



Introduction
The treacle ribosome biogenesis factor 1 (TCOF1) gene is located on the long arm of chromosome 5 at the 5q32-33.3 locus and encodes treacle phosphoprotein [1, 2]. TCOF1 was initially found as a gene related to Treacher Collins syndrome (TCS), a rare genetic disorder characterized by severe craniofacial defects, external ear deformation, and hearing impairment [3, 4]. The role of TCOF1 in TCS has been extensively studied [5–7], and our previous works have identified novel TCOF1 mutations in TCS [8, 9]. Recently, TCOF1 has been reported to play crucial roles in multiple processes, including ribosome biogenesis [10], deoxyribonucleic acid (DNA) damage response (DDR) [11, 12], mitotic regulation [13], and telomere integrity [14, 15]. However, little is known about its role in carcinogenesis. Given that TCOF1 participates in several key cellular processes, in this study we aimed to investigate the part it plays in human cancers.
Cancer remains a major public-health problem worldwide and has been a leading cause of death in the past several decades [16, 17]. Emerging therapies that include chemotherapy, radiotherapy, and immune checkpoint blockade targeting programmed death-1 (PD-1) and cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) have shown great success in the treatment of several cancer types [18–20]. However, a considerable number of patients benefit little from available immunotherapies, and their prognoses remain poor. This dilemma emphasizes the importance of deeply understanding the mechanism underlying tumorigenesis. With the emergence of large-scale, multi-omics, and publicly accessible databases containing sample data from different types of cancer, it is now possible to analyze and evaluate the role of certain genes of interest, namely TCOF1 in this study, at the pan-cancer level.
In this work, we visualized the expression and prognostic landscapes of TCOF1 in pan-cancer based on published data. In addition, mutations to TCOF1 and the gene’s relationships with certain genomic signatures were also explored. We further evaluated TCOF1’s relationships with immune cell infiltration, immune checkpoints, and immunotherapy responses. Finally, potential pathways involving TCOF1 in tumor pathogenesis were also investigated.
Results
Expression level of TCOF1 in various cancers
We examined TCOF1 mRNA expression levels in various cancer types by analyzing TCGA data via Oncomine. The results showed that TCOF1 expression in tumors was significantly higher than in normal tissues in many cancers, including bladder, breast, cervical, colorectal, esophageal, gastric, head and neck, liver, lung, and ovarian, as well as in melanoma and lymphoma. However, in certain studies, TCOF1 was less expressed in brain, central nervous system (CNS), head and neck, kidney, and lung cancers and in lymphoma (Figure 1A).
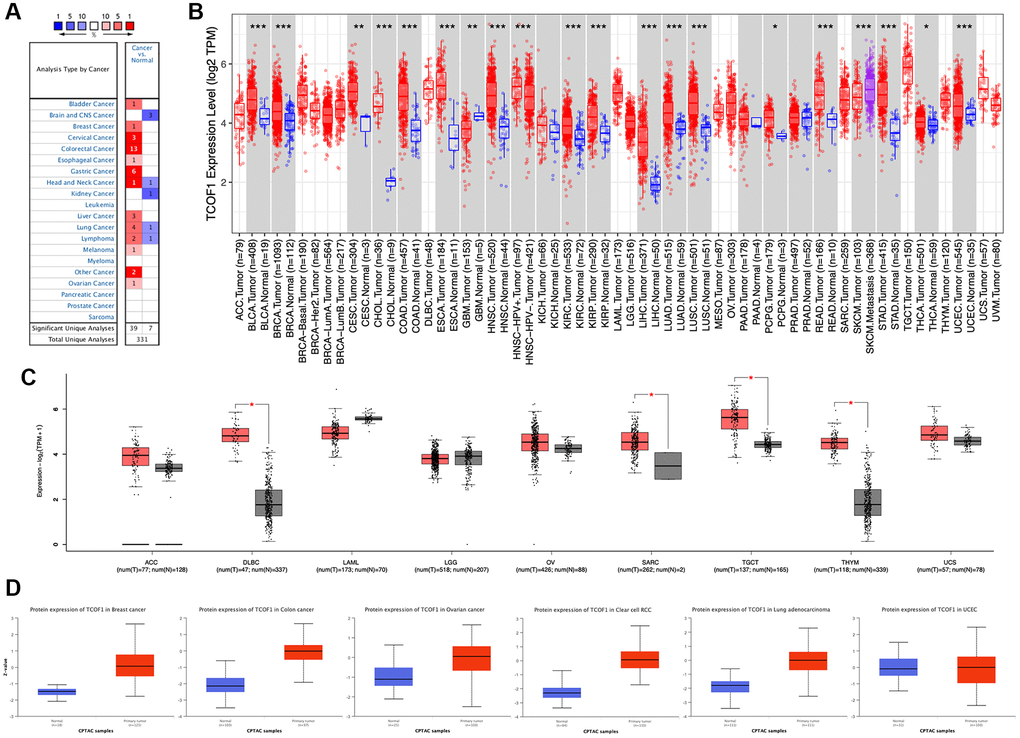
Figure 1. TCOF1 expression levels in cancers. (A) Differential-expression data for TCOF1 in various types of cancer, compared with corresponding normal tissues, in Oncomine. (B) TCOF1 expression levels in different tumor types from TCGA were analyzed using TIMER2. (C) Comparisons of TCOF1 expression levels between tumor tissues from TCGA and normal tissues from GTEx. (D) TCOF1 protein (treacle) expression levels in BC, CC, OC, CCRCC, LUAD, and UCEC. *P < 0.05; **P < 0.01; ***P < 0.001.
To further evaluate differential expression of TCOF1 in pan-cancer, we compared RNA sequencing data from TCGA using TIMER. As shown in Figure 1B, TCOF1 expression was significantly higher in bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous-cell carcinoma (CESC), endocervical adenocarcinoma (ECA), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), esophageal carcinoma (ESCA), head and neck squamous-cell carcinoma (HNSC), clear-cell renal-cell carcinoma (CCRCC), papillary renal-cell carcinoma (PRCC), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous-cell carcinoma (LUSC), pheochromocytoma and paraganglioma (PCPG), rectal adenocarcinoma (READ), stomach adenocarcinoma (STAD), thyroid carcinoma (THCA), and uterine corpus endometrial carcinoma (UCEC). Notably, TCOF1 expression in skin cutaneous melanoma (SKCM) metastatic tissue was remarkably higher than in respective primary tumor tissue. Lower expression of TCOF1 in tumor was found only in glioblastoma multiforme (GBM). For certain tumors lacking normal-tissue data in TCGA, we used corresponding normal tissues from the GTEx dataset as controls and compared differences in TCOF1 expression using GEPIA2. As shown in Figure 1C, expression of TCOF1 was upregulated in diffuse large B-cell lymphoma (DLBCL), sarcoma (SARC), testicular germ cell tumor (TGCT), and thymoma (THYM). However, we did not see significant differences in other tumors, including adrenocortical carcinoma (ACC), acute myeloid leukemia (LAML), brain lower-grade glioma (LGG), ovarian serous cystadenocarcinoma (OV), and uterine carcinosarcoma (UCS).
