



Introduction
Alzheimer’s disease (AD) is a chronic, neurodegenerative disorder and the most common cause of dementia, characterised by mental and functional impairment [1]. It is associated with accumulation of neuronal amyloid plaques and early lesions primarily in hippocampus. It is estimated that AD prevalence doubles every 5 years in individuals over the age of 65. The World Health Organization has stated that AD constitutes a growing universal public health issue with enormous consequences on both individuals and communities [2].
AD severely affects the life of the patient, causing dependency, disability and subsequent fatality [2, 3]. There are two basic types of AD: a) familial or early onset AD which is responsible for more than 5% of the disease incidence and; b) sporadic or late onset AD that accounts for 79% of the disease burden. Late onset AD is highly heritable and it is etiologically heterogeneous originating from a mixture of multiple genetic and environmental risk factors.
Some of the genes that have been associated with the risk of sporadic AD include the ABCA7, APOE, BIN1, CD2AP, CD33, CLU, CR1, EPHA1, MS4A4A/MS4A4E/MS4A6E, PICALM, and SORL1 genes [4]. More than 40 genes/loci have been associated with the risk of AD through the last 10 years, based on genome-wide association studies (GWAS) [5].
The most important genetic factor for AD is the Apolipoprotein E gene (APOE). The APOE gene codes for a 35 kDa glycoprotein, the apolipoprotein E (ApoE), which is strongly expressed in the brain [6]. There are three most common allelic variants in the APOE gene that alter the protein sequence leading to the formation of three different APOE isoforms: APOE2 (cys112, cys158), APOE3 (cys112, arg158), and APOE4 (arg112, arg158) [6, 7] arising from 3 alleles, respectively, ε2, ε3 and ε4. These alleles are associated with different ApoE roles [7]. The ε4 allele is the strongest risk factor for late-onset AD [8, 9], due to its association with increased amyloid deposition and is a known risk factor for cardiovascular disease (CVD) [10]. Individuals with one ε4 allele have a 2 to 3-fold elevated risk of developing AD, while those with two ε4 alleles have about 12-fold increased risk compared to individuals who do not have the ε4 allele. On the other hand, the ε2 allele of the APOE gene appears to display a protective role, as it is associated with reduced risk for AD [7], but remains a risk factor for Type III hyperlipidemia [11]. Thus, this common polymorphism is an excellent candidate to study in the development of genetic risk prediction models for AD.
The human APOE gene is situated on the long arm of chromosome 19q13.1, an AD-associated zone as reported by GWAS [5]. The lipolysis-stimulated lipoprotein receptor (LSR) gene is also located in the same region and encodes the lipolysis-stimulated lipoprotein receptor (LSR) which recognizes ApoE as ligand [12]. As an ApoE receptor, LSR is involved in the process of managing and maintaining lipid balance in the peripheral and central nervous system [13, 14] Recently, our team identified significant epistatic interactions between two LSR gene single nucleotide polymorphisms (SNPs) and APOE in AD patients [12], namely the rs34259399 and the rs916147 SNPs. The former is located on exon 6 and the latter is located in a splicing junction between intron 5 and exon 6. Both these SNPs have been studied by our group and preliminary results indicate a functionality in terms of gene expression modification, while the rs916147 was also associated with lipids in a population of obese individuals (Yen et al., unpublished results).
The vascular endothelial growth factor A (VEGF-A) is also considered as a risk factor for chronic diseases, including AD. The VEGF family plays important roles in angiogenic regulation, neurogenesis and neuronal survival [15]. Although an inverse relationship has also been demonstrated [16], decreased levels of VEGF-A in serum and cerebrospinal fluid have been linked with increased risk for AD and cognitive impairment [17, 18].
Our group has focused on the study of VEGF-A for many years and has been involved in two GWAS by which ten genetic variants have been identified explaining more than 50% of the individual variability of VEGF-A levels [19, 20]. This exceptionally high percentage of variability explained by these variants makes them optimal target SNPs to be used in the candidate genes association studies as determinants of VEGF-A levels. In several previous studies we have demonstrated associations of these polymorphisms with intermediate phenotypes of CVD and other chronic diseases, such as autoimmune thyroid disease and depression [21–25], where VEGF-A is involved in several of their pathophysiology pathways. We have initiated and we are coordinating the Vascular Endothelial growth factor European Genomic Federation Consortium - VEGF Consortium (Sophie Visvikis-Siest coordinator, http://www.vegfconsortium.org) for the study of VEGF-A in chronic diseases and personalized medicine [26].
The precise detection of individuals at high risk for AD is very critical for early diagnosis and appropriate management enabling closer monitoring, enhanced care, as well as closer supervision of targeted risk factors-based interventions [27]. Even though late-onset AD is known to be a multifactorial disease with a strong genetic component, the use of common genetic variations identified in GWAS in disease prediction modelling has been of limited value so far, given that such polymorphisms explain a small relative risk and proportion of the underlying genetic contribution. Thus, it has been proposed that the predictive ability of models would be improved with the inclusion of true functional variants, the incorporation of epistatic effects and the combination with nongenetic biomarkers [28].
In the present study, a machine-learning predictive model for AD risk was developed, using a case-control population of late-onset AD patients and combining novel variants on candidate molecules for AD that have not been assessed before (VEGF-A related variants), as well as their epistatic interactions with known functional polymorphisms (APOE and LSR).
Results
The characteristics of the final population used in the analysis are presented in Table 1. The mean age of participants was 74.59 and 69.94 years old in patients and non-patients, respectively. Men constituted 37.36% of the control population, whereas they contributed to almost half in the cases group (i.e., 47.58%).
Table 1. Populations’ characteristics.
| Age | Sex (male) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mean | SD | n | % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Controls (n = 182) | 74.59 | 8.39 | 68 | 37.36 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Patients (n = 145) | 69.94 | 8.66 | 69 | 47.58 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviation: SD: standard deviation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The SNPs included in the analysis, their minor allele frequencies and their annotation on the genome are presented in Table 2. Since the rs114694170 SNP did not abide by the Hardy-Weinberg equilibrium law, it was removed from the analysis.
