



Introduction
Coronary heart disease (CHD) remains the leading cause of death worldwide [1] and is characterized by the formation of coronary atherosclerotic plaques, causing coronary artery stenosis and finally leading to episodic or persistent angina pectoris (AP). Plaques are mainly composed of lipids, calcium, and inflammatory cells. When plaque rupture results in thrombosis, it can cause myocardial infarction (MI) and even death in severe cases. Although great progress has been made in the treatment of CHD, including reperfusion therapy such as percutaneous coronary intervention (PCI) and secondary prevention treatment of anti-platelet, lipid-lowering, management of hypertension, diabetes and other risk factors, its pathogenic factors and pathophysiology are still not completely clear, and the prevention and treatment situation is still very serious. Therefore, it is of great practical significance to search for effective therapeutic targets for CHD and improve cardioprotection strategies.
Placental growth factor (PlGF) is a member of the VEGF family, which is mainly expressed in placenta, heart and lung tissues [2]. PlGF is a polypeptide hormone with various physiological effects and is involved in the immune response, vascular homeostasis, angiogenesis and other physiological activities, and it plays a key role in cellular metabolic activities [3]. Studies have shown that PlGF can promote angiogenesis, growth and survival of endothelial cells [4, 5]. It can also promote vascular inflammation by activating the expression of adhesion molecules and chemokines in endothelial cells, which plays key roles in the occurrence and development of cardiovascular diseases, including CHD [6].
Clinical studies have shown that PlGF is elevated quickly in myocardial infarcted tissue of patients with MI, and the serum PlGF level is positively correlated with the improvement of left ventricular function [7]. Signs of vascular ageing occurred in women with low PlGF levels in the second trimester of pregnancy in the next 6 to 9 years, supporting that PlGF is required for maintaining normal cardiovascular function [8]. However, it has also been found that PlGF, released by heart tissue during ischaemia [9], is associated with mortality after acute coronary events and has potential value in predicting mortality after acute coronary syndrome (ACS) [10, 11]. In addition, a high baseline plasma PlGF level was associated with an increased risk of cardiovascular death, MI, and stroke, but these associations disappeared or attenuated after adjusting for known cardiovascular risk factors [12]. PlGF may be a long-term biomarker for the risk of coronary heart disease. There is a moderate correlation between plasma PlGF levels and the risk of coronary heart disease in women, and PlGF levels can predict myocardial infarction events several years in advance [1].
The causal association between PlGF level and the risk of CHD is not clear, as traditional observational studies are prone to bias due to residual confounding effects and reverse causality. Therefore, it is necessary to conduct a Mendelian randomization (MR) study to evaluate the causal association between PlGF level and the risk of CHD. The basic principle of MR design is that genetic variation is fixed at conception and randomly assigned to individuals. MR must meet the following three basic principles: (1) the instrumental variables must be associated with the exposure factors to be studied; (2) Instrumental variables must be independent of confounding factors; (3) Instrumental variables can only be related to the outcome by influencing the exposure factors to be studied. Therefore, MR design can be conceptualized as a natural experiment and overcome the limitations of traditional observational studies (Figure 1) [13, 14]. Therefore, this study aimed to investigate the potential causal association between PlGF levels and CHD using MR analyses.
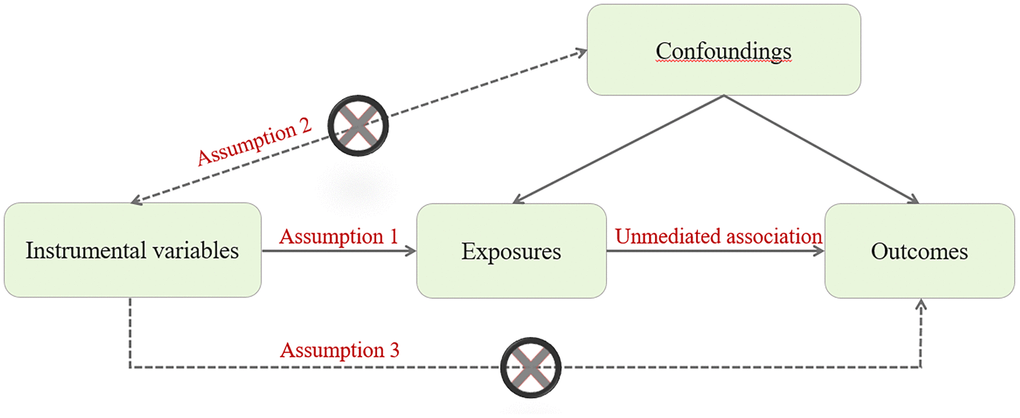
Figure 1. MR assumptions for a two-sample MR study.