Based on the CPTAC dataset, we then evaluated protein expression of TCOF1 in pan-cancer via the UALCAN portal. Compared with normal tissues, expression of TCOF1 total protein was higher in breast cancer (BC), colon cancer (CC), ovarian cancer (OC), CCRCC, and LUAD, but not in UCEC (Figure 1D). We also investigated TCOF1 protein expression in 20 types of cancer using the HPA cohort. IHC staining results showed that high expression of TCOF1 could be observed in colorectal (36.4%), testicular (16.67%), pancreatic (11.11%), urothelial (10%), stomach (9.09%), liver (8.33%), endometrial (8.33), ovarian (8.33%), renal (8.33%), and skin cancers (8.33%; Supplementary Figure 1).
TCOF1 expression and cancer patients’ prognoses
To understand how TCOF1 affects the prognoses of cancer patients, we analyzed the relationship between survival outcomes and TCOF1 expression levels via PrognoScan. TCOF1 expression was significantly correlated with prognosis in seven cancer types: BC, uveal melanoma (UVM), liposarcoma, renal-cell carcinoma (RCC), glioma, meningioma, and colorectal cancer (CRC; Figure 2A). Of these, high expression levels of TCOF1 were detrimental to patient prognosis in BC, UVM, liposarcoma, glioma, and meningioma, but they played a protective role in RCC and CC. Detailed data are shown in a forest plot (Supplementary Figure 2).
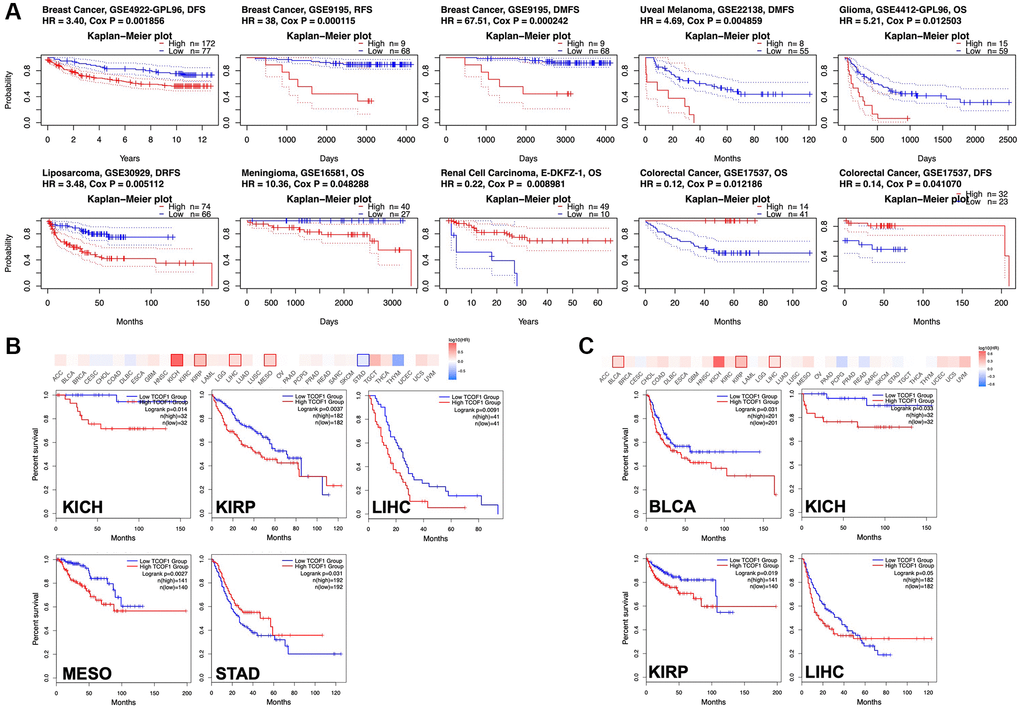
Figure 2. Survival analysis based on expression level of TCOF1. (A) Survival curves with significance in eight cohorts from the GEO dataset (GSE4922-GPL96, GSE9195, GSE22138, GSE4412-GPL96, GSE30929, GSE16581, E-DKFZ-1, and 17537). (B, C) Survival heatmap and Kaplan–Meier curves with positive results from (B) OS and (C) DFS analyses of different tumors in TCGA based on TCOF1 expression.
We further investigated the association between TCOF1 expression and cancer patients’ prognoses in TCGA databases via GEPIA2. As shown in Figure 2B, high expression levels of TCOF1 were related to poor prognosis for overall survival (OS) in kidney chromophobe (KICH; P = 0.014), PRCC (P = 0.0027), LIHC (P = 0.0037), and mesothelioma (MESO, P = 0.0091) in TCGA datasets. Disease-free survival (DFS) analysis results, shown in Figure 2C, revealed that high expression of TCOF1 was correlated with poor prognosis in BLCA (P = 0.031), KICH (P = 0.033), PRCC (P = 0.019), and LIHC (P = 0.05) in TCGA datasets. However, lower expression levels of TCOF1 were linked to poor OS for STAD (P = 0.031, Figure 2A).
Mutation profiles and genome-wide association of TCOF1
Using cBioPortal, we investigated the mutation frequency of TCOF1 in 10,967 samples from 32 TCGA studies. As shown in Figure 3A, the highest alteration frequency of TCOF1 (>6%) appeared in UCEC patients, with “mutation” as the primary type. The “amplification” type of copy number alteration (CAN) was the primary type in CCRCC cases, showing an alteration frequency of >5%. Notably, all UVM cases with genetic alterations (~2% frequency) had copy number deletion of TCOF1 (Figure 3A). We detected 196 mutations (including 164 missense, 22 truncating, 2 inframe, 4 fusion, and 2 duplicate mutations in patients with multiple samples) and located their sites between amino acids 0 and 1412. Of these, L221F/I (from 4 UCEC samples) was the most frequent mutation site (Figure 3B). The details of all mutation profiles are summarized in Supplementary Table 1.