Table 2. Characteristics of the genotyped polymorphisms.
| Variants | Genes | All | Controls | Patients | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MAF | HW p-value | MAF | HW p-value | MAF | HW p-value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs10761741 | JMJD1C | 0.45 | 0.77 | 0.47 | 0.77 | 0.44 | 0.77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs6921438 | LOC100132354 and C6orf223 | 0.42 | 0.43 | 0.41 | 0.43 | 0.42 | 0.43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs7043199 | VLDLR-AS1 | 0.24 | 0.90 | 0.26 | 0.90 | 0.23 | 0.90 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs6993770 | ZFPM2 | 0.33 | 0.71 | 0.33 | 0.71 | 0.34 | 0.71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs114694170 | MEF2C | 0.45 | 0.00 | 0.47 | 0.00 | 0.44 | 0.00 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs1740073 | POLR1C | 0.38 | 0.33 | 0.38 | 0.33 | 0.38 | 0.33 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2375981 | KCNV2 and VLDLR | 0.47 | 0.26 | 0.44 | 0.26 | 0.49 | 0.26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs34528081 | VEGF-A | 0.39 | 0.93 | 0.41 | 0.93 | 0.36 | 0.93 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs916147 | LSR | 0.37 | 0.29 | 0.38 | 0.29 | 0.36 | 0.29 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs34259399 | LSR | 0.14 | 0.76 | 0.12 | 0.76 | 0.15 | 0.76 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs4782371 | ZFPM1 | 0.30 | 0.96 | 0.34 | 0.96 | 0.26 | 0.96 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2639990 | ZADH2 | 0.09 | 0.94 | 0.12 | 0.94 | 0.07 | 0.94 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs429358 | APOE | 0.22 | 0.77 | 0.22 | 0.77 | 0.22 | 0.77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs7412 | APOE | 0.06 | 0.43 | 0.06 | 0.43 | 0.06 | 0.43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: MAF: Minor allele frequency; HW: Hardy-Weinberg. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
After applying the EN method to the data, the model with the highest accuracy included the variables presented in Figure 1. As expected, the presence of 2 ε4 alleles (homozygotes e4/e4) was the first variable to be linked with increased risk for AD, while the absence of ε4 allele was shown to be associated with decreased risk.
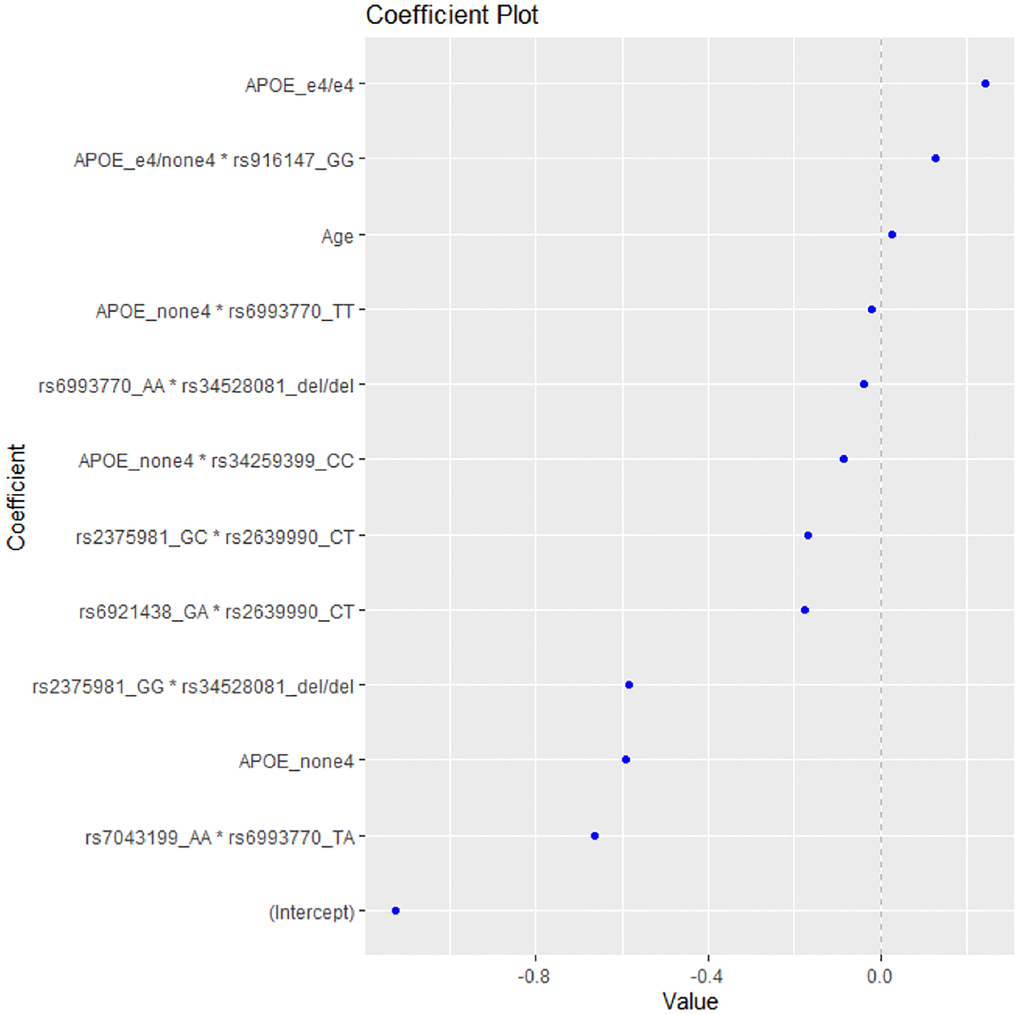
Figure 1. The coefficient plot of the EN model with the highest accuracy.
The epistatic interaction of ε4 allele heterozygotes with the rs916147 variant of LSR gene was also associated with higher risk for AD, followed by age as the third risk factor for AD (Table 3).
Table 3. Coefficients of the prediction model.
| Coefficient | ||
| Risk factor variants | APOE_ε4/ε4 | 0.24 |
| APOE_ε4/non ε4*rs916147 | 0.13 | |
| Age | 0.03 | |
| Protective factor variants | rs7043199*rs6993770 | −0.66 |
| APOE_non ε4 | −0.59 | |
| rs2375981*rs34528081 | −0.58 | |
| rs6921438*rs2639990 | −0.18 | |
| rs2375981*rs2639990 | −0.17 | |
| APOE_non ε4*rs34259399 | −0.09 | |
| rs6993770*rs34528081 | −0.04 | |
| APOE_non ε4*rs6993770 | −0.02 |
The model included five epistatic interactions between VEGF-A-related variants, one APOE*LSR (rs34259399) interaction and one APOE*VEGF-A-related polymorphism interaction (for the rs6993770 SNP), associated with decreased risk for AD (Table 3). The strongest predictive factor of the model appears to be the interaction rs7043199*rs6993770, which decreases the risk of AD, thus playing a protective role, followed by the absence of the e4 APOE allele and the interaction rs2375981*rs34528081.
