



Introduction
Head and neck squamous cell carcinoma (HNSCC), as one of the most common malignant carcinomas, has a poor prognosis, as indicated by its high recurrence rate and metastasis risk. The 5-year overall survival (OS) of patients with HNSCC is 40–50%, while that of patients with advanced stage disease is 30–40% [1, 2]. Local and distant tumor recurrence are the main causes of death in patients with locally advanced HNSCC.
Angiogenesis, the process by which pre-existing blood vessels form new capillaries, is a crucial biological process in normal physiology, for example in healing wounds. In addition, angiogenesis is important in pathological conditions, for example in accelerating the growth, progression, and metastasis of tumors [3, 4]. Various pro-angiogenic signaling pathways drive angiogenesis, among which the vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) pathway is the most important [5, 6].
Apatinib is an oral tyrosine kinase inhibitor of VEGFR2, which exhibited promising anti-neoplastic and antiangiogenic and activities in certain tumors, such as breast carcinoma [7], sarcoma [8], hepatocellular carcinoma [9], non-small-cell lung cancer [10], and recurrent epithelial ovarian cancer [11]. Although apatinib reverses multidrug resistance of chemotherapeutic agents, apatinib resistance can occur, making its full use challenging [8]. For some VEGFR inhibitors, the duration of response is short before the drug resistance occurs, resulting in only limited improvements in progression-free survival (PFS) and OS. Given apatinib’s central role in targeted therapy and the unsatisfactory clinical outcome resulting from apatinib resistance, new methods to ameliorate apatinib resistance are required [12].
In the present study, overexpression of the oncogene ERBB2 (encoding Erb-B2 receptor tyrosine kinase 2), and low expression of the antioncogene STING (encoding stimulator of interferon response cGAMP interactor), were observed in apatinib resistant (AR) cells. Accordingly, we hypothesized that STING and ERBB2 might regulate apatinib resistance in HNSCC via unknown pathway. The relationship between prognosis and VEGFR2 expression was first evaluated. The expression and function of STING and ERBB2 in apatinib resistance of HNSCC were further evaluated. The STING/ERBB2 pathway provides a potential target to overcome apatinib resistance during VEGFR2 inhibition therapy.
Results
Highly expressed VEGFR2 induces angiogenesis in HNSCC
According to the analysis of the TGCA-HNSC dataset, VEGFR2 (also known as KDR) expression was increased in cancer tissue compared with that in normal tissue. (Supplementary Figure 1A). Compared with that of the patients with low VEGFR2 expression, the survival time of patients with high VEGFR2 expression was shorter (Supplementary Figure 1B). According to the qRT-PCR and western blotting results, HNSCC cells had higher VEGFR2 levels than HIOECs. VEGFR2 expression was lower in HN6 cells than in HN30 and CAL27 cells (Figure 1A, 1B). In accordance with the in vitro results, subcutaneous tumors formed by HN30 showed a greater degree of angiogenesis than of those formed by HN6 cells (Figure 1C).
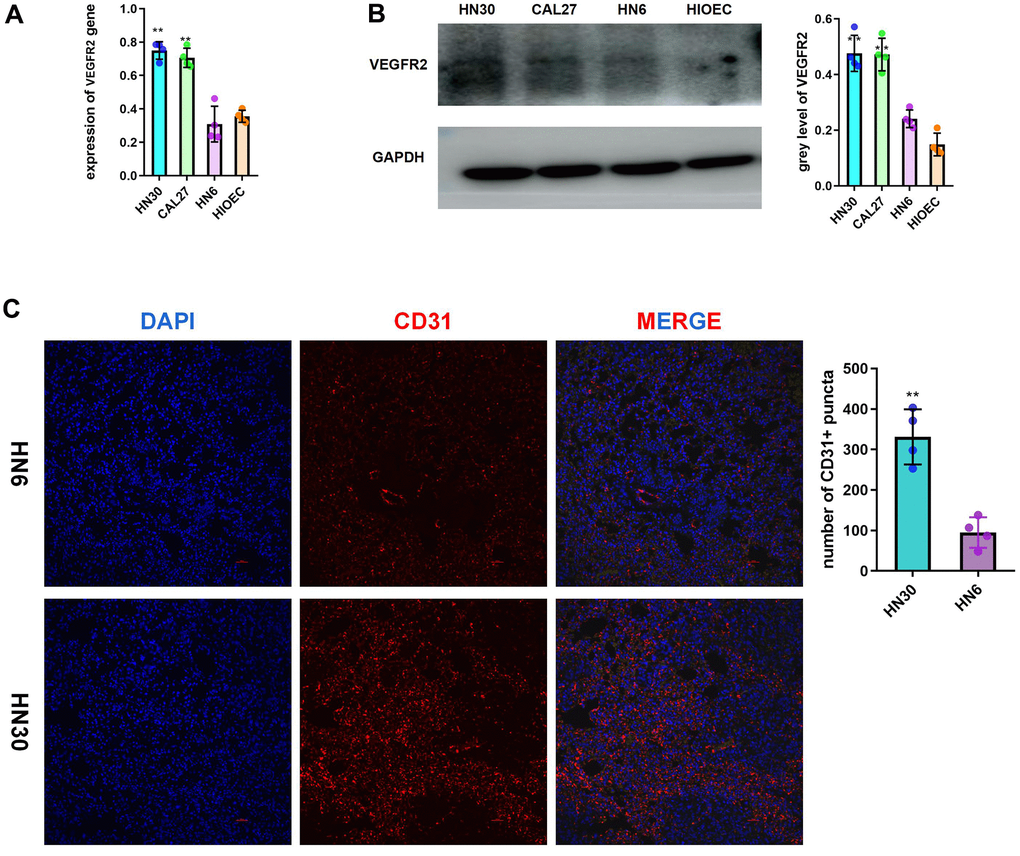
Figure 1. Highly expressed VEGFR2(KDR) induces angiogenesis in HNSCC. (A, B) qRT-PCR and western blotting results for VEGFR2 in HIOEC and HNSCC cell lines (HN30, CAL27, and HN6) (C) Representative images of HN6 and HN30 subcutaneous tumors using immunofluorescence staining against CD31 and DAPI staining of nuclei. Higher angiogenesis was observed in the HN30 group, which has a higher VEGFR2 expression level. *P < 0.05, **P < 0.01, ***P < 0.001 versus the control.
