



Introduction
Osteoarthritis (OA) was considered as a prototypical arthropathy, which is irrelevant to inflammation [1–3]. However, increasing studies have documented that synovitis is observed in a many patients with OA [4]. It is convincing that there is a close relation between the pathogenesis of OA and joint inflammation. The mechanism of OA is quite complex, many cell types such as articular chondrocytes, synovial cells, and other cells of diarthrodial joints, are involved in OA development [1]. These cells express inflammatory mediators, pro-inflammatory cytokines, and matrix degrading enzymes, which are crucial molecules for the progression of OA in synovial joints [4, 5]. Therefore, targeting inflammation pathways could be a novel therapeutic approach for OA.
NF-κB signaling has essential roles in plenty of cellular processes, especially in inflammatory response [6–8]. Importantly, NF-κB pathway induces various genes expression, which could induce further activation of other signaling cascades [9, 10]. There is evidence that NF-κB signaling is widely involved in the pathophysiology of OA and is confirmed as a potential target [11].
NOD-like receptor family (NLR) is a large protein family that act as pro-inflammatory receptors to participate in many biological processes [12–14]. Accumulating evidence has indicated that a member of NLR family, NLRC5, is a critical mediator of inflammatory response [15, 16]. Additionally, NLRC5 inhibits the activation of NF-κB signaling induced by LPS, TNF-α or IL-1β [16]. Based on these evidences, we speculated that NLRC5 may be involved in the pathogenesis of OA.
Here, we used IL-1β to induce inflammation in chondrocytes isolated from OA patients. Then the potential roles of NLRC5 in chondrocytes was investigated and the underlying mechanism was explored.
Materials and Methods
Cell culture of primary human OA chondrocytes and IL-1β treatment
Cartilage samples from 9 OA patients who underwent total knee arthroplasty were collected at Central Hospital Affiliated to Shen Yang Medical Collage (Shenyang, China). Informed consent was obtained from all the patients involved in this study, which was approved by the Ethics Committee of the Central Hospital Affiliated to Shen Yang Medical Collage. The cartilage samples cut with scissors and then digested with 0.25% trypsin-EDTA solution for 30 min, followed by digestion with 0.4% collagenase II (Sigma-Aldrich, St. Louis, MO, USA) for 24 h. Cells were cultured in DMEM/F12 growth medium containing 10% FBS (Hyclone) and 1% penicillin/streptomycin (Sigma). The OA chondrocytes were maintained at 37° C in a humidified atmosphere. Chondrocytes were stimulated by IL-1β (10 ng/ml; Peprotech Asia, Rocky Hill, NJ, USA) for 24 h to induce inflammation. PDTC (5 μM; Sigma) was used to block NF-κB activation.
Quantitative real-time PCR (qRT-PCR)
Total RNA samples were extracted from chondrocytes using Qiazol (Qiagen, Hilden, Germany). RNA was quantified and then applied for the generation of cDNA using a cDNA Synthesis Kit. The mRNA levels of target genes were measured using a SYBR Green qPCR master mix on a 7500 Real-Time PCR System. Results were calculated using the 2-ΔΔCT method.
Cell transfection
Chondrocytes were inoculated into a six-well plate and incubated for 24 h before transfection. Then the cells were transfected with NLRC5 siRNA (si-NLRC5) or negative control siRNA (si-NC), which were obtained from GenePharma (Shanghai, China). The NLRC5 overexpressing plasmid was constructed by inserting the open reading frame of NLRC5 into the pcDNA3.1 expression vector. Chondrocytes were transfected with siRNAs or vectors using RNAiMAX Reagent or Lipo2000 transfection reagent (Invitrogen).
Western blot
Chondrocytes were lysed in RIPA Lysis Buffer containing PMSF and phosphatase inhibitor (Beyotime), and the lysates were obtained from centrifugation. Proteins were separated on 12% SDS-PAGE gels. Then the proteins on the gels were transferred to PVDF membranes (Thermo) and blocked with 5% skimmed milk powder in TBST buffer for 1 h. The membranes were sequentially incubated with primary antibodies (anti-NLRC5, iNOS, COX-2; were obtained from Abcam, Cambridge, MA, USA; anti-p-p65, p65, IĸBα, or β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) at 4° C overnight and HRP-conjugated secondary antibodies (Santa Cruz) for 2 h. The targeted protein bands were developed using ECL reagent (Thermo).
Cell viability assay
Chondrocytes (1 × 104 cells/well) were seeded into 96-well plates and subjected with indicated transfections and treatments. After that, MTT (5 mg/ml) was added to the cells to evaluate the cell viability. After incubation for 4 h, the products were dissolved by incubating with dimethyl sulfoxide (DMSO). Then the OD value at 490 nm was measured using a microplate reader.
Measurement of NO
Twenty-four hours post indicated treatments, the NO production in cell culture samples was measured by a nitrate/nitrite colorimetric assay kit.
ELISA
The secretion of prostaglandin E2 (PGE2), TNF-α, IL-6, MMP-3 and MMP-13 in the supernatants were measured using corresponding ELISA kits purchased from R&D Systems.