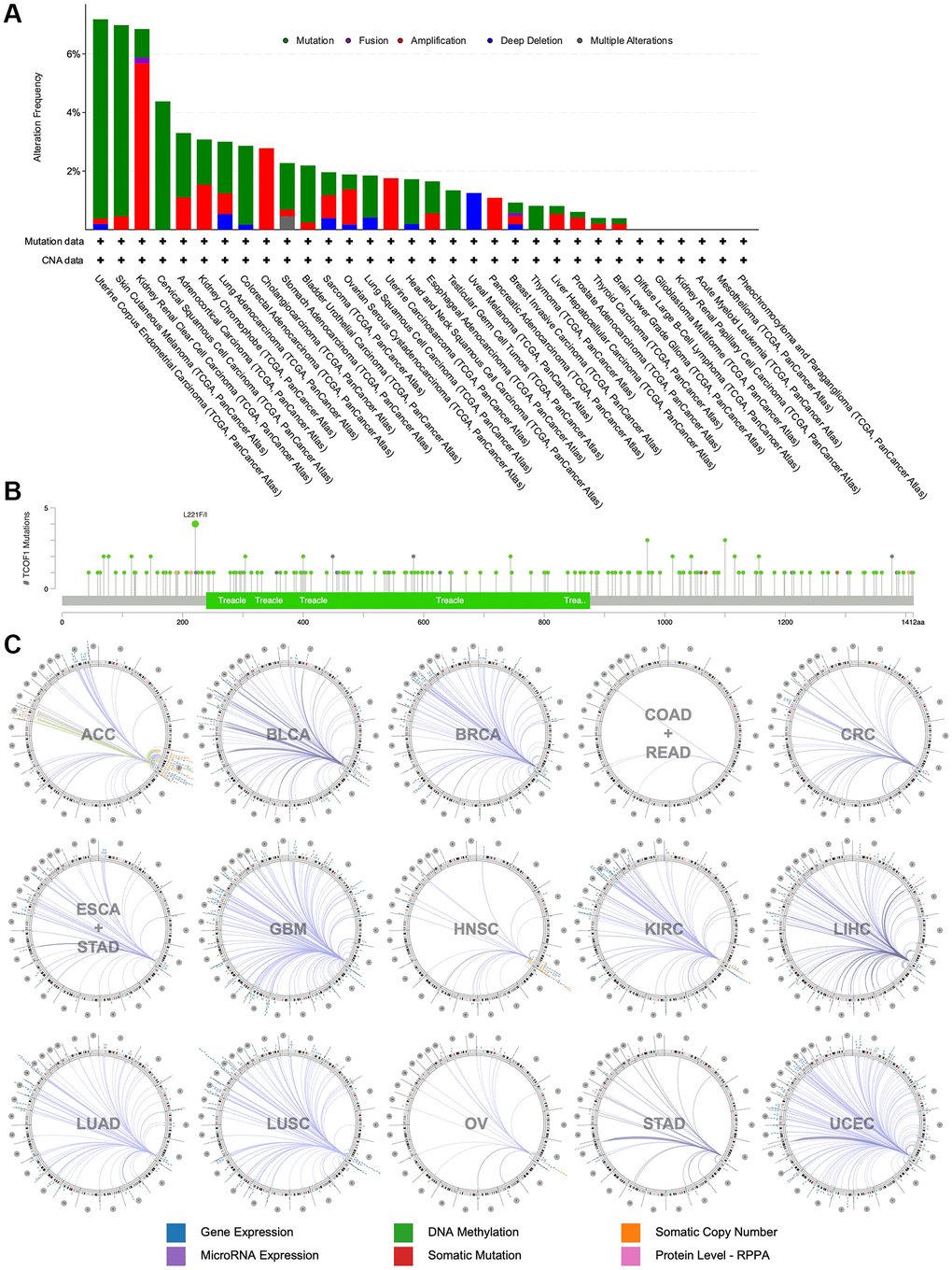
Figure 3. Mutational landscape and genome-wide association of TCOF1. (A) Alternation types and frequency of TCOF1 in different types of cancer. (B) Mutation sites of TCOF1 across cancers. (C) The genome-wide correlation between TCOF1 and other signatures from TCGA, visualized using Regulome Explorer.
We then used the Regulome Explorer to further inspect the genome-wide association between TCOF1 and certain genomic signatures. Expression of TCOF1 and its correlations with other variables in cancers on the chromosomal level (including DNA methylation, somatic copy number, microRNA expression, somatic mutation, and protein level) is displayed in Circos plots (Figure 3C). Based on data from TCGA, associations could be detected between TCOF1 and other signatures in ACC, BLCA, BRCA, COAD, READ, CRC, ESCA, STAD, GBM, HNSC, CCRCC, LIHC, LUAD, LUSC, OV, STAD, and UCEC within the context of genomic coordinates. Detailed data are listed in Supplementary Table 2.
We next examined the relationship between TCOF1 expression and expression levels of four DNA-methyltransferases (DNMTs) in pan-cancer. As shown in Supplementary Figure 3, we observed a significant and positive correlation between TCOF1 and DNMTs in almost all types of cancer except UCS and CHOL. This indicated that upregulated expression of TCOF1 in different cancers might contribute to DNA methylation.
Relationship between TCOF1 expression and immune cell infiltration
We further investigated the correlation between infiltration levels of different immune cells in pan-cancer and TCOF1 expression level. The results indicated that high TCOF1 expression was positively related to tumor purity in six types of cancer (Figure 4A). Additionally, TCOF1 expression level was significantly associated with infiltration levels of Cluster of Differentiation 8–positive (CD8+) T cells, CD4+ T cells, B cells, neutrophils, macrophages, and dendritic cells (DCs) in 14, 16, 12, 20, 13, and 17 cancer types, respectively (Supplementary Figure 4). Cancer-associated fibroblasts (CAFs) are among the most abundant stromal components of the tumor microenvironment (TME), and they can build up and remodel the extracellular-matrix (ECM) structure to facilitate tumor invasion [21]. We observed a significantly positive correlation of TCOF1 expression and CAF infiltration level in ESCA, PRCC, and THCA, but noted a negative correlation in BRCA, TGCT, and THYM (Figure 4B). In addition, abundance of myeloid- derived suppressor cells (MDSCs), which can inhibit T-cell function and thus contribute to the pathogeneses of various diseases, was found to be positively correlated with TCOF1 in almost all cancer types (Figure 4C). Finally, hematopoietic stem cells (HSCs), had a negative relationship with TCOF1 expression in most types of cancer (Figure 4D).
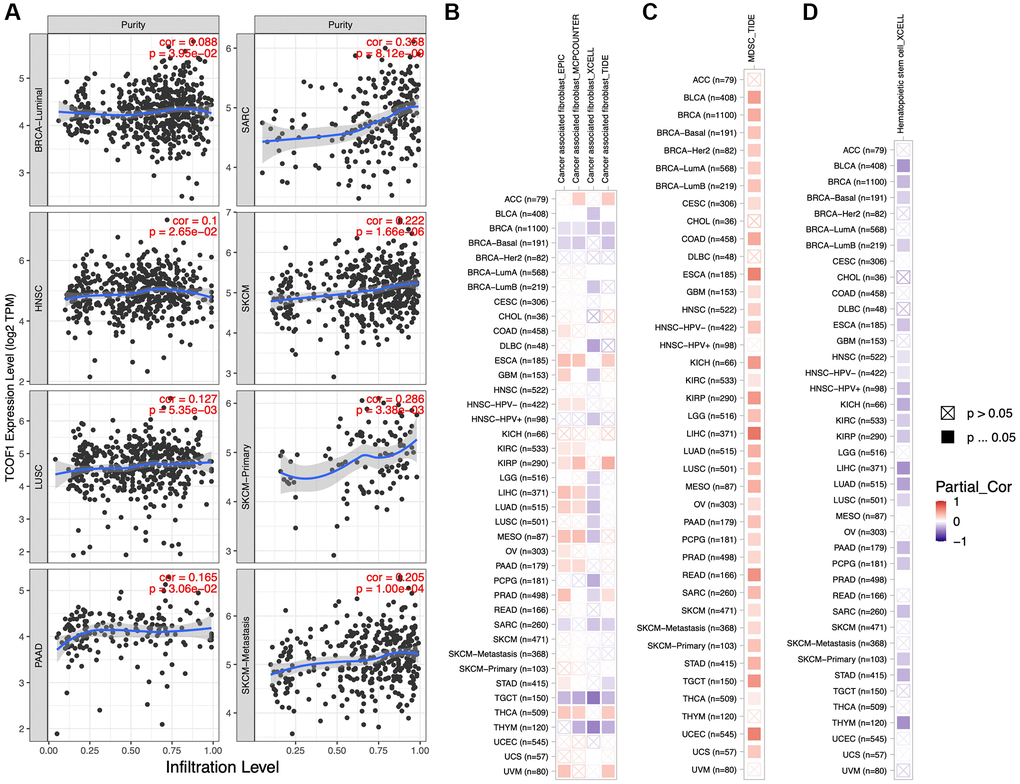
Figure 4. Associations of TCOF1 expression with tumor purity and immune infiltration. (A) TCOF1 expression was positively correlated with tumor purity in BRCA-Luminal, HNSC, LUSC, pancreatic adenocarcinoma (PAAD), SARC, and SKCM based on TCGA data. (B–D) Correlation between TCOF1 expression level and infiltration levels of (B) CAFs, (C) MDSCs, and (D) HSCs across all types of cancer in TCGA.