The accuracy of the model is 72% with a confidence interval CI = (0.6, 0.8) for 95%. The above accuracy has been calculated under a model with P-value: 0.009. The area under the curve (AUC) of the model is 81%, which indicates the detection of true positives (true cases predicted as cases) versus false positives (true controls predictive as cases).
Based on the annotation of the identified polymorphisms of the prediction model, we identified the following list of genes involved in AD risk: VLDLR-AS1, APOE, KCNV2, ZADH2, C6orf223, LSR, ZFPM2, and VEGF-A. After uploading the list to the IPA tool, the top five diseases found to be associated with the aforementioned were CVD, ophthalmic diseases, organismal injury and abnormalities, neurological diseases, and cancer.
Subsequently, a network linking most of these genes and other mediators was developed using IPA tool (Figure 2). The genes and mediators of this network have functions that correspond to cancer, dermatological diseases and conditions, organismal injury and abnormalities.
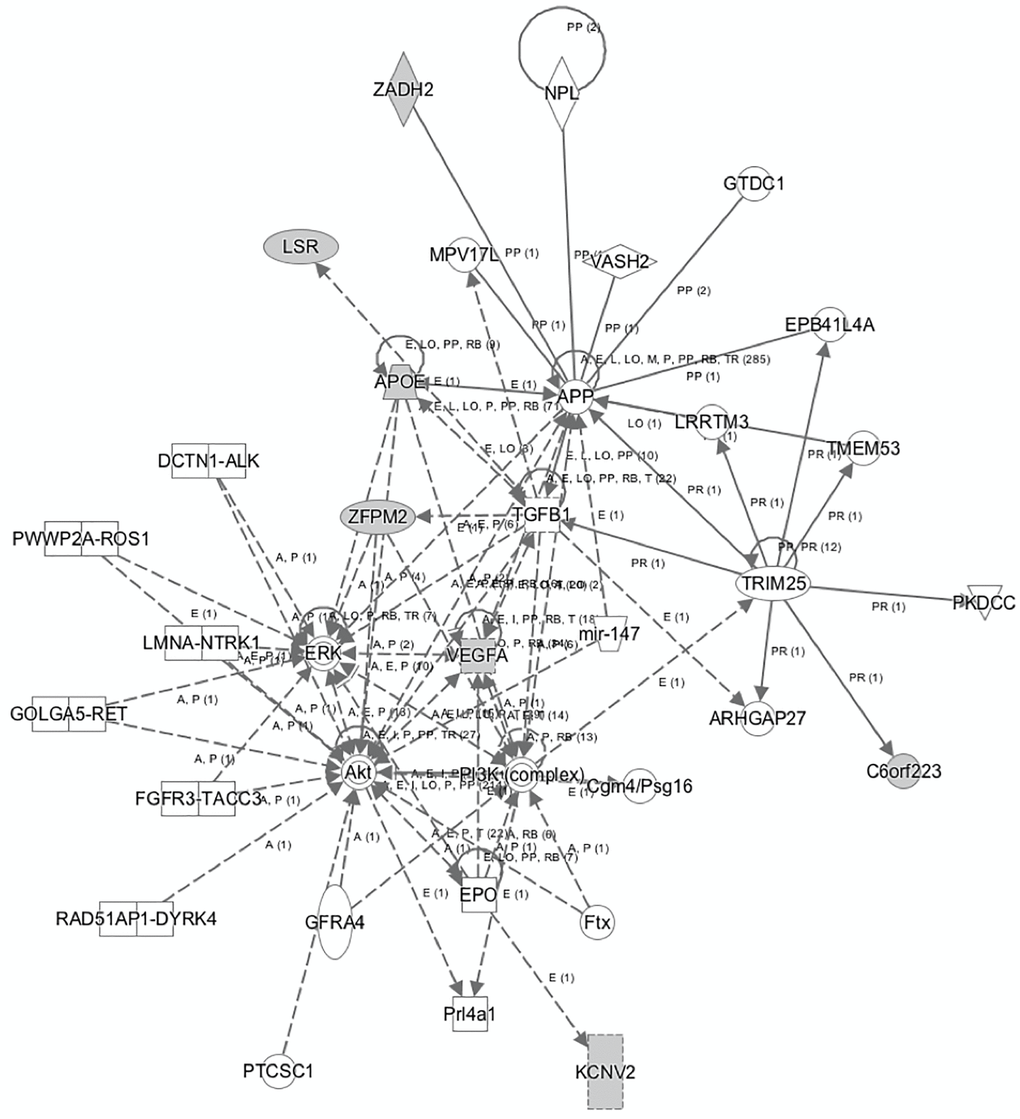
Figure 2. The network that links most of the identified genes that predict AD as generated by IPA tool.
The network displayed in Figure 2 shows the multiple associations identified from the analyses, with the VEGF-A gene located at the center of many of the observed relationships involved in several metabolic pathways connected to cancer and inflammation. In this context, VEGF-A appears to interact with the EPO, AKT, ERK, TGFB1 genes and the PI3K complex, indicating a potential effect in cellular activities, such as proliferation and apoptosis, where the AKT, PI3K, ERK and TGFB1 cascades and pathways play a significant role. The identified network further enhances the aforementioned notions, by highlighting respective relationships between the ERK, AKT, PI3K, TGFB1, APP and the APOE gene. In addition, IPA analysis shows direct association between APP and the VEGF-A gene, among other associations including the TGFB1, MPV17L, ZADH2, NPL, VASH2, GTDC1, EPB41L4A, LRRTM3, TRIM25, and ARHGAP27 genes.
Discussion
Early detection of AD is important for the prognosis of patients, especially to initiate treatments during its pre-symptomatic phase and before the pathological amyloid and tau protein accumulation and the extensive brain damage, which can significantly decrease patient autonomy. Multiple approaches have been used for the early identification of AD-linked parameters including medical assessment, as well as cognitive evaluations and imaging analyses that exploit multimodal biological and molecular features present in AD. Moreover, studies that use regression analyses have demonstrated the relation between AD and variables including clinical examination and cognitive test scores [27]. More recently, artificial intelligence approaches have been used, including supervised predictive analytics tools such as support vector machines, random forests, and artificial neural networks to distinguish AD cases from controls, and for identifying individuals with higher risk of AD in a given period of time [27]. In the present study, we used EN [29], a supervised machine learning method, to assess the role of genetic variants of common AD biomarkers on the prediction of AD risk.
