



Introduction
Hepatocellular carcinoma (HCC) is known as one of the most aggressive and frequent malignant diseases to bring about global attention, which is ranking as the third of mortality and the sixth of morbidity in all malignant neoplasms [1, 2]. The onset of HCC is usually undetectable and subtle bringing about developing into middle and advanced stage [3]. The therapeutic result of this malignant tumor is unsatisfied resulting in the 5 years survival rate is less than 19% [4]. Accordingly, proper clinical diagnosis and exploration of precise molecular markers are essential to improve the prognosis of HCC patients.
Fatty acids (FA) play a vital part in human organisms, releasing energy with effect from adenosine triphosphate (ATP), being a significant component of bio-membranes as same as taking pate in metabolic pathways such as beta-oxidation, hydrolysis, activation, esterification and synthesis [5, 6]. The activation of FAs into serious metabolic pathways through the formation of high-energy CoA intermediates by acyl-CoA synthetases (ACSs) enzymes is required for their synthesis and degradation [7]. ACSs, which are located in the endoplasmic reticulum, mitochondria, and microsomal membranes, are enzymes that convert nonpolar hydrophilic FA substrates into acyl-CoA [6, 8, 9]. On the basis of the substrate preference and sequence identity regarding the chain lengths of fatty acids, the human genome includes 26 ACS genes distributed into 6 distinct families: the ACS short-chain family (ACS), ACS medium-chain family (ACSM), ACS long-chain family (ACSL), ACS very long-chain family (ACSVL), ACS bubblegum family (ACSBG) and ACSF family (ACSF) [10, 11]. This division indicates the chain length of these genes preferred substrate. In mammals, previous research confirmed four distinct ACSF clades, including ACSF1(AACS), ACSF2, ACSF3, ACSF4(AASDH). As one of the novel ACSs isozymes, AACS has been participated in regulation of ketone body utilization on fatty acids and/or cholesterol biosynthesis in rats [12]. ACSF3 is a candidate gene for human metabolism-related disease [13]. Furthermore, Xin et al. indicated that ACSF2 gene may act as an original biomarker for estimating prognosis of breast cancer patients [14].
However, the detailed analysis of the ACSF gene family expression profiles and functions in cancer needs to be further confirmation. Along with the establishment of amounts of bioinformatics databases and the express development of second-generation gene sequencing technology, we could clarify the ACSF family clearer in HCC pathogenesis and treatment based on a extensive analysis of the ACSF family. In this study, we first conducted a detailed and integrated biological function of the ACSF family (including AACS ACSF2-3 and AASDH) in HCC. Additionally, on the strength of multiple public bioinformatics databases, we explored the potential of ACSF family as prognostic biomarkers and therapeutic targets. The purpose of this research is to attempt to select a more appropriate therapy and precise prediction for the long-term prognosis of HCC patients (Supplementary Table 1).
Results
Aberrant expression of ACSF gene family in HCC patients
We adopted ACSF gene both GEPIA and Ualcan database to evaluate the expression levels of this family members. The data revealed that all ACSF gene family members were significantly increased in most of the malignant neoplasms (Supplementary Figure 1). Then, we used Ualcan databases to detect the mRNA expression levels of ACSF gene family members in HCC tissues and adjacent normal tissues, as well as the levels of mRNA expression in HCC tissues and normal tissues were evaluated by the Wanderer database of ACSF gene family members. The results showed that the expression level of AACS (p <1E-12), ACSF2 (p = 4.59E-09) and AASDH (p = 1.18E-06) were significantly higher expressions in patients of HCC (Figure 1A). Then, the expression level of AACS (p < 0.05) and ACSF2 (p < 0.05) were remained significantly raised in HCC tissues according to GEPIA database, but the expression level of AASDH was reduced in tumor tissues than in normal tissues (Figure 1B). Moreover, we used the HCCDB database to analyze the expression levels of ACSF gene family members in HCC tissues and adjacent tissues. The analysis of data revealed that the expression of AACS (p = 7.850E-34), ACSF2 (p = 1.900E-10), and AASDH (p = 0.001596) in dataset of HCCDB4 and the expression of AACS (p = 5.510E-26), ACSF2 (p = 5.920E-14), and AASDH (p = 1.410E-11) in dataset of HCCDB18 in HCC tissues were both upregulated, separately (Table 1).
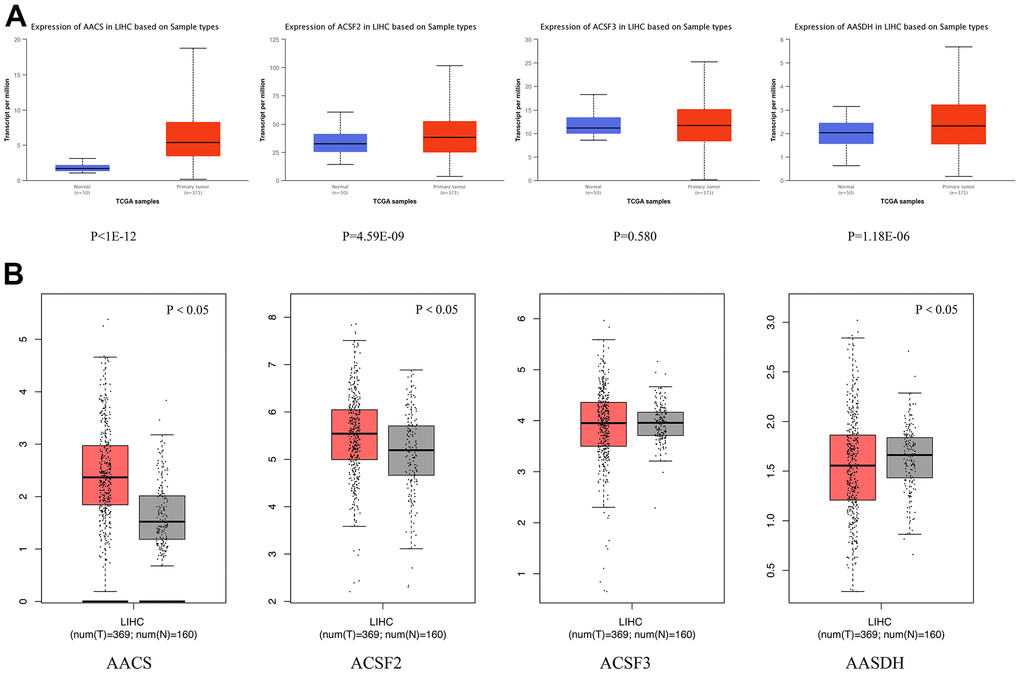
Figure 1. Differential mRNA expression analysis of the ACSF gene family in HCC and normal tissues. (A) The expression profiles were collected from the Ualcan databases. The red and blue graphs indicated the high-regulation and low-regulation, respectively. (B) The expression profiles were collected from the GEPIA databases. The red and gray graphs indicated the high-regulation and low-regulation, respectively.
