



Introduction
Major photoreceptor degenerative diseases are primarily age-related eye disorders, leading to severe vision impairment or irreversible vision loss. These include age-related macular degeneration [1], diabetic retinopathy [2, 3] and retinal detachment (RD) [4, 5]. Despite distinct differences between these diseases, the separation of photoreceptors from the underlying retinal pigment epithelium (RPE) [4–7] or a loss of functional RPE [1, 8, 9], and eventual photoreceptor death is common to all of them. Since photoreceptors are highly metabolic [10], nutrient deprivation from the separated RPE induces the pathological responses that result in permanent neuronal loss. Fortunately, the separation of photoreceptors from the RPE (RD) can be well-modeled in animals [11–13]. By using the RD model, we and others have previously identified apoptosis [14, 15], regulated necrosis [13], oxidative stress [13, 16], and inflammation [17–19] as major pathophysiological changes after the separation. However, currently, no pharmacological approaches have been proven effective in treating photoreceptor degeneration in human clinical trials.
Nicotinamide adenine dinucleotide (NAD+) is a co-factor in redox metabolism that is critical to energy production [20]. In recent years, NAD+ decline has been identified in various diseases. Enhancing NAD+ biosynthesis is beneficial in age-associated metabolic and neurodegenerative disorders [21–23].
In recent years, several papers have reported NAD+ biosynthesis in eye diseases. Mutations in NMNAT1, an enzyme in the NAD+ salvage pathway, is found to cause Leber congenital amaurosis [24, 25]. Restoration of NAD+ appears to be important in vision, and that NMN has been reported to protect photoreceptors against light-induced retinal damage in rodent models [26, 23]. The NAD+-dependent silent information regulator 2 (Sir2) is among the major effectors for NAD+ beneficiaries and plays fundamental roles in aging, metabolism, cancer, stress response, neuronal function, inflammation, apoptosis and DNA repair [27–30]. SIRT1 has recently been found essential in retinal development and survival. Alteration of SIRT1 activity has been found in aged retina [30, 31], diabetic retinopathy [32, 33], light-induced retinal degeneration [34] and oxygen-induced ischemic retinopathy (OIR) [35], though much has to be done to validate these findings.
To date, however, little is known about the role of NAD+ when photoreceptors are separated from the RPE and whether the NAD+ and SIRT1 may play a role in protecting photoreceptors during their separation from the underlying RPE. In this study, we evaluated the effects of boosting NAD+ on the retina and photoreceptors after the induction of RD and explored the mechanisms of observed actions.
Results
NMN supplementation reduces photoreceptor cell death in the early phase of RD
The in vivo experimental procedures of RD were successfully induced in mouse eyes (Figure 1A, 1B), resulting in photoreceptor cell death which was attenuated after NMN administration at 24 hours after RD (Figure 1C, 1E, 1F). NMN at 250 mg/kg reduced cell death numbers by 52.7% (2292 ± 690 cells/mm2, p<0.001) whereas NMN at 500 mg/kg decreased cell death by 71.0% (1405 ± 290 cells/mm2, p<0.001).
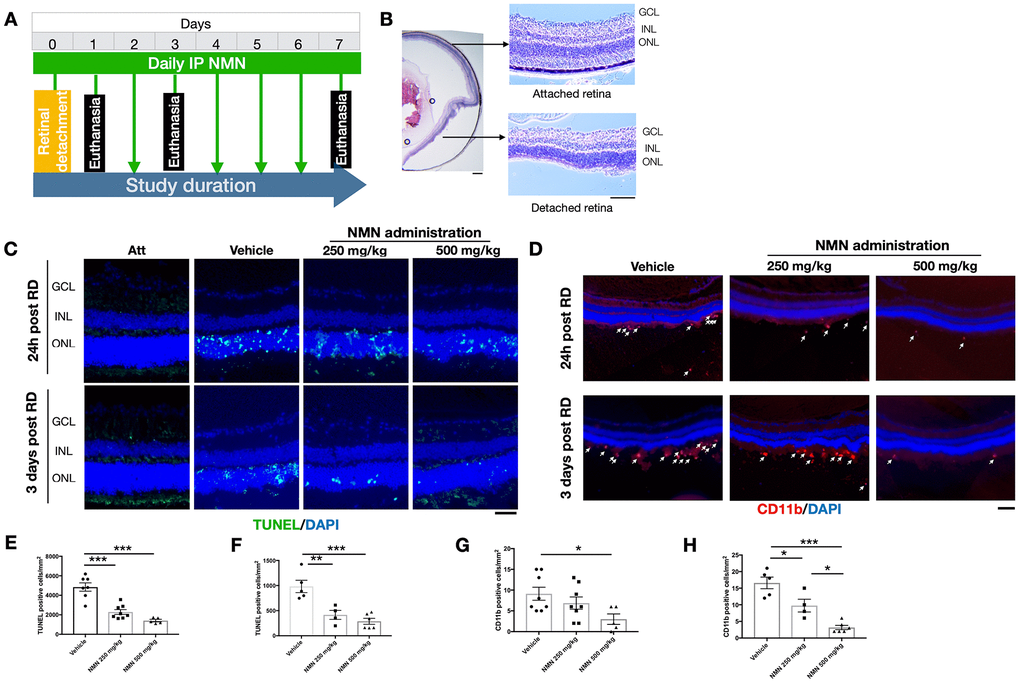
Figure 1. Protective effects of NMN administration in the early phase of retinal detachment (RD). (A) A flow chart for the in vivo experiments. (B) schematic anatomy of a mouse eye by hematoxylin and eosin (HE) staining showing a bullous after successful RD surgery. Scale bar: 50 μm. (C, E, F) TUNEL+ cells (green) were seen with the highest numbers in vehicle-treated retinas and lowest in the 500 mg/kg NMN-treated retinas at both 24h (E) and three days (F) post RD. Nuclei were counterstained with DAPI (blue). N = 4 to 8 eyes per group. Scale bar: 50 μm. (D, G, H) CD11b+ macrophages (red) infiltrated in the subretinal space after RD, NMN administration significantly reduced the number as soon as 24h post RD (G), and in a dose-dependent manner after three days of RD (H). Nuclei were counterstained with DAPI (blue). N = 5 to 8 eyes per group. Scale bar: 100μm. Statistical significance was analyzed with one-way ANOVA followed by Tukey-Kramer adjustments. *p<0.05, **p<0.01, ***p<0.001. Data are mean ± SEM.