OA model in rats and animal treatment
Six-week-old male Sprague-Dawley rats (200 ± 20 g) were purchased from the Animal Center of Chinese Academy of Sciences (Shanghai, China). The OA model was established as described previously [17]. The rats were randomly divided into control group: rats received a sham-operation; model group: rats received an operation; treatment group (NLRC5 group): 1-week after operation, the rats were intraarticularly injected with 50 μl solution (10 ng/ml) every 7 days. After 6 weeks of indicated treatments, the knee joint tissues were collected and stored in 4% paraformaldehyde solution for further histological analysis. The animal experiments were approved by the Animal Care and Use Committee of Central Hospital Affiliated to Shen Yang Medical Collage.
Histological analysis
Knee joint tissues were embedded in paraffin and then cut into sections (5 μm) using a rotary microtome. Then, the sections were stained with hematoxylin-eosin (H&E) and Safranin O. The images were captured using a light microscope. Cartilage destruction was examined by Safranin O staining, finally, the score was evaluated using the Osteoarthritis Research Society International (OARSI) grading system.
Statistical analysis
The data were expressed as the mean ±SD. Two-group comparisons were evaluated by Student’s t test. Multiple-group comparisons were analyzed by one-way analysis of variance (ANOVA). P < 0.05 was considered to indicate a statistically significant difference.
Results
NLRC5 expression was down-regulated in IL-1β-induced chondrocytes
IL-1β signaling plays constructive roles in the pathogenesis of OA [18, 19]. IL-1β is frequently used for inducing inflammation in chondrocytes to simulate OA in vitro [20–22]. Firstly, we investigated NLRC5 expression in IL-1β-induced chondrocytes. Results showed that the mRNA level of NLRC5 was markedly down-regulated in chondrocytes after induction with IL-1β (Figure 1A). Consistently, western blot analysis revealed that NLRC5 is lowly expressed in chondrocytes stimulated with IL-1β (Figure 1B).
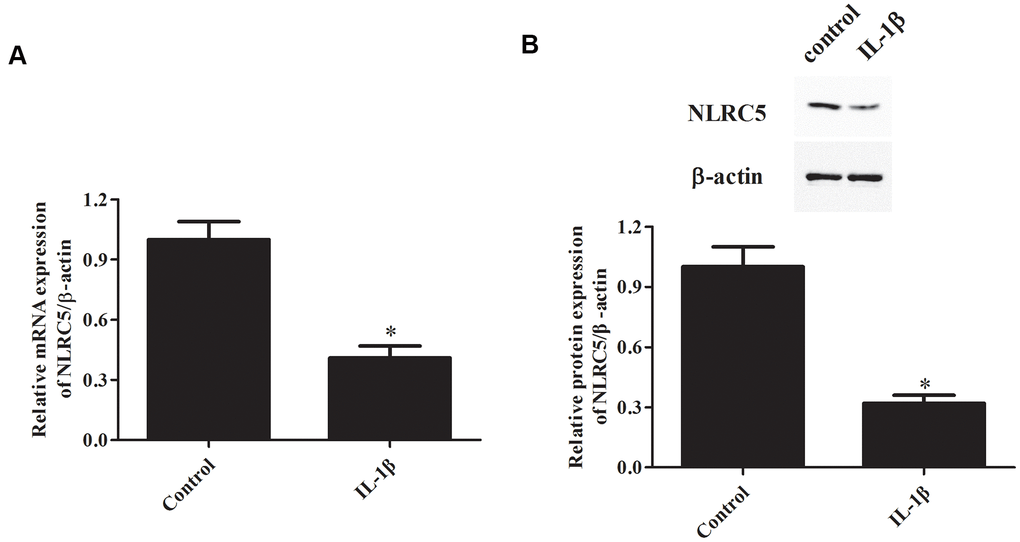
Figure 1. NLRC5 expression was down-regulated by IL-1β induction in chondrocytes. Chondrocytes were stimulated by IL-1β (10 ng/ml) for 24 h to induce inflammation. The mRNA and protein levels of NLRC5 were measured using RT-PCR (A) and western blot analysis (B). *p < 0.05.
Overexpression of NLRC5 increased chondrocytes viability and inhibited inflammatory mediators’ production
To further explore the role of NLRC5, pcDNA3.1-NLRC5 was transfected into chondrocytes to overexpress NLRC5. As shown in Figure 2A, a dramatical increase of the protein level of NLRC5 was observed in the pcDNA3.1-NLRC5-transfected chondrocytes. As shown in Figure 2B, overexpression of NLRC5 significantly increased cell viability in OA chondrocytes.