The EN revealed one model with an adequate accuracy of 72% that mostly included epistatic interactions between the assessed variants as predictors of AD risk. The first and strongest genetic predictor was the APOE e4 allele, which is in agreement with a large volume of scientific results that support this finding [9, 28]. Despite this, the precise processes by which ApoE affects AD remains unclear. AD is characterized by two main features encompassing the existence of extracellular deposition of Aβ generating amyloid plaques, and intracellular occurrence of neurofibrillary tangles (NFT) consisting of clusters of hyper-phosphorylated tau protein. ApoE may be associated with AD through its direct synergism with Aβ proteins. Indeed, the ApoE has been found in Aβ plaques of AD brains [30], and knock-out studies of APOE gene in mice have revealed that ApoE is critical for the initiation and retention of Aβ plaques. Furthermore, the direct synergistic link between ApoE and tau protein may also contribute to the association of ApoE and AD. Previously, the existence of ApoE was detected in Tau-NFT mediated deposits [31]. In addition, up-regulation of APOE4 in neuronal cells of genetically engineered mice triggered a rise in tau hyper-phosphorylation induced by Erk stimulation [6].
Using the EN model, we observed the significant role of the interactions between APOE e4 allele polymorphism and the 2 SNPs of the LSR gene which we have identified in a previous study [12]. LSR is present in the central nervous system [13] and the lsr+/− mice display increased memory deficits following intracerebroventricular injection of the oligomeric soluble form of the β-amyloid peptide [32]. Animal studies have shown the presence of LSR gene transcripts in endothelial cells (ECs) of the blood-brain barrier (BBB) [33]. In lsr−/− knockout mice, the BBB does not appear to seal during embryogenesis [34], highlighting LSR’s critical role in maintaining BBB integrity. Furthermore, given its role as an ApoE receptor, LSR is involved in lipid metabolism in the brain [13]; Herzine et al, manuscript in preparation) and could, therefore, play a role in AD development, with the present genetic associations further supporting this hypothesis.
The novel results of the present model include five epistatic interactions between VEGF-A-related variants and one APOE*VEGF-A-related polymorphism interaction (rs6993770) associated with decreased risk for AD.
VEGF-A has been proposed as a promising novel therapeutic approach for AD [35]. Transplantation of mesenchymal stem cells into the double transgenic AD mouse model (APPswe/PS1dE9 mutations) leads to improvement of cognitive function [36]. Furthermore, higher VEGF-A concentration in the cerebrospinal fluid has been associated with slower cognitive decline in patients with AD risk [17]. Thus, VEGF-A could be considered as a protective factor for individuals having high risk of AD.
Concerning the interactions between APOE and VEGF-A, this is consistent with studies showing that VEGF-A exerts a neuroprotective effect in humanized APOE ε4 mice, where treatment with VEGF-A leads to improvements of behavioral deficits [37]. A recent study also demonstrated that APOE ε4 interacts with VEGF-A gene expression in the brain to affect cognitive performance [38]. Therefore, VEGF-A alone or in interaction with ApoE, seems to play an important role in AD risk, consistent with the results of this study.
The polymorphism rs6993770 is involved in 2 epistatic interactions with other VEGF-A-related SNPs and one interaction with APOE ε4 allele. This constitutes an intronic variant of the ZFPM2 (zinc finger protein, FOG family member 2) gene. The latter codes for the FOG family member 2, which is linked with repression of GATA mediated transcriptional activation [39, 40] and thus with hematopoiesis. The T minor allele has been associated with decreased VEGF-A levels [19, 20]. In the present model, this allele interacted with the non-ε4 alleles of APOE gene to decrease the risk for AD. Also, the TA genotype of rs6993770 interacted with the AA genotype of rs7043199 to decrease the risk for AD. The rs7043199 SNP in an intronic variant of VLDLR-AS1 gene and is located close to the VLDLR gene and its A allele has been associated with decreased VEGF-A levels [19, 20]. The VLDL receptor is a member of the low-density lipoprotein receptor family and binds ApoE. It is involved in pathways essential for the development of laminated structures and for the synaptic plasticity of the brain and is, thus, considered as a receptor that could be involved in the development of AD [41]. It is important to note that this interaction presented the highest coefficient, thus is the strongest predictive factor of the model, with a protective role against AD higher than that of the absence of e4 APOE allele. Our team was among the first to identify the allele 4 of the APOE gene as the strongest genetic risk factor for AD [8] and in this investigation we are proposing the rs7043199*rs6993770 interaction as a strong protective factor against AD. Finally, the A major allele of the rs6993770 (AA genotype) interacted with the deletion genotype of the rs34528081, which is an intergenic SNP close to VEGF-A gene, with the overall result of decreasing the risk for AD. Both alleles of these 2 SNPs have been associated with increased levels of VEGF-A [19, 20]. Thus, this interaction could be linked with further increase in VEGF-A levels that have been shown to have a protective effect against AD [17, 18]. The same deletion genotype of rs34528081 also interacted with the GG genotype of rs2375981, which is an intergenic SNP between KCNV2 and VLDLR gene and whose G allele has been associated with decreased levels of VEGF-A [19, 20]. This interaction that is associated with decreased risk of AD could either be explained by a modification of VEGF-A levels or through an effect on the VLDLR gene. It has a very high coefficient in the model, similar to the absence of APOE ε4 allele, and thus has a strong protective effect. Furthermore, the GC genotype of rs2375981 interacted with the CT genotype of rs2639990 to decrease the risk of AD. This is an intronic variant of ZADH2 gene and the T allele has been associated with increased levels of VEGF-A [19, 20]. Hypomethylation of differentially methylated positions located on ZADH2 has been observed in AD patients and have been associated with memory performance and cerebrospinal fluid levels of Aβ and tau [42], thus indicating a role of this gene on AD. Also, the same genotype of this SNP interacted with the GA genotype of rs6921438 to decrease the risk of AD. The rs6921438 is an intergenic SNP located between LOC100132354 (lnc-RNA) and the C6orf223 gene (encoding an uncharacterized protein) and is near the VEGF-A gene. The A allele is associated with decreased levels of VEGF-A [19, 20] and explains the highest percentage of VEGF-A levels variability (41.19%). This SNP (A allele) has also been associated with decreased HDL and increased LDL [22, 43]. It is thus a marker linked with both VEGF-A levels and lipid metabolism, which could mediate its relationship with AD.
The strengths of the present study include the use of the EN method in the development of the prediction model, which is a machine learning approach more powerful than classical statistics methodologies. Furthermore, the identified model showed a sufficient accuracy of 72% and an AUC of 81%. The accuracy of the model is comparable to other tools [44–46] and this is very important as our model uses genetic factors. In fact, in other studies, the addition of a genetic score led to a small improvement of prediction of the classical variables model [47]. In a recent review, 61 papers describing dementia risk models were identified and most of them had moderate-to-high predictive ability (AUC > 0.70). The highest AUC value was 0.932 [48] and our AUC is 81%. All these data highlight the satisfactory accuracy of the identified prediction model.