Association between TCOF1 expression and immunotherapy
We analyzed correlations between TCOF1 and certain immune checkpoints, including B- and T-lymphocyte attenuator (BTLA), leukocyte-associated immunoglobulin-like receptor 1 (LAIR1), CD244, lymphocyte activation gene 3 (LAG3), inducible T-cell costimulator (ICOS), CD40 ligand (CD40LG), CTLA4, CD48, CD28, CD200 receptor 1 (CD200R1), CD80, programmed cell death protein 1 (PDCD1), transmembrane and immunoglobulin domain containing 2 (TMIGD2), programmed cell death 1 ligand 2 (PDCD1LG2), CD27, TIGIT, CD86, and tumor necrosis factor receptor superfamily 9 (TNFRSF9). As shown in Figure 5A, TCOF1 had significant correlations with most of the immune checkpoints in KICH, CCRCC, and LIHC. Notably, in 20 of 33 types of cancer, CD276 expression was remarkably associated with TCOF1. Based on these results, we hypothesized a synergistic effect of TCOF1 and immune checkpoints in cancers. However, based on the data from TISIDB, we did not observe any significant difference in TCOF1 expression level between immunotherapy responders and non-responders (Supplementary Table 3).
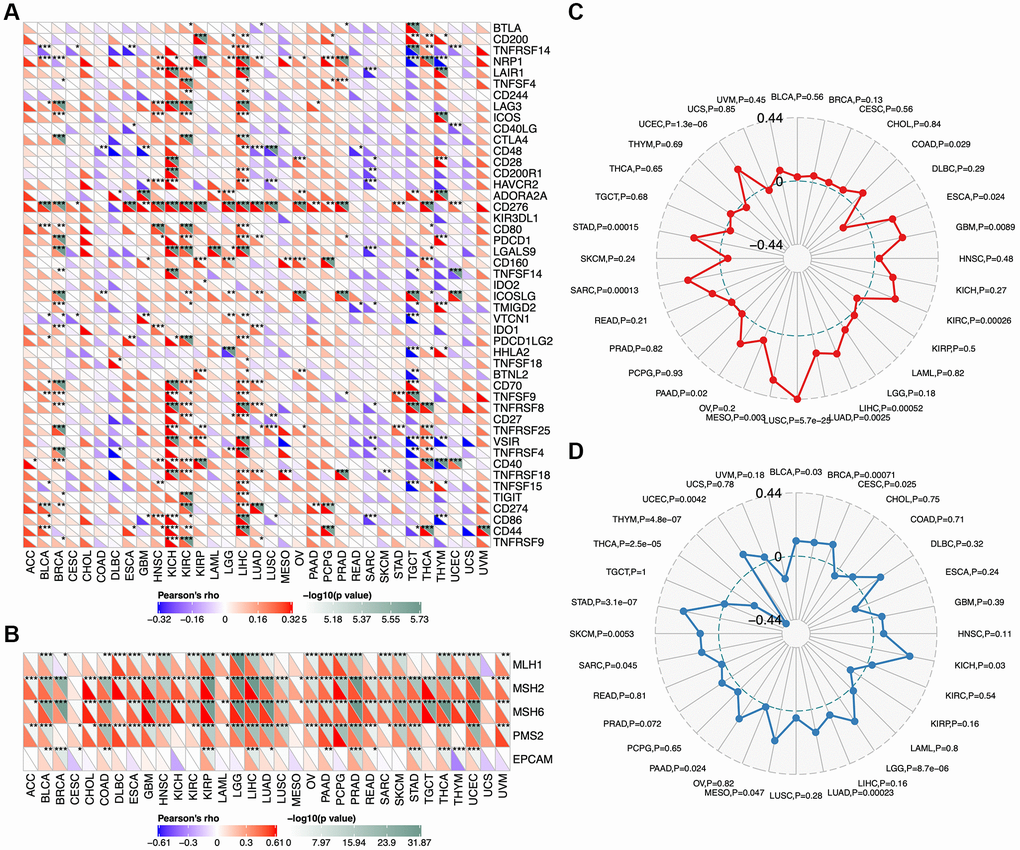
Figure 5. Relationship of TCOF1 and immunotherapy. (A) Correlations between TCOF1 and confirmed immune checkpoints across cancers. (B) Correlations between TCOF1 and five essential genes for MMR in multiple cancers. (C, D) Correlations of TCOF1 with (C) MSI and (D) TBM. *P < 0.05; **P < 0.01; ***P < 0.001.
Microsatellites are short tandem repeat (STR) DNA sequences distributed throughout the human genome and prone to replication errors, which can normally be repaired by the mismatch repair (MMR) system [22]. The MMR system is usually dependent on four key genes: mutL homologue 1 (MLH1), postmeiotic segregation increased 2 (PMS2), mutS homologue 2 (MSH2), and mutS 6 (MSH6). The epithelial-cell adhesion molecule (EPCAM) gene has also been identified as a key MMR gene [23]. We analyzed the correlation between TCOF1 expression and MSI, a strong mutator phenotype with a deficient MMR system, in different types of cancers and found that TCOF1 was significantly and positively correlated with MLH1, MSH2, MSH6, and PMS2 in almost all types but negatively correlated with EPCAM in CESC, KICH, and THYM (Figure 5B). In addition, high-MSI tumors appeared to express higher levels of TCOF1 than genetically stable ones in CCRCC, LIHC, LUSC, SARC, STAD, and UCEC (P < 0.001; Figure 5C). TMB, the total number of mutations per coding area of a tumor genome, is a biomarker of sensitivity to ICIs. We analyzed the correlation between TCOF1 expression and TMB across various cancer types. The results showed that TCOF1 expression was positively correlated with TMB in BRCA, LGG, LUAD, and STAD (P < 0.001) but negatively correlated with TMB in THCA and THYM cohorts (P < 0.001; Figure 5D). However, all significant correlations coefficients of TCOF1 with MSI or TBM were <0.5, which is insufficient to predict a cancer patient’s response to immunotherapy (Supplementary Table 3).
TCOF1-related genes and potential pathways in cancer
To further understand the mechanism of TCOF1 in tumor pathogenesis, we investigated genes and proteins related to TCOF1 expression and conducted pathway enrichment analyses thereof. Using STRING, we identified TCOF1-binding proteins supported by experimentally available evidence. The interaction network of these proteins and TCOF1 is shown in Figure 6A. Then, we used GEPIA2 to determine the top 100 genes correlated with TCOF1 expression in tumor data from TCGA projects. As shown in Figure 6B, TCOF1 expression was most positively significantly correlated to expression of deleted in azoospermia-associated protein 1 (DAZAP1), heterogeneous nuclear ribonucleoprotein A/B (HNRNPAB), Ly1 antibody reactive (LYAR), DNA topoisomerase II binding protein 1 (TOPBP1), interacting checkpoint and replication regulator (TICRR, also known as C15orf42), and polo-like kinase 1 (PLK1). The corresponding detailed heatmap data of the various cancer types is displayed in Figure 6C. Intersection analysis of STRING-based TCOF1-binding proteins and GEPIA2-based TCOF1-correlated genes showed three common members: dyskerin pseudouridine synthase 1 (DKC1), nucleolar protein 56 (NOP56), and NOP58 (Figure 6D).