Similar effects were seen 3 days after RD with NMN reducing the number of TUNEL+ cells by 57.8% in the 250 mg/kg NMN group (984 ± 274 cells/mm2 in vehicle vs. 415 ± 179 cells/mm2 in NMN; p<0.01) and 70.8% in the 500 mg/kg group (287 ± 147 cells/mm2; p<0.001).
These results show that daily supplementation of the NAD+ precursor NMN is effective in inhibiting cell death at the acute phase of photoreceptor injury, with the most protective results achieved at the dose of 500 mg/kg.
NMN supplementation suppresses retinal inflammation
Our results suggested that 24 hours after RD induction, there was an accumulation of CD11b+ cells in the subretinal space in the vehicle RD group (9 ± 4 cell/mm2) (Figure 1D–1G). The number was reduced by an increasing dose of NMN from 250 mg/kg (7 ± 4 cell/mm2) to 500 mg/kg (3 ± 3 cells/mm2; p<0.05 compared to vehicle group).
The number of CD11b+ cells peaked three days post RD with the highest number in the vehicle group (17 ± 4 cells/mm2) followed by fewer cells in the 250 mg/kg NMN group (10 ± 4 cells/mm2; p<0.05 compared to vehicle group) and the lowest number in the 500 mg/kg NMN group (3 ± 2 cell/mm2, p< 0.001 compared to vehicle group) (Figure 1D–1H). Taken together, these results suggest that NMN supplementation is associated with suppressed inflammation after RD in a dose-dependent manner.
NMN normalizes oxidative stress and upregulates antioxidant HO-1 after RD
We have previously identified elevated reactive oxygen species (ROS) as major pathophysiological changes after RD [13, 16]. We, therefore, investigated the effect of NMN supplementation on oxidative stress in the retina. Consistent with our previous results, there was a significant increase in oxidative stress as measured by protein carbonyl content (PCC) in the detached retinas (0.81 ± 0.1 nmol/mg) compared to the attached retinas (0.55 ± 0.15 nmol/mg; p<0.01) three days after RD (Figure 2A). NMN administration completely abolished this consequence (0.55 ± 0.1 nmol/mg, p<0.01 compared to vehicle) and maintained the tissue PCC comparable to the normal level as in the attached retina.
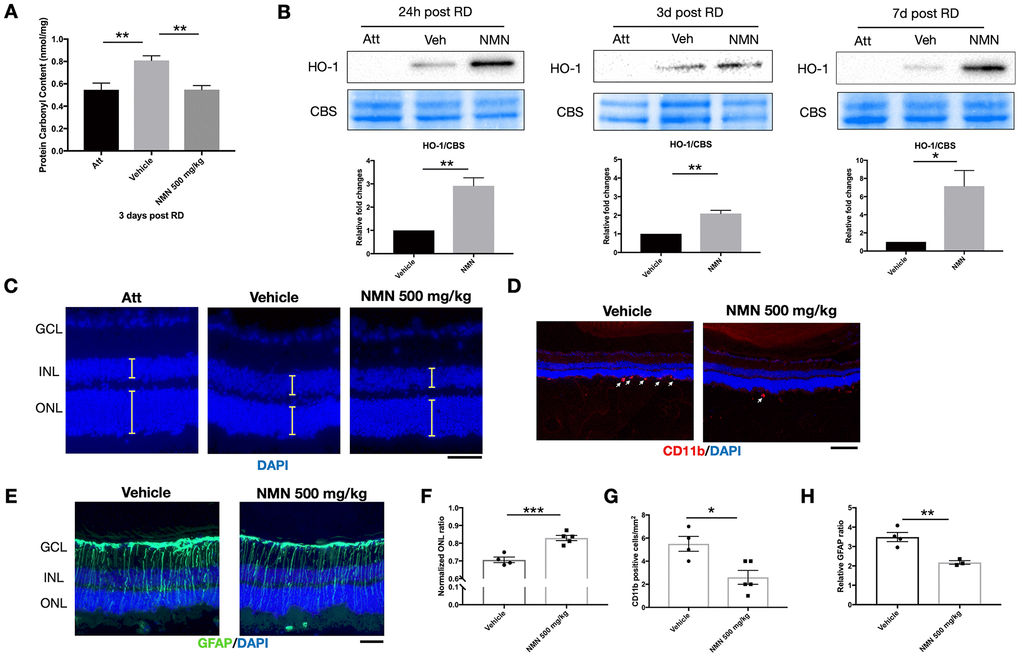
Figure 2. NMN administration attenuates oxidative stress and is protective in the late phase of retinal detachment (RD). (A) Protein carbonyl content (PCC) was significantly higher after RD while NMN treatment normalized PCC level comparable to the attached retinas at three days post RD. N = 6 eyes per group. (B) Heme oxygenase 1 (HO-1) was significantly up-regulated in the NMN groups as soon as 24h and remained highly expressed throughout the experimental period. N = 4 to 5 eyes per group. (C–F) Preservation of outer nuclear layer (ONL) thickness after NMN administration was seen 7 days after RD. N = 4 to 5 eyes per group. Scale bar=50 μm. The thickness of the layers was measured as the yellow lines indicated. (D–G) Inhibition of neuroinflammation seen by a reduced number of CD11b+ infiltrating macrophages (red) in NMN-treated retinas compared to vehicle-treated retinas. N = 4 to 5 eyes per group. Scale bar: 100 μm. (E–H) Reduced reactive gliosis represented by GFAP (green) staining in NMN-treated retinas compared to vehicle-treated retinas. N = 3 to 4 eyes per group. Scale bar: 50 μm. Statistical significance was analyzed with one-way ANOVA followed by Tukey-Kramer adjustments or the unpaired Student's t-test. *p<0.05. **p<0.01. ***p<0.001. Data are mean ± SEM.
To elucidate the antioxidant effect of NMN after RD, we further found that HO-1, an inducible isoform of heme oxygenase, was significantly upregulated in the NMN-treated retinas (Figure 2B). HO-1 expression was barely detectable in the attached retinas. In contrast, under stress induced by RD, HO-1 expressions were detectable in protein level, and NMN administration highly upregulated HO-1 levels as soon as 24 hours and throughout the whole experimental period. Our results demonstrate that NMN administration can counteract excessive oxidative stress in the detached retina, and this effect is possibly executed through the upregulation of HO-1.