A few limitations of the study, however, include the relatively small sample size and the lack of VEGF-A levels’ measurements in the studied populations.
In summary, the prediction model proposed in the present study consists of 8 epistatic interactions that, in combination with the APOE ε4 allele, directly affect the risk for AD. These interactions involve 9 polymorphisms in 8 genes: VLDLR-AS1, APOE, KCNV2, ZADH2, C6orf223, LSR, ZFPM2, and VEGF-A.
IPA analysis highlighted relationships between the identified genes and neurological diseases, within the first top five disorders associated with said genes, including CVD and cancer. This finding indicates that the genetic determinants of the selected biomarkers (VEGF-A, LSR and APOE) could act as common links between important chronic diseases. In fact, most of these genes are shown to be linked in the context of an enlarged common network, the functions of which correspond to cancer, dermatological diseases and conditions, organismal injury and abnormalities.
In conclusion, these novel epistatic interactions between APOE, LSR and VEGF-A related polymorphisms allow for prediction of AD risk, constituting not only a useful prediction model, but also providing new insights about molecular mechanisms that can be involved in AD development which could be useful as biomarkers and/or treatment targets. We are also proposing two epistatic interactions (rs7043199*rs6993770 and the rs2375981*rs34528081) between VEGF-A-related polymorphisms as strong protective factors against AD.
Methods
Population and data collection
The study population consisted of 1078 (602 controls and 476 cases) unrelated adults of European origin, recruited during the period 1996–1998. The study was approved by the related ethics committees and all participants provided written consent prior to their enrollment in the study.
The recruitment and data collection procedures of the present population have been previously extensively described [9]. The clinical diagnosis of AD was based on 5 criteria: the National Institute of Neurological and Communicative Disorders and Stroke and the AD and Related Disorders Association classification of probable AD [28]; the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition [28]; the International Classification of Diseases and Related Health Problems, 10th Edition [28]; the Mini Mental State Examination score equal to or less than 23 (Mini Mental State Examination >23 for controls) [28]; and, lastly the modified ischemic scale less than three [28]. Secondary causes of dementia were excluded by computerized tomography scan of the brain. Individuals with other chronic or neurological diseases, such as cancer and Parkinson's diseases, respectively, were excluded from the study. Participants serving as controls followed similar assessment and interviews as cases, except for the computerized tomography scan. All controls presented a Mini Mental State Examination >23 and were free of dementia.
Genotyping data were available in a subsample of the population. The final sample size after quality control for genotyping and outliers exclusion was 323 individuals (143 AD cases and 180 controls).
Genotyping analyses
DNA was extracted from peripheral blood [49] and all samples were stored in biobanks of the BRC IGE-PCV (Biological Resources Center ‘Interactions Gène-Environnement en Physiopathologie Cardio-Vasculaire’ BB-0033-00051). Two SNPs of the LSR gene (rs34259399 and rs916147) previously identified as candidate variants for AD were genotyped, along with the 10 VEGF-A-related polymorphisms (rs6921438, rs7043199, rs6993770, rs2375981, rs34528081, rs4782371, rs2639990, rs10761741, rs114694170, rs1740073) and the common APOE variants. The genotyping analyses were performed in LGC genomics (http://www.lgcgroup.com) using the competitive allele-specific PCR (KASP) chemistry coupled with a Förster resonance energy transfer-based genotyping system (http://www.kbioscience.co.uk/reagents/KASP/KASP.html). Two of the APOE common polymorphisms rs429358 (Cys112Arg) and rs7412 (Arg158Cys) were genotyped as previously described [50].
Statistical analyses
The agreement of the frequencies of genotypes with the Hardy-Weinberg equilibrium was tested using the chi-squared test.
For the purposes of developing the prediction model, a machine learning method was applied, entitled “Elastic Net” (EN) [29], with the aim of identifying the strongest predictors for the risk of AD combining all genotyping data (direct effects and epistatic interactions), as well as age and sex. In short, the method tests hundreds of logistic regression models and penalizes each of them, in order to reach the final, optimal one. The former constitutes an extensively used method, especially in the development of disease prediction models with special attention to the integration of omics data [51–54]. Furthermore, the method can allow accurate predictions with smaller sample sizes as it tolerates a big number of predictors [29] and it is also used in analyses entailing genetic data, as well as GWAS studies [55, 56] and it is considered to function better in cases where gene * environment interactions are involved in the prediction of a disease [57]. Comparison studies have shown that EN is a powerful tool, especially when additive gene effect is expected [58]. It has also been previously used in AD risk prediction with clinical factors, imaging and omics [59–61] but not to study the effect of candidate genes as predicting factors.
In the context of the present analyses, APOE genotypes were divided into 3 groups according to the potential presence of ε4 allele (coded as 2 = ε4/4; 1 = ε2/4, ε3/4; and 0 = ε2/2, ε3/2, ε3/3). The complete dataset was split into two separate datasets, the “train” dataset that included the 80% of the whole population (AD patients + controls) and the “test” dataset that included the remaining 20% of the whole population (AD patients + controls). This selection was random and was performed by the R command ‘createDataPartition’ that is specific to this. The EN analysis was performed using the R software.
Pathway analysis
All genes identified to participate in AD risk prediction were further assessed using the QIAGEN Ingenuity Pathway Analysis (IPA) tool, in order to examine potential relationships and identify causal links, with the overall aim of proposing mechanisms to explain the results deriving from the prediction model.
Author Contributions
VSS conceived hypothesis and with PAM the design of the study. PAM designed and performed the statistical analysis. MC, MH, LJ and FP contributed to data collection and genotyping. PAM, SMG, KM, DG, FTY and VSS contributed to data interpretation. PAM drafted the first version of the manuscript. All authors critically contributed to the final version of the manuscript. VSS supervised and assured project implementation.
Acknowledgments
We would like to thank the “Agence Nationale de la Recherche - Programme d’Investissements d’Avenir” - PIA (grant number ANR-15RHU-0004), the “Fondation Plan Alzheimer”- DYSLIP-AD project – 2019, the “Region Grand Est” - project “Gutenberg Chair” (Georges DEDOUSSIS) – 2018 and the VEGF Consortium (http://vegfconsortium.org).
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
The present investigation was funded in part by the “Agence Nationale de la Recherche - Programme d’Investissements d’Avenir” - PIA (grant number ANR-15RHU-0004), the “Fondation Plan Alzheimer”- DYSLIP-AD project - 2019 and the “Region Grand Est_ Gutenberg Chair” project (Georges DEDOUSSIS) – 2018.