High ERBB2 expression and low STING expression were observed in AR cells
The colony formation assay and CCK-8 results for the AR cells compared with the PC cells showed that proliferation occurred in a fold-change manner (Figure 2A). (**P < 0.01). The viability of the AR cells higher than that of the PC controls. To analyze the signaling pathways related to apatinib resistance, RNA seq of AR and PC cells was performed. A total of 198 downregulated and 277 upregulation mRNAs were obtained for differential analysis (Figure 2B). STING and ERBB2 showed the highest fold change. To evaluate the relationship between VEGFR2, STING, and ERBB2, the TIMER database was used (http://timer.cistrome.org/), which identified a negative correlation between VEGFR2 and STING (also known as TMEM173) (Figure 2C). Meanwhile, a positive correlation was confirmed between VEGFR2 and ERBB2 (Figure 2C). In accordance with bioinformatic results, qRT-PCR and western blotting results showed upregulation of ERBB2 levels and downregulation of STING levels in AR cells compared with those in PC cells (Figure 2D).
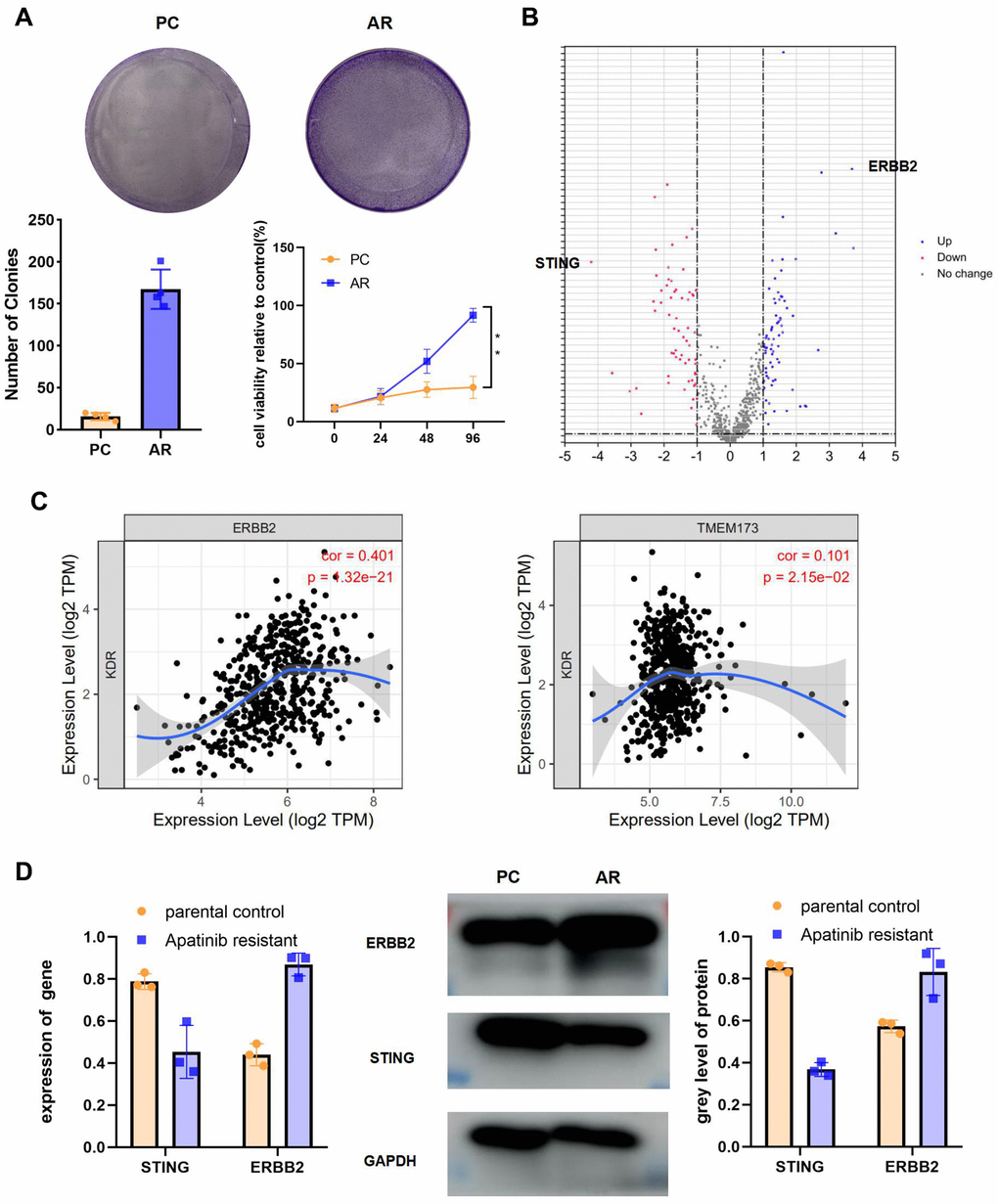
Figure 2. High ERBB2 expression and low STING expression were observed in AR cells. (A) Cell proliferation after treatment with apatinib for different times, as assessed using an MTT assay (*P < 0.05, **P < 0.01). The successful establishment of AR HN30 cells was demonstrated by their insensitivity to apatinib. Colony formation of AR cells was enhanced compared with that of the control. (B) Venn diagram of predicted up or downregulated mRNAs for AR cells compared with PC controls. A total of 198 downregulated and 277 upregulated mRNAs were obtained for differential analysis. STING and ERBB2 were identified as having the highest fold change. (C) A correlation was determined among VEGFR2, ERBB2, and STING using TIMER correlation analysis (http://timer.cistrome.org/). (D) AR and PC cells were treated with apatinib (20 μM) for 24 hours. Then, qRT-PCR and western blotting were performed to assess expression of ERBB2 and STING in AR and PC cells. *P < 0.05, **P < 0.01, ***P < 0.001 versus the control.
An ERBB2 inhibitor, lapatinib, and apatinib in combination re-sensitized AR cells to apatinib
To further explore the function of ERBB2 in apatinib resistance, TIMER analysis was used, which revealed the negative correlation between ERBB2 and immune cell infiltration (Figure 3A). This led us to speculate that apatinib combined with an ERBB2 inhibitor might increase apatinib sensitivity effectively. Lapatinib is an FDA approved ERBB2 inhibitor. According to a previous report [13], phosphorylation of ERBB2 is greatly downregulated by lapatinib. To address the efficacy of ERBB2 inhibition in apatinib resistance, AR cells were treated with lapatinib combined with apatinib. The results showed that the combined treatment suppressed AR cell proliferation in a synergistic manner (Figure 3B, 3C). Lapatinib has been reported to inhibit the growth of cancer via the ERBB2/AKT/mTOR [14] and RAF/MEK/ERK [15, 16] signaling pathways. The western blotting results showed that levels of p-ERBB, p-ERK, and p-AKT in AR cells decreased after 24 h of treatment with apatinib and lapatinib (Figure 3D). The amount of total AKT or ERBB2 protein in AR cells did not change after treatment.