NMN supplementation preserves ONL thickness and has an overall protection to the retina
To evaluate the long-term effect of NMN administration on photoreceptors after RD, we examined the overall impact on photoreceptor cell survival by measuring ONL thickness on Day 7 post RD. Administration of NMN at 500 mg/kg significantly preserved the retinal thickness seen by normalized ONL ratio (0.83) as compared to the vehicle-treated group (0.71, p<0.001) (Figure 2C–2F). Along with the increased survival of photoreceptors, NMN had a continued inhibitory effect of infiltrating CD11b+ macrophages (3 ± 1 cell/mm2) compared to the vehicle-treated retina (6 ± 1 cell/mm2, p<0.05) (Figure 2D–2G), and a significant suppression of gliosis as measured by GFAP immunofluorescence (p<0.01) (Figure 2E–2H). These results further demonstrated the beneficial effect of NAD+-boosting molecules on regulating retinal homeostasis after injury.
NMN increases NAD+ levels and SIRT1 expression after RD
NAD+ deficiency has been identified among the primary causes leading to many neurodegenerative diseases [20, 22, 23], and the NAD+ precursors have beneficial effects in protecting neurons by replenishing the NAD+ pool in the majority of studies. However, we did not observe any change of NAD+ level in the vehicle-treated detached retina at neither 24h (NAD ratio of vehicle group 101.5% vs. attached retina 100%, p>0.05) or three days (NAD ratio of vehicle group 97.5% vs. attached retina 100%, p>0.05) post RD compared to the attached normal retina (Figure 3A, 3B). Whereas NMN administration significantly increased the NAD+ above normal levels in both time points (24h: 119.5%, p=0.005 compare to attached; 3 days: 119.7%, p=0.008 compare to attached). Similar to the results from the retina, NMN administration significantly increased NAD+ levels compared to both vehicle and the attached RPE/choroid (Supplementary Figure 1).
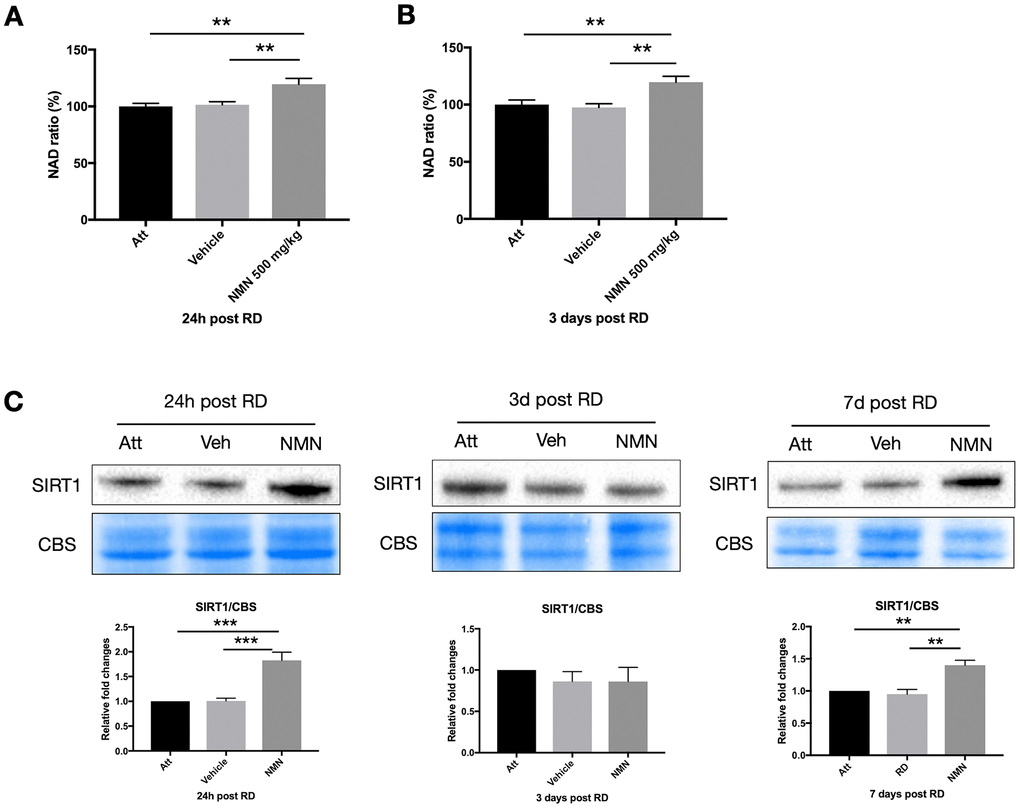
Figure 3. Increased NAD+ and SIRT1 levels after retinal detachment (RD) with NMN supplementation. Significant increase of NAD+ levels in the NMN-treated detached retinas compared to the vehicle and the attached retinas at 24h (A) and three days (B) after RD. NAD+ levels in vehicle-treated detached retinas were not lower compared to untreated controls. N = 8 to 11 eyes per group. (C) Upregulation of SIRT1 protein levels in NMN-treated retinas seen at 24h and 7 days post RD. SIRT1 levels were comparable among groups at three days post RD. N = 3 to 7 eyes per group. Statistical significance was analyzed with one-way ANOVA followed by Tukey-Kramer adjustments. *p<0.05. **p<0.01. ***p<0.001. Data are mean ± SEM.
To further elucidate the mechanism of NMN protection in the retina after increasing NAD+ levels, we examined the sirtuins, and found an upregulation of SIRT1 expression in the NMN-treated retinas at 24h and seven days after RD (Figure 3C). Retinal detachment alone did not induce the upregulation of SIRT1. This expression pattern was very similar to NAD+ content. SIRT1 expression did not differ significantly among groups three days after RD. Thus, SIRT1 may be necessary at different time points to confer protection after NMN administration, or they may have distinct and differing roles in photoreceptor survival.
In vitro study replicated the protection of NMN and revealed an association to be in part with the SIRT1/HO-1 signaling
Since we observed a very high oxidative stress status in the detached retina, we next tested the NMN mechanism of action in vitro using the photoreceptor-like cell line 661W in an oxidative stress model. A significant increase in cell survival was seen in the NMN administration group after ROS insult (Figure 4A). SRT2104, a direct SIRT1 activator, was also found protective to 661W cells after ROS insult (Figure 4B). However, the baseline level of NAD+ was also declined in the oxidative stress model (Supplementary Figure 2A). To examine if SIRT1 plays a role in mediating the effects of NMN supplementation, we transfected the 661W cells with SIRT1 siRNAs (Figure 4C). Our results showed a significantly lower level of cell survival in the SIRT1 siRNA transfection groups compared to the scramble siRNA control groups under NMN administration after ROS (Figure 4D). These results suggested that SIRT1 is at least partially responsible for the antioxidant property of NMN.