Results
MR analyses
There was a suggestive causal association between the genetically predicted PlGF level and the risk of CHD overall. The higher the PlGF level was, the lower the risk of CHD (OR = 0.79, 95% CI: 0.66–0.95, P = 0.011).
Subsequently, we also analysed the CHD subgroup. We observed that there was no statistical association between the genetically predicted PlGF level and the risk of AP, but there was still a negative correlation trend (OR = 0.89, 95% CI: 0.79–1.01, P = 0.067). There was a significant negative association between the genetically predicted PlGF level and the risk of UAP (OR = 0.78, 95% CI: 0.64–0.94, P = 0.008). We found that there was a significant causal association between the genetically predicted PlGF level and the risk of MI. The higher the PlGF level was, the lower the risk of MI (OR = 0.83, 95% CI: 0.72–0.95, P = 0.007). In addition, a suggestive negative association was found between the genetically predicted PlGF level and the adverse events of CHD (OR = 0.89, 95% CI: 0.80–0.99, P = 0.046) (Figure 2, Supplementary Table 1). The scatter plot shows the impact of circulating PlGF levels on the risk of different types of CHD (Supplementary Figure 1).
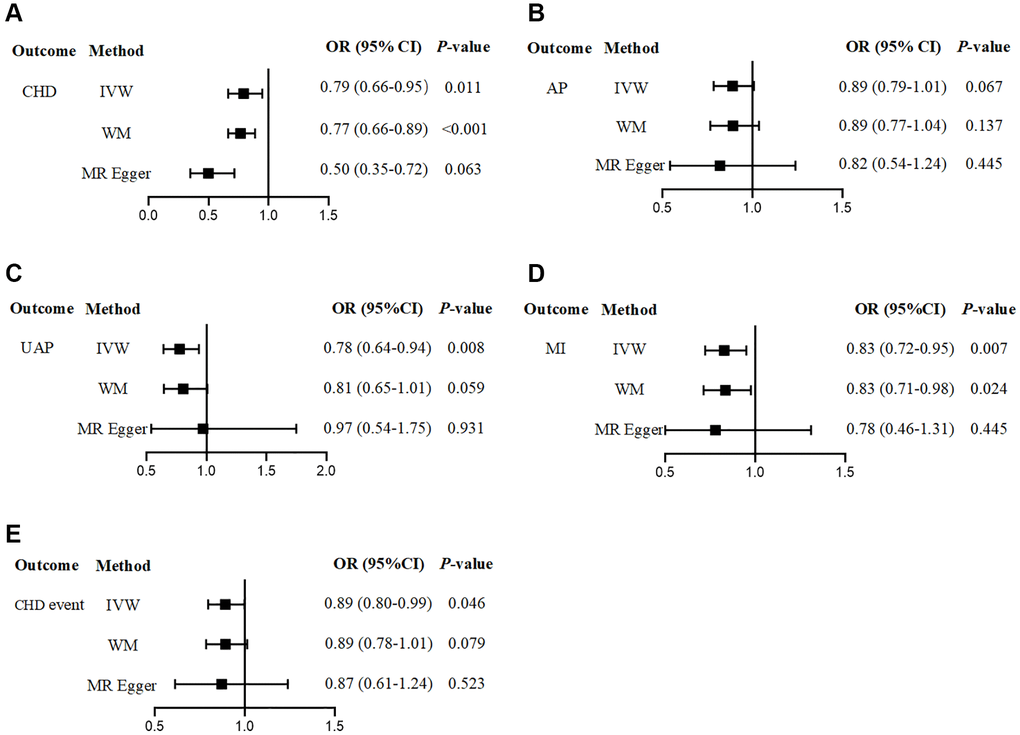
Figure 2. The effect of genetically determined circulating PlGF level on the risk of CHD. (A) CHD; (B) AP; (C) UAP; (D) MI; (E) CHD event. Abbreviations: CHD: coronary artery disease; AP: angina pectoris; UAP: unstable angina pectoris; MI: myocardial infarction.
