



Introduction
Periodontitis, which is a chronic multifactorial inflammatory disease related to microbial dysfunction, is characterized by the progressive destruction of periodontal tissues. It is a worldwide public health problem and has a measurable impact on overall health of patients [1]. It is widely believed that the occurrence and development of periodontitis depends on the presence of toxic microorganisms, which are capable of causing periodontitis. Although bacteria are initiator of periodontitis, and response of host to pathogenic infections plays a critical role in the progression of periodontitis [2, 3]. After the onset of periodontitis, periodontitis will progress rapidly with the loss of collagen fibers and attachment to cementum surface, migration of the apex of connective epithelium, deepening of periodontal pockets, and absorption of alveolar bone [4]. If not treated in time, periodontitis will continue to develop, leading to bone destruction, tooth movement and subsequent tooth loss. In the United States, more than half of adults have periodontal disease, and about 10 % have severe periodontal disease and early tooth loss. Traditional periodontal diagnostic parameters include, for example, bleeding detection, clinical attachment degree, plaque index, and x-ray film to assess alveolar bone level [5], which have the advantages of easy-to-use, cost-effectiveness, and less invasiveness. Clinical reading of tooth attachment with periodontal probes and radiological assessments of alveolar bone loss is used to evaluate the extent of damage from past destructive events, and this requires a 2-3 mm threshold change to determine whether a site underwent a significant anatomic event [6]. Therefore, effective molecular diagnostic approach, medical care, and periodontal management are essential to reducing adverse outcomes of periodontitis.
DNA methylation is a heritable epigenetic modification of cells to control gene expression without changing gene sequence [7, 8–11]. With the flexibility of epigenomic modifications, methylation events can respond to nutritional and environmental influences and regulate gene expression patterns accordingly [12, 13], and may also serve as potential biomarkers for early cell transformation [14]. A growing number of articles have focused specifically on epigenetic changes in DNA, highlighting the importance of "epigenetic phenotypes" in many diseases. Methylation is defined as enrichment in the area of CpG island (CGI), representing an area of at least 200 bp, where the ratio of guanine to cytosine is greater than 50%, and CpG ratio is greater than 0.6 [15]. CGIs mainly focuses on gene promoters and is the first candidate for the study of gene expression methylation [16]. Evidence of methylation in affecting gene expression, especially in cancers, has been found. Serum DNA methylation is used as a biomarker for early detection of cancer [17–20]. DNA methylation of specific genes (SEPT9, RASSF1A, APC, GADD45a) has been proposed as a diagnostic and prognostic biomarker for colorectal cancer [21–23] and breast cancer [24].
In this study, abnormal expression of methyltransferase in periodontitis patients was observed, pointing to different methylation patterns in periodontitis patients. Simultaneous activation of multiple immunomodulation-related pathways at the transcriptional level indicated that these abnormal methylation patterns may be related to the dysregulation of immune pathways. The purpose of this study was to integrate high-throughput methylation profile and expression profile data of a large number of patients, so as to study the altered DNA methylation pattern, especially the promoter methylation of inflammation-related genes, between periodontitis patients and healthy people. This study aimed to identify specific DNA methylation sites as potential biomarkers and to establish a diagnostic classifier for periodontitis.
Results
The translational disorders in the periodontitis were related to immunity and epigenetic inheritance
A total of 18 disordered KEGG pathways were identified by GSEA analysis on abnormal KEGG pathway in periodontitis samples (Supplementary Table 1). Noticeably, these 18 pathways were activated in periodontitis, many of them were related to immune regulation-related pathways (Figure 1A), indicating that the abnormal expression of immune-related genes played an important role in the regulation of periodontitis. The expression distribution of methyltransferase-related genes was further analyzed, and it was observed that EZH2, DNMT1 and DNMT3B were significantly upregulated in normal samples (Figure 1B, 1C, 1E), whereas that of DNMT3A was sharply downregulated (Figure 1D), indicating that histone methylation and DNA methylation were abnormal in periodontitis. Gene correlation analysis demonstrated that EZH2 gene was highly positively correlated with DNMT1 and DNMT3N in periodontitis samples (FDR<0.01) (Supplementary Figure 1). These results indicated that apart from the important functions of DNA methylation in the progression of periodontitis, the co-expression relationship between EZH2 and DNMT was related to histone methylation and DNA methylation in periodontitis.
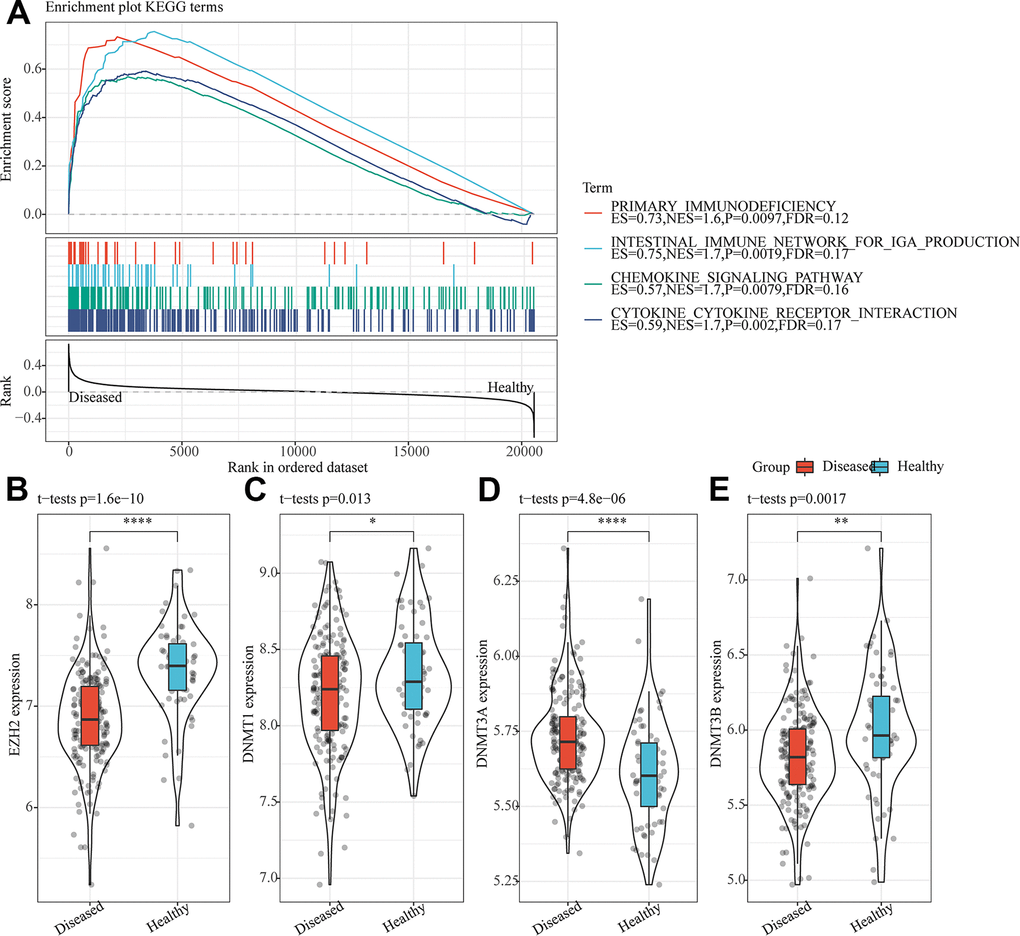
Figure 1. The translational disorders in the periodontitis were related to immunity and epigenetic inheritance. (A) Differences between four immune-related pathways significantly enriched by GSEA in periodontitis and normal samples. (B) Expression of EZH2 in periodontitis and normal samples. (C) Expression of DNMT1 in periodontitis and normal samples. (D) Expression of DNMT3A in periodontitis and normal samples. (E) Expression of DNMT3B in periodontitis and normal samples. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Identification of DMPs between periodontitis and healthy control samples
Considering that periodontitis transcriptional dysregulation is related to immunity and epigenetics, we analyzed the difference in methylation between normal people and patients with periodontitis, and identified a total of 8029 different CpG sites using Limma (Supplementary Figure 2). By annotating these CpG sites to genes, a total of 4940 genes were obtained, of which 275 were determined to be immune-related genes (Figure 2A). Methylation distribution of 275 immune gene promoter differential CpG sites in periodontitis and normal samples showed that most of these genes were downregulated, which was consistent with the downregulation of methyltransferase at transcription level (Figure 2B). 275 immune genes were mainly distributed in the process of Antimicrobials, Cytokines (Figure 2C), which were consistent with CYTOKINE_CYTOKINE_RECEPTOR_INTERACTION pathways in the transcription activation, suggesting that hypomethylation of promoters of immune-related genes upregulated the expression of immune genes and activated immune-related pathways.