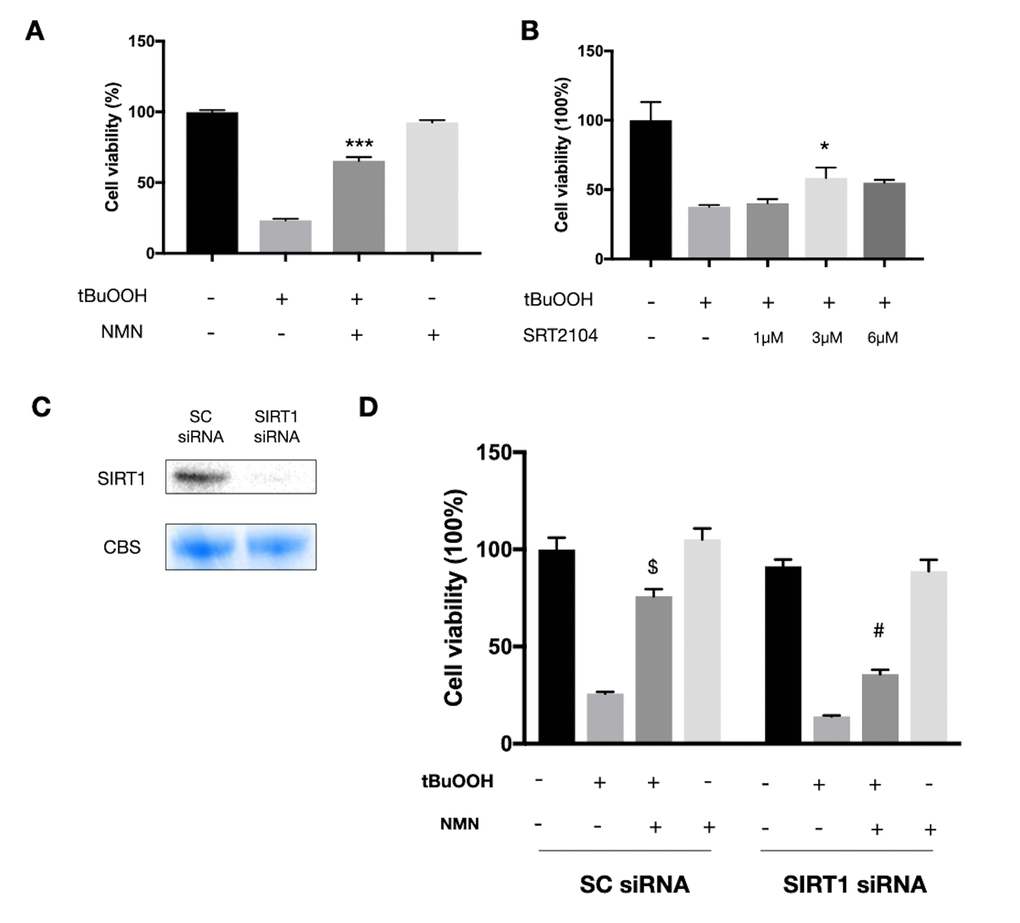
Figure 4. Protected effects of NMN on 661W cells under oxidative stress are partially SIRT1 dependent. (A) NMN administration significantly increased 661W cell viability after ROS insult. ***p<0.001 to tBuOOH only group. N = 4 to 5 per group. (B) SRT2104, a SIRT1 direct activator, had a similar effect as NMN in protecting 661W cells from ROS insult. *p<0.05 to tBuOOH only group. N = 3 to 4 per group. (C) SIRT1 was knocked down by siRNAs. (D) Silencing SIRT1 by siRNAs attenuated the protective effect of NMN after ROS insult compared to the scrambled (SC) siRNAs control group. $p<0.001 to tBuOOH only group in SC siRNA condition. #p<0.001 to tBuOOH and NMN treated group in SC siRNA condition. N = 4 to 8 per group. Statistical significance was analyzed with one-way ANOVA followed by Tukey-Kramer adjustments. Data are mean ± SEM.
Using western blot analysis, we found an upregulation of HO-1 protein expression after both NMN (Figure 5A) and SRT2104 treatment (Figure 5B) in the oxidative stress model. Knocking down of SIRT1 almost totally inhibited the induction of HO-1 with NMN administration (Figure 5C), whereas knocking down HO-1 didn’t alter the expression of SIRT1 under the NMN treated conditions (Figure 5D) suggesting that HO-1 upregulation after NMN was the downstream of SIRT1 upregulation.
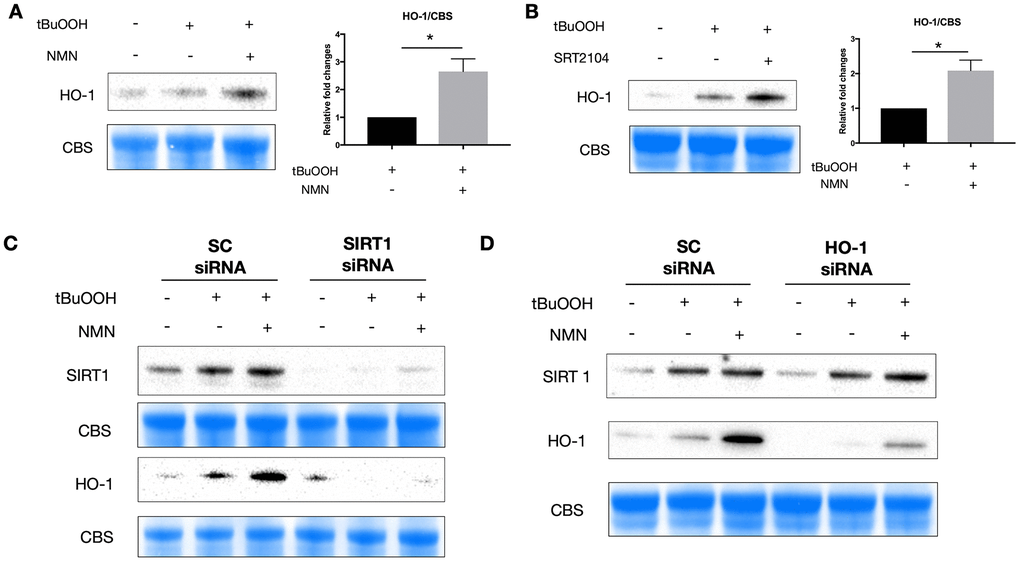
Figure 5. A potential SIRT1/HO-1 signaling under the NMN-treated oxidative stress model. Both NMN (A) and SRT2104 (B) administration significantly increased HO-1 protein levels after ROS insult. N = 4 per group. Statistical significance was analyzed with the unpaired Student's t-test. *p<0.05. **p<0.01. ***p<0.001. Data are mean ± SEM. (C) Silencing SIRT1 by siRNA almost abolished the induced HO-1 expression by NMN after ROS insult. (D) HO-1 siRNA didn’t alter the expression of SIRT1 by NMN after ROS insult.