Sensitivity analysis
As shown in Table 1, in all MR analyses in this study, Cochran’s Q statistics did not find significant heterogeneity, and MR Egger regression analysis did not find evidence of significant pleiotropy. The estimated values obtained after removing a single SNP had no significant change in the leave-one-out test, indicating that no single SNP had a significant effect on the overall estimation (Supplementary Figure 2).
Table 1. Heterogeneity test and horizontal pleiotropy test of PlGF associated SNPs.
| Outcome | Heterogeneity test | Horizontal pleiotropy test | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IVW Cochran’s Q | IVW P | MR-Egger Cochran’s Q | MR-Egger P | Intercept | SE | P for intercept | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CHD | 7.179 | 0.066 | 0.114 | 0.944 | 0.04 | 0.015 | 0.117 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AP | 2.429 | 0.488 | 2.241 | 0.326 | 0.007 | 0.017 | 0.721 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| UAP | 0.693 | 0.875 | 0.07 | 0.966 | −0.019 | 0.024 | 0.513 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MI | 3.275 | 0.351 | 3.18 | 0.204 | 0.005 | 0.021 | 0.829 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CHD event | 1.286 | 0.732 | 1.268 | 0.530 | 0.002 | 0.014 | 0.906 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: PlGF: placental growth factor; IVW: inverse variance weighted; CHD: coronary artery disease; AP: angina pectoris; UAP: unstable angina pectoris; MI: myocardial infarction; SE: standard error. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Discussion
In this study, we investigated the association between the genetically predicted level of PlGF and the risk of CHD. Overall, there is a suggestive, negative causal association between the PlGF level and the risk of CHD. A significant negative causal association was found between the PlGF level and the risk of MI and UAP. A suggestive negative association exists between the level of PlGF and the adverse events of CHD. The PlGF level was not statistically related to the risk of AP, but there was still a trend towards a negative association. These results indicate that PlGF may be a protective factor for cardiovascular disease.
Clinical studies have shown that PlGF may be a long-term biomarker for the risk of coronary heart disease. There is a moderate correlation between plasma PlGF levels and the risk of coronary heart disease in women, and PlGF levels can predict myocardial infarction events several years in advance [15]. The increase of PlGF is a new independent predictor of incidence rate and mortality of long-term cardiovascular disease in patients with Type 1 diabetes nephropathy [16]. Lenderink et al. found that elevated plasma levels of PlGF are associated with adverse cardiac outcomes during long-term follow-up (a median follow-up period of four years) in ACS patients [17]. Bui et al. also reported that higher concentration of PlGF is associated with long-term risk of recurrent cardiovascular events independent of traditional risk factors in ACS patients [18]. In addition, PIGF may also be a short-term prognostic biomarker of coronary heart disease risk. Elevated PlGF concentration has become an important independent biomarker for short-term adverse outcomes in patients with acute chest pain and known or suspected ACS [19].
Oxidative stress and apoptosis play key roles in myocardial ischaemia-reperfusion injury [20, 21]. PlGF is a selective ligand of VEGFR1 (vascular endothelial growth factor receptor 1). In heterozygous VEGFR1 knockout mice, the protective effect of ischaemic preconditioning on the heart was significantly inhibited [20, 22], suggesting that VEGFR1 plays an important role in cardioprotection. Zhang et al. found that pretreatment with PlGF could significantly improve ischaemia/reperfusion injury, reduce the infarct area, improve cardiac function, and reduce the degree of cardiomyocyte apoptosis using a mouse heart ischaemia/reperfusion model. They also confirmed that PlGF can inhibit the production of reactive oxygen species (ROS) in the mitochondria of cardiomyocytes after the activation of VEGFR1. Further studies showed that pretreatment with PlGF could activate the phosphorylation of Akt and GSK-3β and inhibit the activation of caspase-3 after reperfusion [23]. This shows that PlGF can activate VEGFR1 to protect the heart from ischaemia-reperfusion injury by inhibiting oxidative stress and reducing cardiomyocyte apoptosis.
Previous studies have shown that PlGF is beneficial to angiogenesis and arteriogenesis of ischaemic myocardium [3, 24, 25]. PlGF can promote the proliferation of endothelial cells and recruit bone marrow cells to target tissues [24]. As a selective ligand of VEGFR1, PlGF can activate VEGFR1 to promote angiogenesis and endothelial cell growth without any related side effects, such as oedema, hypotension and the occurrence of haemangioma [24, 25]. Takeda et al. found that exogenous recombinant PlGF treatment not only improved the survival rate after MI and improved cardiac function but also significantly increased the number of CD31-positive cells and α-smooth muscle actin-positive vessels in the infarcted area and mobilized endothelial progenitor cells into the peripheral circulation [26]. The above research shows that PlGF can reduce the infarct area after MI and improve cardiac function by enhancing angiogenesis and arteriogenesis.