Table 1. The expression of ACSF gene family members in HCC tissues and adjacent tissues from the HCCDB database.
| Dataset | Gene | Type | Nums | Mean | STD | P-value | IQR |
| HCCDB4 | AACS | HCC | 240 | 6.476 | 0.3388 | 7.850e-34 | 0.4992 |
| Adjacent | 193 | 6.143 | 0.1577 | 0.2309 | |||
| ACSF2 | HCC | 240 | 7.877 | 0.5510 | 1.900e-10 | 0.7366 | |
| Adjacent | 193 | 7.601 | 0.3149 | 0.4096 | |||
| ACSF3 | HCC | 240 | 6.985 | 0.3381 | 0.2203 | 0.4525 | |
| Adjacent | 193 | 6.949 | 0.2843 | 0.3756 | |||
| AASDH | HCC | 240 | 6.559 | 0.3469 | 0.001596 | 0.5615 | |
| Adjacent | 193 | 6.468 | 0.2476 | 0.3966 | |||
| HCCDB18 | AACS | HCC | 212 | 1.893 | 0.7057 | 5.510e-26 | 0.8800 |
| Adjacent | 177 | 1.237 | 0.4046 | 0.4600 | |||
| ACSF2 | HCC | 212 | 4.549 | 0.7931 | 5.920e-14 | 1.030 | |
| Adjacent | 177 | 4.035 | 0.4890 | 0.6200 | |||
| ACSF3 | HCC | 212 | 2.842 | 0.6466 | 0.9923 | 0.8525 | |
| Adjacent | 177 | 2.842 | 0.4040 | 0.5300 | |||
| AASDH | HCC | 212 | 1.900 | 0.3491 | 1.410e-11 | 0.4425 | |
| Adjacent | 177 | 1.703 | 0.1963 | 0.2500 |
Then, the correlations between the pathological stages of patients with HCC and the expression profiles of ACSF gene family were analyzed. From Figure 2A–2D, the expression of AACS (p = 0.00178), ACSF1 (p = 0.00981) and AASDH (p = 0.0305) indicated a significant difference with four clinical stage subgroups. These results demonstrated that abnormal expression of ACSF gene family members might involve in tumor progression in patients with HCC.
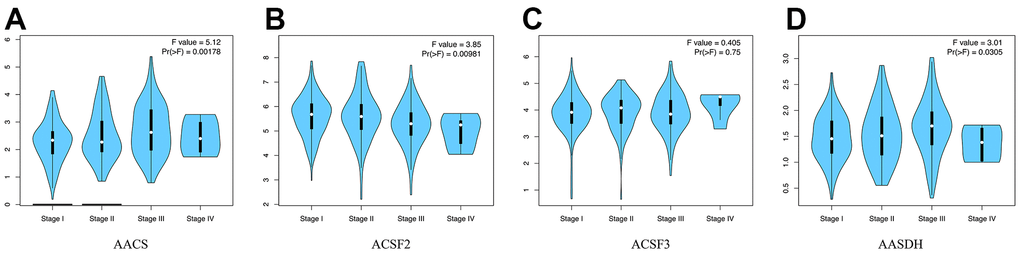
Figure 2. The relationship between the expression of the ACSF gene family and the pathological stage of HCC patients (GEPIA). (A–D) GEPIA databases were used to evaluate the correlations of the ACSF gene family with the pathological stage of HCC patients.
The prognostic value of the ACSF gene family in HCC patients
To explore the prognostic value of the ACSF gene family in patients with HCC, the association between the survival endpoints and expression of ACSF gene members, such as overall survival (OS), disease-free survival (DFS) and relapse-free survival (RFS) were further explored through Kaplan-Meier plotter. We found that HCC patients with depressed expression of AACS (p=0.0032) was correlated with a better OS, while elevated transcriptional level of ACSF2 (p=0.013) and ACSF3 (p=0.0015) were greatly related to a better OS time (Figure 3A). In the meantime, the prognostic significance of ACSF gene family on the DFS of HCC patients were evaluated. As shown in Figure 3B, we found that high levels of ACSF2 (p = 0.013) and ACSF3 (p = 0.00015) have an intimate correlation with a longer DFS time. Similarly, low expression of AACS (p=0.014), high expression of ACSF2 (p=0.012) and ACSF3 (p=0.01) that meaning a better RFS (Figure 3C).
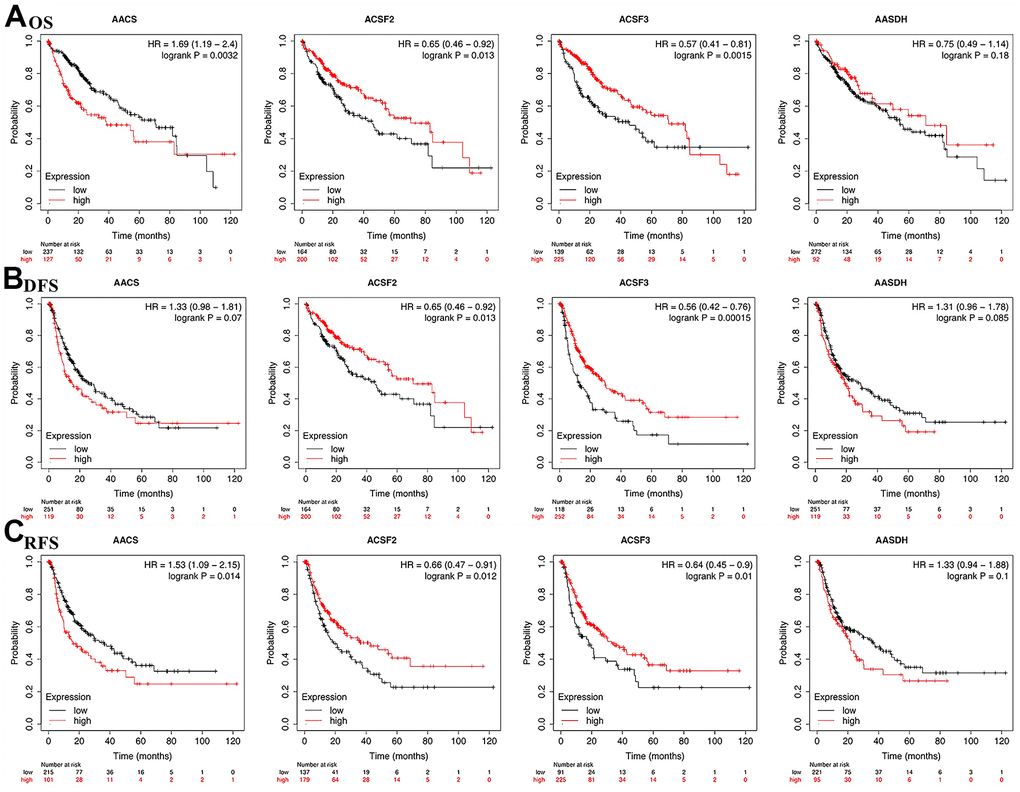
Figure 3. The correlations of ACSF gene family expression with OS and PFS in HCC patients. (A–C) Kaplan-Meier plotter was used to assess the correlation of ACSF gene family members with the patients’ OS (A), DFS (B) and RFS (C).