NMN and SIRT1 exerted protective effects directly on 661W cells not through action on immune cells in an in vitro model of immune mediated toxicity to photoreceptor cells
Our in vivo results have shown a significant reduction of infiltrating macrophages in the subretinal space after the NMN supplement. To determine whether this result is a direct anti-inflammatory effect of NMN or indirectly from the reduced photoreceptor cell death, we examined the role of NMN and SIRT1 in a RAW264.7 macrophage conditioned media (CM) toxicity model. CM samples from RAW264.7 cells treated or without the LPS were both toxic to 661W cells. NMN administration significantly increased the viability of 661W cells in both the LPS treated and untreated RAW264.7 CM (Figure 6A). The direct SIRT1 activator SRT1720 also showed a protective effect on 661W cells in the LPS treated RAW264.7 CM (Figure 6B).
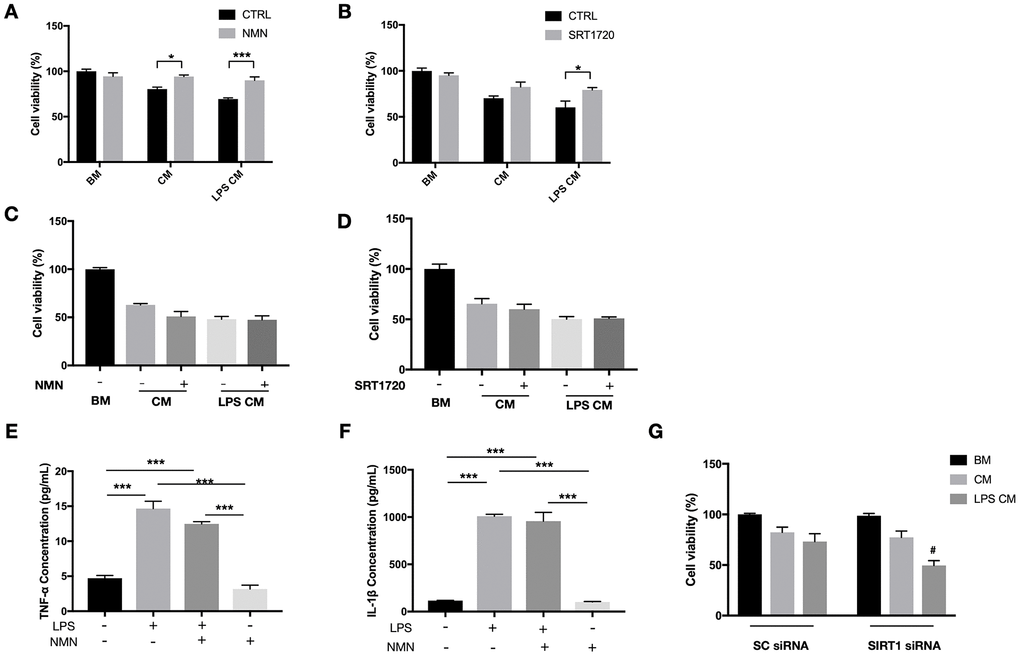
Figure 6. Protective effects of NMN and SIRT1 activation to 661W cells in RAW264.7 conditioned media (CM). (A) NMN administration significantly increased the cell viability of 661W cells in both the LPS treated and untreated RAW264.7 CM. N = 3 to 4 per group. (B) SRT1720 also showed a protective effect to 661W cells in the LPS treated RAW264.7 CM. N = 4 per group. The CM from neither NMN (C) or SRT1720 (D) treated RAW264.7 cells had protective effects to 661W cells. N = 4 to 6 per group. LPS can induce significant increases in both TNF-α (E) and IL-1β (F) expressions, which were not altered by NMN administration. N = 3 per group. (G) SIRT1 siRNA in 661W cells significantly reduced cell viability in the LPS stimulated RAW264.7 CM. #p<0.05 to LPS CM treated group in SC siRNA condition. N = 3 to 4 per group. Statistical significance was analyzed with the unpaired Student's t-test between each treatment and its control, or with one-way ANOVA followed by Tukey-Kramer adjustments. *p<0.05. **p<0.01. ***p<0.001. Data are mean ± SEM.
Next, we administrated NMN or SRT1720 directly to RAW264.7 cells (stimulated with or without LPS), and subsequently, CM was collected and used to treat 661W cells. Neither NMN- nor SRT1720-treated RAW264.7 cells resulted in any protective effects on 661W cells, in contrast to those 661W cells that were directly treated with either molecules (Figure 6C, 6D). These results indicated that the protective effects of NMN and SRT2104 were probably acting directly on 661W cells, rather than by altering the toxicity of RAW264.7 CM. In order to see whether NMN administration has effect on the inflammatory cytokines, we treated RAW264.7 cells with LPS, in the presence or absence of NMN. The CM samples were collected, and the expression of neurotoxic cytokines TNF-α and IL-1β were assessed by ELISA. These results showed that LPS can induce significant increases in both TNF-α (Figure 6E) and IL-1β (Figure 6F). However, NMN administration at 1 mM didn’t significantly alter the levels of both cytokines.
Lastly, we used SIRT1 siRNAs to knockdown SIRT1 in 661W cells before treating them with RAW264.7 CM with or without LPS. Knocking down SIRT1 subjected 661W cells to a higher sensitivity to the neurotoxic effects of LPS stimulated CM from RAW264.7 cells, as can be seen by the significantly lower cell viability compared to the control group (Figure 6G).
Delayed NMN supplementation continues to exert protection to the retina after RD
To be more relevant to clinical settings, we administrated NMN at Day 3 post RD and evaluated the effects on the retina at Day 7 post RD (Figure 7A). Our results showed a preserved ONL thickness in comparison to the vehicle group (Figure 7B–7E), along with an inhibitory effect of infiltrating CD11b+ macrophages compared to the vehicle group (Figure 7C–7F). GFAP was unaltered (Figure 7D–7G). These results showed that delayed NMN administration can still exert protective effect after RD.