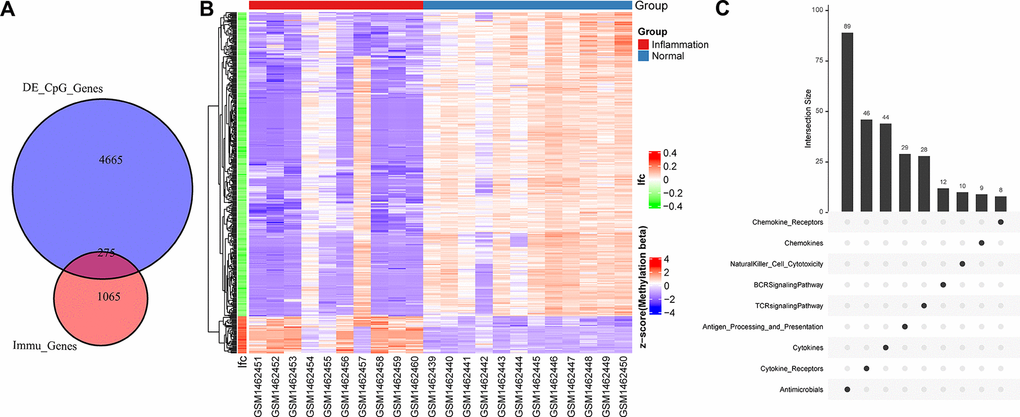
Figure 2. The relationship between different methylation sites and immune genes. (A) Venn diagram of promoter methylation differential gene and immune gene. (B) Heat map of methylation sites of immune gene promoters. The horizontal axis is the sample, the vertical axis is the CpGs, and the color is the methylation level. (C) Distribution of immune pathways of differentially promoter methylated immune genes.
Weighted co-expression analysis identified immune-related co-expression of DMPs
To further screen immune-related co-DMPs, the methylation profiles of 275 immune-related genes at the promoter difference CpG sites were selected, and the co-expression network was constructed using WGCNA. The power of n = 20 (scale-free R^2 = 0.87) was the soft threshold to ensure the scale-free network (Figure 3A, 3B). A total of 2 modules were identified here (Figure 3C). We first calculated the spearman correlation coefficient between CpG site methylation and the occurrence of periodontitis in each module, both modules were found to have a high correlation with periodontitis, and most of the CpGs in the blue module showed a higher correlation with periodontitis (Figure 3D). Furthermore, the correlation coefficient between the eigenvectors of each module and the CpG methylation in the corresponding module was analyzed, and we found that CpGs in blue and turquoise modules were highly correlated with the corresponding feature vectors (Figure 3E). Based on these two methods, we selected CpG loci with higher correlation with the module than the median of the turquoise module and higher correlation with the periodontitis than the median of the blue module, and finally 23 CpG loci were obtained.
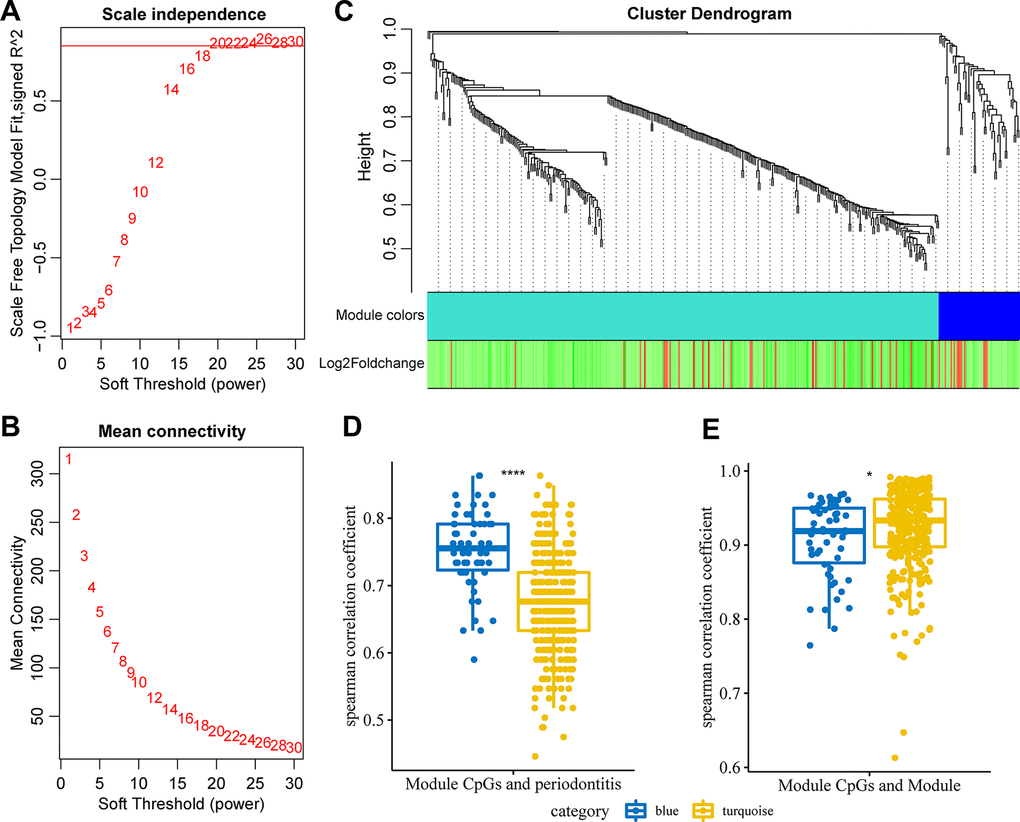
Figure 3. Weighted co-expression analysis identified immune-related co-expression of DMPs. (A) Analysis of the scale-free fit index for various soft-thresholding powers (β). (B) Analysis of the mean connectivity for various soft-thresholding powers. (C) Dendrogram of all differentially expressed genes clustered based on a dissimilarity measure (1-TOM). (D) The correlation distribution of CpG site methylation in the module and periodontitis. (E) The correlation distribution of CpG site methylation in the module and module.