Genetic alteration and functional enrichment analyses of ACSF gene family in HCC patients
In the ACSF gene family in LIHC, the frequency and types of alteration profiles were acquired through the cBioPortal tool and TCGA database. As shown in Figure 4A, the results showed that the alternation rate of ACSF2 and AASDH was the highest in 8% of cases, whereas the other family members are 5% (for AACS and ACSF3). In the matter of genetic alterations types, mRNA high alteration was the main genetic alterations in all ACSF gene family. However, in several HCC patients, gene amplification alteration was also observed in ACSF2, and mRNA low alteration were found in ACSF3 and AASDH (Figure 4A).
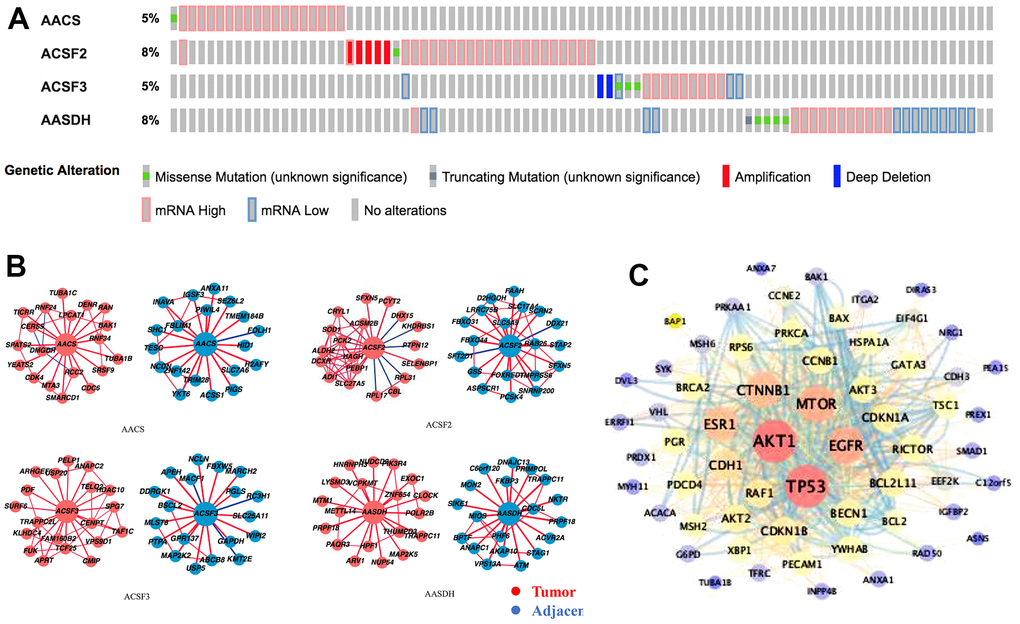
Figure 4. Genetic alterations and interaction analyses of the ACSF gene family in HCC patients. (A) Genetic alteration of the ACSF gene family in HCC patients analyzed with cBioPortal. (B) The HCCDB database was used to analyze the ACSF gene family co-expressed genes in HCC and adjacent tissue samples. (C) The most frequently altered genes identified from cBioPortal that are linked to the ACSF gene family in HCC patients.
Secondly, the associated molecules of ACSF gene family were evaluated by using HCCDB database indicating that the positive molecules in all ACSF gene family had varied significantly between the adjacent normal tissues and neoplasm tissues, respectively (Figure 4B). Further, we abstracted 202 most frequently altered genes which were significantly associated with ACSF gene family in HCC from the cBioPortal tool (Supplementary Table 2). Then, the analysis results proved that many hub genes, such as AKT1, TP53, MTOR, MAPK1, EGFR and CTNNB1, were closely participated in the biological behavior prognosis in ACSF gene family (Figure 4C). These results demonstrated that the ACSF gene family involved in the adjustment of varieties of signaling pathways influenced the LIHC pathophysiology.
To further understand the potential biological functions of ACSF gene family, on the basis of the 202 ACSF-associated genes, we selected the WebGestalt database to perform the functional enrichment analysis. Accordingly, ACSF gene family members were most highly concentrated in biological function (BF) category like metabolic process, biological regulation, response to stimulus, developmental process and multicellular organismal process. In the aspect of categories of cellular component (CC)cytosol, nucleus, membrane, protein-containing complex and membrane-enclosed lumen were extremely enriched. Moreover, the ACSF gene family members were mainly enriched in the following categories of molecular function (MF), including transferase activity, ion binding, nucleic acid binding, nucleotide binding and protein binding (Figure 5A). Simultaneously, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was implemented to suggest that the enriched pathways were mainly involved in results displayed in Figure 5B. Obviously, some cancer-related pathways, including long-term potentiation, phospholipase D signaling pathway and purine metabolism, which we can demonstrate, were significantly related to the feasible biological functions of ACSF gene family involved in the occurrence and development of HCC.