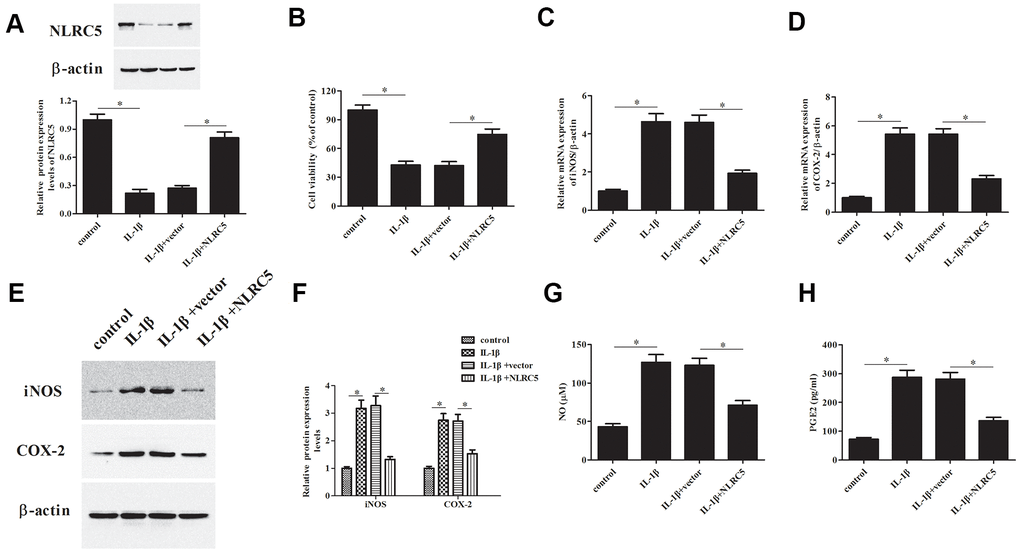
Figure 2. Overexpression of NLRC5 attenuated IL-1β-induced inflammatory injury in human OA chondrocytes. The pcDNA3.1-NLRC5 or pcDNA3.1 vector was transfected into chondrocytes, followed by IL-1β (10 ng/ml) stimulation for 24 h. (A) The expression levels of NLRC5 in chondrocytes were measured using western blot after transfection. (B) Cell viability of chondrocytes was detected using MTT assay. (C–F) The mRNA and protein levels of iNOS and COX-2 were measured using RT-PCR and western blot analysis. (G, H) The production of NO and PGE2 in chondrocytes. *p < 0.05.
The iNOS and COX-2 are two important enzymes, which are responsible for the production of NO and PGE2 [23]. Results indicated that NLRC5-overexpressing cells exhibited markedly reduced expression levels of iNOS and COX-2, as compared to the chondrocytes exposed to IL-1β (Figure 2C–2F). Besides, NLRC5 repressed the IL-1β-induced levels of NO and PGE2 in the culture supernatants of chondrocytes (Figure 2G, 2H).
Knockdown of NLRC5 reduced cell viability and increased inflammatory mediators’ production
Additionally, si-NLRC5 was transfected into chondrocytes to knock down NLRC5. Transfection efficiency assay showed that NLRC5 protein expression was dramatically decreased after transfection with si-NLRC5 (Figure 3A). Knockdown of NLRC5 significantly enhanced the IL-1β-induced reduction in chondrocytes viability (Figure 3B). Transfection with si-NLRC5 induced iNOS and COX-2 expression in IL-1β-stimulated chondrocytes (Figure 3C–3F). Meanwhile, the production levels of NO and PGE2 were also elevated by NLRC5 knockdown in IL-1β-induced chondrocytes (Figure 3G, 3H).
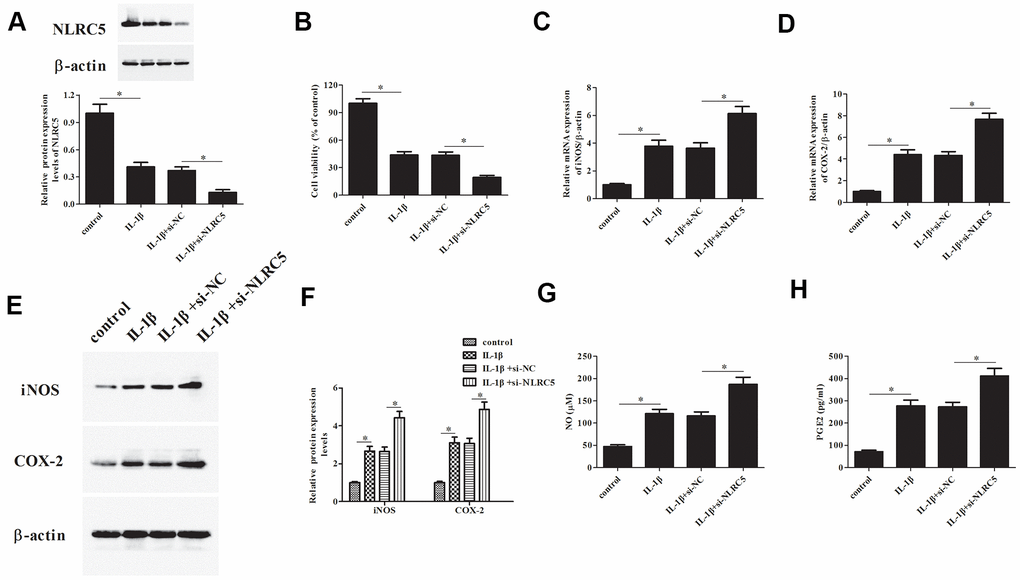
Figure 3. Knockdown of NLRC5 promoted IL-1β-induced inflammatory injury in human OA chondrocytes. The si-NLRC5 or si-NC was transfected into chondrocytes, followed by IL-1β (10 ng/ml) stimulation for 24 h. (A) The expression levels of NLRC5 in chondrocytes were measured using western blot after transfection. (B) Cell viability of chondrocytes was detected using MTT assay. (C–F) The mRNA and protein levels of iNOS and COX-2 were measured using RT-PCR and western blot analysis. (G, H) The production of NO and PGE2 in chondrocytes. *p < 0.05.