Screening of genomic characteristics of immune-related co-DMPs
To further screen immune gene-related CpG markers, the distribution of 23 CpG sites differentially co-expressed in the patients and healthy groups was analyzed (Figure 4A). As expected, these CpG sites were significantly hypomethylated in periodontitis. 23 genes were annotated to the promoter regions of 20 immune genes, and we observed significant differential expression in 15 genes (79%), 13 of them were noticeably overexpressed in the samples of patients with periodontitis (Figure 4B). Furthermore, according to the methylation level of these 23 CpG sites in the samples, those below 0.2 were defined as unmethylated, while those above 0.8 were defined as hypermethylated, and the distribution of hypermethylated and unmethylated CpG sites in each sample was analyzed (Figure 4C). We observed that 13 CPGs (cg01930477, cg02786267, cg03732055, cg04774620, cg09443479, cg10836855, cg14662728, cg16386158, cg17907057, cg19301273, cg19503731, cg24116886, cg26594503) were generally hypermethylated in the healthy group, and some of them were methylated in the majority of patients. The 13 CpG sites was mainly distributed in the upstream of the transcription starting site between 200-1300bp (Figure 4D).
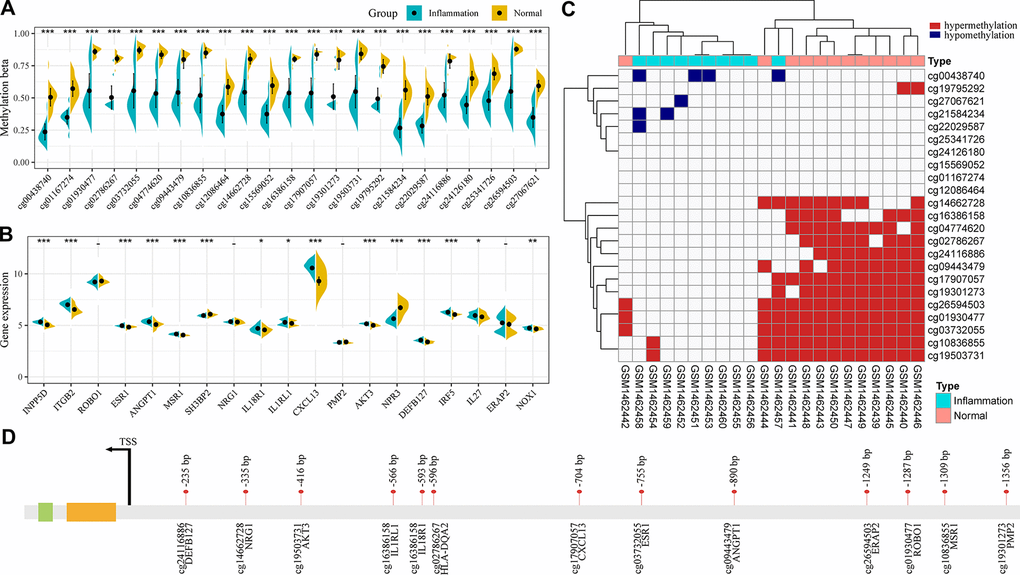
Figure 4. Genomic characteristics of immune-related co-DMPs. (A) The methylation distribution of 23 co-DMPs in patients and healthy groups. (B) The 20 immune genes annotated by 23 co-DMPs were differentially expressed in patients and healthy groups. (C) Hypermethylated and unmethylated distributions of 23 co-DMPs in each sample. (D) Distribution of 13 potential disease-specific markers in the promoter region.
Construction and testing of diagnostic model
In training dataset GSE59932 (N=22, Inflammation=10, Normal=12), to examine the effect of different combinations of diagnostic markers on diagnostic efficiency, all 8178 combinations of 13 Co-DMPs were calculated, and these combinations were used to construct a support vector machine classification model to analyze the prediction accuracy distribution of each combination. The results showed that all have a high prediction accuracy rate, with an average accuracy rate of 90% or higher (Figure 5A). The final 5 CpGs combinations (cg10836855, cg14662728, cg19301273, cg19503731, cg26594503) were determined by stepwise regression and were used to construct the classification model with support vector machine, and the model test was conducted by the ten-fold cross validation method. The results demonstrated that the classification accuracy was 95.5%, as 21 out of 22 samples were correctly classified, and that the sensitivity and specificity of the model were 90% and 100%, respectively (Figure 5B), area under the ROC curve (AUC) was 0.95 (Figure 5C).
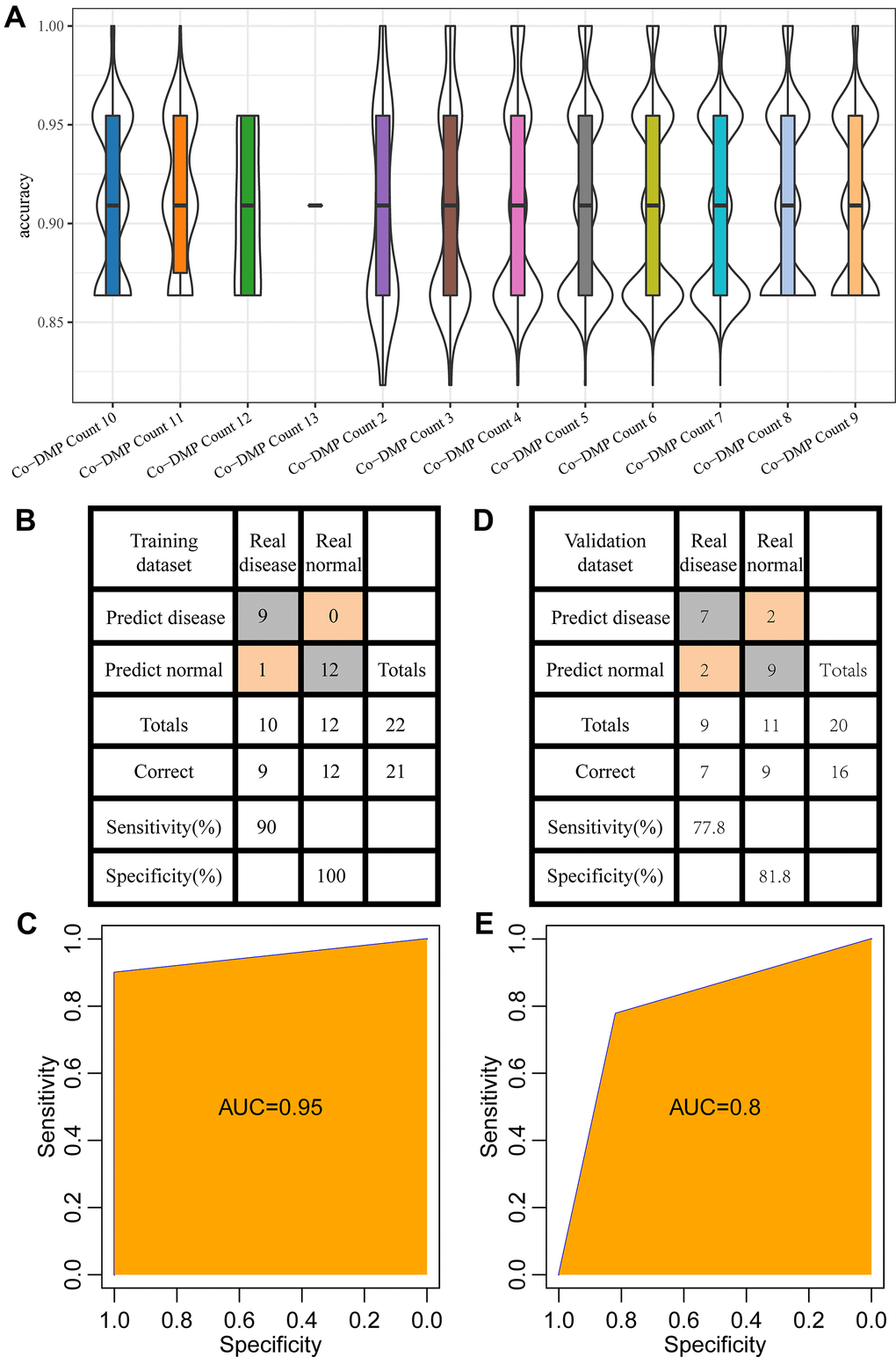
Figure 5. Establishment of diagnostic model. (A) The accuracy of different combinations of co-DMPs in periodontitis prediction. (B) The classification of samples in the training data set by the diagnostic model. (C) The ROC curve of the diagnostic model in the training data set. (D) The classification of samples in the validation data set by the diagnostic model. (E) The ROC curve of the diagnostic model in the validation data set.