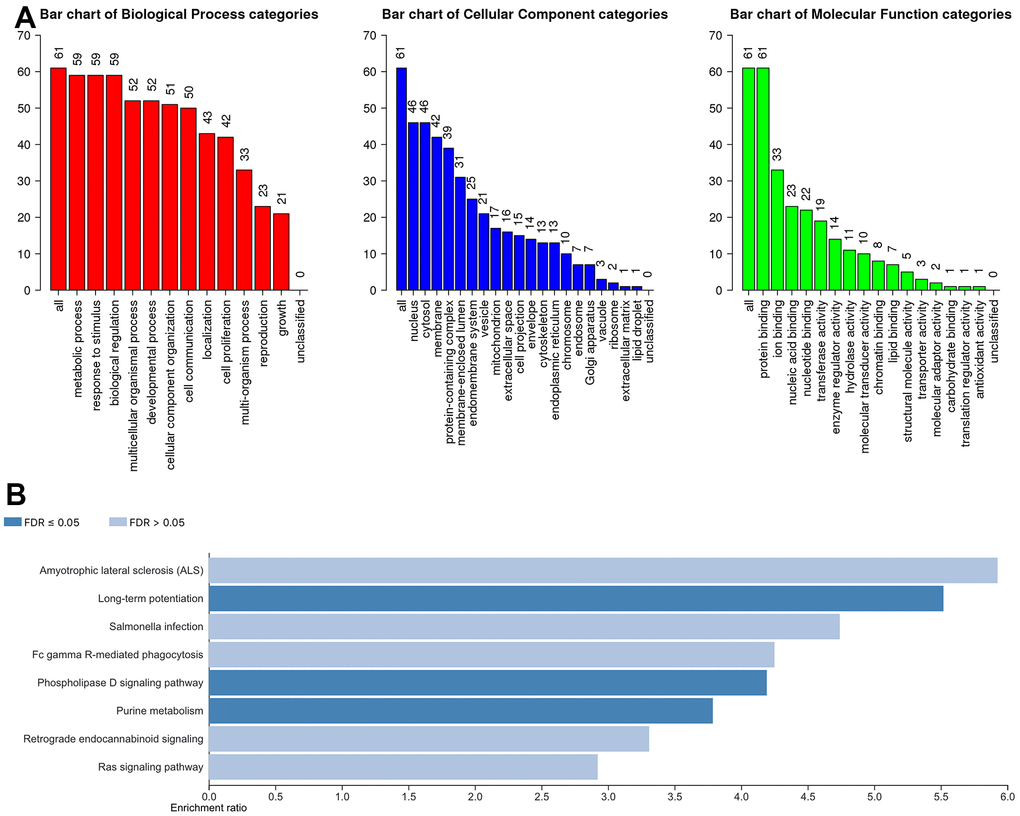
Figure 5. The biological pathways of the ACSF gene family were evaluated by the WebGestalt database. (A) Bar plot of GO enrichment in cellular components, biological processes, and molecular functions. (B) The bar plot of KEGG enrichment.
Correlation between immune cell infiltration and each ACSF gene family
The relationship between immune cell infiltration and each ACSF gene family was explored through the TIMER database. The AACS expression was affirmatively associated with the infiltration of B cells (Cor = 0.363, p = 3.81e-12), CD8+ T cells (Cor = 0.247, p = 3.66e-06), CD4+ T cells (Cor = 0.375, p = 6.68e-13), macrophages (Cor = 0.452, p = 1.37e-18), neutrophils (Cor =0.42, p = 3.69e-16) and dendritic cells (Cor =0.384, p = 2.26e-13) (Figure 6A). Meanwhile, the expression of ACSF2 was positively correlated with the infiltration of B cells (Cor = 0.157, p = 3.45e-03), CD8+ T cells (Cor = 0.153, p = 4.53e-03), CD4+ T cells (Cor = 0.292, p = 3.33e-08), macrophages (Cor = 0.33, p = 4.15e-10), neutrophils (Cor =0.402, p = 8.27e-15) and dendritic cells (Cor =0.316, p = 2.42e-09) (Figure 6B). Conversely, negative correlations between AASDH and the infiltration of B cells (Cor = -0.202, p = 1.68e-04), CD8+ T cells (Cor = -0.108, p = 4.62e-02), CD4+ T cells (Cor =- 0.222, p = 3.32e-05), macrophages (Cor = -0.204, p = 1.51e-04), neutrophils (Cor =-0.161, p = 2.78e-03) and dendritic cells (Cor =-0.182, p = 7.67e-04) were showed in Figure 6D. Nevertheless, the relationship between ACSF3 and immune cells was uncorrelated (Figure 6C). Surprisingly, above all six types of immune cells which were analyzed were positively linked to AACS and ACSF2. Furthermore, the Cox proportional hazard model proved that B cells (p = 0.016), CD8+ T cells (p = 0.019), macrophage cells (p = 0.032), dendritic cells (p = 0.001) and AACS (p = 0.014) correlated with the prognosis of HCC patient significantly (Table 2).
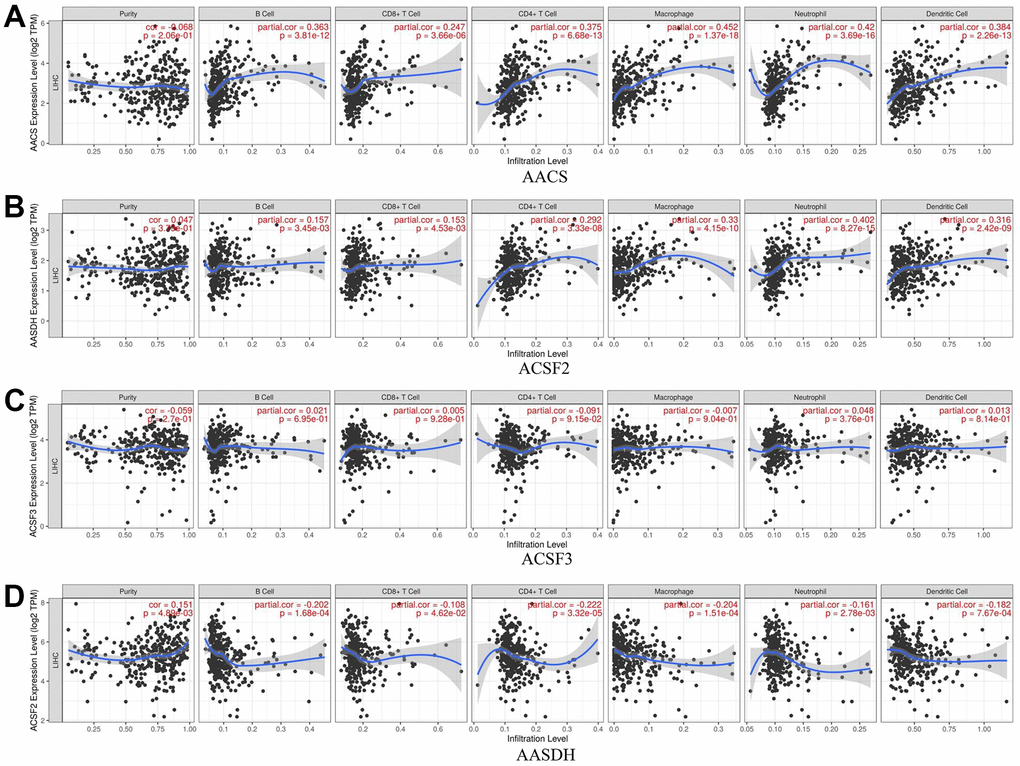
Figure 6. The relationship between immune cell infiltration and the expression of the ACSF gene family. (A–D) The Timer database was used to analyze the effect of (A) AACS, (B) ACSF2, (C) ACSF3, (D) AASDH on the abundance of immune cells in HCC patients.