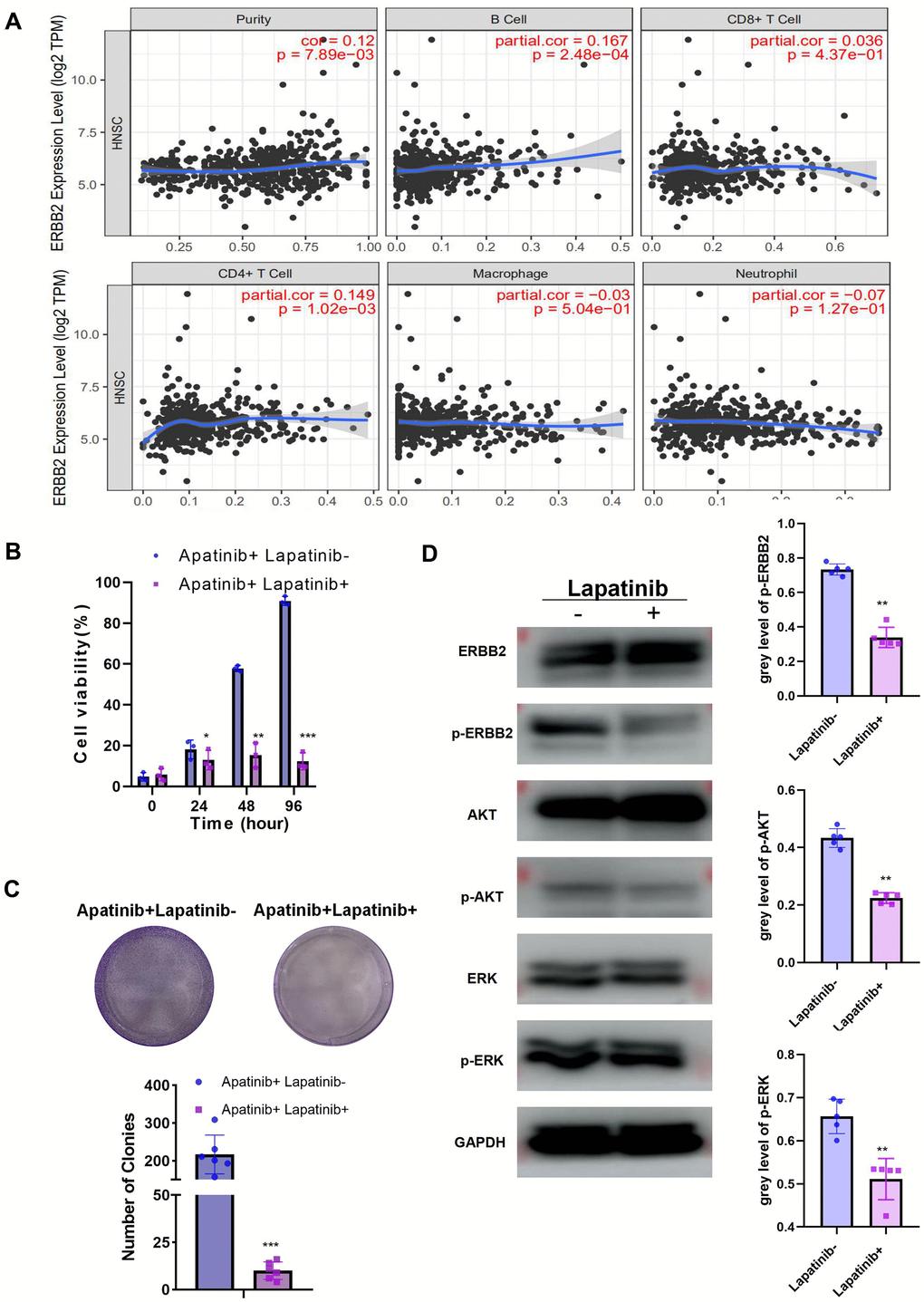
Figure 3. The combination of lapatinib and apatinib re-sensitizes AR cells to apatinib (A) The negative correlation between ERBB2 and TILs was confirmed using TIMER 2.0. (B) Cell viability after apatinib (20 μM) treatment, with or without lapatinib (20 μM), for different times, as assessed using an MTT assay. The combination of lapatinib and apatinib re-sensitized AR cells to apatinib. (C) Colony formation was inhibited in the combination group compared with that in the groups treated with each drug alone. (D) Western blotting illustrating the abundance of related signaling pathway proteins after apatinib (20 μM) treatment, with or without lapatinib(20 μM). Lapatinib treatment suppressed the levels of ERRB2, AKT and ERK phosphorylation. *P < 0.05, **P < 0.01, ***P < 0.001 versus the control.
Phosphorylation of ERBB2 could be inhibited by a STING agonist, vadimenzan
We observed a negative correlation between ERBB2 and STING (Figure 4A). Moreover, STING could stimulate immune cell infiltration. To explore the mechanism of STING in ERBB2-mediated apatinib resistance, a plasmid overexpressing STING was constructed and transfected into AR cells (Figure 4B). STING overexpression was confirmed using western blotting and qRT-PCR. Overexpression of STING resulted in inhibition of the phosphorylation of ERBB2, AKT, and ERK (Figure 4C). Considering the significance of STING in apatinib resistance, we hypothesized that the combination of apatinib with a STING agonist, Vadimenzan, would effectively enhance apatinib sensitivity. Combined treatment with vadimenzan and apatinib displayed a synergistic effect by markedly inhibiting AR cell proliferation (Figure 4D, 4E).