Furthermore, the established model was applied to predict the samples in the validation data set (GSE59939, (N=20, Inflammation=9, Normal=11)) to determine the prediction performance of the model. 16 out of the 20 samples were correctly classified, with an accuracy of 80%. The sensitivity and specificity of the model were 77.8% and 81.8%, respectively (Figure 5D), and area under the ROC curve (AUC) was 0.8 (Figure 5E). These results indicated that the established diagnostic prediction model could effectively distinguish the patients with periodontitis from the normal control.
Validation of the diagnostic model
A set of independent data set GSE53849 containing 23 samples was selected, and the methylation matrix of 5 CpG was extracted. Here, 8 out of 11 normal samples were predicted as normal, and 10 out of the 12 patients were predicted as periodontitis samples, showing an accuracy rate of 82% (Figure 6A), and area under the ROC curve (AUC) of 0.78 (Figure 6B). In general, the prediction performance of the model was high across the data platform. To analyze the model and immune genetic relationship, 5 CpG loci were annotated to the 5 immune genes (MSR1, NRG1, PMP2, AKT3, ERAP2). The expression profiles of these 5 genes were extracted from the dataset GSE10334. We observed the expression of the 5 genes was significantly higher in patients with periodontitis than healthy controls (Figure 6C). Considering the small number of validation samples, we supplemented and combined two sets of validation data sets (GSE59939 and GSE53849) to form a larger data set incorporating 43 samples in total. Based on this data set, we verified the CpG methylation model again, and found that 20 out of the 22 normal samples were predicted as normal, and that 20 out of 21 patients were predicted as periodontitis patients, showing an accuracy of 93% and the area under the ROC curve of 0.93 (Figure 6D, 6E). Furthermore, the expression profiles of these 5 genes were used to establish a diagnostic model with support vector machine for prediction. Among them, 27 out of 64 normal patients were predicted as normal samples, and 177 out of 183 patients were predicted as periodontitis patients, showing an accuracy rate of 82.6% (Figure 6F) and area under the ROC curve (AUC) of 0.69 (Figure 6G). The above results indicated a high consistency between transcription level and methylation level.
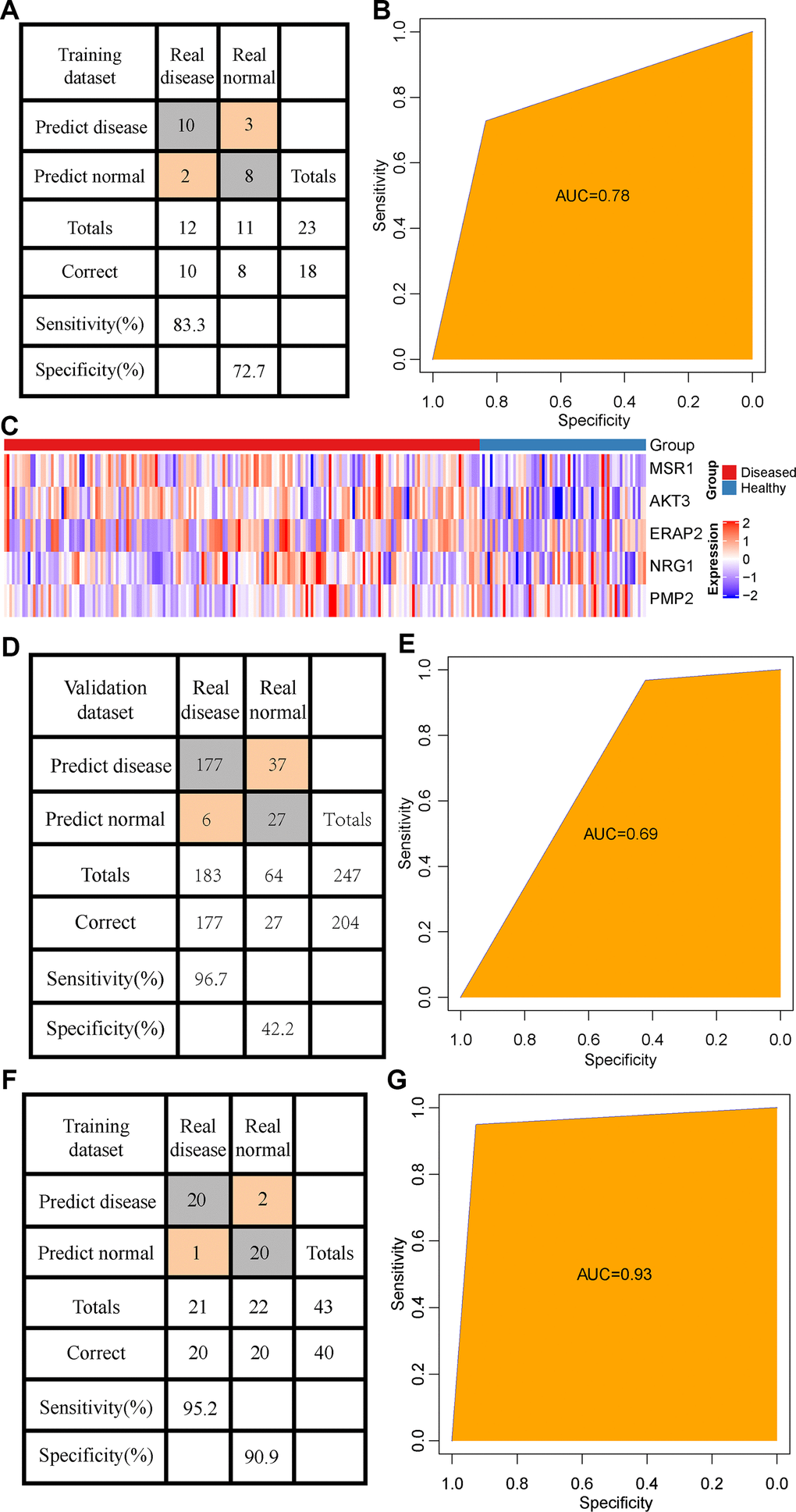
Figure 6. Validation of the diagnostic model. (A) The classification of samples in the GSE53849 data set by the diagnostic model. (B) The ROC curve of the diagnostic model in the GSE53849 data set. (C) Heat map of the expression distribution of 5 immune genes annotated by 5 CpG in disease and normal samples. (D) The classification of samples in the combined validation data sets (GSE59939 and GSE53849) by the diagnostic model. (E) The ROC curve of the diagnostic model in the combined validation data sets (GSE59939 and GSE53849) data set. (F) The classification result of the sample by the diagnostic model constructed by immune genes. (G) ROC curve of the diagnostic model constructed by immune genes.