Table 2. The Cox proportional hazard model of ACSF domain family and seven types of immune cells in HCC patients from timer database.
| Coef | HR | 95%CI_l | 95%CI_u | P.value | Sig | |
| Purity | 1.154 | 3.170 | 1.079 | 9.318 | 0.036 | * |
| B_cell | -8.868 | 0.000 | 0.000 | 0.186 | 0.016 | * |
| CD8_Tcell | -5.989 | 0.003 | 0.000 | 0.373 | 0.019 | * |
| CD4_Tcell | -6.418 | 0.002 | 0.000 | 1.389 | 0.062 | |
| Macrophage | 5.610 | 273.246 | 1.605 | 46529.528 | 0.032 | * |
| Neutrophil | 0.626 | 1.870 | 0.000 | 255364.856 | 0.917 | |
| Dendritic | 6.102 | 446.559 | 13.316 | 14975.279 | 0.001 | ** |
| AACS | 0.300 | 1.349 | 1.063 | 1.713 | 0.014 | * |
| ACSF2 | 0.022 | 1.023 | 0.838 | 1.247 | 0.825 | |
| ACSF3 | -0.228 | 0.796 | 0.608 | 1.042 | 0.097 | ** |
| AASDH | -0.115 | 0.892 | 0.598 | 1.329 | 0.573 |
Methylation level of ACSF gene family in HCC patients
In addition, the methylation levels of ACSF gene family in HCC patients were analyzed through the Diseasemeth database. The analysis data showed that the DNA methylation levels of AACS (p=3.476E-08) and AASDH (p=2.293E-12) were lower in HCC tissues than in normal tissues, but ACSF2 (p=2.046E-04) was higher in HCC than in normal cases (Figure 7A–7D). These DNA methylation changes could indicate possibly the difference of ACSF gene family in expression levels to a certain extent in HCC patients.
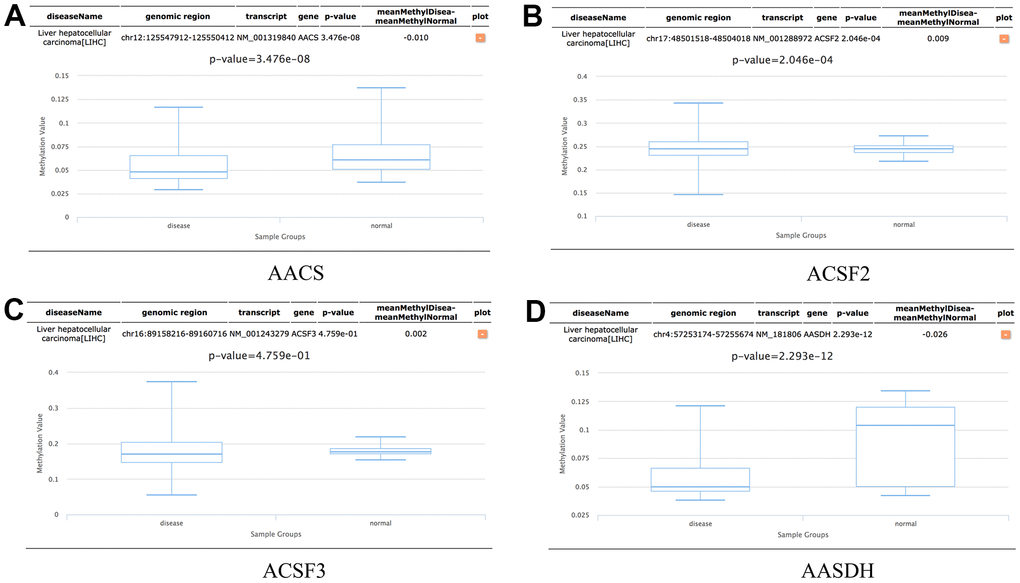
Figure 7. The methylation levels of the ACSF gene family in HCC tissues. (A–D) The methylation values of (A) AACS, (B) ACSF2, (C) ACSF3, (D) AASDH were evaluated using the Diseasemeth database.
Discussion
The ACSF gene family catalyzes the fundamental, initial reaction in fatty acid metabolism of mammals [10]. Whereas, for the moment, little knowledge of ACSF family in cancer has been reported until now. Hasegawa et al. [15, 16] demonstrate that the limited proteolysis of AACS can encode the ketone body-utilizing enzyme, and is adjusted transcriptionally by SREBP-2. Meanwhile, the ketone body metabolism regulated by AACS plays a significant role in cholesterol metabolism. In literature, the data reveals that the ketone body is an fundamental element of cellular function and lipid metabolism, and exploration of transcriptional regulation of AACS could facilitate to clarify the role of ketone body utilization in neurological disorders [17]. To explain the possible mechanism of regulating the gene expression of human AACS and to elucidate the biological characteristics, Francesca et al. [18] isolated that the human promoter is a PPARγ (peroxisome-proliferator-activated receptor γ) target gene and PPARγ is employed to AACS promoter through the direct interaction with Sp1 (stimulating protein-1). Liu et al. [19] initially presented that the AACS may serve as new biomarker for a better recognition to the development, progression, and recurrence of GBM. Acyl-CoA synthetase family member 3 (ACSF3) is verified as the premier enzyme in the mtFAS pathway. The study found that deletion of ACSF3 enzymes could lead to combined malonic and methylmalonic aciduria (CMAMMA) [13]. Recent report emphasized that pharmacological activation of the SIRT3/ACSF3 pathway was an effective approach to alleviate NAFLD and protocatechuic acid was a novel candidate therapy for non-alcoholic fatty liver disease [20]. In addition, high expression of circ-AASDH was found to participate in the tumor size, clinical stage and regulate the expression level of E2F7 by sponging miR-140-3p, which involves in the malignant progression of LUAD [21].