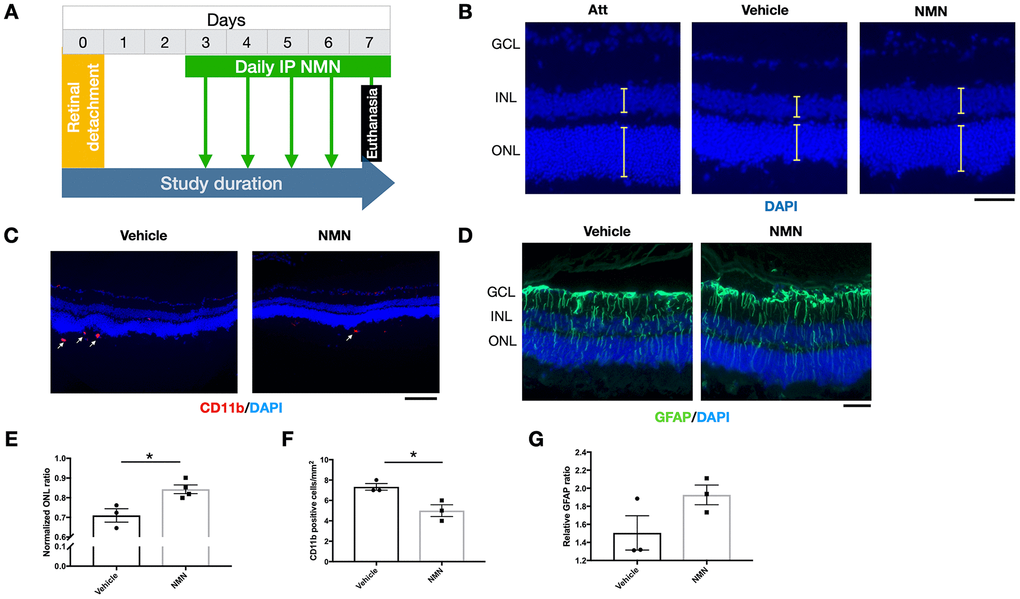
Figure 7. Protective effects of delayed NMN administration to retinal detachment (RD). (A) A flow chart for the in vivo experiments. (B–E) Delayed NMN administration still have protective effect to outer nuclear layer (ONL) thickness 7 days post RD. N = 3 to 4 eyes per group. Scale bar=50 μm. (C–F) Reduced number of CD11b+ infiltrating macrophages (red) in delayed NMN-treated groups compared to vehicle. N = 3 eyes per group. Scale bar: 100 μm. (D–G) No significance was found between NMN and vehicle treated retinas of the GFAP (green). N = 3 eyes per group. Scale bar: 50 μm. Statistical significance was analyzed with the unpaired Student's t-test. *p<0.05. **p<0.01. ***p<0.001. Data are mean ± SEM.
Discussion
Our study provides evidence of neuroprotective effects of NMN, a NAD+-boosting molecule, in photoreceptor degeneration after RD. NMN administration can attenuate neuroinflammation by reducing CD11b+ infiltrating macrophages, decreasing oxidative stress, upregulating HO-1 expression, and inhibiting photoreceptor cell death by reducing TUNEL+ cells, thus achieving an overall protection of the retina. Although RD was not associated with NAD+ deficiency, NMN administration increased retinal NAD+ levels, upregulated SIRT1, and HO-1 at various time points. In vitro studies revealed a neuroprotective effect of NMN in ROS-induced 661W cell death probably through the SIRT1/HO-1 signaling. Moreover, NMN and SIRT1 protected 661W cells cultured in the RAW264.7 CM by a direct protection to the cells rather than altering the toxicity of the inflammatory CM. Lastly, delayed NMN administration continues to show protection to the retina after RD. Taken together, our results suggest a potential therapeutic value of NMN administration in treating photoreceptor degeneration in clinical settings.
We have shown previously that photoreceptor separation from underlying RPE triggers ROS production [13, 16]. Here, we show that NMN treatment in vivo normalized excessive ROS production and increased the antioxidant responses. In particular, increasing NAD+ level above normal was associated with a significant upregulation of HO-1 protein level. HO-1 (also named HMOX1 or HSP32) is a stress-inducible heat shock protein that catalyzes heme into biliverdin, carbon monoxide, and iron, exerting anti-oxidative and anti-inflammatory functions [36–40]. HO-1 is induced in the detached retina but is insufficient to completely counteract the damage of RD. The higher level of HO-1 induced by NMN enabled neuroprotective outcomes. Several other studies have also suggested activation of antioxidant response after NAD+ precursors [41–43], with one of the studies showing that increased recruitment of Nrf2 to HO-1 gene promoter after NMN supplement [43].
Contrary to most studies showing a decline of NAD+ levels in neurodegenerative models, our results did not show NAD+ levels to be significantly altered after RD. However, the NAD+-boosting molecule NMN was associated with neuroprotective effects with the NAD+ levels above physiologic. Nonetheless, if this supraphysiologic elevation of NAD+ is a mere association or partially causative remains unknown. Our finding is similar to a recent paper reporting the toxicity of SOD1 loss in an astrocyte cell line was not associated with reduced NAD+ levels or sirtuins, yet NMN administration increased survival and depended on sirtuin presence [43]. It is possible that young (7-8 week) adult mice used in our study may have the capacity to restore NAD+ level after RD, whereas older animals used in other neurodegenerative models may have limited capacity for NAD+ restoration under stress. Besides, NAD+ levels of 661W cells under oxidative stress was indeed declined (Supplementary Figure 2A) and the SIRT1 protein level increased (Supplementary Figure 2B). The differences in the observations can be attributed to the fact that the complex tissue of the retina responses to noxious stimuli differ from cell models. It may be that compensatory mechanism in cells other than photoreceptors counteract a potential NAD depletion in photoreceptors. Based on our results, we think that although NAD+ levels do not decline after experimental RD, NMN administration boosts NAD+ levels beyond physiologic, and this may help activate enzymes, such as SIRT1, that may play a protective role in the process.