References
- 1. Soria Lopez JA, González HM, Léger GC. Alzheimer's disease. Handb Clin Neurol. 2019; 167:231–55. https://doi.org/10.1016/B978-0-12-804766-8.00013-3 [PubMed]
- 2. Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol. 2018; 25:59–70. https://doi.org/10.1111/ene.13439 [PubMed]
- 3. Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012; 2:a006239. https://doi.org/10.1101/cshperspect.a006239 [PubMed]
- 4. Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012; 2:a006296. https://doi.org/10.1101/cshperspect.a006296 [PubMed]
- 5. Institute NHGR. GWAS Catalog. 2021. https://www.ebi.ac.uk/gwas/.
- 6. Muñoz SS, Garner B, Ooi L. Understanding the Role of ApoE Fragments in Alzheimer's Disease. Neurochem Res. 2019; 44:1297–305. https://doi.org/10.1007/s11064-018-2629-1 [PubMed]
- 7. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009; 63:287–303. https://doi.org/10.1016/j.neuron.2009.06.026 [PubMed]
- 8. Lucotte G, David F, Visvikis S, Leininger-Müller B, Siest G, Babron MC, Couderc R. Apolipoprotein E-epsilon 4 allele and Alzheimer's disease. Lancet. 1993; 342:1309. [PubMed]
- 9. Siest G, Bertrand P, Qin B, Herbeth B, Serot JM, Masana L, Ribalta J, Passmore AP, Evans A, Ferrari M, Franceschi M, Shepherd J, Cuchel M, et al. Apolipoprotein E polymorphism and serum concentration in Alzheimer's disease in nine European centres: the ApoEurope study. ApoEurope group. Clin Chem Lab Med. 2000; 38:721–30. https://doi.org/10.1515/CCLM.2000.102 [PubMed]
- 10. Liu S, Liu J, Weng R, Gu X, Zhong Z. Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc Disord. 2019; 19:213. https://doi.org/10.1186/s12872-019-1194-0 [PubMed]
- 11. Geisel J, Bunte T, Bodis M, Oette K, Herrmann W. Apolipoprotein E2/E2 genotype in combination with mutations in the LDL receptor gene causes type III hyperlipoproteinemia. Clin Chem Lab Med. 2002; 40:475–9. https://doi.org/10.1515/CCLM.2002.082 [PubMed]
- 12. Xie T, Akbar S, Stathopoulou MG, Oster T, Masson C, Yen FT, Visvikis-Siest S. Epistatic interaction of apolipoprotein E and lipolysis-stimulated lipoprotein receptor genetic variants is associated with Alzheimer's disease. Neurobiol Aging. 2018; 69:292.e1–e5. https://doi.org/10.1016/j.neurobiolaging.2018.04.013 [PubMed]
- 13. Stenger C, Pinçon A, Hanse M, Royer L, Comte A, Koziel V, Olivier JL, Pillot T, Yen FT. Brain region-specific immunolocalization of the lipolysis-stimulated lipoprotein receptor (LSR) and altered cholesterol distribution in aged LSR+/- mice. J Neurochem. 2012; 123:467–76. https://doi.org/10.1111/j.1471-4159.2012.07922.x [PubMed]
- 14. Yen FT, Roitel O, Bonnard L, Notet V, Pratte D, Stenger C, Magueur E, Bihain BE. Lipolysis stimulated lipoprotein receptor: a novel molecular link between hyperlipidemia, weight gain, and atherosclerosis in mice. J Biol Chem. 2008; 283:25650–9. https://doi.org/10.1074/jbc.M801027200 [PubMed]
- 15. Beazley-Long N, Hua J, Jehle T, Hulse RP, Dersch R, Lehrling C, Bevan H, Qiu Y, Lagrèze WA, Wynick D, Churchill AJ, Kehoe P, Harper SJ, et al. VEGF-A165b is an endogenous neuroprotective splice isoform of vascular endothelial growth factor A in vivo and in vitro. Am J Pathol. 2013; 183:918–29. https://doi.org/10.1016/j.ajpath.2013.05.031 [PubMed]
- 16. Tarkowski E, Issa R, Sjögren M, Wallin A, Blennow K, Tarkowski A, Kumar P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer's disease and vascular dementia. Neurobiol Aging. 2002; 23:237–43. [PubMed]
- 17. Hohman TJ, Bell SP, Jefferson AL, and Alzheimer’s Disease Neuroimaging Initiative. The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: exploring interactions with biomarkers of Alzheimer disease. JAMA Neurol. 2015; 72:520–9. https://doi.org/10.1001/jamaneurol.2014.4761 [PubMed]
- 18. Huang L, Jia J, Liu R. Decreased serum levels of the angiogenic factors VEGF and TGF-β1 in Alzheimer's disease and amnestic mild cognitive impairment. Neurosci Lett. 2013; 550:60–3. https://doi.org/10.1016/j.neulet.2013.06.031 [PubMed]
- 19. Debette S, Visvikis-Siest S, Chen MH, Ndiaye NC, Song C, Destefano A, Safa R, Azimi Nezhad M, Sawyer D, Marteau JB, Xanthakis V, Siest G, Sullivan L, et al. Identification of cis- and trans-acting genetic variants explaining up to half the variation in circulating vascular endothelial growth factor levels. Circ Res. 2011; 109:554–63. https://doi.org/10.1161/CIRCRESAHA.111.243790 [PubMed]
- 20. Choi SH, Ruggiero D, Sorice R, Song C, Nutile T, Vernon Smith A, Concas MP, Traglia M, Barbieri C, Ndiaye NC, Stathopoulou MG, Lagou V, Maestrale GB, et al. Six Novel Loci Associated with Circulating VEGF Levels Identified by a Meta-analysis of Genome-Wide Association Studies. PLoS Genet. 2016; 12:e1005874. https://doi.org/10.1371/journal.pgen.1005874 [PubMed]
- 21. Azimi-Nezhad M, Stathopoulou MG, Bonnefond A, Rancier M, Saleh A, Lamont J, Fitzgerald P, Ndiaye NC, Visvikis-Siest S. Associations of vascular endothelial growth factor (VEGF) with adhesion and inflammation molecules in a healthy population. Cytokine. 2013; 61:602–7. https://doi.org/10.1016/j.cyto.2012.10.024 [PubMed]
- 22. Stathopoulou MG, Bonnefond A, Ndiaye NC, Azimi-Nezhad M, El Shamieh S, Saleh A, Rancier M, Siest G, Lamont J, Fitzgerald P, Visvikis-Siest S. A common variant highly associated with plasma VEGFA levels also contributes to the variation of both LDL-C and HDL-C. J Lipid Res. 2013; 54:535–41. https://doi.org/10.1194/jlr.P030551 [PubMed]