According to our understanding, there is no systematic analysis aiming at the particular possibly functions and mechanisms of ACSF gene family in HCC. In this study, on the basis of the Ualcan, GEPIA, Kaplan-Meier plotter and HCCDB database, we demonstrated that the ACSF gene family expression levels are different between HCC tissues and normal cases. Our study explored the expression levels and function profiles of ACSF gene family in cancer for the first time. The data displayed that the AACS, ACSF2 and AASDH were strongly upregulated in HCC tissues and cells, meaning the potential to be oncogenes. Moreover, we implemented the relationship between ACSF family expression levels and HCC pathological stage. We detected that the expression level of AACS, ACSF2 and AASDH were raised positively with HCC stage progressed, indicating that these three proteins might involve in HCC progression. Additionally, only AACS was found to possess the prognostic value for HCC patients since the patients with high expression of AACS had a shorter OS and RFS time, while patients with high expression of ACSF2 and ACSF3 had a higher OS, DFS and RFS value in LIHC. These databases imply that AACS might be more valuable to be a crucial biomarker and therapeutic target for the patients with HCC. While, more specific evidence of studies and systemic elucidation of the mechanism of ACSF gene family in HCC needs to be further studied.
In addition, to better realize the potential mechanism of ACSF gene family in HCC, we used the GO and KEGG pathway enrichment analysis to examine the possible function of ACSF gene family. In HCC, the functional enrichment analysis in HCC proved that the amyotrophic lateral sclerosis (ALS), long-term potentiation and salmonella infection were the most relevant pathways in which ACSF family were involved. As reported by previous studies, serine biosynthesis pathway and tyrosine relative kinase inhibitors play critical roles in the regulation of HCC progression and immunotherapy [22, 23]. These findings suggest that ACSF gene family might participate in HCC progression through regulating signaling pathways.
Immune cell infiltration has been acknowledged as a critical role which is highly lined to the clinical outcomes, tumor responses and efficacy of immunotherapy [24–26]. A large amount of researches have concentrated on the correlation between tumors and immune cells [27, 28] and testified that immune cell infiltration could play important roles on the cancer immunotherapy and clinical outcomes [29, 30]. In this study, we found the expression of AACS and ACSF2 were greatly corelated with the infiltration of immune cells, including B cells, CD8+ T cells, CD4+ T cells, macrophages, neutrophils and dendritic cells, however, there was a negative correlation between AASDH and immune cell infiltration. These results highlighted that the ACSF gene family might regulate the immune cell infiltration, which could serve as the crucial indicators for the clinical outcomes of HCC patients.
To our knowledge, as an epigenetic modification, DNA methylation could regulate gene transcription and maintains genome stability. Meanwhile, methylation-associated gene silencing, which is a key component of post-transcriptional modification, plays an important role in tumor progression [31, 32]. In this study, the data revealed that the DNA methylation level of the AACS and AASDH gene levels are significantly reduced in HCC tissues, while the ACSF2 was upregulated, indicating the potential impact on the DNA methylation in the ACSF gene family.
In our study, some limitations can not be neglected. First, all of data and information we studied were mostly obtained from the bioinformatics databases. Consequently, additional and deep in vitro and in vivo experiments are needed to prove the functions and mechanisms of ACSF gene family in HCC. Next, in this study, the follow-up study of HCC patients was not performed concerning the therapeutic outcome. Therefore, the exploration of prognostic effects of the ACSF gene family on HCC patients is greatly imperative to strengthen the application values in clinical therapeutics.
To sum up, we comprehensively and systematically analyzed the molecular profiles of ACSF gene family in HCC from the aspect of bioinformatics, containing the gene expression levels, prognostic values and immune response. The results demonstrated that the ACSF gene family, particularly AACS, dominated enormous potential to be potential prognostic biomarkers and essential therapeutic targets in HCC. Therefore, our achievements might benefit of establishing more precise therapeutic strategies to modify the clinical outcomes and prognosis of HCC patients.
Materials and Methods
Data acquisition
GEPIA and Ualcan
Both of the Gene Expression Profiling Interactive Analysis (GEPIA) and Ualcan are databases which could support comprehensive expression analyses based on TCGA and GTEx data [33, 34]. We used these two databases to estimate the expression profiles of ACSF gene family in tumor and normal tissues. The p-value was a cutoff of 0.05.
HCCDB
HCCDB could provide the visualization to the results of several computational analyses, containing differential expression level analysis, tumor-specific and tissue-specific expression analysis, survival analysis, and co-expression analysis [35]. In our study, we selected HCCDB4, HCCDB18 to identify whether there existed significant difference of protein expression between tumor tissues and adjacent tissues of ACSF gene family in HCC. Meanwhile, we showed co-expressed genes of ACSF gene family in HCC and adjacent tissues, respectively.
Kaplan-Meier plotter
Kaplan-Meier plotter is a database exploring the correlation between gene expression with the prognostic role of multiple cancer patients [36]. In this study, we appraised the prognosis of patients with HCC through means of overall survival (OS) and progression free survival (PFS) curves. The data showed statistical significance if the p-value was a cutoff of 0.05.
cBioPortal
cBioPortal provides analysis of genomic alteration data on more than 200 cancer patients [37]. In this study, the genetic alterations and co-expression profiles of ACSF gene family in HCC tissues were analyzed by searching cBioPortal.
STRING
STRING is performed to evaluate potential protein-protein interactions (PPIs), and a database supporting an interactive network among numerous proteins [38]. Simultaneously, the STRING and Cytoscape were applied to analyze the ACSF gene family member-associated PPI network [39].
GeneMANIA
GeneMANIA provides a convenient approach to identify protein-protein interactive networks [40]. We successfully identified the ACSF gene family associated genes by using the GeneMANIA.
WebGestalt
WebGestalt is applied to provide a better understanding of gene interpretation, which can support enrichment results from this database [41]. In our study, we implemented Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analysis linked to the ACSF gene family in HCC.