Overexpression of NLRC5 suppressed the production of TNF-α, IL-6, and MMP-3/13
TNF-α and IL-6 are majorly increased in the process of OA [24, 25], thus, we examined the effects of NLRC5 on TNF-α and IL-6 production. Results in Figure 4A, 4B revealed that overexpression of NLRC5 suppressed the production of TNF-α and IL-6 in chondrocytes. It has been demonstrated that MMPs expression are upregulated with the increased proinflammatory cytokines levels. MMPs, especially MMP-3 and MMP-13, are implicated in the pathogenesis of OA [26]. Our results showed that overexpression of NLRC5 also inhibited the production of MMP-3 and MMP-13 (Figure 4C, 4D).
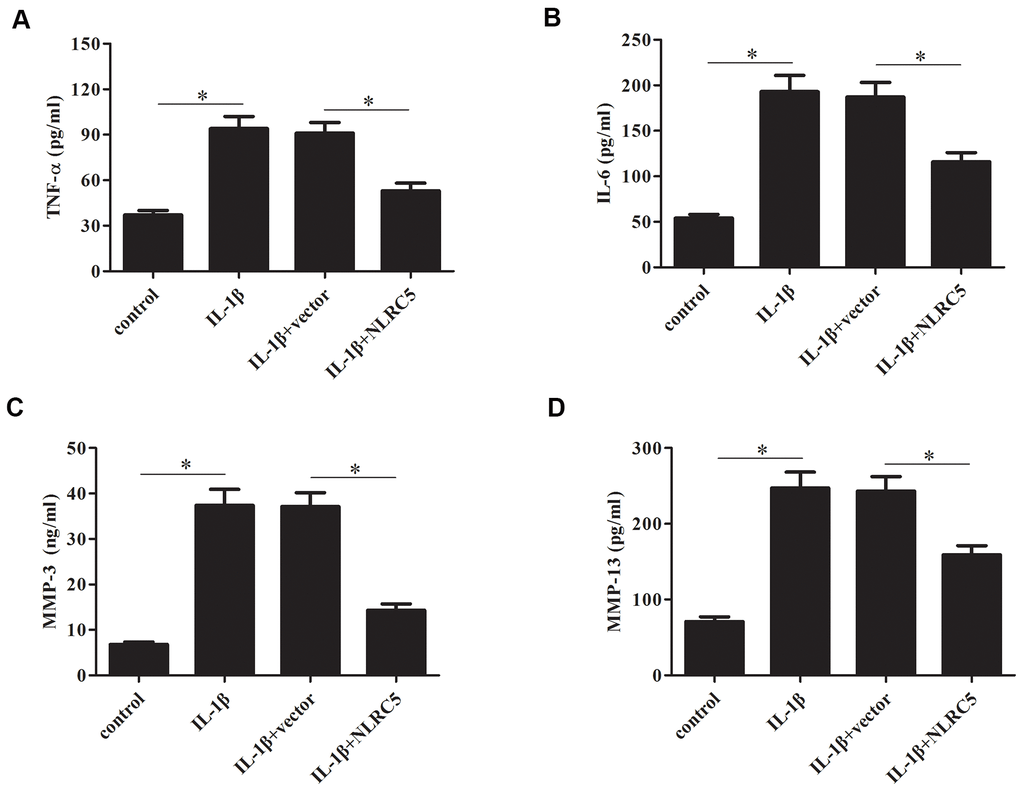
Figure 4. Overexpression of NLRC5 suppressed the production of inflammatory cytokines in IL-1β-stimulated human OA chondrocytes. After transfection with pcDNA3.1-NLRC5 or pcDNA3.1 vector and the following incubation with IL-1β, (A–D) the production of TNF-α, IL-6, MMP-3 and MMP-13 were detected using ELISA. *p < 0.05.
Knockdown of NLRC5 increased the production of TNF-α, IL-6, MMP-3/13
In contrast to the effects of NLRC5 overexpression, knockdown of NLRC5 significantly induced the production of TNF-α and IL-6 (Figure 5A, 5B). The levels of MMP-3 and MMP-13 in cell culture of IL-1β-stimulated chondrocytes were increased after transfection with si-NLRC5 (Figure 5C, 5D).