Discussion
Periodontal disease is caused by bacteria at the tooth biofilm. To eliminate bacteria, immune system cells release substances that may cause inflammation and damage the gums, periodontal ligament, or alveolar bone, thereby causing swelling and bleeding of the gums, which is a sign of gingivitis. Damage from periodontal disease may also lead to tooth relaxation [25]. In this study, differences in gene expression between periodontitis patients and healthy control samples were compared. We found that there were various immune pathway disorders, such as PRIMARY_IMMUNODEFICIENCY, AUTOIMMUNE_THYROID_DISEASE, B_CELL_RECEPTOR_SIGNALING_PATHWAY, etc., in periodontitis patients. This again proved that the inflammation-related pathway was more active in periodontitis [26]. Interestingly, we also found that the TYPE_II_DIABETES_MELLITUS pathway was significantly activated in patients with periodontitis, showing a certain relationship between periodontitis patients and diabetes, and that periodontitis may affect diabetes and diabetes-related complications [27]. Furthermore, by analyzing the expression characteristics of methyltransferase-related genes such as EZH2 and DNMT, we found that these genes were significantly altered in periodontitis patients, and that EZH2 was highly positively correlated with DNMT1 and DNMT3N, indicating that both DNA methylation and histone methylation may play an important role in periodontitis patients [28, 29]. In addition, we preliminarily evaluated methyltransferase molecule EZH2 in periodontitis, and examined its role in PDLSCs, PDLSCs+LPS, PDLSCs+LPS+GSK126. RT-pPCR data showed a lower expression of EZH2 in inflammatory cells, which was consistent with the results of our data analysis and was also confirmed by Western Blot.
DNA methylation is an important epigenetic modification that suppresses gene transcription by inhibiting the binding of specific transcription factors [30]. New evidence showed that epigenetics plays a key role in human pathology, including in inflammation and cancer development. Epigenome is influenced by environmental factors throughout life. Nutritional factors have profound effects on the expression of specific genes through epigenetic modifications and may be passed on to offspring. Many cancers are associated with epigenetic changes, which will lead to changes in the expression of genes involved in cell growth or differentiation. The incidence of autoimmune diseases and tumors increases with age, and epigenetic disorders are considered as a potential explanation for differences in CpG methylation status, single allele silencing, and other epigenetic regulatory mechanisms observed in key inflammatory response genes [31]. In this study, CpGs of 8,029 differentially promoter regions were screened, and 4,940 genes were annotated, among which, immune genes showed stronger promoter methylation differences than random ones, indicating that DNA methylation plays an important role in the transcriptional regulation of immune inflammatory genes in chronic periodontitis.
In addition, if patients with periodontitis are left untreated, periodontitis can continue to develop, leading to bone destruction, tooth movement and subsequent tooth loss. Therefore, early diagnosis of periodontitis and personalized medical intervention are of great significance. Some studies have been conducted to screen biomarkers for periodontitis. JANET S. KINNEY et al. [32] determined that pyridinoline cross-linked carboxy-terminal peptides can predict future disease activity from oral fluids. Giannobile WV screened gingival sulcus fluid for biomarkers indicative of bone loss [33]. Frodge BD et al. [34] identified a bone remodeling biomarker for periodontal disease from saliva. Yoon AJ et al. [35] assessed the association between diabetes and periodontal status and oral inflammatory burden, and identified inflammatory biomarkers in saliva. In this study, 5 CpGs were determined based on the differences in promoter methylation of immune-related genes, and the classifier was constructed and verified. The high accuracy of the classifier in the training set and the external data set indicated that these genes had a strong ability to classify periodontitis and were applicable to different data platforms. Despite reduced accuracy, different methods of data standardization and batch effect as well as environmental factors, different regions, races, diets and some other factors could also the accuracy of external dataset validation. Furthermore, we used the transcriptome data set and the 5 immune genes annotated by the 5 CpGs to build a diagnostic model, which showed an accuracy of periodontitis prediction of 82.6%. These results indicated that these 5 CpGs and 5 immune genes can be used as diagnostic markers for periodontitis, and provide targets and references for clinicians and biological experimentalists.
The 5 CpGs were annotated to 5 immune genes including MSR1, NRG1, PMP2, AKT3, ERAP2. MSR1 is an important marker of macrophages and its abnormal expression is associated with multiple diseases including Porphyromonas gingivalis [36–38]. NRG1, which controls the formation of excitatory and inhibitory synapses in cortical circuits, is a schizophrenia risk gene [39], and is associated with multiple relapsing disorders. In addition, NRG1 plays an important role in parabens of fibroblasts and macrophages [40]. Overexpression of mutant PMP2 also leads to CMT1 phenotype [41], and aberrant AKT3 expression helps form M2 macrophage specificity [42]. These genes are directly or indirectly associated with periodontal disease, and multiple gene expressions will increase the possibility of macrophage abnormalities, suggesting that aberrant methylation of these CpGs may be involved in the development and progression of periodontitis via macrophages.
Although bioinformatics techniques were used to identify potential candidate genes involved in periodontitis in large samples, some limitations of this study should be noted. Firstly, the samples lacked clinical follow-up information, especially some diagnostic details, therefore we did not differentiate diagnostic biomarkers for periodontitis by taking into account factors such as the presence of other patient health conditions. Secondly, the results obtained by bioinformatics analysis alone were not convincing enough, and experimental verification is required to confirm the current results. Therefore, further genetic and experimental studies with larger sample sizes and experimental validation should be performed.
By correlating the expression of inflammation-related genes and the methylation relationship of their promoter regions and combining with the co-expression network, this study identified and screened diagnostic markers for periodontitis. A diagnostic model for the prediction and prevention of periodontitis was established based on the pattern recognition of support vector machine (SVM). We determined the expression and methylation characteristics of epigenetic key genes in periodontitis, and found that these gene promoter methylation was closely related to the occurrence and development of periodontitis. Although our gene expression profile still lacks high specificity required for immediate diagnostic application, CpG methylation in oral samples could predict periodontitis with high accuracy (AUC = 0.95), providing a target and reference for clinicians and biological experimentalists.
Materials and Methods
Data collection
We screened three sets of gene methylation data and one set of gene expression data from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) [43]. The methylation data came from the GPL13534 platform (Illumina HumanMethylation450 BeadChip). The gene expression profile data was from GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array) numbered GSE10334 [44]. The data set contained a total of 183 periodontitis patient samples and 64 healthy control samples. The sample distribution of each data set is shown in Table 1. The work flow chart is shown in Figure 7.