SIRT1 has recently been found essential in retinal development and various pathological conditions [30–35, 44, 45]. Notably, we found that silencing SIRT1 didn’t completely abolish the protective effect of NAD+. Here are several possible reasons: 1) other sirtuin family members may also participate in the neuroprotection after the increase of NAD+. For example, both SIRT1 and SIRT2 can mediate neural stem and/or progenitor cell fate decisions into oligodendrocytes [46]. SIRT3 was shown to protect against mitochondrial fragmentation and neuronal cell death induced by SOD1 G93A overexpression [47]. SIRT6 is shown to extend lifespan by modulating telomeric chromatin [48, 49]. SIRT7 increases the stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice [50]. 2) Antioxidant defenses may be regulated by other sirtuins. Accumulating studies revealed a complex relationship between sirtuins and ARE defense systems, with several sirtuins (SIRT1, 2, 3, 5 and 6) participating in the regulation of various antioxidant genes in multiple cells, tissues and disease models [42, 51–53]. Besides, a disruption of redox homeostasis is also capable of affecting the expression level, post-translation, and interactions of sirtuins [36]. 3) NAD+ metabolism alone is sufficient to mediate various biological processes independent of sirtuins [54]. For example, changes in the NAD+/NADH ratio affect the production of superoxide by complex I [55–57], and directly influenced ROS and the antioxidant defenses. Thus, it is critical to determine whether the protective effects via boosting NAD+ levels are mediated via changes to the NAD+/NADH ratio directly or via activation of sirtuins [54–58].
We observed a significant reduction of infiltrating macrophages in the subretinal space after the NMN supplement in vivo. The subsequent in vitro outcome revealed a direct protective effect to 661W cells instead of indirectly altering the toxicity of RAW264.7 CM by NMN or SIRT1 activator. The NAD+ biosynthesis pathway and SIRT1 are important regulators in the immune system. NAD+ and SIRT1 have been reported to suppress inflammation in various diseases such as in autoimmune encephalomyelitis (EAE) [59], liver ischemia-reperfusion injury [39], high-fat-diet obesity [60], etc. It is reported that SIRT1 can reduce pro-inflammatory responses by deacetylating the p65 subunit of nuclear factor kappa B (NF-κB) [61] The NAD biosynthesis pathways regulates the macrophages and act as the metabolic switch that determine the immune responses under specific settings [62, 63].
The limitations of our study that can also serve as future directions are the following: 1) although we have shown a benefit of NMN administration in the acute phase of RD, the long-term effect and safety of NMN administration in the retina of mice and humans needs to be evaluated. A study of one-year oral administration of NMN showed safe and well-tolerated effects in wild-type C57BL/6 mice, and human clinical studies have been performed with no apparent negative effects [64]; 2) more work is needed to elucidate the exact molecular role of SIRT1 and identify its substrate in regulating the antioxidant pathways and mitochondrial homeostasis; 3) other sirtuin family members, as well as other NAD+-dependent enzymes, such as PARPs, CD38 may also play a role in the retinal homeostasis, which requires further investigation. 4) the investigation of the pharmacokinetics of NMN and the metabolism of NAD+ in photoreceptor cells will be fundamental to understanding retinal neuroprotection and how to dose the molecule in humans. 5) a further elucidation of the anti-inflammatory effects of NMN and SIRT1 in the retina after RD is needed.
In conclusion, our study demonstrates the neuroprotective effects of NMN supplementation in rescuing photoreceptor degeneration after RD. NMN treatment was associated with an increase in NAD+ and SIRT1 levels in the injured retina, leading to anti-apoptotic, anti-inflammatory, and antioxidant effects. The therapeutic effect of NMN may occur at least partially through the activation of SIRT1/HO-1 signaling. These results provide an impetus for studies in larger animals and humans with photoreceptor degeneration in a clinical setting.
Materials and Methods
Animals
Wildtype C57BL/6 mice (7-10 weeks old) were purchased (Jackson Laboratory, Bar Harbor, ME, USA) and fed ad libitum on standard laboratory chow and water in an air-controlled room with a 12-h light/12-h dark cycle. All animal experiments adhered to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research under the approval by the Animal Care Committee of Massachusetts Eye and Ear Infirmary.
Induction of experimental retinal detachment
The induction of our rodent retinal detachment model has been addressed in detail in our previously published paper from our lab [64]. We used this model to achieve reproducible and sustainable bullous of RD. Briefly, after general anesthesia, pupils were dilated and proparacaine eye drops (Sandoz Inc., Princeton, NJ, USA) were applied. The conjunctiva was opened, and a self-sealing scleral tunnel was created followed by a corneal paracentesis to lower intraocular pressure. Next, a 34-gauge needle connected to a 10μl syringe (Hamilton Company, Reno, NV, USA) was inserted through the scleral tunnel and 2 μl of 1% sodium hyaluronate (Provisc; Alcon, Fort Worth, TX, USA) were injected to separate the neurosensory retina from RPE. The scleral wound was sealed by Webglue (Patterson Companies, Mendota Heights, MN, USA). Antibiotic ointment (Bacitracin Zinc Ointment; Fougera Pharmaceuticals Inc, Melville, NY, USA) was applied followed by analgesia. Mice were kept on a heating pad with careful monitoring until fully awake.
NMN administration and experimental groups
We administrated NMN (Bontac Bio-engineering (Shenzhen) CO.,LTD) at doses of 250 mg/kg/d and 500 mg/kg/d by diluting in sterile PBS for our study based on the effective dose in previous studies ranges from 62.5 mg/kg to 500 mg/kg by intraperitoneal injections [23, 65–67].
Mice were divided into the following groups: normal attached control group (Att), RD with PBS injection group (Vehicle), RD with 250 mg/kg NMN group and RD with 500 mg/kg NMN group. On the day of surgery, mice were undergoing the induction of RD followed by NMN or vehicle injections half an hour later. Daily injections of NMN or vehicle were then performed at the end of the light phase. Retina tissues were harvested at 3 time points: 24 hours, 3 days and 7 days post RD. A separate group of mice received NMN administration at Day 3 post RD, and then with daily injections of NMN till the Day 7 post RD.
Hematoxylin and eosin (HE) staining
Sections were prefixed in 4% paraformaldehyde (PFA) and then stained with Gill’s I hematoxylin (Sigma) and eosin (Sigma).
TUNEL assay and normalized ONL thickness ratio
A TdT-dUTP terminal nick-end labeling (TUNEL) assay was performed following the manufacturer’s protocol (ApopTag Fluorescein In Situ Apoptosis Detection Kit; MilliporeSigma, Burlington, MA, USA). Pictures were taken with an upright AXIO Imager.M2 Zeiss fluorescence microscope under a 10×/0.3 lens (Zeiss EC-PLAN NEOFLUAR, Carl Zeiss Inc., Thornwood, NY, USA). TUNEL positive cells were quantified by using the Image J software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD, USA). Any samples with hemorrhage, cataract, or shrunk part of the retina were excluded.
The thickness ratio of the ONL and corresponding inner nuclear layer (INL) can be measured in DAPI stained slides in the detached and attached part of the retina by masked observers. The ‘‘normalized ONL thickness ratio’’ is defined as the (ONL thickness/neuroretina thickness in the detached retina)/(ONL thickness/neuroretina thickness in the attached retina) as previously described [13].