TIMER
TIMER is a public resource employed to evaluate infiltration of immune cells in various types of cancer, providing more authoritative programs for an modified therapeutic response and prognosis [42]. Here, we primarily implemented correlation analysis between the expression of ACSF gene family with different types of immune cells.
DiseaseMeth2.0
DiseaseMeth2.0 is a professional online database supporting DNA methylation information in various kinds of human diseases [43]. In our study, we explored the correlation between the ACSF gene family expression and DNA methylation levels. The p-value was a cutoff of 0.05.
Author Contributions
Conception and design: Zhao Z and Liu M. Writing, review, and/or revision of the manuscript: Xu Z, Cai Y, Peng B, Yan Y, Zeng S and Peng J. Administrative, technical, or material support: Liu W, Kang F, He Q, Hong Q Zhang W and Li J. All authors approved final version of manuscript.
Conflicts of Interest
This research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study is supported by grants from the National Natural Science Foundation of China (82104225), the Fundamental Research Funds for the Central Universities of Central South University (2022ZZTS0986) and the Natural Science Foundation of Hunan Province (2020JJ5953 and 2022JJ80044).
References
- 1. Huang X, Qin F, Meng Q, Dong M. Protein tyrosine phosphatase receptor type D (PTPRD)-mediated signaling pathways for the potential treatment of hepatocellular carcinoma: a narrative review. Ann Transl Med. 2020; 8:1192. https://doi.org/10.21037/atm-20-4733 [PubMed]
- 2. Zhang J, Huang H, Bian J, Sang X, Xu Y, Lu X, Zhao H. Safety, feasibility, and efficacy of associating liver partition and portal vein ligation for staged hepatectomy in treating hepatocellular carcinoma: a systematic review. Ann Transl Med. 2020; 8:1246. https://doi.org/10.21037/atm-20-2214 [PubMed]
- 3. Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J Hepatol. 2020; 72:250–61. https://doi.org/10.1016/j.jhep.2019.08.025 [PubMed]
- 4. Feng D, Wang M, Hu J, Li S, Zhao S, Li H, Liu L. Prognostic value of the albumin-bilirubin grade in patients with hepatocellular carcinoma and other liver diseases. Ann Transl Med. 2020; 8:553. https://doi.org/10.21037/atm.2020.02.116 [PubMed]
- 5. Yan S, Yang XF, Liu HL, Fu N, Ouyang Y, Qing K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol. 2015; 21:3492–8. https://doi.org/10.3748/wjg.v21.i12.3492 [PubMed]
- 6. Lopes-Marques M, Cunha I, Reis-Henriques MA, Santos MM, Castro LF. Diversity and history of the long-chain acyl-CoA synthetase (Acsl) gene family in vertebrates. BMC Evol Biol. 2013; 13:271. https://doi.org/10.1186/1471-2148-13-271 [PubMed]
- 7. Zhao H, Kosma DK, Lü S. Functional Role of Long-Chain Acyl-CoA Synthetases in Plant Development and Stress Responses. Front Plant Sci. 2021; 12:640996. https://doi.org/10.3389/fpls.2021.640996 [PubMed]
- 8. Ellis JM, Frahm JL, Li LO, Coleman RA. Acyl-coenzyme A synthetases in metabolic control. Curr Opin Lipidol. 2010; 21:212–7. https://doi.org/10.1097/mol.0b013e32833884bb [PubMed]
- 9. Tian W, Zheng H, Yang L, Li H, Tian Y, Wang Y, Lyu S, Brockmann GA, Kang X, Liu X. Dynamic Expression Profile, Regulatory Mechanism and Correlation with Egg-laying Performance of ACSF Gene Family in Chicken (Gallus gallus). Sci Rep. 2018; 8:8457. https://doi.org/10.1038/s41598-018-26903-6 [PubMed]
- 10. Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res. 2007; 48:2736–50. https://doi.org/10.1194/jlr.M700378-JLR200 [PubMed]
- 11. Watkins PA, Ellis JM. Peroxisomal acyl-CoA synthetases. Biochim Biophys Acta. 2012; 1822:1411–20. https://doi.org/10.1016/j.bbadis.2012.02.010 [PubMed]
- 12. Yamasaki M, Hasegawa S, Suzuki H, Hidai K, Saitoh Y, Fukui T. Acetoacetyl-CoA synthetase gene is abundant in rat adipose, and related with fatty acid synthesis in mature adipocytes. Biochem Biophys Res Commun. 2005; 335:215–9. https://doi.org/10.1016/j.bbrc.2005.07.053 [PubMed]
- 13. Alfares A, Nunez LD, Al-Thihli K, Mitchell J, Melançon S, Anastasio N, Ha KC, Majewski J, Rosenblatt DS, Braverman N. Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. J Med Genet. 2011; 48:602–5. https://doi.org/10.1136/jmedgenet-2011-100230 [PubMed]
- 14. Wang D, Wei G, Ma J, Cheng S, Jia L, Song X, Zhang M, Ju M, Wang L, Zhao L, Xin S. Identification of the prognostic value of ferroptosis-related gene signature in breast cancer patients. BMC Cancer. 2021; 21:645. https://doi.org/10.1186/s12885-021-08341-2 [PubMed]
- 15. Hasegawa S, Noda K, Maeda A, Matsuoka M, Yamasaki M, Fukui T. Acetoacetyl-CoA synthetase, a ketone body-utilizing enzyme, is controlled by SREBP-2 and affects serum cholesterol levels. Mol Genet Metab. 2012; 107:553–60. https://doi.org/10.1016/j.ymgme.2012.08.017 [PubMed]
- 16. Hasegawa S, Inoue D, Yamasaki M, Li C, Imai M, Takahashi N, Fukui T. Site-specific cleavage of acetoacetyl-CoA synthetase by legumain. FEBS Lett. 2016; 590:1592–601. https://doi.org/10.1002/1873-3468.12197 [PubMed]
- 17. Hasegawa S, Imai M, Yamasaki M, Takahashi N, Fukui T. Transcriptional regulation of acetoacetyl-CoA synthetase by Sp1 in neuroblastoma cells. Biochem Biophys Res Commun. 2018; 495:652–8. https://doi.org/10.1016/j.bbrc.2017.11.068 [PubMed]
- 18. Aguiló F, Camarero N, Relat J, Marrero PF, Haro D. Transcriptional regulation of the human acetoacetyl-CoA synthetase gene by PPARgamma. Biochem J. 2010; 427:255–64. https://doi.org/10.1042/BJ20090851 [PubMed]