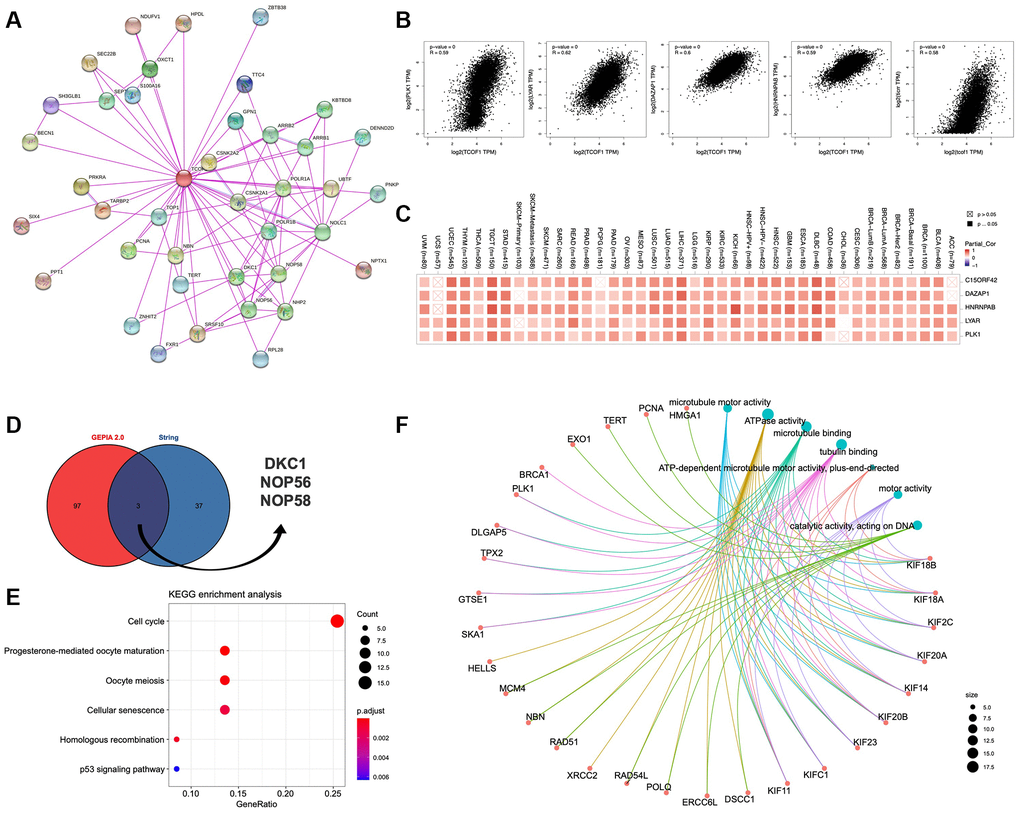
Figure 6. Pathway enrichment analysis of TCOF1. (A) Available experimentally determined TCOF1-binding proteins discovered using the STRING tool. (B) Correlation of TCOF1 expression with that of the top 5 related genes via GEPIA2 (DAZAP1, HNRNPAB, LYAR, TICRR/C15orf42, and PLK1). (C) The corresponding heatmap data of these top 5 genes in detailed cancer types. (D) DKC1, NOP56, and NOP58 are three common genes (proteins) of STRING-based TCOF1-binding proteins and GEPIA2-based TCOF1-correlated genes. (E) KEGG pathway analysis based on TCOF1-binding and -interacting genes. (F) The cnetplot of Molecular Function data in GO analysis.
Next, we used the identified genes to perform KEGG and GO enrichment analyses. KEGG results (Figure 6E) indicated that TCOF1 might influence tumorigenesis through “cell cycle” and “cellular-senescence” pathways. GO Molecular Function analysis suggested most of the TCOF1-related genes were associated with adenosine triphosphatase (ATPase) activity, microtubule binding, tubulin binding, catalytic activity (acting on DNA), and other functions (Figure 6F).
Discussion
The TCOF1 gene, located on the long arm of chromosome 5 at the 5q32-33.3 locus, is composed of 28 exons [2, 24]. In transcription, TCOF1 is alternatively transcribed and spliced into several mRNA isoforms. Most of these isoforms contain exon 6A/treacle, a translational product of exon 6A– contained TCOF1 mRNA isoform [24], which is a nucleolar phosphoprotein with 1488 amino acids and a low-complexity three-domain structure [25]. TCOF1 and treacle are reported to regulate multiple key cellular processes, including ribosome biogenesis, mitosis, proliferation, cellular response to DNA damage, and apoptosis [26]. While TCOF1-related mechanisms in TCS have been extensively studied, research focused on the role of TCOF1 in malignancies is limited.
In this work, we used integrated bioinformatics methods to comprehensively analyze the role of TCOF1 and its corresponding protein treacle in different types of cancers. According to the results of expression analysis, TCOF1 was upregulated in most cancer types when compared to corresponding normal tissues (Figure 1A–1C). The expression level of treacle protein was significantly higher in BC, CC, OC, CCRCC, and LUAD, but not in UCEC (Figure 1D). As subsequently confirmed in mutation profiles of TCOF1, UCEC patients had the highest alternation frequency, with “mutation” as the primary type (Figure 3A). Survival analysis results then indicated that TCOF1 was correlated with prognosis in several cancers, including BC, UVM, liposarcoma, RCC, glioma, meningioma, and CRC (Figure 2). Multifaceted, even contradictory, prognostic values of TCOF1 in different types of cancer might be attributable to distinct underlying mechanisms in certain tumors; heterogeneous data collection approaches; and other clinical factors such as gender, ethnicity, and tumor stage. These results indicated the potential prognostic value of TCOF1 in different types of cancer.
By using different algorithms, we found significant correlations between TCOF1 and infiltration levels of several types of immune cells, including CAFs and MDSCs (Figure 5). Immune cells interact with tumor cells in the TME and therefore play vital roles in anti- or pro-tumor incidents. For example, CAFs, the prominent components of stromal cells, are reportedly associated with poor outcomes, therapy resistance, and tumor recurrence in various types of cancer [27]. Our findings indicated that TCOF1 might exert an essential effect on cancer development and it might serve as a potential therapy target. However, a cause–effect relationship could not be established in this study. Additionally, pathway enrichment analyses showed that TCOF1 might act mainly on ATPase activity, microtubule binding, tubulin binding, and catalytic activity (on DNA) and participate in tumorigenesis through “cell cycle” and “cellular-senescence” pathways (Figure 6). This finding was partially in line with those of existing TCOF1-related studies, as further discussed below.
Treacle acts as a key regulator in the biogenesis of ribosomes, one of the most important cell processes and essential for nearly 95% of total transcription [28]. Ribosome biogenesis has three main stages: transcription of ribosomal DNA (rDNA) into precursor ribosomal RNA (pre-rRNA), post-transcriptional processing from pre-rRNA to mature rRNA, and ribosome assembly [28, 29]. The first stage starts with the formation of the pre-initiation complex around the rDNA promoter region in the nucleolus. The latter consists of upstream binding factor (UBF), transcription initiation factor 1 alpha (TIF1-α), selectivity factor 1 (SL1, also known as TIF1-β), and DNA polymerase I (Pol I) [30, 31]. Treacle plays a crucial role in ribosome biogenesis in that its different domains can bind and recruit UBF, Pol I, and Nopp140 to the rDNA promoter [10]. In addition, treacle is an activator of UBF, an important regulator in the transcription of rDNA [32]. Insufficient treacle leads to the dispersion and dysfunction of UBF and Pol I and the resulting inhibition of rRNA transcription [11, 32]. During the second stage, treacle interacts with ribonucleoprotein NOP56 (Figure 6D) to regulate post-transcriptional pre-rRNA modifications, including methylation; this ensures the flexibility of RNA strands, protects RNA from hydrolysis, and regulates translation in cells [33, 34]. In the cervical-carcinoma HeLa cell line, silencing of TCOF1 leads to inhibition of rRNA transcription and attenuated cell proliferation [32]. It also causes dysfunction of an RNA helicase called DEAD-box RNA helicase 21 (DDX21) by relocating it from the nucleolus to the nucleoplasm [35]. DDX21 has been reported to promote gastric-cancer (GC) proliferation and tumor growth [36].