Table 1. Sample distribution of data set.
| GEO Accession | No. of Normal | No. of Periodontitis |
| GSE59932 | 12 | 10 |
| GSE59939 | 11 | 9 |
| GSE53849 | 11 | 12 |
| GSE10334 | 64 | 183 |
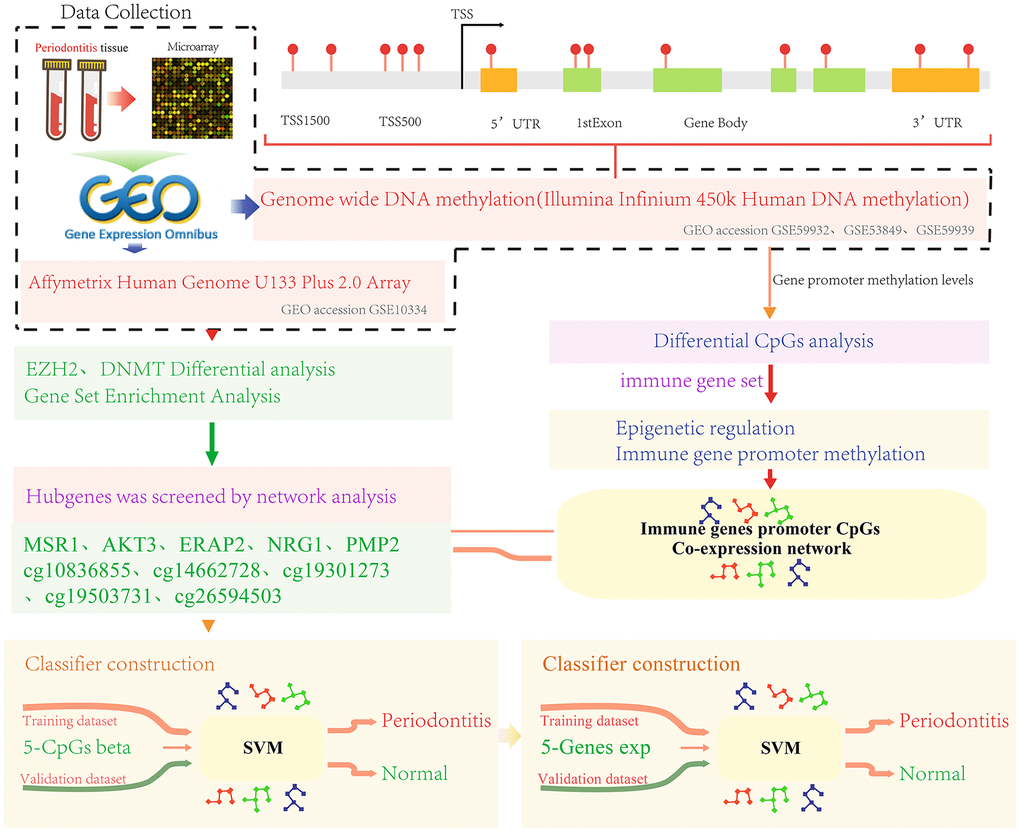
Figure 7. Work flow chart.
Methylation data processing
The methylation β values of the standardized CpG sites were downloaded, and the missing values of CpG sites greater than 20% in each sample were removed. The missing value completion was performed using the R package impute [45]. Probes binding to sex chromosomes, cross-hybridizing to multiple locations, or targeting a single-nucleotide polymorphism (SNP) were removed, according to previous annotation [46, 47]. By referring to the processing method of Zhang et al. [48], the methylation site of the non-promoter region was further removed, resulting in 232189 probes for DNA methylation analysis. All analysis was performed using β values to improve the statistical calculation of differential methylation [49, 50]. β values are also included in the tables for biological interpretation.
Gene chip data processing
For gene expression data, we first downloaded the standardized chip data. R package hgu133plus2.db was used for probe annotation. Probes that match multiple genes were removed. When multiple probes matched to one gene, the median of these probes were used as the expression value of the modified gene. Finally, the expression profiles of 20549 genes were obtained. In addition, we also downloaded 2498 immune genes from InnateDB [51].
Identification of differential methylation
The R software package limma [52] was used to detect different methylation sites between periodontitis patients and normal samples. Difference multiple greater than 20% and FDR <0.05 was the threshold to include more CpG sites with statistical differences.
Gene set enrichment analysis
Gene Set Enrichment Analysis(GSEA) [53] was performed by the JAVA program (http://software.broadinstitute.org/gsea/downloads.jsp) using the MSigDB [54] C2 Canonical pathways gene set collection with 1320 gene sets. After performing 1000 permutations, significantly enriched gene set was defined as having a p value less than 0.05.
Co-expression network construction
CpG methylation data profile of DMPs was assessed to evaluate whether the samples and CpGs included were qualified. Then, we used the weighted gene co-expression network analysis (WGCNA) [55] package in R to construct scale-free co-expression network for the DMPs. Pearson's correlation matrices and average linkage method were performed for all pair-wise CpGs. Then, a weighted adjacency matrix was constructed using a power function Amn = |Cmn|β (Cmn = Pearson's correlation between CpG m and CpG n; Amn = adjacency between CpG m and CpG n). β was a soft-thresholding parameter that could emphasize strong correlations between CpGs and penalize weak correlations. After choosing the power of β, the adjacency was transformed into a topological overlap matrix (TOM) to measure the network connectivity of a CpG, which is defined as the sum of its adjacency to all other CpGs for network CpG ration. The corresponding dissimilarity (1-TOM) was also calculated. To classify CpGs with similar expression profiles into CpG modules, average linkage hierarchical clustering was conducted according to the TOM-based dissimilarity measured with a minimum size (CpG group) of 30 for the CpGs dendrogram. To further analyze the module, we calculated the dissimilarity of module eigenCpGs, determined a cut line for module dendrogram and merged some modules.
Construction of diagnostic prediction model and evaluation of model prediction performance
A diagnostic prediction model was constructed using feature genes based on support vector machine (SVM) classification [56]. SVM is a supervised learning model of machine learning algorithm, and can analyze data and identify patterns. A support vector mechanism creates a hyperplane in a high or infinite dimensional space and can be used for classification and regression. All the samples were randomly and uniformly divided into training data set and verification data set. The model was constructed in the training data set, and the classification ability of the model was verified by ten-fold cross validation method. The established model was then used to predict the samples in the validation data set. The predictive performance of the model was assessed with the area under the ROC curve (AUC), and the sensitivity and specificity of the model for predicting periodontitis were analyzed.
Clinical effectiveness of the model
Two sets of Illumina HumanMethylation450 BeadChip platform data sets GSE59939 [57] and GSE53849 were selected as independent external verification data sets. After downloading the standardized data, the methylation level of characteristic CpGs was extracted and substituted into the model to assess the prediction ability of the model. Furthermore, a set of expression profile data set GSE10334 [58] was used to extract the expression profile of immune genes from the characteristic CpGs annotation to the promoter. A diagnostic model was established to distinguish normal healthy samples from periodontitis.