Studies have shown that PlGF may also have a direct protective effect on cardiomyocytes [26–28]. Roncal et al. found that exogenous PlGF treatment can induce compensatory hypertrophy of cardiomyocytes in noninfarcted myocardium and improve cardiac recovery after MI by using a mouse MI model [28]. PlGF also promotes angiogenesis at the infarct border and vascular dilatation in the distal myocardium, increasing the vascular perfusion area and improving the adaptive remodelling of the heart after MI [28]. These results suggest that PlGF can directly improve cardiac function and promote adaptive remodelling after MI.
In addition, some studies have shown that PlGF is proatherogenic, attributed to its ability to activate endothelial adhesion molecule expression and monocyte recruitment to the arterial wall [6, 29, 30]. However, experimental studies have not found any evidence for a pathogenic role of PlGF in more advanced stages of atherosclerosis [31]. In contrast, PlGF may be beneficial to maintain the stability of advanced atherosclerotic plaques because it could stimulate endothelial cell proliferation [32], which provides an important basis for the protective role of PlGF in CHD.
This study explored the causal association between PlGF levels and different types of CHD, including AP, UAP, MI and CHD events. The effect estimates of different data sources all point to the same direction, further strengthening the negative causal association between genetically predicted PlGF level and CHD risk. In particular, the PlGF level was significantly associated with ACS, including UAP and MI, which suggests that PlGF may be a potential predictor and potential effective target of ACS, and its mechanism is worthy of further investigation. Sensitivity analysis was also carried out in this paper, and the trend of the results did not change, thus increasing the reliability of this study.
This study also has some limitations. First, the number of SNPs of IV used in this study was relatively small, which limited the power to detect associations. Second, the data of this study are all from European populations. Although it reduces the deviation caused by population stratification, further testing is needed to determine whether it can be extended to other populations. Third, the available data in the study are summary statistics, without data at the individual level. Therefore, it may bring inevitable deviation to the study. Fourth, in the leave-one-out method, several SNPs removed, the influence of the remaining SNPs on the outcome was inconsistent with the overall outcome, and the data was biased.
In conclusion, this study used a two-sample MR method to explore the causal association between genetically predicted PlGF levels and CHD. The results showed that the genetically predicted PlGF level was negatively correlated with the risk of CHD, especially ACS. PlGF is a potential effective target for the prevention or treatment of CHD. Future research needs to explore the mechanism of PlGF in the occurrence and development of CHD, especially ACS.
Materials and Methods
Study design
We performed 2-sample MR analyses to evaluate the causal association between PlGF and CHD. All data were obtained from the currently published genome-wide association study (GWAS) and Finn Gen consortium. Additional ethical approval or informed consent was not needed, as ethical informed consent and approval were completed in the original studies. The flowchart of the study design overview is shown in Figure 3. Single nucleotide polymorphisms (SNPs) associated with PlGF were selected as genetic instrumental variables (IVs). Pooled GWAS statistics of outcomes related to CHD, MI, AP, unstable angina pectoris (UAP) and major CHD events were selected from published large-scale GWAS meta-analyses and the FinnGen consortium. Genetic outcome associations were extracted and analysed by MR analyses, and corresponding sensitivity analyses were performed.