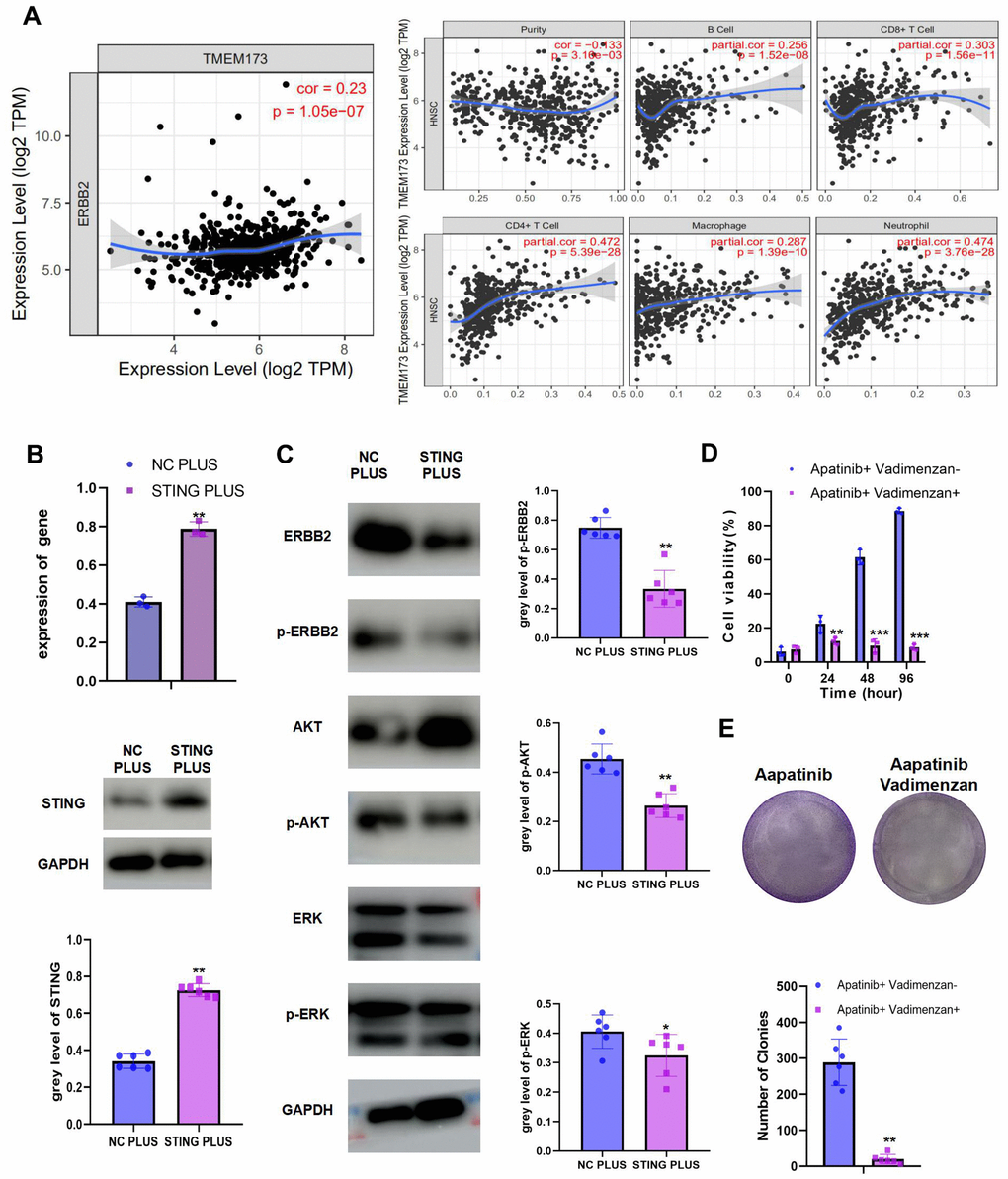
Figure 4. The combination of STING and apatinib re-sensitizes AR cells to apatinib (A) The positive correlation between STING and TILs was confirmed using TIMER 2.0. (B) AR cells were transfected with a STING overexpression plasmid, qRT-PCR and western blot result confirmed the successful establishment of STING PLUS AR cells. (C) Levels of ERBB2, AKT, ERK, p-ERBB2, p-AKT, and p-ERK were detected using western blotting. Compared with NC PLUS cells, the levels of ERBB2, p-ERBB2, p-AKT, and p-ERK were downregulated in STING PLUS cells. (D) MTT assay illustrating the cell viability of NC PLUS and STING PLUS AR cells after apatinib (20 μM) treatment for different times. The combination of STING and apatinib re-sensitized AR cells to apatinib. (E) Colony formation was inhibited in the combination group compared with that in the groups treated with each drug alone. *P < 0.05, **P < 0.01, ***P < 0.001 versus the control.
The combination of lapatinib and vadimenzan re-sensitizes AR cells to apatinib in vivo
We used HNSCC xenografts in nude mice to verify the in vitro results. The tumor volume in the combined treatment group was significantly smaller than that in the groups treated with each single agent (Figure 5A). Moreover, immunohistochemistry indicated decreased Ki67 levels, which suggested inhibition of proliferation in the combined treatment group (Figure 5B). Correspondingly, the subcutaneous tumors of the combination group showed less angiogenesis than those from the apatinib only group (Figure 5C).
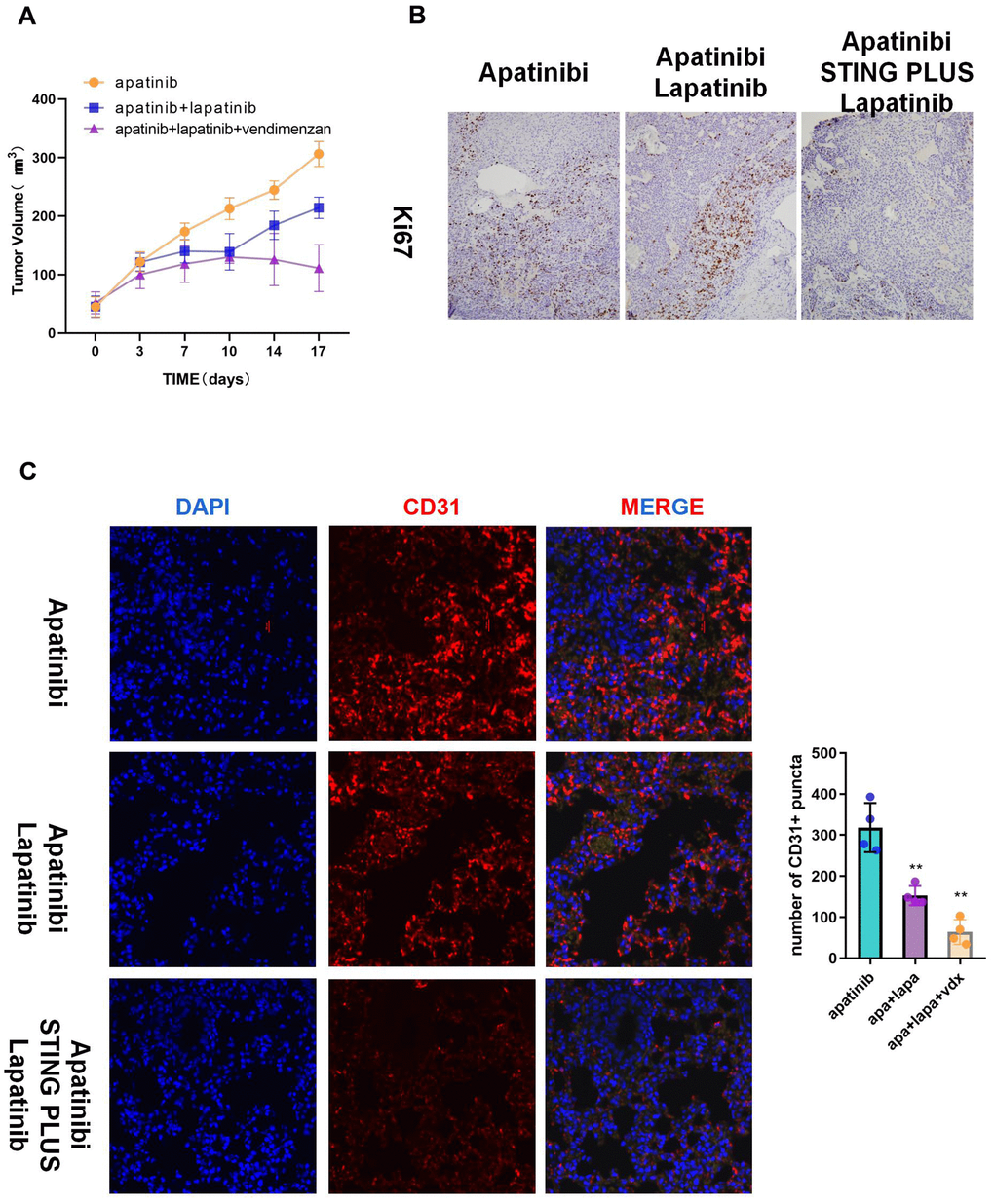
Figure 5. The combination of vadimenzan and lapatinib re-sensitizes AR cells to apatinib in vivo. (A) After washing with PBS three times, AR cells (total of 1 × 106) suspended in 50 μL of DMEM were injected in each injection site. The tumor volumes of the mice were evaluated among the three groups. (B) Ki67 staining images of cancer samples in the different groups. (C) Representative images of subcutaneous tumors using immunofluorescence staining against CD31 and DAPI nuclear staining. *P < 0.05, **P < 0.01, versus the control.
