, Lin J, Gao W, Chen J, Zeng S, Wu G, Li L, et al. Circular RNA circ_001422 promotes the progression and metastasis of osteosarcoma via the miR-195-5p/FGF2/PI3K/Akt axis. J Exp Clin Cancer Res. 2021; 40:235. https://doi.org/10.1186/s13046-021-02027-0 [PubMed]
- 29. Meng XF, Liu AD, Li SL. SNHG1 promotes proliferation, invasion and EMT of prostate cancer cells through miR-195-5p. Eur Rev Med Pharmacol Sci. 2020; 24:9880–8. https://doi.org/10.26355/eurrev_202010_23198 [PubMed]
- 30. Zhou C, Zhu S, Li H. miR-195-5p Targets CDK1 To Regulate New DNA Synthesis and Inhibit the Proliferation of Hepatocellular Carcinoma Cells. Appl Biochem Biotechnol. 2023; 195:3477–90. https://doi.org/10.1007/s12010-022-04279-8 [PubMed]
- 31. Xu Q, Xu JL, Chen WQ, Xu WX, Song YX, Tang WJ, Xu D, Jiang MP, Tang J. Roles and mechanisms of miR-195-5p in human solid cancers. Biomed Pharmacother. 2022; 150:112885. https://doi.org/10.1016/j.biopha.2022.112885 [PubMed]
- 32. Bing Z, Han J, Zheng Z, Liang N. FOXO3-induced oncogenic lncRNA CASC9 enhances gefitinib resistance of non-small-cell lung cancer through feedback loop. Life Sci. 2021; 287:120012. https://doi.org/10.1016/j.lfs.2021.120012 [PubMed]
- 33. Yang R, Xing L, Zheng X, Sun Y, Wang X, Chen J. The circRNA circAGFG1 acts as a sponge of miR-195-5p to promote triple-negative breast cancer progression through regulating CCNE1 expression. Mol Cancer. 2019; 18:4. https://doi.org/10.1186/s12943-018-0933-7 [PubMed]
- 34. Zhong C, Yu Q, Peng Y, Zhou S, Liu Z, Deng Y, Guo L, Zhao S, Chen G. Novel LncRNA OXCT1-AS1 indicates poor prognosis and contributes to tumorigenesis by regulating miR-195/CDC25A axis in glioblastoma. J Exp Clin Cancer Res. 2021; 40:123. https://doi.org/10.1186/s13046-021-01928-4 [PubMed]
- 35. Bayat A, Raad M, Sharafshah A, Ahmadvand M, Aminian H. Identification of miR-195-5p as a novel prognostic biomarker for colorectal cancer. Mol Biol Rep. 2022; 49:6453–7. https://doi.org/10.1007/s11033-022-07462-6 [PubMed]
- 36. Liu Y, Shen Z, Zhu T, Lu W, Fu Y. Curcumin enhances the anti-cancer efficacy of paclitaxel in ovarian cancer by regulating the miR-9-5p/BRCA1 axis. Front Pharmacol. 2023; 13:1014933. https://doi.org/10.3389/fphar.2022.1014933 [PubMed]
- 37. Hang C, Yan HS, Gong C, Gao H, Mao QH, Zhu JX. MicroRNA-9 inhibits gastric cancer cell proliferation and migration by targeting neuropilin-1. Exp Ther Med. 2019; 18:2524–30. https://doi.org/10.3892/etm.2019.7841 [PubMed]
- 38. Li SM, Zhao YQ, Hao YL, Liang YY. Upregulation of miR-504-3p is associated with favorable prognosis of acute myeloid leukemia and may serve as a tumor suppressor by targeting MTHFD2. Eur Rev Med Pharmacol Sci. 2019; 23:1203–13. https://doi.org/10.26355/eurrev_201902_17013 [PubMed]
- 39. Liu Z, Zhang W, Zhang B, Chen S, Ling C. MiR-504-3p Has Tumor-Suppressing Activity and Decreases IFITM1 Expression in Non-Small Cell Lung Cancer Cells. Genet Test Mol Biomarkers. 2022; 26:351–9. https://doi.org/10.1089/gtmb.2021.0158 [PubMed]