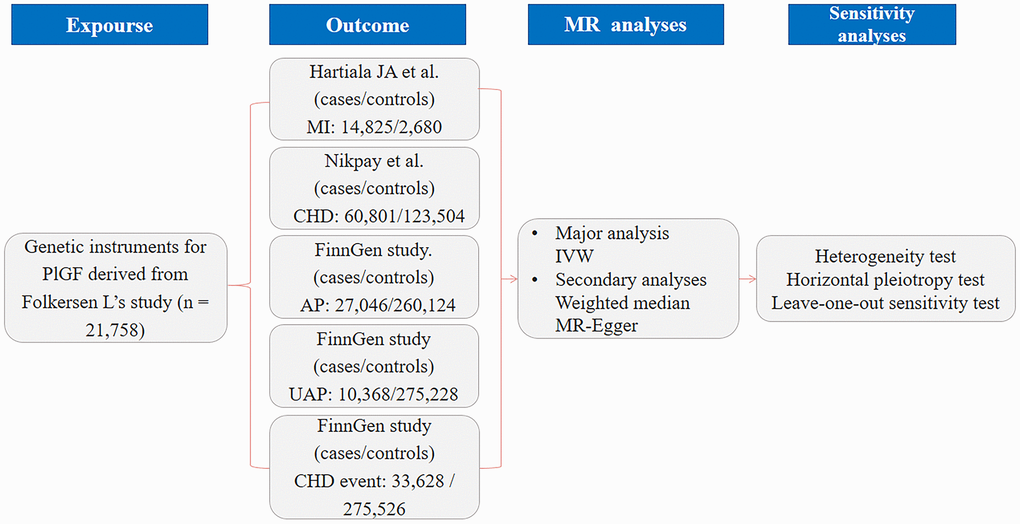
Figure 3. Flowchart of the study design overview. Abbreviations: PlGF: placental growth factor; CHD: coronary artery disease; AP: angina pectoris; UAP: unstable angina pectoris; MI: myocardial infarction; IVW: inverse variance weighted; MR: Mendelian randomization.
Selection of instrumental variables
The genome-wide significant SNPs (P < 5 × 10−7) after linkage disequilibrium clustering were selected from the study by Folkersen et al. as the instrumental variable of PlGF for this 2-sample MR study. The study included 30931 participants of predominantly European descent and mapped and replicated protein quantitative trait loci (pQTL) for 90 cardiovascular proteins, resulting in 451 pQTLs for 85 proteins [33]. Linkage disequilibrium among all SNPs was performed based on the European 1000 Genome Project reference panel. The independent SNP was selected by clumping (r2 ≤ 0.001), and the SNP with the smallest P value was retained. The SNP characteristics related to PlGF are shown in Table 2, Supplementary Table 2.
Table 2. The characteristics of the selected PlGF associated SNPs.
| SNP ID | Chr | Position | EA | EAF | Beta | SE | P | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs184474 | 1 | 7954463 | G | 0.3974 | 0.0538 | 0.0106 | 3.93E-07 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs10182686 | 2 | 40610830 | G | 0.4541 | −0.0517 | 0.0103 | 4.76E-07 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9551468 | 13 | 28985316 | G | 0.5227 | 0.0835 | 0.0102 | 2.97E-16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs175510 | 14 | 75524839 | A | 0.4587 | 0.1149 | 0.0102 | 1.27E-29 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: PlGF: placental growth factor; SNP: Single nucleotide polymorphism; EA: effect _ allele; EAF: effect allele frequency; Chr: chromosome; SE: standard error. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Outcome GWAS dataset selection
The participants of the outcome-related GWASs selected in this study were mainly of European descent. The summary data for CHD were obtained from a large GWAS that conducted a meta-analysis of 48 studies, collecting 60801 cases and 123504 controls [34]. The summary data for MI came from a large GWAS that conducted a meta-analysis of 48 studies, collecting 61000 cases and 577000 controls [35]. The outcome summary data on AP, UAP, and major CHD events were obtained from the FinnGen consortium (data freeze 7). For the definition of major CHD events, please refer to Supplementary Table 3 and the website: https://risteys.finregistry.fi/endpoints/I9_CHD. Among them, 27046 cases and 260124 controls were collected from the summary data of AP, and 10368 cases and 275228 controls were collected from the data of UAP. The major CHD adverse events were collected in 33628 cases and 275526 controls. Relevant participants and statistical analysis, gene platform for detailed information, please visit FinnGen website (https://www.finngen.fi/en/).