- 19. Liu Z, Zhang H, Hu H, Cai Z, Lu C, Liang Q, Qian J, Wang C, Jiang L. A Novel Six-mRNA Signature Predicts Survival of Patients With Glioblastoma Multiforme. Front Genet. 2021; 12:634116. https://doi.org/10.3389/fgene.2021.634116 [PubMed]
- 20. Sun R, Kang X, Zhao Y, Wang Z, Wang R, Fu R, Li Y, Hu Y, Wang Z, Shan W, Zhou J, Tian X, Yao J. Sirtuin 3-mediated deacetylation of acyl-CoA synthetase family member 3 by protocatechuic acid attenuates non-alcoholic fatty liver disease. Br J Pharmacol. 2020; 177:4166–80. https://doi.org/10.1111/bph.15159 [PubMed]
- 21. Wang Y, Wo Y, Lu T, Sun X, Liu A, Dong Y, Du W, Su W, Huang Z, Jiao W. Circ-AASDH functions as the progression of early stage lung adenocarcinoma by targeting miR-140-3p to activate E2F7 expression. Transl Lung Cancer Res. 2021; 10:57–70. https://doi.org/10.21037/tlcr-20-1062 [PubMed]
- 22. Sun L, Song L, Wan Q, Wu G, Li X, Wang Y, Wang J, Liu Z, Zhong X, He X, Shen S, Pan X, Li A, et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015; 25:429–44. https://doi.org/10.1038/cr.2015.33 [PubMed]
- 23. Sangro B, Sarobe P, Hervás-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021; 18:525–43. https://doi.org/10.1038/s41575-021-00438-0 [PubMed]
- 24. Yamaguchi O, Imai H, Minemura H, Suzuki K, Wasamoto S, Umeda Y, Osaki T, Kasahara N, Uchino J, Sugiyama T, Ishihara S, Ishii H, Naruse I, et al. Efficacy and safety of immune checkpoint inhibitor monotherapy in pretreated elderly patients with non-small cell lung cancer. Cancer Chemother Pharmacol. 2020; 85:761–71. https://doi.org/10.1007/s00280-020-04055-7 [PubMed]
- 25. Schett A, Rothschild SI, Curioni-Fontecedro A, Krähenbühl S, Früh M, Schmid S, Driessen C, Joerger M. Predictive impact of antibiotics in patients with advanced non small-cell lung cancer receiving immune checkpoint inhibitors : Antibiotics immune checkpoint inhibitors in advanced NSCLC. Cancer Chemother Pharmacol. 2020; 85:121–31. https://doi.org/10.1007/s00280-019-03993-1 [PubMed]
- 26. Bhoopalan SV, Cross SJ, Panetta JC, Triplett BM. Pharmacokinetics of alemtuzumab in pediatric patients undergoing ex vivo T-cell-depleted haploidentical hematopoietic cell transplantation. Cancer Chemother Pharmacol. 2020; 86:711–7. https://doi.org/10.1007/s00280-020-04160-7 [PubMed]
- 27. Ju Q, Li XM, Zhang H, Zhao YJ. BRCA1-Associated Protein Is a Potential Prognostic Biomarker and Is Correlated With Immune Infiltration in Liver Hepatocellular Carcinoma: A Pan-Cancer Analysis. Front Mol Biosci. 2020; 7:573619. https://doi.org/10.3389/fmolb.2020.573619 [PubMed]
- 28. Ju Q, Li X, Zhang H, Yan S, Li Y, Zhao Y. NFE2L2 Is a Potential Prognostic Biomarker and Is Correlated with Immune Infiltration in Brain Lower Grade Glioma: A Pan-Cancer Analysis. Oxid Med Cell Longev. 2020; 2020:3580719. https://doi.org/10.1155/2020/3580719 [PubMed]
- 29. Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017; 26:49–70. https://doi.org/10.1016/j.cmet.2017.06.004 [PubMed]
- 30. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017; 17:209–22. https://doi.org/10.1038/nrc.2016.154 [PubMed]
- 31. Hu C, Liu X, Zeng Y, Liu J, Wu F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin Epigenetics. 2021; 13:166. https://doi.org/10.1186/s13148-021-01154-x [PubMed]
- 32. Simpson DJ, Chandra T. Epigenetic age prediction. Aging Cell. 2021; 20:e13452. https://doi.org/10.1111/acel.13452 [PubMed]
- 33. Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019; 47:W556–60. https://doi.org/10.1093/nar/gkz430 [PubMed]
- 34. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, Varambally S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017; 19:649–58. https://doi.org/10.1016/j.neo.2017.05.002 [PubMed]
- 35. Lian Q, Wang S, Zhang G, Wang D, Luo G, Tang J, Chen L, Gu J. HCCDB: A Database of Hepatocellular Carcinoma Expression Atlas. Genomics Proteomics Bioinformatics. 2018; 16:269–75. https://doi.org/10.1016/j.gpb.2018.07.003 [PubMed]
- 36. Gyorffy B, Gyorffy A, Tulassay Z. A “multiple testing” problémája és a genomiális kísérletekre alkalmazott megoldások [The problem of multiple testing and solutions for genome-wide studies]. Orv Hetil. 2005; 146:559–63. Hungarian. [PubMed]
- 37. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–4. https://doi.org/10.1158/2159-8290.CD-12-0095 [PubMed]
- 38. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019; 47:D607–13. https://doi.org/10.1093/nar/gky1131 [PubMed]
- 39. Otasek D, Morris JH, Bouças J, Pico AR, Demchak B. Cytoscape Automation: empowering workflow-based network analysis. Genome Biol. 2019; 20:185. https://doi.org/10.1186/s13059-019-1758-4 [PubMed]
- 40. Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010; 38:W214–20. https://doi.org/10.1093/nar/gkq537 [PubMed]
- 41. Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019; 47:W199–205. https://doi.org/10.1093/nar/gkz401 [PubMed]
- 42. Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020; 48:W509–14. https://doi.org/10.1093/nar/gkaa407 [PubMed]
- 43. Xiong Y, Wei Y, Gu Y, Zhang S, Lyu J, Zhang B, Chen C, Zhu J, Wang Y, Liu H, Zhang Y. DiseaseMeth version 2.0: a major expansion and update of the human disease methylation database. Nucleic Acids Res. 2017; 45:D888–95. https://doi.org/10.1093/nar/gkw1123 [PubMed]