Discussion
Therapeutic strategies targeting vascular endothelial growth factor receptors (VEGFRs) have been studied extensively because of the important roles of VEGFRs in carcinogenesis [17]. Apatinib is an oral tyrosine kinase inhibitor of VEGFR-2 that can induce autophagy [18] and apoptosis, and suppresses tumor proliferation in anaplastic thyroid cancer [8], hepatocellular carcinoma [19], and osteosarcoma [20]. However, effective treatment is challenged by apatinib resistance [21]. The acquired resistance involves the activation of pathways such as JAK/STAT, PI3K/AKT, and MAK/ERK signaling [22, 23]. Thus, new approaches are required to enhance apatinib’s efficacy [7, 24]. However, most related research has focused on microRNAs and circular RNAs, which are unlikely to be transformed into clinical application in coming years. ERBB2 is a well-known oncogene, and in preclinical studies of HER2-positive advanced solid tumors, ERBB2 inhibitors have displayed very good antitumor activity, both in vivo and in vitro [25, 26]. A previous study showed that an ERBB2 inhibitor combined with apatinib was effective against HER2-positive gastric cancer and acquired resistance against the ERBB2 inhibitor [27]. In HNSCC, ERBB2 is upregulated in primary and metastatic tumors, which is related to poor prognosis [28].
In this study, we confirmed that upregulation of ERBB2 enhances apatinib resistance through PI3K/AKT and MAK/ERK signaling, which is consistent with the results of previous studies [22, 23]. We speculated that phosphorylation of ERBB2 could increase apatinib resistance by activating AKT and ERK. Inhibition of ERBB2 phosphorylation by the TKI inhibitor, lapatinib, effectively re-sensitized AR cells to apatinib. Furthermore, cell viability was significantly decreased under the combined treatment of apatinib and lapatinib.
STING expression correlates negatively with that of many oncogenes and is thus believed to be a tumor suppressor [29, 30]. Within the tumor microenvironment (TME), STING pathway activation in antigen-presenting cells leads to increased production of type I interferons and promotes tumor-mediated cross priming of CD8+ T cells, finally resulting in adaptive anticancer immune responses [31, 32]. STING shows strong expression in HPV+ HNSCC cancer cells, but not in HPV− HNSCC cancer cells [33]. STING ligands administered locally to the tumor led to non-autonomous activation of STING in non-cancer cells in the TME, suggesting that such therapy might be effective to treat STING− and STING+ tumors [33, 34]. According to these activities, STING agonists were demonstrated to synergize cancer treatment by promoting CAR T cells or overcoming tumor resistance to PD-1 blockade [35–37]. Our research has shown that the antitumor effort of STING acts via proliferation and this proliferation sensitizes AR cells to apatinib. To the best of our knowledge, this is the first report of the effect of STING on apatinib resistance.
In HPV- HNSCC, STING expression is suppressed, and is further downregulated in AR cells; therefore, we investigated whether treatment promoting STING combined with apatinib has therapeutic potential in HNSCC. Our results demonstrated that ERBB2, AKT, and ERK confer apatinib desensitization. In addition, vadimenzan, a STING agonist, attenuated apatinib desensitization and decreased the proliferation on AR cells in vitro. In vivo, vadimenzan treatment resulted in xenografts becoming sensitive to apatinib therapy. Thus, targeting the ERBB2/AKT/ERK axis by stimulating STING represents a feasible method to restore the apatinib sensitivity of HNSCC cells.
Overall, the results of the present study indicated that the ERBB2/AKT/ERK axis regulates apatinib desensitization. Importantly, upregulation of STING expression overcame apatinib resistance effectively by inhibiting ERBB2 phosphorylation. Apatinib combined with a STING agonist, e.g., vadimenzan, could be used to ameliorate apatinib resistance in HNSCC.
Materials and Methods
Cells and chemicals
The American Type Culture Collection (ATCC) (Manassas, VA, USA) provided the CAL27, HN6, and HN30 cells. Human immortalized oral epithelial cells (HIOECs) were grown in defined keratinocyte serum-free medium (Invitrogen, Waltham, MA, USA). CAL27, HN6, and HN30 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) containing 1% penicillin-streptomycin, 1% glutamine, and 10% fetal bovine serum. Apatinib was obtained from Hengrui Medicine Co., Ltd. (Jiangsu, China). The STING agonist, vadimenzan, was purchased from Selleck CO., Ltd. (Shanghai, China). In vitro, AR CAL27 cells were established by treating the cells with 5 μM apatinib initially and the increasing the concentration incrementally to 20 μM once a week for 3 months. To verify the successful establishment of AR cells, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed.
Bioinformatics
The mRNA profiles (Normal: 44, Tumor: 520) were obtained from The Cancer Genome Atlas (TCGA) database as the TCGA-HNSC dataset (https://portal.gdc.cancer.gov/). Kaplan–Meier analysis of OS and PFS was performed based on the TCGA-HNSC dataset.