Data extraction and MR analysis
Two-sample MR analysis should be performed according to the identified exposure factor SNPS, and the information of SNPS in the outcome should be extracted. We extracted gene-outcome association information from the corresponding outcome of GWAS data using the four SNPs identified by the exposure instrumental variables. All MR analyses were performed using the “Two Sample MR” package in R software (version 4.1.2 with packages, R Foundation for Statistical Computing, Vienna, Austria). To ensure the validity of our conclusions, we used Bonferroni correction for P values in the primary analysis with a threshold of P < 0.01 (α = 0.01 (0.05/5)). We considered P < 0.01 to indicate statistical significance, while 0.01 ≤ P < 0.05 indicated suggestive significance. Three methods, including inverse variance weighted (IVW), weighted median (WM) method and MR-Egger regression analysis, were used for MR analysis [13]. The random effects IVW method was the main MR method in this study. The IVW estimate can be acquired by an IVW meta-analysis of the ratio estimates for the individual variants. Considering that traditional IVW-MR methods are susceptible to the effects of imbalanced level pleiotropy. Therefore, we used additional MR methods, including WM and MR Egger. The WM method may provide robust estimates, even if only half of SNPs meet the requirements of valid instruments. The MR Egger method can identify and control biases caused by directional pleiotropy. Even if all variants are ineffective, as long as the association between individual variants and exposures are independent of the corresponding pleiotropic effects, the MR Egger method will still produce effective estimates. The F statistics of the remaining SNPs were acquired by the following formula: F = R2 × (N–k–1)/((1–R2) × k), where R2 = 2 × β2 × (1–EAF) × EAF, N is the sample size of PIGF, k is the number of SNPs, β is the estimate of genetic effect on PIGF, and EAF is the frequency of the effect allele. The SNPs with an F statistic >10 was considered strong IVs of PIGF [36].
Sensitivity analysis
Finally, we further performed a sensitivity analysis. Cochran’s Q statistic was used to test heterogeneity, and P < 0.05 was considered statistically significant. MR-Egger regression (intercept term) was used to test pleiotropy, and P < 0.05 was considered statistically significant. In addition, to assess whether the MR estimates could be driven by a single SNP with significant level pleiotropy, we conducted a leave-one-out test. We performed MR analyses separately for each outcome database and then pooled effect values for atherosclerosis outcomes at different sites using a random-effects model [14].
Availability of data and material
All data used in the present study are based on publicly available summary data from the GWAS databases. Data generated during this study are available from the corresponding author on reasonable request.
Author Contributions
BZ designed the study, performed data analysis and drafted the article. GTZ and HYB conducted data acquisition, and SZ contributed to the guidance of the research and manuscript revision. All authors read and approved the final manuscript.
Acknowledgments
Data used in this study were downloaded from the IEU Open GWAS project and FinnGen consortium. We thank all GWAS participants and investigators for their contributions to the summary statistics data. The authors thank all investigators for sharing these data.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
This study was supported by the National Natural Science Foundation of China (No. 82000328).
References
- 1. GBD 2017 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018; 392:1859–922. https://doi.org/10.1016/S0140-6736(18)32335-3 [PubMed]
- 2. Persico MG, Vincenti V, DiPalma T. Structure, expression and receptor-binding properties of placenta growth factor (PlGF). Curr Top Microbiol Immunol. 1999; 237:31–40. https://doi.org/10.1007/978-3-642-59953-8_2 [PubMed]
- 3. Dewerchin M, Carmeliet P. PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harb Perspect Med. 2012; 2:a011056. https://doi.org/10.1101/cshperspect.a011056 [PubMed]