- 23. Xie T, Stathopoulou MG, de Andrés F, Siest G, Murray H, Martin M, Cobaleda J, Delgado A, Lamont J, Peñas-LIedó E, LLerena A, Visvikis-Siest S. VEGF-related polymorphisms identified by GWAS and risk for major depression. Transl Psychiatry. 2017; 7:e1055. https://doi.org/10.1038/tp.2017.36 [PubMed]
- 24. Rancier M, Zaaber I, Stathopoulou MG, Chatelin J, Saleh A, Marmouch H, El Shamieh S, Masson C, Murray H, Lamont J, Fitzgerald P, Mahjoub S, Said K, et al. Pro- and anti-angiogenic VEGF mRNAs in autoimmune thyroid diseases. Autoimmunity. 2016; 49:366–72. https://doi.org/10.1080/08916934.2016.1199019 [PubMed]
- 25. Zaaber I, Rancier M, Stathopoulou MG, Saleh A, Marmouch H, Masson C, Murray H, Kurth MJ, Lamont J, Fitzgerald P, Mahjoub S, Said K, Bel Hadj Jrad Tensaout B, et al. Plasma VEGF-related polymorphisms are implied in autoimmune thyroid diseases. Autoimmunity. 2016; 49:229–35. https://doi.org/10.3109/08916934.2016.1151005 [PubMed]
- 26. Stathopoulou MG, Xie T, Ruggiero D, Chatelin J, Rancier M, Weryha G, Kurth MJ, Aldasoro Arguinano AA, Gorenjak V, Petrelis AM, Dagher G, Dedoussis G, Deloukas P, et al, and VEGF Consortium. A transnational collaborative network dedicated to the study and applications of the vascular endothelial growth factor-A in medical practice: the VEGF Consortium. Clin Chem Lab Med. 2018; 56:83–6. https://doi.org/10.1515/cclm-2017-0838 [PubMed]
- 27. Marinescu RV, Oxtoby NP, Young AL, Bron EE, Toga AW, Weiner MW, Barkhof F, Fox NC, Golland P, Klein S, Alexander DC. TADPOLE Challenge: Accurate Alzheimer's disease prediction through crowdsourced forecasting of future data. Predict Intell Med. 2019; 11843:1–10. https://doi.org/10.1007/978-3-030-32281-6_1 [PubMed]
- 28. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016; 18:421–30. https://doi.org/10.1038/gim.2015.117 [PubMed]
- 29. Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc Ser B (Stat Methodol). 2005; 67:301–20. https://doi.org/10.1111/j.1467-9868.2005.00503.x
- 30. Burns MP, Noble WJ, Olm V, Gaynor K, Casey E, LaFrancois J, Wang L, Duff K. Co-localization of cholesterol, apolipoprotein E and fibrillar Abeta in amyloid plaques. Brain Res Mol Brain Res. 2003; 110:119–25. https://doi.org/10.1016/s0169-328x(02)00647-2 [PubMed]
- 31. Yamaguchi H, Ishiguro K, Sugihara S, Nakazato Y, Kawarabayashi T, Sun X, Hirai S. Presence of apolipoprotein E on extracellular neurofibrillary tangles and on meningeal blood vessels precedes the Alzheimer beta-amyloid deposition. Acta Neuropathol. 1994; 88:413–9. https://doi.org/10.1007/BF00389492 [PubMed]
- 32. Pinçon A, Thomas MH, Huguet M, Allouche A, Colin JC, Georges A, Derrien A, Lanhers MC, Malaplate-Armand C, Oster T, Corbier C, Pillot T, Olivier JL, Yen FT. Increased susceptibility of dyslipidemic LSR+/- mice to amyloid stress is associated with changes in cortical cholesterol levels. J Alzheimers Dis. 2015; 45:195–204. https://doi.org/10.3233/JAD-142127 [PubMed]
- 33. Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010; 5:e13741. https://doi.org/10.1371/journal.pone.0013741 [PubMed]
- 34. Sohet F, Lin C, Munji RN, Lee SY, Ruderisch N, Soung A, Arnold TD, Derugin N, Vexler ZS, Yen FT, Daneman R. LSR/angulin-1 is a tricellular tight junction protein involved in blood-brain barrier formation. J Cell Biol. 2015; 208:703–11. https://doi.org/10.1083/jcb.201410131 [PubMed]
- 35. Shim JW, Madsen JR. VEGF Signaling in Neurological Disorders. Int J Mol Sci. 2018; 19:275. https://doi.org/10.3390/ijms19010275 [PubMed]
- 36. Garcia KO, Ornellas FL, Martin PK, Patti CL, Mello LE, Frussa-Filho R, Han SW, Longo BM. Therapeutic effects of the transplantation of VEGF overexpressing bone marrow mesenchymal stem cells in the hippocampus of murine model of Alzheimer's disease. Front Aging Neurosci. 2014; 6:30. https://doi.org/10.3389/fnagi.2014.00030 [PubMed]
- 37. Salomon-Zimri S, Glat MJ, Barhum Y, Luz I, Boehm-Cagan A, Liraz O, Ben-Zur T, Offen D, Michaelson DM. Reversal of ApoE4-Driven Brain Pathology by Vascular Endothelial Growth Factor Treatment. J Alzheimers Dis. 2016; 53:1443–58. https://doi.org/10.3233/JAD-160182 [PubMed]
- 38. Moore AM, Mahoney E, Dumitrescu L, De Jager PL, Koran MEI, Petyuk VA, Robinson RA, Ruderfer DM, Cox NJ, Schneider JA, Bennett DA, Jefferson AL, Hohman TJ. APOE ε4-specific associations of VEGF gene family expression with cognitive aging and Alzheimer's disease. Neurobiol Aging. 2020; 87:18–25. https://doi.org/10.1016/j.neurobiolaging.2019.10.021 [PubMed]
- 39. Svensson EC, Tufts RL, Polk CE, Leiden JM. Molecular cloning of FOG-2: a modulator of transcription factor GATA-4 in cardiomyocytes. Proc Natl Acad Sci U S A. 1999; 96:956–61. https://doi.org/10.1073/pnas.96.3.956 [PubMed]
- 40. Lin AC, Roche AE, Wilk J, Svensson EC. The N termini of Friend of GATA (FOG) proteins define a novel transcriptional repression motif and a superfamily of transcriptional repressors. J Biol Chem. 2004; 279:55017–23. https://doi.org/10.1074/jbc.M411240200 [PubMed]
- 41. Dlugosz P, Nimpf J. The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor. Int J Mol Sci. 2018; 19:3090. https://doi.org/10.3390/ijms19103090 [PubMed]