Quantitative real-time reverse transcription PCR (qRT-PCR)
The total mRNAs were extracted from HIOEC and HNSCC cells lines. A NanoDrop 2000/2000C spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA) was used to assess the RNA purity and concentration at wavelengths of 260/280 nm. The RNA was transcribed into cDNA using a PrimeScript™ RT Reagent Kit (Takara Biotechnology, Dalian, China). A TB Green® Premix Ex TaqTM Kit (Takara Biotechnology) master mix was used to perform the qPCR reactions using the cDNAs as templates on a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). GAPDH (encoding glyceraldehyde-3-phosphate dehydrogenase) expression was detected as an internal control. The primers used for qPCR of human genes were:
| Gene name | Type | Sequence 5'-3' |
| ERBB2 | Forward | TGCAGGGAAACCTGGAACTC |
| Reverse | ACAGGGGTGGTATTGTTCAGC | |
| STING | Forward | CGCTTCCTGGATAAACTGCC |
| Reverse | GCCCACAGTAACCTCTTCCT | |
| VEGFR2 | Forward | GGCCCAATAATCAGAGTGGCA |
| Reverse | CCAGTGTCATTTCCGATCACTTT | |
| GAPDH | Forward | AATCCCATCACCATCTTCCAG |
| Reverse | GAGCCCCAGCCTTCTCCAT |
Western blotting
Cells were treated with 20 μM apatinib, with or without lapatinib (20 μM) and vadimenzan (20 μM), for 24 h. Then, at various time points, we extracted total cellular proteins. The proteins were separated electrophoretically and then electrotransferred onto a membrane. Next, 5% skim milk in 1% TBST was used to block the membrane for 1 h at 4° C. The proteins on the membrane were then reacted with primary antibodies that recognized STING (catalog number 13647S; 1:1,000); ERBB2 (catalog number 2165S; 1:1,000); phosphorylated (p)-ERBB2 (catalog number 6942; 1:1,000); VEGFR2 (catalog number 9698; 1:1,000); protein kinase B (AKT) (catalog number 4685S; 1:1,000); p-AKT (catalog number 4060S; 1:1,000); extracellular regulated kinase (ERK) (catalog number 9194S; 1:1,000); p-ERK (catalog number 4370S; 1:1,000); and GAPDH (catalog number 174S; 1:1,000). All primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
Immunohistochemistry and immunofluorescence
To evaluate angiogenesis, an anti-CD31 primary antibody was incubated with tumor sections, followed by reaction with an Alexa 488-conjugated goat anti-mouse secondary antibody according to the manufacturer’s protocol. 4′,6-diamidino-2-phenylindole (DAPI) was used to stain the cell nuclei. The stained sections were observed under a confocal microscope (SP5, Leica, Wetzlar, Germany). To evaluate cell apoptosis, marker of proliferation Ki-67 (Ki67) staining was performed using anti-Ki67 primary antibodies.
Colony formation and cell viability assays
To assess cytotoxicity, parental control (PC), AR, and CAL27 cells were seeded at a density of 1 × 104 cells/ml in 96-well flat-bottom plates in triplicate and cultured in 100 μL medium for 12 h before being exposed to apatinib, with or without lapatinib and vadimenzan. Then, Cell Counting Kit 8 (CCK-8) solution (10% in 100 μL of medium; Dojindo, Japan) was added to each well at different timepoints. The absorbance at 450 nm was measured after 2 h of incubation. For the colony formation assay, PC, AR, and CAL27 cells were seeded in six-well plates at 1 × 104 cells per well. Ten days later, neutral paraformaldehyde was used to fix the cells, followed by staining with a crystal violet solution. Formed colonies comprising 50 to 100 cells were counted.
RNA sequencing (RNA-seq)
For apatinib resistance studies, the HiseqXTen system (Genomeditech Co. Ltd., Shanghai, China) was used to perform RNA-seq. Differentially expressed mRNAs were identified based on their fold-change in expression and their P-values, which were determined using one-way analysis of variance. Differentially expressed genes (DEGs) were displayed on a volcano plot according the set as a fold-change of X and a P-value of Y, and the DEGs were displayed used a volcano plot.
Tumor xenografts
The Chongqing experimental animal center supplied 4-week-old specific pathogen free male BALB/c nude mice (nu/nu), weighing 16.31 ± 0.9 g. All laboratory procedures were approved by the laboratory animal care and use committee of the hospital. HN6/HN30 cells (1 × 106 cells) were washed in PBS three times before being suspended in 50 μL of DMEM per injection site. The cells were injected subcutaneously into the back of the right rear leg in each group of mice (n = 5). The sample size was calculated according to previous research [38, 39]. At 10 days after injection, the average tumor volume was nearly 200 mm3. At this point, the mice were euthanized humanely. In another experiment, 1 × 106 AR cells (3rd passage) were washed in PBS three times before being suspended in 50 μL of DMEM per injection site. The cells were injected subcutaneously into the back of the right rear leg of the mice. Seven days later, the average tumor volume was nearly 125 mm3. The exact sizes of the tumors on each animal before treatment are shown in Supplementary Table 1. Then, the mice were randomly separated into three groups (n = 5 per group): the apatinib group (orally-delivered apatinib at 150 mg/kg per day), the apatinib+lapatinib group (the same dose and schedule of apatinib plus lapatinib solution (100 mg/kg)), and the apatinib+lapatinib+vendimenzan group (the same oral doses and schedule of apatinib and lapatinib plus i.p. administered vadimenzan (50 mg/kg) twice a week). The development and progression of solid tumors were monitored every two days until the tumor reached greater than 1.5 cm in length. At this point, the mice were euthanized humanely. The tumor volume (V) was calculated as: V = L x W2/2, where L is the tumor length and W is the tumor width. Immunohistochemistry and immunofluorescence analyses were performed on xenograft samples.
Statistical analysis
The mean ± SD was used to represent continuous variables. The clinical and histological data were analyzed using the chi-squared test or Pearson’s chi-squared test. All statistical data analysis was carried out using GraphPad Prism version 7 (GraphPad Software Inc., San Diego, CA, USA). The statistical significance of differences was assessed using Student's t-test and one-way analysis of variance. In the figures, statistical significance is indicated using: *p < 0.05, **p < 0.01 and ***p < 0.001.
Ethics approval and consent to participate
The Ethical Committee of Chongqing University Cancer Hospital approved the study design.
Author Contributions
Chengyao Zhang was responsible for manuscript revision. Junbin Zhang performed the animal experiment and collected the samples. Guo Ye conceived the study, carried out the in vitro experiments, collected and analyzed the data, and is critically revised the article for important intellectual content. All the authors read and approved the final copy of the manuscript for publication.
Acknowledgments
We are grateful to the staff of the Chongqing Key Laboratory of Translational Research for Cancer Metastasis and Individualized Treatment, Chongqing, China for their support and comments during the preparation of this manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by the Cancer Research Youth Science Foundation of Chinese Anti-Cancer Association [grant number CAYC18A49]; and Chongqing Science and Technology Commission Research Institute Performance Incentive Program of China [grant number 2017jxjl30022].