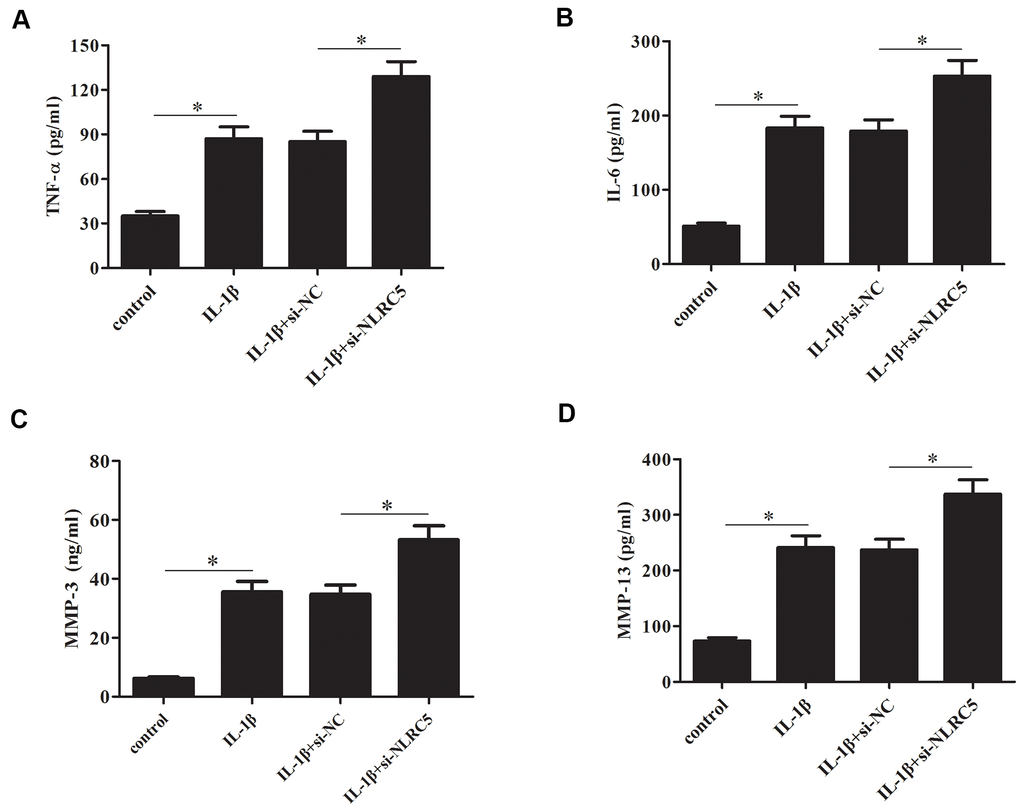
Figure 5. Knockdown of NLRC5 increased the production of inflammatory cytokines in IL-1β-stimulated human OA chondrocytes. After transfection with si-NLRC5/si-NC and the following incubation with IL-1β, (A–D) the production of TNF-α, IL-6, MMP-3 and MMP-13 were detected using ELISA. *p < 0.05.
NLRC5 inhibited IL-1β-induced NF-κB activation in chondrocytes
The levels of p-p65, p65, and IκBα were determined to explore the involvement of NF-κB signaling in inflammatory response. The results implied that the p-p65 expression was decreased, while IκBα expression was increased by NLRC5 overexpression (Figure 6A–6C), indicating that NLRC5 blocked the activation of NF-κB pathway.
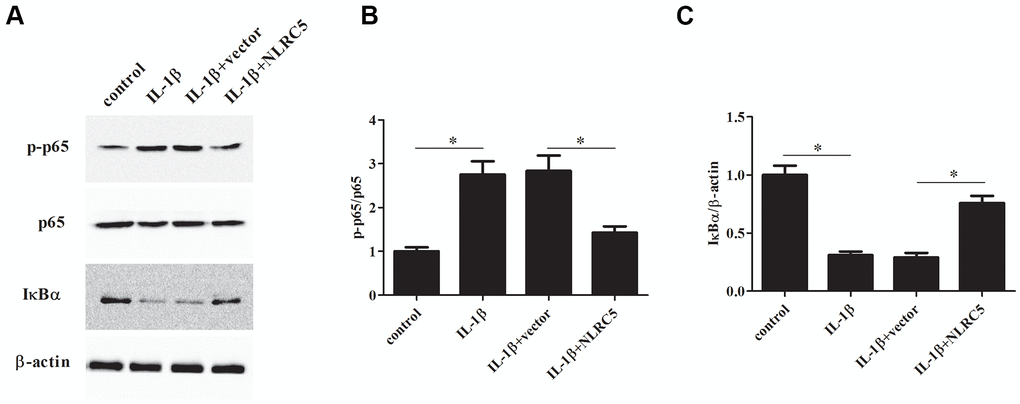
Figure 6. NLRC5 inhibited IL-1β-induced NF-κB activation in chondrocytes. After transfection with pcDNA3.1-NLRC5 or pcDNA3.1 vector, the chondrocytes were stimulated by IL-1β (10 ng/ml) for 24 h. (A–C) The expression levels of p-p65, p65, IĸBα were determined using western blot analysis. *p < 0.05.
Inhibition of NF-κB partially reversed the si-NLRC5-mediated inflammatory injury
Subsequently, chondrocytes were treated with pyrrolidinedithiocarbamate (PDTC, an NF-κB pathway inhibitor) to prevent the NF-κB activation. The deceased cell viability caused by si-NLRC5 was mitigated by PDTC (Figure 7A). Additionally, the si-NLRC5-mediated increases in expression levels of iNOS and COX-2, and production of TNF-α, IL-6, MMP-3/13 were attenuated in PDTC-treated cells (Figure 7B–7G).