- 42. Madrid A, Hogan KJ, Papale LA, Clark LR, Asthana S, Johnson SC, Alisch RS. DNA Hypomethylation in Blood Links B3GALT4 and ZADH2 to Alzheimer's Disease. J Alzheimers Dis. 2018; 66:927–34. https://doi.org/10.3233/JAD-180592 [PubMed]
- 43. Belhassan K, Ouldim K, Sefiani AA. Genetics and genomic medicine in Morocco: the present hope can make the future bright. Mol Genet Genomic Med. 2016; 4:588–98. https://doi.org/10.1002/mgg3.255 [PubMed]
- 44. Danso SO, Zeng Z, Muniz-Terrera G, Ritchie CW. Developing an Explainable Machine Learning-Based Personalised Dementia Risk Prediction Model: A Transfer Learning Approach With Ensemble Learning Algorithms. Front Big Data. 2021; 4:613047. https://doi.org/10.3389/fdata.2021.613047 [PubMed]
- 45. Qiu S, Joshi PS, Miller MI, Xue C, Zhou X, Karjadi C, Chang GH, Joshi AS, Dwyer B, Zhu S, Kaku M, Zhou Y, Alderazi YJ, et al. Development and validation of an interpretable deep learning framework for Alzheimer's disease classification. Brain. 2020; 143:1920–33. https://doi.org/10.1093/brain/awaa137 [PubMed]
- 46. Wang L, Li P, Hou M, Zhang X, Cao X, Li H. Construction of a risk prediction model for Alzheimer's disease in the elderly population. BMC Neurol. 2021; 21:271. https://doi.org/10.1186/s12883-021-02276-8 [PubMed]
- 47. Chouraki V, Reitz C, Maury F, Bis JC, Bellenguez C, Yu L, Jakobsdottir J, Mukherjee S, Adams HH, Choi SH, Larson EB, Fitzpatrick A, Uitterlinden AG, et al, and International Genomics of Alzheimer’s Project. Evaluation of a Genetic Risk Score to Improve Risk Prediction for Alzheimer's Disease. J Alzheimers Dis. 2016; 53:921–32. https://doi.org/10.3233/JAD-150749 [PubMed]
- 48. Hou XH, Feng L, Zhang C, Cao XP, Tan L, Yu JT. Models for predicting risk of dementia: a systematic review. J Neurol Neurosurg Psychiatry. 2019; 90:373–9. https://doi.org/10.1136/jnnp-2018-318212 [PubMed]
- 49. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988; 16:1215. https://doi.org/10.1093/nar/16.3.1215 [PubMed]
- 50. Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990; 31:545–8. [PubMed]
- 51. Li J, Guasch-Ferré M, Chung W, Ruiz-Canela M, Toledo E, Corella D, Bhupathiraju SN, Tobias DK, Tabung FK, Hu J, Zhao T, Turman C, Feng YA, et al. The Mediterranean diet, plasma metabolome, and cardiovascular disease risk. Eur Heart J. 2020; 41:2645–56. https://doi.org/10.1093/eurheartj/ehaa209 [PubMed]
- 52. Xiao J, Ding R, Xu X, Guan H, Feng X, Sun T, Zhu S, Ye Z. Comparison and development of machine learning tools in the prediction of chronic kidney disease progression. J Transl Med. 2019; 17:119. https://doi.org/10.1186/s12967-019-1860-0 [PubMed]
- 53. Das J, Gayvert KM, Bunea F, Wegkamp MH, Yu H. ENCAPP: elastic-net-based prognosis prediction and biomarker discovery for human cancers. BMC Genomics. 2015; 16:263. https://doi.org/10.1186/s12864-015-1465-9 [PubMed]
- 54. Millstein J, Budden T, Goode EL, Anglesio MS, Talhouk A, Intermaggio MP, Leong HS, Chen S, Elatre W, Gilks B, Nazeran T, Volchek M, Bentley RC, et al, and AOCS Group. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann Oncol. 2020; 31:1240–50. https://doi.org/10.1016/j.annonc.2020.05.019 [PubMed]
- 55. Cho S, Kim H, Oh S, Kim K, Park T. Elastic-net regularization approaches for genome-wide association studies of rheumatoid arthritis. BMC Proc. 2009 (Suppl 7); 3:S25. https://doi.org/10.1186/1753-6561-3-s7-s25 [PubMed]
- 56. Waldmann P, Mészáros G, Gredler B, Fuerst C, Sölkner J. Evaluation of the lasso and the elastic net in genome-wide association studies. Front Genet. 2013; 4:270. https://doi.org/10.3389/fgene.2013.00270 [PubMed]
- 57. Frost HR, Shen L, Saykin AJ, Williams SM, Moore JH, and Alzheimers Disease Neuroimaging Initiative. Identifying significant gene-environment interactions using a combination of screening testing and hierarchical false discovery rate control. Genet Epidemiol. 2016; 40:544–57. https://doi.org/10.1002/gepi.21997 [PubMed]
- 58. Momen M, Mehrgardi AA, Sheikhi A, Kranis A, Tusell L, Morota G, Rosa GJM, Gianola D. Predictive ability of genome-assisted statistical models under various forms of gene action. Sci Rep. 2018; 8:12309. https://doi.org/10.1038/s41598-018-30089-2 [PubMed]
- 59. Bouts MJR, Möller C, Hafkemeijer A, van Swieten JC, Dopper E, van der Flier WM, Vrenken H, Wink AM, Pijnenburg YAL, Scheltens P, Barkhof F, Schouten TM, de Vos F, et al. Single Subject Classification of Alzheimer's Disease and Behavioral Variant Frontotemporal Dementia Using Anatomical, Diffusion Tensor, and Resting-State Functional Magnetic Resonance Imaging. J Alzheimers Dis. 2018; 62:1827–39. https://doi.org/10.3233/JAD-170893 [PubMed]
- 60. Liu Y, Thalamuthu A, Mather KA, Crawford J, Ulanova M, Wong MWK, Pickford R, Sachdev PS, Braidy N. Plasma lipidome is dysregulated in Alzheimer's disease and is associated with disease risk genes. Transl Psychiatry. 2021; 11:344. https://doi.org/10.1038/s41398-021-01362-2 [PubMed]
- 61. de Vos F, Schouten TM, Hafkemeijer A, Dopper EG, van Swieten JC, de Rooij M, van der Grond J, Rombouts SA. Combining multiple anatomical MRI measures improves Alzheimer's disease classification. Hum Brain Mapp. 2016; 37:1920–9. https://doi.org/10.1002/hbm.23147 [PubMed]