Due to recombination between rDNA sequences from different chromosomes, the genes encoding rRNA are unstable and prone to damage [37]. DDR is a signal transduction pathway involving multiple repair mechanisms in cells, and it is closely associated with tumorigenesis [38, 39]. The DDR repair process for the reconstruction of double-stranded breaks is triggered by ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) kinases, and transcriptional silencing is then induced to save energy and to prevent collision between catalyzing complexes of transcription and repair [40]. Treacle plays an essential role in DDR mechanisms by recruiting nibrin (NBS1) and TOPBP1, the key adaptor proteins of ATM and ATR kinases, respectively [12, 37, 41]. Accumulation of NBS1 mediated by treacle is crucial for stopping rRNA transcription in the DDR process, and NBS1 overexpression is reportedly associated with chemoresistance and with tumor development and metastasis [11, 42–44]. Treacle can interact with NBS1 and thereby contribute to cancer development. Furthermore, overexpression of TCOF1 has been associated with radioresistance in acinar progenitor cells of rat salivary glands, but attenuated TCOF1 expression can sensitize human osteosarcoma cells to irradiation [41, 45]. Since treacle recruits TOPBP1 in the DDR process and the latter confers radioresistance in osteosarcoma [12], TOPBP1 might mediate the effect of treacle on sensitivity to radiotherapy. However, the potential contribution of TCOF1 to cancer progression and resistance to therapy needs to be elucidated in future studies.
Although we integrated information from different public databases to present a broad view of TCOF1 in pan-cancer, our study had some limitations. First, we analyzed tumor tissue information mainly based on microarray and sequencing data, meaning that cellular-level analysis results might be systemically biased. Future studies using high-resolution methods, such as single-cell RNA sequencing [46], should be performed to overcome or minimize such bias. Second, via multiple databases, we conducted bioinformatic analyses of TCOF1 expression and patient prognosis only, but in vivo/vitro experimental evidence on the cellular or molecular level is still needed to help clarify the role of TCOF1 in tumors. Third, although we found significant correlations among immune cell infiltration levels, survival outcomes, potential pathways, and TCOF1, we failed to identify TCOF1 as friend or foe in cancers due to conflicting results from different databases. Since tumorigenesis is a complex process involving multiple pathways, our study provided only preliminary findings on the oncogenic role of TCOF1; its exact role in certain type of cancers should be evaluated and validated more precisely and comprehensively.
In summary, we presented a broad view of TCOF1’s role in pan-cancer. TCOF1 was upregulated in most types of cancers, and we believe it might serve as a prognostic biomarker depending on cancer type. Infiltration levels of several types of immune cells, including CAFs and MDSCs, were highly correlated with TCOF1 expression, suggesting underlying mechanisms involving TCOF1 and immunity in tumorigenesis that should be further explored. Future studies should focus on potential regulation of TCOF1 by multiple oncogenic-signaling pathways.
Materials and Methods
Databases
The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov) is a prestigious cancer genomics project funded by the National Cancer Institute (NCI; Bethesda, MD, USA), which has characterized >20,000 primary cancer and matched normal-tissue samples from various cancer types [47]. The Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo) is a publicly available genomics-data repository containing array- and sequence-based data [48]. The Genotype-Tissue Expression Project (GTEx; http://commonfund.nih.gov/GTEx) is a commonly funded data resource and tissue bank containing tissue-specific gene expression data [49]. The Clinical Proteomic Tumor Analysis Consortium (CPTAC; https://proteomics.cancer.gov/programs/cptac), also funded by the NCI, is a comprehensive database that aims to identify proteins in cancer genomes and related biological processes and that provides genomic and proteomic data from >1100 cancer patients [50]. Analyses in our study were conducted based on the data from TCGA, GEO, GTEx, and CPTAC.
Expression analysis
Oncomine is a cancer microarray database and web-based data-mining platform aimed at facilitating discovery from genome-wide expression analyses [51]. In this study, we recorded differential-expression data of TCOF1 between various cancer samples and corresponding normal tissues from Oncomine. Thresholds of P-values and fold change (FC) were 0.01 and 1.5, respectively. Next, we used the “Gene_DE” module of Tumor IMmune Estimation Resource 2 (TIMER2; http://timer.cistrome.org), an online tool for systematical analysis of immune infiltrates across diverse cancer types [52], to visualize expression differences of TCOF1 in pan-cancer from TCGA data. For certain cancer types without normal-tissue data, we matched and compared them with corresponding normal-tissue data from GTEx via the “Expression DIY” panel of Gene Expression Profiling Interactive Analysis 2 (GEPIA2; http://gepia2.cancer-pku.cn), a portal for analyzing ribonucleic acid (RNA) sequencing expression data from the TCGA and GTEx projects [53]. Cutoffs of P-values and |Log2FC| were 0.01 and 1, respectively. TCOF1 protein expression analysis in six cancer types based on CPTAC data was conducted on UALCAN (http://ualcan.path.uab.edu), a web portal for analyzing cancer omics data [54].
The Human Protein Atlas (HPA; https://www.proteinatlas.org/) is an online portal that contributes to the mapping of human proteins in tissues. We obtained protein expression levels and immunohistochemical (IHC) staining results of TCOF1 protein in 20 types of cancer samples from HPA.
Survival analysis based on TCOF1
PrognoScan http://dna00.bio.kyutech.ac.jp/PrognoScan/index.html is a publicly accessible and powerful platform for evaluating the association between a gene and clinical outcome in cancers [55]. We conducted survival analysis of different cancer types and explored its relationship with TCOF1 expression on PrognoScan. We used the “forestplot” package in R studio (version 1.4.1103; R Foundation for Statistical Computing, Vienna, Austria) to summarize the results and drew a forest plot of them. In addition, given that PrognoScan is a collection of cancer microarray datasets, we then performed survival analysis based on TCGA data using the “Survival Analysis” panel of GEPIA2. The significance level was set to 0.05.
TCOF1’s mutational landscape and correlation with genomic signatures
We employed cBioPortal (http://www.cbioportal.org), an open-access resource for interactive exploration of multidimensional cancer genomics datasets [56], to investigate the mutation profiles of TCOF1 in different tissues. We chose data from 10,967 samples in 32 studies from TCGA and determined the frequency, types, and sites of TCOF1 mutations in multiple kinds of cancer. Regulome Explorer (http://explorer.cancerregulome.org) is an online tool to search, filter, and visualize analytical results generated from TCGA data. We used this tool to explore the correlation between TCOF1 expression and certain genomic signatures. The filter of associations was set as follows: −Log10(p) ≥2; Correlation ≥0.4; Max results = 200. We calculated the relationship between TCOF1 expression and four DNA-methyltransferases (DNMTs) and presented the results as a circular plot using SangerBox (http://www.sangerbox.com/tool), a powerful computerized online tool for bioinformatics analysis.