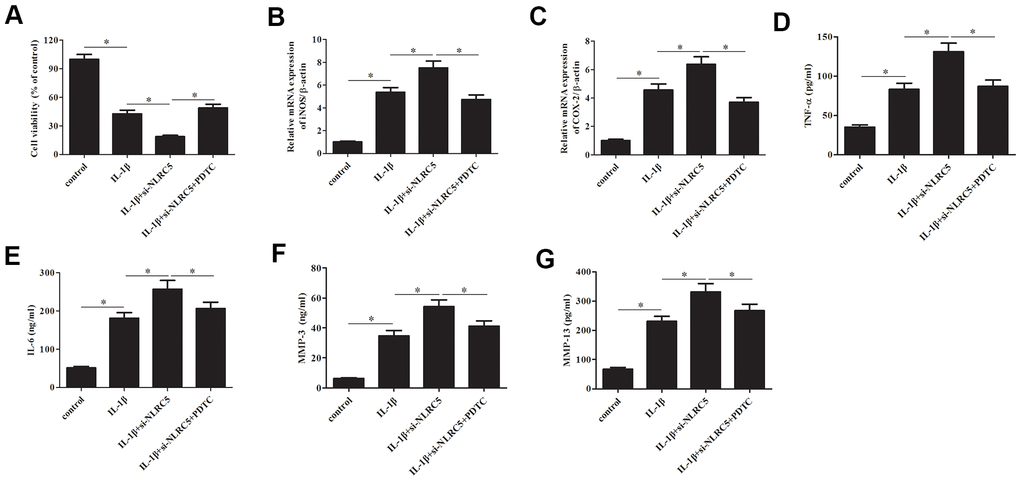
Figure 7. Inhibition of NF-κB partially reversed the si-NLRC5-mediated promotion of IL-1β-induced inflammatory injury in chondrocytes. Chondrocytes were treated with PDTC (5 ;μM) to prevent the activation of NF-κB signaling pathway. (A) Cell viability of chondrocytes was detected using MTT assay. (B, C) The mRNA levels of iNOS and COX-2 were measured using RT-PCR. (D–G) The production of TNF-α, IL-6, MMP-3/13 in chondrocytes. *p < 0.05.
NLRC5 treatment ameliorated cartilage degeneration in an OA rat model
The role of NLRC5 in OA was further examined in vivo using an OA rat model. Cartilage degeneration in the rats were evaluated by H&E and Safranin O staining. As compared with the sham group, rats in the OA model group presented obvious hypocellularity, extensive proteoglycan loss, cartilage erosion, and superficial cartilage destruction. Compared to the OA model group, rats with intra-articular injection of NLRC5 exhibited remarkable alleviation in cartilage destruction (Figure 8A, 8B).
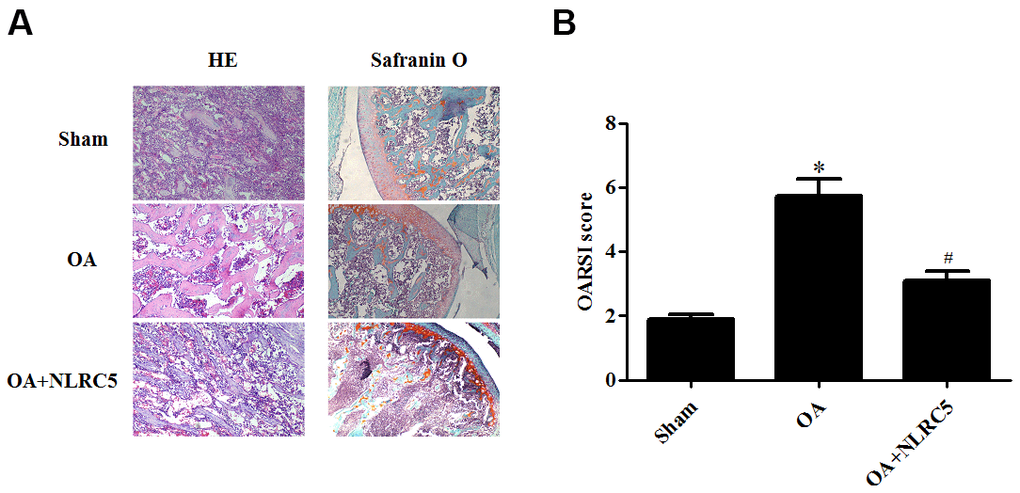
Figure 8. NLRC5 treatment ameliorated cartilage degeneration in an OA model in rats. (A) HE and Safranin O staining in each group (100×). (B) OARSI scores of each group to assess cartilage degeneration. *p < 0.05 vs sham group, #p < 0.05 vs OA group.
Discussion
NLRC5 has been demonstrated to be a critical mediator of inflammatory response. Here, we found that NLRC5 expression was down-regulated in IL-1β-stimulated chondrocytes. NLRC5 inhibited IL-1β-induced inflammatory response. In contrast, NLRC5 knockdown exhibits opposite effect. The underlying mechanism was found to be attributed to the regulation of NF-κB signaling. Finally, NLRC5 treatment ameliorated cartilage degeneration in vivo in an OA model.