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019; 69:7–34. https://doi.org/10.3322/caac.21551 [PubMed]
- 2. Chinn SB, Myers JN. Oral Cavity Carcinoma: Current Management, Controversies, and Future Directions. J Clin Oncol. 2015; 33:3269–76. https://doi.org/10.1200/JCO.2015.61.2929 [PubMed]
- 3. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002; 29:15–18. https://doi.org/10.1053/sonc.2002.37263 [PubMed]
- 4. Tulotta C, He S, van der Ent W, Chen L, Groenewoud A, Spaink HP, Snaar-Jagalska BE. Imaging Cancer Angiogenesis and Metastasis in a Zebrafish Embryo Model. Adv Exp Med Biol. 2016; 916:239–63. https://doi.org/10.1007/978-3-319-30654-4_11 [PubMed]
- 5. Hill MA, Jaisser F, Sowers JR. Role of the vascular endothelial sodium channel activation in the genesis of pathologically increased cardiovascular stiffness. Cardiovasc Res. 2020. [Epub ahead of print]. https://doi.org/10.1093/cvr/cvaa326 [PubMed]
- 6. Lee SH, Jeong D, Han YS, Baek MJ. Pivotal role of vascular endothelial growth factor pathway in tumor angiogenesis. Ann Surg Treat Res. 2015; 89:1–8. https://doi.org/10.4174/astr.2015.89.1.1 [PubMed]
- 7. Liu J, Liu Q, Li Y, Li Q, Su F, Yao H, Su S, Wang Q, Jin L, Wang Y, Lau WY, Jiang Z, Song E. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: an open-label phase II trial. J Immunother Cancer. 2020; 8:e000696. https://doi.org/10.1136/jitc-2020-000696 [PubMed]
- 8. Yang QK, Chen T, Wang SQ, Zhang XJ, Yao ZX. Apatinib as targeted therapy for advanced bone and soft tissue sarcoma: a dilemma of reversing multidrug resistance while suffering drug resistance itself. Angiogenesis. 2020; 23:279–98. https://doi.org/10.1007/s10456-020-09716-y [PubMed]
- 9. Xu J, Shen J, Gu S, Zhang Y, Wu L, Wu J, Shao G, Zhang Y, Xu L, Yin T, Liu J, Ren Z, Xiong J, et al. Camrelizumab in Combination with Apatinib in Patients with Advanced Hepatocellular Carcinoma (RESCUE): A Nonrandomized, Open-label, Phase II Trial. Clin Cancer Res. 2021; 27:1003–11. https://doi.org/10.1158/1078-0432.CCR-20-2571 [PubMed]
- 10. Zhou C, Wang Y, Zhao J, Chen G, Liu Z, Gu K, Huang M, He J, Chen J, Ma Z, Feng J, Shi J, Yu X, et al. Efficacy and Biomarker Analysis of Camrelizumab in Combination with Apatinib in Patients with Advanced Nonsquamous NSCLC Previously Treated with Chemotherapy. Clin Cancer Res. 2021; 27:1296–304. https://doi.org/10.1158/1078-0432.CCR-20-3136 [PubMed]
- 11. Miao M, Deng G, Luo S, Zhou J, Chen L, Yang J, He J, Li J, Yao J, Tan S, Tang J. A phase II study of apatinib in patients with recurrent epithelial ovarian cancer. Gynecol Oncol. 2018; 148:286–90. https://doi.org/10.1016/j.ygyno.2017.12.013 [PubMed]
- 12. Yu J, Wang N, Gong Z, Liu L, Yang S, Chen GG, Lai PB. Cytochrome P450 1A2 overcomes nuclear factor kappa B-mediated sorafenib resistance in hepatocellular carcinoma. Oncogene. 2021; 40:492–507. https://doi.org/10.1038/s41388-020-01545-z [PubMed]
- 13. Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, Li S, Chen T, Poteete A, Estrada-Bernal A, Le AT, Truini A, Nilsson MB, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med. 2018; 24:638–46. https://doi.org/10.1038/s41591-018-0007-9 [PubMed]
- 14. Bui T, Rennhack J, Mok S, Ling C, Perez M, Roccamo J, Andrechek ER, Moraes C, Muller WJ. Functional Redundancy between β1 and β3 Integrin in Activating the IR/Akt/mTORC1 Signaling Axis to Promote ErbB2-Driven Breast Cancer. Cell Rep. 2019; 29:589–602.e6. https://doi.org/10.1016/j.celrep.2019.09.004 [PubMed]
- 15. Jaiswal BS, Durinck S, Stawiski EW, Yin J, Wang W, Lin E, Moffat J, Martin SE, Modrusan Z, Seshagiri S. ERK Mutations and Amplification Confer Resistance to ERK-Inhibitor Therapy. Clin Cancer Res. 2018; 24:4044–55. https://doi.org/10.1158/1078-0432.CCR-17-3674 [PubMed]
- 16. Li S, Ma YM, Zheng PS, Zhang P. GDF15 promotes the proliferation of cervical cancer cells by phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J Exp Clin Cancer Res. 2018; 37:80. https://doi.org/10.1186/s13046-018-0744-0 [PubMed]
- 17. Zhang Q, Yu C, Peng S, Xu H, Wright E, Zhang X, Huo X, Cheng E, Pham TH, Asanuma K, Hatanpaa KJ, Rezai D, Wang DH, et al. Autocrine VEGF signaling promotes proliferation of neoplastic Barrett’s epithelial cells through a PLC-dependent pathway. Gastroenterology. 2014; 146:461–72.e6. https://doi.org/10.1053/j.gastro.2013.10.011 [PubMed]
- 18. Zhou ZH, Zhao TC, Liang SY, Zhang ZY, Zhu DW, Ju WT, Zhong LP. A therapeutic approach with combination of interferon-gamma and autophagy inhibitor for oral squamous cell carcinoma. Am J Cancer Res. 2021; 11:1503–21. https://doi.org/10.21203/rs.3.rs-30028/v1 [PubMed]
- 19. Zhang H, Cao Y, Chen Y, Li G, Yu H. Apatinib promotes apoptosis of the SMMC-7721 hepatocellular carcinoma cell line via the PI3K/Akt pathway. Oncol Lett. 2018; 15:5739–43. https://doi.org/10.3892/ol.2018.8031 [PubMed]
- 20. Liu K, Ren T, Huang Y, Sun K, Bao X, Wang S, Zheng B, Guo W. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. 2017; 8:e3015. https://doi.org/10.1038/cddis.2017.422 [PubMed]
- 21. Zhou ZH, Liang SY, Zhao TC, Chen XZ, Cao XK, Qi M, Huang YY, Ju WT, Yang M, Zhu DW, Pang YC, Zhong LP. Overcoming chemotherapy resistance using pH-sensitive hollow MnO2 nanoshells that target the hypoxic tumor microenvironment of metastasized oral squamous cell carcinoma. J Nanobiotechnology. 2021; 19:157. https://doi.org/10.1186/s12951-021-00901-9 [PubMed]