Author Contributions
Pengcheng Wang and Zuomin Wang designed the study. Zheng Zhang and Bingbing Wang contributed to the literature search. Pengcheng Wang wrote the initial draft of the manuscript. Zuomin Wang reviewed and edited the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
Authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Funding
This study was supported by National Natural Science Foundation of China (grant# 81870763 to Z.W.), Natural Science Foundation of Beijing, China (grant# 7192073 to Z.W.), and the Youth Research Fund of Beijing Shijitan Hospital, Capital Medical University (2019-q08 to P.W.).
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence
References
- 1. Barros SP, Fahimipour F, Tarran R, Kim S, Scarel-Caminaga RM, Justice A, North K. Epigenetic reprogramming in periodontal disease: Dynamic crosstalk with potential impact in oncogenesis. Periodontol 2000. 2020; 82:157–72. https://doi.org/10.1111/prd.12322 [PubMed]
- 2. Socransky SS, Haffajee AD. The bacterial etiology of destructive periodontal disease: current concepts. J Periodontol. 1992; 63:322–31. https://doi.org/10.1902/jop.1992.63.4s.322 [PubMed]
- 3. Genco RJ. Host responses in periodontal diseases: current concepts. J Periodontol. 1992; 63:338–55. https://doi.org/10.1902/jop.1992.63.4s.338 [PubMed]
- 4. Offenbacher S. Periodontal diseases: pathogenesis. Ann Periodontol. 1996; 1:821–78. https://doi.org/10.1902/annals.1996.1.1.821 [PubMed]
- 5. Armitage GC. The complete periodontal examination. Periodontol 2000. 2004; 34:22–33. https://doi.org/10.1046/j.0906-6713.2002.003422.x [PubMed]
- 6. Goodson JM. Conduct of multicenter trials to test agents for treatment of periodontitis. J Periodontol. 1992; 63:1058–63. https://doi.org/10.1902/jop.1992.63.12s.1058 [PubMed]
- 7. Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005; 6:597–610. https://doi.org/10.1038/nrg1655 [PubMed]
- 8. Bird AP. DNA methylation versus gene expression. J Embryol Exp Morphol. 1984; 83:31–40. [PubMed]
- 9. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012; 13:484–92. https://doi.org/10.1038/nrg3230 [PubMed]
- 10. Lim YC, Li J, Ni Y, Liang Q, Zhang J, Yeo GS, Lyu J, Jin S, Ding C. A complex association between DNA methylation and gene expression in human placenta at first and third trimesters. PLoS One. 2017; 12:e0181155. https://doi.org/10.1371/journal.pone.0181155 [PubMed]
- 11. Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, Lin S, Lin Y, Jung I, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015; 523:212–16. https://doi.org/10.1038/nature14465 [PubMed]
- 12. Yuen RK, Chen B, Blair JD, Robinson WP, Nelson DM. Hypoxia alters the epigenetic profile in cultured human placental trophoblasts. Epigenetics. 2013; 8:192–202. https://doi.org/10.4161/epi.23400 [PubMed]
- 13. Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J Nutr Biochem. 2012; 23:853–59. https://doi.org/10.1016/j.jnutbio.2012.03.003 [PubMed]
- 14. Van Neste L, Herman JG, Otto G, Bigley JW, Epstein JI, Van Criekinge W. The epigenetic promise for prostate cancer diagnosis. Prostate. 2012; 72:1248–61. https://doi.org/10.1002/pros.22459 [PubMed]
- 15. Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using hidden Markov models. Biostatistics. 2010; 11:499–514. https://doi.org/10.1093/biostatistics/kxq005 [PubMed]
- 16. Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986; 321:209–13. https://doi.org/10.1038/321209a0 [PubMed]
- 17. Tang Q, Cheng J, Cao X, Surowy H, Burwinkel B. Blood-based DNA methylation as biomarker for breast cancer: a systematic review. Clin Epigenetics. 2016; 8:115. https://doi.org/10.1186/s13148-016-0282-6 [PubMed]
- 18. Wang J, Han X, Sun Y. DNA methylation signatures in circulating cell-free DNA as biomarkers for the early detection of cancer. Sci China Life Sci. 2017; 60:356–62. https://doi.org/10.1007/s11427-016-0253-7 [PubMed]
- 19. Visvanathan K, Fackler MS, Zhang Z, Lopez-Bujanda ZA, Jeter SC, Sokoll LJ, Garrett-Mayer E, Cope LM, Umbricht CB, Euhus DM, Forero A, Storniolo AM, Nanda R, et al. Monitoring of Serum DNA Methylation as an Early Independent Marker of Response and Survival in Metastatic Breast Cancer: TBCRC 005 Prospective Biomarker Study. J Clin Oncol. 2017; 35:751–58. https://doi.org/10.1200/JCO.2015.66.2080 [PubMed]
- 20. Tahara T, Arisawa T. DNA methylation as a molecular biomarker in gastric cancer. Epigenomics. 2015; 7:475–86. https://doi.org/10.2217/epi.15.4 [PubMed]
- 21. Grützmann R, Molnar B, Pilarsky C, Habermann JK, Schlag PM, Saeger HD, Miehlke S, Stolz T, Model F, Roblick UJ, Bruch HP, Koch R, Liebenberg V, et al. Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. PLoS One. 2008; 3:e3759. https://doi.org/10.1371/journal.pone.0003759 [PubMed]
- 22. deVos T, Tetzner R, Model F, Weiss G, Schuster M, Distler J, Steiger KV, Grützmann R, Pilarsky C, Habermann JK, Fleshner PR, Oubre BM, Day R, et al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin Chem. 2009; 55:1337–46. https://doi.org/10.1373/clinchem.2008.115808 [PubMed]
- 23. Reis IM, Ramachandran K, Speer C, Gordian E, Singal R. Serum GADD45a methylation is a useful biomarker to distinguish benign vs malignant prostate disease. Br J Cancer. 2015; 113:460–68. https://doi.org/10.1038/bjc.2015.240 [PubMed]
- 24. Müller HM, Fiegl H, Widschwendter A, Widschwendter M. Prognostic DNA methylation marker in serum of cancer patients. Ann N Y Acad Sci. 2004; 1022:44–49. https://doi.org/10.1196/annals.1318.008 [PubMed]
- 25. Tsuchida S, Satoh M, Takiwaki M, Nomura F. Current Status of Proteomic Technologies for Discovering and Identifying Gingival Crevicular Fluid Biomarkers for Periodontal Disease. Int J Mol Sci. 2018; 20:86. https://doi.org/10.3390/ijms20010086 [PubMed]
- 26. Liu YJ, Liu Y, Xu Y. [Developments of neutrophil function and the relationship between neutrophils dysfunction and periodontitis]. Hua Xi Kou Qiang Yi Xue Za Zhi. 2016; 34:210–14. https://doi.org/10.7518/hxkq.2016.02.021 [PubMed]
- 27. Chee B, Park B, Bartold PM. Periodontitis and type II diabetes: a two-way relationship. Int J Evid Based Healthc. 2013; 11:317–29. https://doi.org/10.1111/1744-1609.12038 [PubMed]