NLRC5, an important member of NLR family, is involved in inflammation. NLRC5 deficiency promotes high fat diet-induced myocardial damage in mice, as evidenced by the accelerated fibrosis and inflammation response [27]. NLRC5 knockout mice exhibit NF-κB activation, indicating NLRC5 might has anti-inflammatory activity via suppressing NF-κB signaling. Additionally, NLRC5 negatively regulates lipoteichoic acid (LTA)-induced inflammatory response via the TLR2/NF-κB pathway in macrophage cells [28]. These findings imply that NLRC5 plays an important role in inflammation, which are attributed to its modulation of inflammatory pathways.
A recent study has shown that NLRC5 expression was increased in synovial tissues of rheumatoid arthritis (RA) rats [29]. Overexpression of NLRC5 also comes with the rise of expression of inflammatory cytokines and exacerbated proliferation of fibroblast-like synoviocytes (FLSs). While NLRC5 silencing exhibits inhibitory effects on cell proliferation and inflammatory cytokine production via inhibiting NF-κB activation [29]. Moreover, NLRC5 expression level was found to be higher in the synovial tissues from adjuvant arthritis rats compared with that from control rats [30]. Increased NLRC5 expression is associated with high levels of inflammatory cytokines and FLSs proliferation. However, our results showed that NLRC5 was down-regulated in IL-1β-stimulated chondrocytes. Overexpression of NLRC5 suppressed IL-1β-induced inflammation through inhibiting the production of multiple inflammatory mediators and MMPs in chondrocytes. Consistently, NLRC5 knockdown enhanced the IL-1β-induced production of these inflammatory mediators in chondrocytes. Furthermore, NLRC5 blocked NF-κB activation in IL-1β-stimulated chondrocytes. These findings suggest that NLRC5 regulates NF-κB signaling can differ profoundly among cell types and experimental conditions.
Additionally, our results showed that NLRC5 caused an increase in IκBα expression, while reduced the expression of p-p65, indicating that NLRC5 inhibited the activation of NF-κB signaling. Moreover, inhibition of NF-κB by PDTC mitigated the si-NLRC5-mediated promotion of inflammatory injury in chondrocytes, suggesting that NLRC5 attenuated IL-1β-mediated inflammatory injury through regulation of NF-κB pathway in chondrocytes. Interestingly, Cui et al. [31] demonstrated that NLRC5 controls innate immunity through inhibiting NF-κB activation. Our results together with the previous study indicated that NLRC5 might exert anti-inflammatory in IL-1β-induced chondrocytes via inhibiting NF-κB signaling.
In conclusion, NLRC5 attenuated IL-1β-induced inflammatory injury in chondrocytes through regulating the NF-κB signaling.
Author Contributions
Qi Li designed this manuscript. Yiping Mu wrote this manuscript. Yiping Mu and Yang Zhang performed experiments. Jie Wu analyzed the data. All the authors approved the manuscript for submission.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by the Shenyang Medical College clinical research foundation in China (No: 20206050).
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence
References
- 1. Shen J, Abu-Amer Y, O’Keefe RJ, McAlinden A. Inflammation and epigenetic regulation in osteoarthritis. Connect Tissue Res. 2017; 58:49–63. https://doi.org/10.1080/03008207.2016.1208655 [PubMed]
- 2. Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015; 11:35–44. https://doi.org/10.1038/nrrheum.2014.162 [PubMed]
- 3. Daghestani HN, Kraus VB. Inflammatory biomarkers in osteoarthritis. Osteoarthritis Cartilage. 2015; 23:1890–96. https://doi.org/10.1016/j.joca.2015.02.009 [PubMed]
- 4. Rahmati M, Mobasheri A, Mozafari M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone. 2016; 85:81–90. https://doi.org/10.1016/j.bone.2016.01.019 [PubMed]
- 5. Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014; 2014:561459. https://doi.org/10.1155/2014/561459 [PubMed]
- 6. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017; 168:37–57. https://doi.org/10.1016/j.cell.2016.12.012 [PubMed]
- 7. Christian F, Smith EL, Carmody RJ. The Regulation of NF-κB Subunits by Phosphorylation. Cells. 2016; 5:12. https://doi.org/10.3390/cells5010012 [PubMed]
- 8. Sokolova O, Naumann M. NF-κB Signaling in Gastric Cancer. Toxins (Basel). 2017; 9:119. https://doi.org/10.3390/toxins9040119 [PubMed]
- 9. Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets. 2010; 11:599–613. https://doi.org/10.2174/138945010791011938 [PubMed]
- 10. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009; 1:a001651. https://doi.org/10.1101/cshperspect.a001651 [PubMed]
- 11. Saito T, Tanaka S. Molecular mechanisms underlying osteoarthritis development: Notch and NF-κB. Arthritis Res Ther. 2017; 19:94. https://doi.org/10.1186/s13075-017-1296-y [PubMed]
- 12. Mótyán JA, Bagossi P, Benkő S, Tőzsér J. A molecular model of the full-length human NOD-like receptor family CARD domain containing 5 (NLRC5) protein. BMC Bioinformatics. 2013; 14:275. https://doi.org/10.1186/1471-2105-14-275 [PubMed]
- 13. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA. 2010; 107:13794–99. https://doi.org/10.1073/pnas.1008684107 [PubMed]
- 14. Neerincx, Heinrich A. Characterization of the human NLR protein NLRC5. PhD thesis, Universität zu Köln. 2013. https://kups.ub.uni-koeln.de/5303/.