- 4. Kim KJ, Cho CS, Kim WU. Role of placenta growth factor in cancer and inflammation. Exp Mol Med. 2012; 44:10–9. https://doi.org/10.3858/emm.2012.44.1.023 [PubMed]
- 5. Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994; 269:25646–54. [PubMed]
- 6. Skoda M, Stangret A, Szukiewicz D. Fractalkine and placental growth factor: A duet of inflammation and angiogenesis in cardiovascular disorders. Cytokine Growth Factor Rev. 2018; 39:116–23. https://doi.org/10.1016/j.cytogfr.2017.12.001 [PubMed]
- 7. Iwama H, Uemura S, Naya N, Imagawa K, Takemoto Y, Asai O, Onoue K, Okayama S, Somekawa S, Kida Y, Takeda Y, Nakatani K, Takaoka M, et al. Cardiac expression of placental growth factor predicts the improvement of chronic phase left ventricular function in patients with acute myocardial infarction. J Am Coll Cardiol. 2006; 47:1559–67. https://doi.org/10.1016/j.jacc.2005.11.064 [PubMed]
- 8. Benschop L, Schalekamp-Timmermans S, Broere-Brown ZA, Roeters van Lennep JE, Jaddoe VWV, Roos-Hesselink JW, Ikram MK, Steegers EAP, Roberts JM, Gandley RE. Placental Growth Factor as an Indicator of Maternal Cardiovascular Risk After Pregnancy. Circulation. 2019; 139:1698–709. https://doi.org/10.1161/CIRCULATIONAHA.118.036632 [PubMed]
- 9. Draker N, Torry DS, Torry RJ. Placenta growth factor and sFlt-1 as biomarkers in ischemic heart disease and heart failure: a review. Biomark Med. 2019; 13:785–99. https://doi.org/10.2217/bmm-2018-0492 [PubMed]
- 10. Marković M, Ignjatović S, Dajak M, Majkić-Singh N. Placental growth factor as short-term predicting biomarker in acute coronary syndrome patients with non-ST elevation myocardial infarction. South Med J. 2010; 103:982–7. https://doi.org/10.1097/SMJ.0b013e3181eda4ef [PubMed]
- 11. Apple FS, Pearce LA, Chung A, Ler R, Murakami MM. Multiple biomarker use for detection of adverse events in patients presenting with symptoms suggestive of acute coronary syndrome. Clin Chem. 2007; 53:874–81. https://doi.org/10.1373/clinchem.2006.080192 [PubMed]
- 12. Chen Y, Nilsson AH, Goncalves I, Edsfeldt A, Engström G, Melander O, Orho-Melander M, Rauch U, Tengryd C, Venuraju SM, Lahiri A, Liang C, Nilsson J. Evidence for a protective role of placental growth factor in cardiovascular disease. Sci Transl Med. 2020; 12:eabc8587. https://doi.org/10.1126/scitranslmed.abc8587 [PubMed]
- 13. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018; 362:k601. https://doi.org/10.1136/bmj.k601 [PubMed]
- 14. Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol. 2013; 178:1177–84. https://doi.org/10.1093/aje/kwt084 [PubMed]
- 15. Cassidy A, Chiuve SE, Manson JE, Rexrode KM, Girman CJ, Rimm EB. Potential role for plasma placental growth factor in predicting coronary heart disease risk in women. Arterioscler Thromb Vasc Biol. 2009; 29:134–9. https://doi.org/10.1161/ATVBAHA.108.171066 [PubMed]
- 16. Tarnow L, Astrup AS, Parving HH. Elevated placental growth factor (PlGF) predicts cardiovascular morbidity and mortality in type 1 diabetic patients with diabetic nephropathy. Scand J Clin Lab Invest Suppl. 2005; 240:73–9. https://doi.org/10.1080/00365510500235970 [PubMed]
- 17. Lenderink T, Heeschen C, Fichtlscherer S, Dimmeler S, Hamm CW, Zeiher AM, Simoons ML, Boersma E, and CAPTURE Investigators. Elevated placental growth factor levels are associated with adverse outcomes at four-year follow-up in patients with acute coronary syndromes. J Am Coll Cardiol. 2006; 47:307–11. https://doi.org/10.1016/j.jacc.2005.08.063 [PubMed]
- 18. Bui AH, Bonaca MP, Sabatine MS, Ray KK, Rifai N, Cannon CP, Morrow DA. Elevated concentration of placental growth factor (PlGF) and long term risk in patients with acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Thromb Thrombolysis. 2012; 34:222–8. https://doi.org/10.1007/s11239-012-0704-z [PubMed]
- 19. Heeschen C, Dimmeler S, Fichtlscherer S, Hamm CW, Berger J, Simoons ML, Zeiher AM, and CAPTURE Investigators. Prognostic value of placental growth factor in patients with acute chest pain. JAMA. 2004; 291:435–41. https://doi.org/10.1001/jama.291.4.435 [PubMed]
- 20. Hausenloy DJ, Yellon DM. Cardioprotective growth factors. Cardiovasc Res. 2009; 83:179–94. https://doi.org/10.1093/cvr/cvp062 [PubMed]