- 40. Lv J, Zhang H, Gao Z, Zhang X, Huang X, Jia X. Prognostic value of miR-892a in gastric cancer and its regulatory effect on tumor progression. Cancer Biomark. 2020; 28:247–54. https://doi.org/10.3233/CBM-191323 [PubMed]
- 41. Li S, Liu Y, Qiu G, Luo Y, Luan L, Xu T, Wang Y, Xia S. Long Non-Coding RNA CAR10 Facilitates Non-Small Cell Lung Cancer Cell Migration and Invasion by Modulating the miR-892a/GJB2 Pathway. Cancer Manag Res. 2021; 13:1967–79. https://doi.org/10.2147/CMAR.S287386 [PubMed]
- 42. Németh K, Darvasi O, Likó I, Szücs N, Czirják S, Reiniger L, Szabó B, Krokker L, Pállinger É, Igaz P, Patócs A, Butz H. Comprehensive Analysis of Circulating miRNAs in the Plasma of Patients With Pituitary Adenomas. J Clin Endocrinol Metab. 2019; 104:4151–68. https://doi.org/10.1210/jc.2018-02479 [PubMed]
- 43. Li Y, Lv Y, Cheng C, Huang Y, Yang L, He J, Tao X, Hu Y, Ma Y, Su Y, Wu L, Yu G, Jiang Q, et al. SPEN induces miR-4652-3p to target HIPK2 in nasopharyngeal carcinoma. Cell Death Dis. 2020; 11:509. https://doi.org/10.1038/s41419-020-2699-2 [PubMed]
- 44. Li T, Ren J, Ma J, Wu J, Zhang R, Yuan H, Han X. LINC00702/miR-4652-3p/ZEB1 axis promotes the progression of malignant meningioma through activating Wnt/β-catenin pathway. Biomed Pharmacother. 2019; 113:108718. https://doi.org/10.1016/j.biopha.2019.108718 [PubMed]
- 45. Kong S, Fang Y, Wang B, Cao Y, He R, Zhao Z. miR-152-5p suppresses glioma progression and tumorigenesis and potentiates temozolomide sensitivity by targeting FBXL7. J Cell Mol Med. 2020; 24:4569–79. https://doi.org/10.1111/jcmm.15114 [PubMed]
- 46. You W, Zhang X, Ji M, Yu Y, Chen C, Xiong Y, Liu Y, Sun Y, Tan C, Zhang H, Li J, Chen W, Li R. MiR-152-5p as a microRNA passenger strand special functions in human gastric cancer cells. Int J Biol Sci. 2018; 14:644–53. https://doi.org/10.7150/ijbs.25272 [PubMed]
- 47. Chang DL, Wei W, Yu ZP, Qin CK. miR-152-5p inhibits proliferation and induces apoptosis of liver cancer cells by up-regulating FOXO expression. Pharmazie. 2017; 72:338–43. https://doi.org/10.1691/ph.2017.7406 [PubMed]
- 48. Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao W, Xu Z, Yang Y, Weng Q, Zhao Z, Chen M, Ji J. Targeted xCT-mediated Ferroptosis and Protumoral Polarization of Macrophages Is Effective against HCC and Enhances the Efficacy of the Anti-PD-1/L1 Response. Adv Sci (Weinh). 2023; 10:e2203973. https://doi.org/10.1002/advs.202203973 [PubMed]
- 49. Mak TK, Li X, Huang H, Wu K, Huang Z, He Y, Zhang C. The cancer-associated fibroblast-related signature predicts prognosis and indicates immune microenvironment infiltration in gastric cancer. Front Immunol. 2022; 13:951214. https://doi.org/10.3389/fimmu.2022.951214 [PubMed]
- 50. Lukhele S, Rabbo DA, Guo M, Shen J, Elsaesser HJ, Quevedo R, Carew M, Gadalla R, Snell LM, Mahesh L, Ciudad MT, Snow BE, You-Ten A, et al. The transcription factor IRF2 drives interferon-mediated CD8+ T cell exhaustion to restrict anti-tumor immunity. Immunity. 2022; 55:2369–85.e10. https://doi.org/10.1016/j.immuni.2022.10.020 [PubMed]