- 15. Ludigs K, Jandus C, Utzschneider DT, Staehli F, Bessoles S, Dang AT, Rota G, Castro W, Zehn D, Vivier E, Held W, Romero P, Guarda G. NLRC5 shields T lymphocytes from NK-cell-mediated elimination under inflammatory conditions. Nat Commun. 2016; 7:10554. https://doi.org/10.1038/ncomms10554 [PubMed]
- 16. Wu Y, Shi T, Li J. NLRC5: A paradigm for NLRs in immunological and inflammatory reaction. Cancer Lett. 2019; 451:92–99. https://doi.org/10.1016/j.canlet.2019.03.005 [PubMed]
- 17. Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006; 14:13–29. https://doi.org/10.1016/j.joca.2005.07.014 [PubMed]
- 18. Jenei-Lanzl Z, Meurer A, Zaucke F. Interleukin-1β signaling in osteoarthritis - chondrocytes in focus. Cell Signal. 2019; 53:212–23. https://doi.org/10.1016/j.cellsig.2018.10.005 [PubMed]
- 19. Daheshia M, Yao JQ. The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol. 2008; 35:2306–12. https://doi.org/10.3899/jrheum.080346 [PubMed]
- 20. Zhuang Z, Ye G, Huang B. Kaempferol Alleviates the Interleukin-1β-Induced Inflammation in Rat Osteoarthritis Chondrocytes via Suppression of NF-κB. Med Sci Monit. 2017; 23:3925–31. https://doi.org/10.12659/msm.902491 [PubMed]
- 21. Wan ZH, Zhao Q. Gypenoside inhibits interleukin-1β-induced inflammatory response in human osteoarthritis chondrocytes. J Biochem Mol Toxicol. 2017; 31:e21926. https://doi.org/10.1002/jbt.21926 [PubMed]
- 22. Pan T, Chen R, Wu D, Cai N, Shi X, Li B, Pan J. Alpha-Mangostin suppresses interleukin-1β-induced apoptosis in rat chondrocytes by inhibiting the NF-κB signaling pathway and delays the progression of osteoarthritis in a rat model. Int Immunopharmacol. 2017; 52:156–62. https://doi.org/10.1016/j.intimp.2017.08.021 [PubMed]
- 23. Amin AR, Attur M, Abramson SB. Nitric oxide synthase and cyclooxygenases: distribution, regulation, and intervention in arthritis. Curr Opin Rheumatol. 1999; 11:202–09. https://doi.org/10.1097/00002281-199905000-00009 [PubMed]
- 24. Wang T, He C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018; 44:38–50. https://doi.org/10.1016/j.cytogfr.2018.10.002 [PubMed]
- 25. Laavola M, Leppänen T, Hämäläinen M, Vuolteenaho K, Moilanen T, Nieminen R, Moilanen E. IL-6 in Osteoarthritis: Effects of Pine Stilbenoids. Molecules. 2018; 24:109. https://doi.org/10.3390/molecules24010109 [PubMed]
- 26. Malemud CJ. Matrix Metalloproteinases and Synovial Joint Pathology. Prog Mol Biol Transl Sci. 2017; 148:305–25. https://doi.org/10.1016/bs.pmbts.2017.03.003 [PubMed]
- 27. Ma SR, Xie XW. NLRC5 deficiency promotes myocardial damage induced by high fat diet in mice through activating TLR4/NF-κB. Biomed Pharmacother. 2017; 91:755–66. https://doi.org/10.1016/j.biopha.2017.03.062 [PubMed]
- 28. Wang M, Wang L, Fang L, Li S, Liu R. NLRC5 negatively regulates LTA-induced inflammation via TLR2/NF-κB and participates in TLR2-mediated allergic airway inflammation. J Cell Physiol. 2019; 234:19990–20001. https://doi.org/10.1002/jcp.28596 [PubMed]
- 29. Liu YR, Yan X, Yu HX, Yao Y, Wang JQ, Li XF, Chen RN, Xu QQ, Ma TT, Huang C, Li J. NLRC5 promotes cell proliferation via regulating the NF-κB signaling pathway in Rheumatoid arthritis. Mol Immunol. 2017; 91:24–34. https://doi.org/10.1016/j.molimm.2017.08.024 [PubMed]
- 30. Liu YR, Yang L, Xu QQ, Lu XY, Ma TT, Huang C, Li J. Long noncoding RNA MEG3 regulates rheumatoid arthritis by targeting NLRC5. J Cell Physiol. 2019; 234:14270–84. https://doi.org/10.1002/jcp.28126 [PubMed]
- 31. Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, Wang RF. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010; 141:483–96. https://doi.org/10.1016/j.cell.2010.03.040 [PubMed]