- 22. Guo W, Li W, Yuan L, Mei X, Hu W. MicroRNA-106a-3p Induces Apatinib Resistance and Activates Janus-Activated Kinase 2 (JAK2)/Signal Transducer and Activator of Transcription 3 (STAT3) by Targeting the SOCS System in Gastric Cancer. Med Sci Monit. 2019; 25:10122–28. https://doi.org/10.12659/MSM.919610 [PubMed]
- 23. Yu R, Bai H, Gao B, Li T, He X, Zhang P, Wang J. Rare case of apatinib acquired resistance induced by point mutation of WRN p.V697F through activation of the PI3K/AKT apoptosis-inhibiting pathway. Thorac Cancer. 2021; 12:128–32. https://doi.org/10.1111/1759-7714.13726 [PubMed]
- 24. Ma L, Wang Z, Xie M, Quan Y, Zhu W, Yang F, Zhao C, Fan Y, Fang N, Jiang H, Wang Q, Wang S, Zhou J, et al. Silencing of circRACGAP1 sensitizes gastric cancer cells to apatinib via modulating autophagy by targeting miR-3657 and ATG7. Cell Death Dis. 2020; 11:169. https://doi.org/10.1038/s41419-020-2352-0 [PubMed]
- 25. Ma F, Li Q, Chen S, Zhu W, Fan Y, Wang J, Luo Y, Xing P, Lan B, Li M, Yi Z, Cai R, Yuan P, et al. Phase I Study and Biomarker Analysis of Pyrotinib, a Novel Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor, in Patients With Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. J Clin Oncol. 2017; 35:3105–12. https://doi.org/10.1200/JCO.2016.69.6179 [PubMed]
- 26. Taira N, Sawaki M, Uemura Y, Saito T, Baba S, Kobayashi K, Kawashima H, Tsuneizumi M, Sagawa N, Bando H, Takahashi M, Yamaguchi M, Takashima T, et al, and RESPECT Study Group. Health-Related Quality of Life With Trastuzumab Monotherapy Versus Trastuzumab Plus Standard Chemotherapy as Adjuvant Therapy in Older Patients With HER2-Positive Breast Cancer. J Clin Oncol. 2021; 39:2452–62. https://doi.org/10.1200/JCO.20.02751 [PubMed]
- 27. Su B, Huang T, Jin Y, Yin H, Qiu H, Yuan X. Apatinib exhibits synergistic effect with pyrotinib and reverses acquired pyrotinib resistance in HER2-positive gastric cancer via stem cell factor/c-kit signaling and its downstream pathways. Gastric Cancer. 2021; 24:352–67. https://doi.org/10.1007/s10120-020-01126-9 [PubMed]
- 28. Martín-Ezquerra G, Salgado R, Toll A, Gilaberte M, Baró T, Alameda Quitllet F, Yébenes M, Solé F, Garcia-Muret M, Espinet B, Pujol RM. Multiple genetic copy number alterations in oral squamous cell carcinoma: study of MYC, TP53, CCDN1, EGFR and ERBB2 status in primary and metastatic tumours. Br J Dermatol. 2010; 163:1028–35. https://doi.org/10.1111/j.1365-2133.2010.09947.x [PubMed]
- 29. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014; 41:830–42. https://doi.org/10.1016/j.immuni.2014.10.017 [PubMed]
- 30. Frémond ML, Crow YJ. STING-Mediated Lung Inflammation and Beyond. J Clin Immunol. 2021; 41:501–14. https://doi.org/10.1007/s10875-021-00974-z [PubMed]
- 31. Motani K, Kosako H. Activation of stimulator of interferon genes (STING) induces ADAM17-mediated shedding of the immune semaphorin SEMA4D. J Biol Chem. 2018; 293:7717–26. https://doi.org/10.1074/jbc.RA118.002175 [PubMed]
- 32. Guo F, Tang L, Shu S, Sehgal M, Sheraz M, Liu B, Zhao Q, Cheng J, Zhao X, Zhou T, Chang J, Guo JT. Activation of Stimulator of Interferon Genes in Hepatocytes Suppresses the Replication of Hepatitis B Virus. Antimicrob Agents Chemother. 2017; 61:e00771–17. https://doi.org/10.1128/AAC.00771-17 [PubMed]
- 33. Baird JR, Feng Z, Xiao HD, Friedman D, Cottam B, Fox BA, Kramer G, Leidner RS, Bell RB, Young KH, Crittenden MR, Gough MJ. STING expression and response to treatment with STING ligands in premalignant and malignant disease. PLoS One. 2017; 12:e0187532. https://doi.org/10.1371/journal.pone.0187532 [PubMed]
- 34. Baird JR, Friedman D, Cottam B, Dubensky TW
Jr , Kanne DB, Bambina S, Bahjat K, Crittenden MR, Gough MJ. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016; 76:50–61. https://doi.org/10.1158/0008-5472.CAN-14-3619 [PubMed] - 35. Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, Trotter BW, Altman MD, Buevich AV, Cash B, Cemerski S, Chang W, Chen Y, et al. An orally available non-nucleotide STING agonist with antitumor activity. Science. 2020; 369:eaba6098. https://doi.org/10.1126/science.aba6098 [PubMed]
- 36. Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, Willis C, Dotti G, Vincent BG, Restifo NP, Serody JS. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med. 2021; 218:e20200844. https://doi.org/10.1084/jem.20200844 [PubMed]
- 37. Lemos H, Ou R, McCardle C, Lin Y, Calver J, Minett J, Chadli A, Huang L, Mellor AL. Overcoming resistance to STING agonist therapy to incite durable protective antitumor immunity. J Immunother Cancer. 2020; 8:e001182. https://doi.org/10.1136/jitc-2020-001182 [PubMed]
- 38. Ma H, Chang H, Yang W, Lu Y, Hu J, Jin S. A novel IFNα-induced long noncoding RNA negatively regulates immunosuppression by interrupting H3K27 acetylation in head and neck squamous cell carcinoma. Mol Cancer. 2020; 19:4. https://doi.org/10.1186/s12943-019-1123-y [PubMed]
- 39. Yang W, Jiang C, Xia W, Ju H, Jin S, Liu S, Zhang L, Ren G, Ma H, Ruan M, Hu J. Blocking autophagy flux promotes interferon-alpha-mediated apoptosis in head and neck squamous cell carcinoma. Cancer Lett. 2019; 451:34–47. https://doi.org/10.1016/j.canlet.2019.02.052 [PubMed]