- 21. Thirunavukkarasu M, Juhasz B, Zhan L, Menon VP, Tosaki A, Otani H, Maulik N. VEGFR1 (Flt-1+/-) gene knockout leads to the disruption of VEGF-mediated signaling through the nitric oxide/heme oxygenase pathway in ischemic preconditioned myocardium. Free Radic Biol Med. 2007; 42:1487–95. https://doi.org/10.1016/j.freeradbiomed.2007.02.011 [PubMed]
- 22. Addya S, Shiroto K, Turoczi T, Zhan L, Kaga S, Fukuda S, Surrey S, Duan LJ, Fong GH, Yamamoto F, Maulik N. Ischemic preconditioning-mediated cardioprotection is disrupted in heterozygous Flt-1 (VEGFR-1) knockout mice. J Mol Cell Cardiol. 2005; 38:345–51. https://doi.org/10.1016/j.yjmcc.2004.11.033 [PubMed]
- 23. Zhang Y, Cao C, Xin J, Lv P, Chen D, Li S, Yang H, Chen C, Liu B, Li Q. Treatment with placental growth factor attenuates myocardial ischemia/reperfusion injury. PLoS One. 2018; 13:e0202772. https://doi.org/10.1371/journal.pone.0202772 [PubMed]
- 24. Kolakowski S
Jr , Berry MF, Atluri P, Grand T, Fisher O, Moise MA, Cohen J, Hsu V, Woo YJ. Placental growth factor provides a novel local angiogenic therapy for ischemic cardiomyopathy. J Card Surg. 2006; 21:559–64. https://doi.org/10.1111/j.1540-8191.2006.00296.x [PubMed] - 25. Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, Bohlen P, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002; 8:831–40. https://doi.org/10.1038/nm731 [PubMed]
- 26. Takeda Y, Uemura S, Iwama H, Imagawa K, Nishida T, Onoue K, Takemoto Y, Soeda T, Okayama S, Somekawa S, Ishigami K, Takaoka M, Kawata H, et al. Treatment with recombinant placental growth factor (PlGF) enhances both angiogenesis and arteriogenesis and improves survival after myocardial infarction. Circ J. 2009; 73:1674–82. https://doi.org/10.1253/circj.cj-08-1067 [PubMed]
- 27. Huang K, Yan ZQ, Zhao D, Chen SG, Gao LZ, Zhang P, Shen BR, Han HC, Qi YX, Jiang ZL. SIRT1 and FOXO Mediate Contractile Differentiation of Vascular Smooth Muscle Cells under Cyclic Stretch. Cell Physiol Biochem. 2015; 37:1817–29. https://doi.org/10.1159/000438544 [PubMed]
- 28. Roncal C, Buysschaert I, Chorianopoulos E, Georgiadou M, Meilhac O, Demol M, Michel JB, Vinckier S, Moons L, Carmeliet P. Beneficial effects of prolonged systemic administration of PlGF on late outcome of post-ischaemic myocardial performance. J Pathol. 2008; 216:236–44. https://doi.org/10.1002/path.2408 [PubMed]
- 29. Khurana R, Moons L, Shafi S, Luttun A, Collen D, Martin JF, Carmeliet P, Zachary IC. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation. 2005; 111:2828–36. https://doi.org/10.1161/CIRCULATIONAHA.104.495887 [PubMed]
- 30. Roncal C, Buysschaert I, Gerdes N, Georgiadou M, Ovchinnikova O, Fischer C, Stassen JM, Moons L, Collen D, De Bock K, Hansson GK, Carmeliet P. Short-term delivery of anti-PlGF antibody delays progression of atherosclerotic plaques to vulnerable lesions. Cardiovasc Res. 2010; 86:29–36. https://doi.org/10.1093/cvr/cvp380 [PubMed]
- 31. Wu M, Pokreisz P, Swinnen M, Caluwe E, Gillijns H, Vanden Driessche N, Casazza A, Verbeken E, Collen D, Janssens S. Sustained Placental Growth Factor-2 Treatment Does Not Aggravate Advanced Atherosclerosis in Ischemic Cardiomyopathy. J Cardiovasc Transl Res. 2017; 10:348–58. https://doi.org/10.1007/s12265-017-9742-4 [PubMed]
- 32. Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, Autiero M, Wyns S, Plaisance S, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007; 131:463–75. https://doi.org/10.1016/j.cell.2007.08.038 [PubMed]
- 33. Folkersen L, Gustafsson S, Wang Q, Hansen DH, Hedman ÅK, Schork A, Page K, Zhernakova DV, Wu Y, Peters J, Eriksson N, Bergen SE, Boutin TS, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab. 2020; 2:1135–48. https://doi.org/10.1038/s42255-020-00287-2 [PubMed]
- 34. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015; 47:1121–30. https://doi.org/10.1038/ng.3396 [PubMed]
- 35. Hartiala JA, Han Y, Jia Q, Hilser JR, Huang P, Gukasyan J, Schwartzman WS, Cai Z, Biswas S, Trégouët DA, Smith NL, Seldin M, Pan C, et al, and INVENT Consortium, and CHARGE Consortium Hemostasis Working Group, and GENIUS-CHD Consortium, and Biobank Japan. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur Heart J. 2021; 42:919–33. https://doi.org/10.1093/eurheartj/ehaa1040 [PubMed]
- 36. Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011; 40:740–52. https://doi.org/10.1093/ije/dyq151 [PubMed]