Ethics statement
The studies involving human participants were reviewed and approved by all the research data based on the bioinformatic analysis of the open resources from the TIMER, Oncomine, TCGA, and GEPIA databases. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Data availability statement
Publicly available datasets were analyzed in this study. The data can be found here: http://timer.cistrome.org, http://gepia2.cancer-pku.cn, http://ualcan.path.uab.edu, http://dna00.bio.kyutech.ac.jp/PrognoScan/index.html, http://www.cbioportal.org, https://hiplot.com.cn, https://string-db.org, http://cis.hku.hk/TISIDB, http://www.sangerbox.com/tool. However, the Oncomine database has been closed and it’s no longer available. The original contributions presented in the study are included in the Supplementary Material. Further inquiries can be directed to the corresponding author.
Supplementary Materials
Author Contributions
JW and XC: project design and administration. WG: software and manuscript writing. WG and LS: statistical analysis and visualization. All authors: contributed to the article and approved the submitted version.
Acknowledgments
Dr. Gu is grateful for the invaluable support received from Professor Jian Wang and Professor Xiaowei Chen over the years. We sincerely thank the public databases, including Oncomine, GEPIA, cBioPortal, TIMER2 and PrognoScan, for providing open access. We thank LetPub (https://www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA1601040201, Jian Wang), and the National Key Research and Development Program of China (Grant No. 2016YFC0901501, Xiaowei Chen).
References
- 1. Dixon J, Edwards SJ, Gladwin AJ, Dixon MJ, Loftus SK, Bonner CA, Koprivnikar K, Wasmuth JJ, and The Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet. 1996; 12:130–6. https://doi.org/10.1038/ng0296-130 [PubMed]
- 2. Wise CA, Chiang LC, Paznekas WA, Sharma M, Musy MM, Ashley JA, Lovett M, Jabs EW. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc Natl Acad Sci U S A. 1997; 94:3110–5. https://doi.org/10.1073/pnas.94.7.3110 [PubMed]
- 3. Sakai D, Trainor PA. Treacher Collins syndrome: unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol. 2009; 41:1229–32. https://doi.org/10.1016/j.biocel.2008.10.026 [PubMed]
- 4. Sakai D, Dixon J, Achilleos A, Dixon M, Trainor PA. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat Commun. 2016; 7:10328. https://doi.org/10.1038/ncomms10328 [PubMed]
- 5. Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006; 103:13403–8. https://doi.org/10.1073/pnas.0603730103 [PubMed]
- 6. Splendore A, Silva EO, Alonso LG, Richieri-Costa A, Alonso N, Rosa A, Carakushanky G, Cavalcanti DP, Brunoni D, Passos-Bueno MR. High mutation detection rate in TCOF1 among Treacher Collins syndrome patients reveals clustering of mutations and 16 novel pathogenic changes. Hum Mutat. 2000; 16:315–22. https://doi.org/10.1002/1098-1004(200010)16:4<315::AID-HUMU4>3.0.CO;2-H [PubMed]
- 7. Vincent M, Geneviève D, Ostertag A, Marlin S, Lacombe D, Martin-Coignard D, Coubes C, David A, Lyonnet S, Vilain C, Dieux-Coeslier A, Manouvrier S, Isidor B, et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet Med. 2016; 18:49–56. https://doi.org/10.1038/gim.2015.29 [PubMed]
- 8. Fan X, Wang Y, Fan Y, Du H, Luo N, Zhang S, Chen X. TCOF1 pathogenic variants identified by Whole-exome sequencing in Chinese Treacher Collins syndrome families and hearing rehabilitation effect. Orphanet J Rare Dis. 2019; 14:178. https://doi.org/10.1186/s13023-019-1136-z [PubMed]
- 9. Zhang X, Fan Y, Zhang Y, Xue H, Chen X. A novel mutation in the TCOF1 gene found in two Chinese cases of Treacher Collins syndrome. Int J Pediatr Otorhinolaryngol. 2013; 77:1410–5. https://doi.org/10.1016/j.ijporl.2013.05.013 [PubMed]
- 10. Lin CI, Yeh NH. Treacle recruits RNA polymerase I complex to the nucleolus that is independent of UBF. Biochem Biophys Res Commun. 2009; 386:396–401. https://doi.org/10.1016/j.bbrc.2009.06.050 [PubMed]
- 11. Larsen DH, Hari F, Clapperton JA, Gwerder M, Gutsche K, Altmeyer M, Jungmichel S, Toledo LI, Fink D, Rask MB, Grøfte M, Lukas C, Nielsen ML, et al. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol. 2014; 16:792–803. https://doi.org/10.1038/ncb3007 [PubMed]
- 12. Mooser C, Symeonidou IE, Leimbacher PA, Ribeiro A, Shorrocks AK, Jungmichel S, Larsen SC, Knechtle K, Jasrotia A, Zurbriggen D, Jeanrenaud A, Leikauf C, Fink D, et al. Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nat Commun. 2020; 11:123. https://doi.org/10.1038/s41467-019-13981-x [PubMed]
- 13. Sakai D, Dixon J, Dixon MJ, Trainor PA. Mammalian neurogenesis requires Treacle-Plk1 for precise control of spindle orientation, mitotic progression, and maintenance of neural progenitor cells. PLoS Genet. 2012; 8:e1002566. https://doi.org/10.1371/journal.pgen.1002566 [PubMed]
- 14. Nie X, Xiao D, Ge Y, Xie Y, Zhou H, Zheng T, Li X, Liu H, Huang H, Zhao Y. TRF2 recruits nucleolar protein TCOF1 to coordinate telomere transcription and replication. Cell Death Differ. 2021; 28:1062–75. https://doi.org/10.1038/s41418-020-00637-3 [PubMed]
- 15. Lalonde M, Chartrand P. TERRA, a Multifaceted Regulator of Telomerase Activity at Telomeres. J Mol Biol. 2020; 432:4232–43. https://doi.org/10.1016/j.jmb.2020.02.004 [PubMed]
- 16. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71:209–49. https://doi.org/10.3322/caac.21660 [PubMed]
- 17. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021; 71:7–33. https://doi.org/10.3322/caac.21654 [PubMed]
- 18. Ferris RL. Immunology and Immunotherapy of Head and Neck Cancer. J Clin Oncol. 2015; 33:3293–304. https://doi.org/10.1200/JCO.2015.61.1509 [PubMed]
- 19. Macleod LC, Tykodi SS, Holt SK, Wright JL, Lin DW, Tretiakova MS, True LD, Gore JL. Trends in Metastatic Kidney Cancer Survival From the Cytokine to the Targeted Therapy Era. Urology. 2015; 86:262–8. https://doi.org/10.1016/j.urology.2015.05.008 [PubMed]
- 20. Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, Viale G, Gelber RD, Goldhirsch A. Cancer immunotherapy efficacy and patients' sex: a systematic review and meta-analysis. Lancet Oncol. 2018; 19:737–46. https://doi.org/10.1016/S1470-2045(18)30261-4 [PubMed]
- 21. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019; 18:99–115. https://doi.org/10.1038/s41573-018-0004-1 [PubMed]
- 22. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010; 7:153–62. https://doi.org/10.1038/nrclinonc.2009.237 [PubMed]
- 23. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017; 357:409–13. https://doi.org/10.1126/science.aan6733 [PubMed]