- 28. Luo Y, Peng X, Duan D, Liu C, Xu X, Zhou X. Epigenetic Regulations in the Pathogenesis of Periodontitis. Curr Stem Cell Res Ther. 2018; 13:144–50. https://doi.org/10.2174/1574888X12666170718161740 [PubMed]
- 29. Martins MD, Jiao Y, Larsson L, Almeida LO, Garaicoa-Pazmino C, Le JM, Squarize CH, Inohara N, Giannobile WV, Castilho RM. Epigenetic Modifications of Histones in Periodontal Disease. J Dent Res. 2016; 95:215–22. https://doi.org/10.1177/0022034515611876 [PubMed]
- 30. Bender J. DNA methylation and epigenetics. Annu Rev Plant Biol. 2004; 55:41–68. https://doi.org/10.1146/annurev.arplant.55.031903.141641 [PubMed]
- 31. Wilson AG. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J Periodontol. 2008; 79:1514–19. https://doi.org/10.1902/jop.2008.080172 [PubMed]
- 32. Kinney JS, Ramseier CA, Giannobile WV. Oral fluid-based biomarkers of alveolar bone loss in periodontitis. Ann N Y Acad Sci. 2007; 1098:230–51. https://doi.org/10.1196/annals.1384.028 [PubMed]
- 33. Giannobile WV. Crevicular fluid biomarkers of oral bone loss. Curr Opin Periodontol. 1997; 4:11–20. [PubMed]
- 34. Frodge BD, Ebersole JL, Kryscio RJ, Thomas MV, Miller CS. Bone remodeling biomarkers of periodontal disease in saliva. J Periodontol. 2008; 79:1913–19. https://doi.org/10.1902/jop.2008.080070 [PubMed]
- 35. Yoon AJ, Cheng B, Philipone E, Turner R, Lamster IB. Inflammatory biomarkers in saliva: assessing the strength of association of diabetes mellitus and periodontal status with the oral inflammatory burden. J Clin Periodontol. 2012; 39:434–40. https://doi.org/10.1111/j.1600-051X.2012.01866.x [PubMed]
- 36. Baer MT, Huang N, Gibson FC 3rd. Scavenger receptor A is expressed by macrophages in response to Porphyromonas gingivalis, and participates in TNF-alpha expression. Oral Microbiol Immunol. 2009; 24:456–63. https://doi.org/10.1111/j.1399-302X.2009.00538.x [PubMed]
- 37. Rose AM, Krishan A, Chakarova CF, Moya L, Chambers SK, Hollands M, Illingworth JC, Williams SM, McCabe HE, Shah AZ, Palmer CN, Chakravarti A, Berg JN, et al. MSR1 repeats modulate gene expression and affect risk of breast and prostate cancer. Ann Oncol. 2018; 29:1292–303. https://doi.org/10.1093/annonc/mdy082 [PubMed]
- 38. Thalji GN, Nares S, Cooper LF. Early molecular assessment of osseointegration in humans. Clin Oral Implants Res. 2014; 25:1273–85. https://doi.org/10.1111/clr.12266 [PubMed]
- 39. Navarro-González C, Huerga-Gómez A, Fazzari P. Nrg1 Intracellular Signaling Is Neuroprotective upon Stroke. Oxid Med Cell Longev. 2019; 2019:3930186. https://doi.org/10.1155/2019/3930186 [PubMed]
- 40. Dugaucquier L, Feyen E, Mateiu L, Bruyns TA, De Keulenaer GW, Segers VF. The role of endothelial autocrine NRG1/ERBB4 signaling in cardiac remodeling. Am J Physiol Heart Circ Physiol. 2020; 319:H443–55. https://doi.org/10.1152/ajpheart.00176.2020 [PubMed]
- 41. Hong YB, Joo J, Hyun YS, Kwak G, Choi YR, Yeo HK, Jwa DH, Kim EJ, Mo WM, Nam SH, Kim SM, Yoo JH, Koo H, et al. A Mutation in PMP2 Causes Dominant Demyelinating Charcot-Marie-Tooth Neuropathy. PLoS Genet. 2016; 12:e1005829. https://doi.org/10.1371/journal.pgen.1005829 [PubMed]
- 42. Gu S, Dai H, Zhao X, Gui C, Gui J. AKT3 deficiency in M2 macrophages impairs cutaneous wound healing by disrupting tissue remodeling. Aging (Albany NY). 2020; 12:6928–46. https://doi.org/10.18632/aging.103051 [PubMed]
- 43. Clough E, Barrett T. The Gene Expression Omnibus Database. Methods Mol Biol. 2016; 1418:93–110. https://doi.org/10.1007/978-1-4939-3578-9_5 [PubMed]
- 44. Lin L, Yu W, Zhang W, Li S, Hu S, Jiang B, Gu Y, Lu E. Expression profile of lipoxygenases in gingival tissues of human periodontitis. Oral Dis. 2021; 27:567–76. https://doi.org/10.1111/odi.13558 [PubMed]
- 45. Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001; 17:520–25. https://doi.org/10.1093/bioinformatics/17.6.520 [PubMed]
- 46. Nordlund J, Bäcklin CL, Wahlberg P, Busche S, Berglund EC, Eloranta ML, Flaegstad T, Forestier E, Frost BM, Harila-Saari A, Heyman M, Jónsson OG, Larsson R, et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013; 14:r105. https://doi.org/10.1186/gb-2013-14-9-r105 [PubMed]
- 47. Price ME, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013; 6:4. https://doi.org/10.1186/1756-8935-6-4 [PubMed]
- 48. Zhang S, Wang Y, Gu Y, Zhu J, Ci C, Guo Z, Chen C, Wei Y, Lv W, Liu H, Zhang D, Zhang Y. Specific breast cancer prognosis-subtype distinctions based on DNA methylation patterns. Mol Oncol. 2018; 12:1047–60. https://doi.org/10.1002/1878-0261.12309 [PubMed]
- 49. Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010; 11:587. https://doi.org/10.1186/1471-2105-11-587 [PubMed]
- 50. Zhuang J, Widschwendter M, Teschendorff AE. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinformatics. 2012; 13:59. https://doi.org/10.1186/1471-2105-13-59 [PubMed]
- 51. Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013; 41:D1228–33. https://doi.org/10.1093/nar/gks1147 [PubMed]
- 52. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. https://doi.org/10.1093/nar/gkv007 [PubMed]
- 53. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007; 23:3251–53. https://doi.org/10.1093/bioinformatics/btm369 [PubMed]
- 54. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011; 27:1739–40. https://doi.org/10.1093/bioinformatics/btr260 [PubMed]
- 55. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559. https://doi.org/10.1186/1471-2105-9-559 [PubMed]
- 56. Sanz H, Valim C, Vegas E, Oller JM, Reverter F. SVM-RFE: selection and visualization of the most relevant features through non-linear kernels. BMC Bioinformatics. 2018; 19:432. https://doi.org/10.1186/s12859-018-2451-4 [PubMed]
- 57. Planello AC, Singhania R, Kron KJ, Bailey SD, Roulois D, Lupien M, Line SR, de Souza AP, De Carvalho DD. Pre-neoplastic epigenetic disruption of transcriptional enhancers in chronic inflammation. Oncotarget. 2016; 7:15772–86. https://doi.org/10.18632/oncotarget.7513 [PubMed]
- 58. Demmer RT, Behle JH, Wolf DL, Handfield M, Kebschull M, Celenti R, Pavlidis P, Papapanou PN. Transcriptomes in healthy and diseased gingival tissues. J Periodontol. 2008; 79:2112–24. https://doi.org/10.1902/jop.2008.080139 [PubMed]