- 24. So RB, Gonzales B, Henning D, Dixon J, Dixon MJ, Valdez BC. Another face of the Treacher Collins syndrome (TCOF1) gene: identification of additional exons. Gene. 2004; 328:49–57. https://doi.org/10.1016/j.gene.2003.11.027 [PubMed]
- 25. Dixon J, Edwards SJ, Anderson I, Brass A, Scambler PJ, Dixon MJ. Identification of the complete coding sequence and genomic organization of the Treacher Collins syndrome gene. Genome Res. 1997; 7:223–34. https://doi.org/10.1101/gr.7.3.223 [PubMed]
- 26. Grzanka M, Piekiełko-Witkowska A. The Role of TCOF1 Gene in Health and Disease: Beyond Treacher Collins Syndrome. Int J Mol Sci. 2021; 22:2482. https://doi.org/10.3390/ijms22052482 [PubMed]
- 27. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. 2019; 18:70. https://doi.org/10.1186/s12943-019-0994-2 [PubMed]
- 28. Zhou X, Liao WJ, Liao JM, Liao P, Lu H. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol. 2015; 7:92–104. https://doi.org/10.1093/jmcb/mjv014 [PubMed]
- 29. Xu X, Xiong X, Sun Y. The role of ribosomal proteins in the regulation of cell proliferation, tumorigenesis, and genomic integrity. Sci China Life Sci. 2016; 59:656–72. https://doi.org/10.1007/s11427-016-0018-0 [PubMed]
- 30. Bell SP, Learned RM, Jantzen HM, Tjian R. Functional cooperativity between transcription factors UBF1 and SL1 mediates human ribosomal RNA synthesis. Science. 1988; 241:1192–7. https://doi.org/10.1126/science.3413483 [PubMed]
- 31. Turi Z, Lacey M, Mistrik M, Moudry P. Impaired ribosome biogenesis: mechanisms and relevance to cancer and aging. Aging (Albany NY). 2019; 11:2512–40. https://doi.org/10.18632/aging.101922 [PubMed]
- 32. Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci U S A. 2004; 101:10709–14. https://doi.org/10.1073/pnas.0402492101 [PubMed]
- 33. Gonzales B, Henning D, So RB, Dixon J, Dixon MJ, Valdez BC. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum Mol Genet. 2005; 14:2035–43. https://doi.org/10.1093/hmg/ddi208 [PubMed]
- 34. Monaco PL, Marcel V, Diaz JJ, Catez F. 2'-O-Methylation of Ribosomal RNA: Towards an Epitranscriptomic Control of Translation? Biomolecules. 2018; 8:106. https://doi.org/10.3390/biom8040106 [PubMed]
- 35. Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, Flynn RA, Swigut T, Chang HY, Attardi LD, Wysocka J. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature. 2018; 554:112–7. https://doi.org/10.1038/nature25449 [PubMed]
- 36. Cao J, Wu N, Han Y, Hou Q, Zhao Y, Pan Y, Xie X, Chen F. DDX21 promotes gastric cancer proliferation by regulating cell cycle. Biochem Biophys Res Commun. 2018; 505:1189–94. https://doi.org/10.1016/j.bbrc.2018.10.060 [PubMed]
- 37. Korsholm LM, Gál Z, Lin L, Quevedo O, Ahmad DA, Dulina E, Luo Y, Bartek J, Larsen DH. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. 2019; 47:8019–35. https://doi.org/10.1093/nar/gkz518 [PubMed]
- 38. Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol. 2011; 3:a000745. https://doi.org/10.1101/cshperspect.a000745 [PubMed]
- 39. Rai R, Peng G, Li K, Lin SY. DNA damage response: the players, the network and the role in tumor suppression. Cancer Genomics Proteomics. 2007; 4:99–106. [PubMed]
- 40. Gorgoulis VG, Pefani DE, Pateras IS, Trougakos IP. Integrating the DNA damage and protein stress responses during cancer development and treatment. J Pathol. 2018; 246:12–40. https://doi.org/10.1002/path.5097 [PubMed]
- 41. Ciccia A, Huang JW, Izhar L, Sowa ME, Harper JW, Elledge SJ. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc Natl Acad Sci U S A. 2014; 111:18631–6. https://doi.org/10.1073/pnas.1422488112 [PubMed]
- 42. Chen YC, Su YN, Chou PC, Chiang WC, Chang MC, Wang LS, Teng SC, Wu KJ. Overexpression of NBS1 contributes to transformation through the activation of phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005; 280:32505–11. https://doi.org/10.1074/jbc.M501449200 [PubMed]
- 43. Yang MH, Chang SY, Chiou SH, Liu CJ, Chi CW, Chen PM, Teng SC, Wu KJ. Overexpression of NBS1 induces epithelial-mesenchymal transition and co-expression of NBS1 and Snail predicts metastasis of head and neck cancer. Oncogene. 2007; 26:1459–67. https://doi.org/10.1038/sj.onc.1209929 [PubMed]
- 44. Wu Z, Li S, Tang X, Wang Y, Guo W, Cao G, Chen K, Zhang M, Guan M, Yang D. Copy Number Amplification of DNA Damage Repair Pathways Potentiates Therapeutic Resistance in Cancer. Theranostics. 2020; 10:3939–51. https://doi.org/10.7150/thno.39341 [PubMed]
- 45. Weber TJ, Qian WJ, Smith JN, Gritsenko MA, Hu D, Chrisler WB, Timchalk C. Stable Acinar Progenitor Cell Model Identifies Treacle-Dependent Radioresistance. Radiat Res. 2019; 192:135–44. https://doi.org/10.1667/RR15342.1 [PubMed]
- 46. Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, Livak KJ. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat Rev Clin Oncol. 2021; 18:244–56. https://doi.org/10.1038/s41571-020-00449-x [PubMed]
- 47. Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015; 19:A68–77. https://doi.org/10.5114/wo.2014.47136 [PubMed]
- 48. Clough E, Barrett T. The Gene Expression Omnibus Database. Methods Mol Biol. 2016; 1418:93–110. https://doi.org/10.1007/978-1-4939-3578-9_5 [PubMed]
- 49. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, et al, and GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45:580–5. https://doi.org/10.1038/ng.2653 [PubMed]
- 50. Ellis MJ, Gillette M, Carr SA, Paulovich AG, Smith RD, Rodland KK, Townsend RR, Kinsinger C, Mesri M, Rodriguez H, Liebler DC, and Clinical Proteomic Tumor Analysis Consortium (CPTAC). Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013; 3:1108–12. https://doi.org/10.1158/2159-8290.CD-13-0219 [PubMed]
- 51. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007; 9:166–80. https://doi.org/10.1593/neo.07112 [PubMed]
- 52. Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020; 48:W509–14. https://doi.org/10.1093/nar/gkaa407 [PubMed]
- 53. Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019; 47:W556–60. https://doi.org/10.1093/nar/gkz430 [PubMed]
- 54. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVS, Varambally S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017; 19:649–58. https://doi.org/10.1016/j.neo.2017.05.002 [PubMed]
- 55. Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genomics. 2009; 2:18. https://doi.org/10.1186/1755-8794-2-18 [PubMed]
- 56. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–4. https://doi.org/10.1158/2159-8290.CD-12-0095 [PubMed]
- 57. Ru B, Wong CN, Tong Y, Zhong JY, Zhong SSW, Wu WC, Chu KC, Wong CY, Lau CY, Chen I, Chan NW, Zhang J. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019; 35:4200–2. https://doi.org/10.1093/bioinformatics/btz210 [PubMed]
- 58. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019; 47:D607–13. https://doi.org/10.1093/nar/gky1131 [PubMed]