Immunofluorescence
Sections were fixed in 4% paraformaldehyde (PFA), blocked with 0.5% bovine serum albumin (BSA) with 0.3% Triton X-100 in PBS, and incubated overnight at 4 ° C with anti-CD11b antibody (BD Pharmingen, BD Biosciences, San Jose, CA, USA) and anti-GFAP antibody (Proteintech, Rosemont, IL, USA). The secondary antibodies used in the study were Alexa-Fluor 647 goat anti-rat antibody and Alexa-Fluor 488 goat anti-rabbit antibody (Molecular Probes, Thermo Fisher Scientific, Waltham, MA, USA). Finally, sections were counterstained with DAPI (AnaSpec/Eurogentec Group) and mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL, USA).
The total number of CD11b+ cells in the subretinal space was counted, and the area of subretinal space was measured. The results were expressed as: CD11b+ cells (cells/mm2) = total cell number/area of subretinal space.
The glial fibrillary acidic protein (GFAP) is a sign of the reactive gliosis including the astrocytes and Müller cells. [68, 69]. We measured the area of GFAP and the total area from ILM to OLM in each section. The results were expressed as: GFAP ratio = (GFAP area /total area of ILM to OLM in the detached retina)/(GFAP area /total area of ILM to OLM in the attached retina).
NAD+ level measurement
NAD+ levels were determined following the instructions of a commercial NAD/NADH Quantification Kit (Sigma, St. Louis, MO, USA).
Protein carbonyl content (PCC)
The amount of PCC in the retina was measured by using a protein carbonyl ELISA kit (Oxiselect; Cell Biolabs, San Diego, CA) according to the manufacturer’s instructions.
Cell culture and treatment
The photoreceptor-derived cell line 661W was kindly provided by Dr Miayyad Al-Ubaidi (University of Houston, TX, USA) [70] and was maintained at 37° C under a humidified atmosphere of 5% CO2.
The oxidative stress model was induced by tert-Butyl hydroperoxide (tBuOOH, ACROS Organics, Geel, Belgium). The cell viability tests were performed in 10 mM tBuOOH to evaluate the following conditions: 1 mM NMN administration, SRT2104 administration, or SIRT1 siRNA transfection with NMN administration. Protein expressions in Figure 5 were examined by western blots after treatment with 5 mM tBuOOH.
In the macrophage CM toxicity model, RAW264.7 cells were cultured and stimulated with or without LPS. The CM samples were collected to culture 661W. Cell viability tests were performed to determine the effects of NMN or SRT1720 administration. Besides, NMN or SRT1720 was also administrated in the RAW264.7 cells with or without LPS stimulation, CM was collected to culture 661W, and the cell viability was tested under these conditions.
Gene silencing by siRNA transfection
Transfection of siRNAs was performed by mixing Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific, Waltham, MA USA) with SIRT1 siRNAs according to the manufacturer’s instruction. A scramble siRNA (sc siRNA) was used separately as a negative control. All siRNAs were purchased from Origene (Rockville, MD, USA).
Cell viability test
We used the Cell Counting Kit (CCK-8, Dojindo, Rockville, Maryland, USA) to perform the cell viability tests according to the manufacturer’s instructions. Briefly, after the treatment, the medium was replaced with fresh culture medium containing 10% (v/v) CCK-8 reagent. The absorption at 450 nm was measured with multiple times using a SpectraMax 190 microplate spectrophotometer (Molecular Device, San Jose, CA). Each experiment has been replicated for three or more times.
Enzyme-linked immunosorbent assay (ELISA)
The expression levels of TNF-α and IL-1β were measured with Mouse TNF-α (R&D Systems, Inc., Minneapolis, MN) and mouse IL-1β/IL-1F2 (R&D Systems, Inc.) ELISA kits, according to the manufacturer’s protocol.
Western blot analysis
After euthanasia, eyes were enucleated and retinas were extracted and homogenized in ice-cold mammalian protein extraction reagent (T-PER, Thermo Fisher, Waltham, MA) with protease/phosphatase inhibitor cocktail (Cell Signaling, Danvers, MA). Protein concentration was measured using a Pierce Coomassie (Bradford) protein assay. Western blots were performed with incubation of primary antibodies: HO-1 and SIRT1 from Proteintech followed by secondary antibodies. The results were visualized with HRP substrate reagent (Genetex, Irvine, CA) and detected with an ECL imaging system ChemiDoc MP (Bio-Rad Laboratories, Hercules, CA).
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis between two groups was performed using an unpaired Student’s t-test. Multiple groups were analyzed by ANOVA followed by Turkey-Kramer adjustments. The significance levels were defined as P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***). Graphs were plotted with Prism GraphPad Prism 7 (La Jolla, CA, USA).
Supplementary Materials
Author Contributions
X.C., J.A., D.G.V. and D.A.S. designed the experiments, X.C., G.A.M., J.L. Z.Y., and K.I. conducted the experiments, X.C., J.A., G.A.M., and J.L. performed data analysis, X.C. and J.A. wrote the manuscript. All authors contributed to the edits of the manuscript.
Acknowledgments
The authors would like to thank the help of the animal facility of Massachusetts Eye and Ear.
Conflicts of Interest
D. A. S. is a founder, equity owner, advisor, director, consultant, investor, and inventor on patents licensed to MetroBiotech, Liberty Biosecurity, Jumpstart Fertility, and Animal Biosciences, working on NAD-based therapeutics. D.A.S. is an inventor on a patent application filed by Harvard Medical School licensed to Elysium Health. https://genetics.med.harvard.edu/sinclair/people/sinclair-other.php. All other authors declare no competing interests.
Funding
This work was supported by the Oversea Study Program of Guangzhou Elite Project for scholarship (to X.C), the Yeatts Family Foundation (to D.G.V.); Monte J. Wallace (to D.G.V.); 2013 Macula Society Research Grant Award (to D.G.V.); a Physician Scientist Award (to D.G.V.); unrestricted grant from the Research to Prevent Blindness Foundation (to J.W.M. and D.G.V.); National Eye Institute (NEI) R21EY023079-01/A1 R01EY025362-01 (to D.G.V.); NEI Core Grant EY014104, P30EY003790 (Massachusetts Eye and Ear Infirmary Core Grant.); R01-DK- 100263-01A1 and R37AG028730- 08 (to D.A.S.); Loeffler Family Fund (to D.G.V.); ARI Young Investigator Award (to D.G.V.).
References
- 1. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, Wong TY. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014; 2:e106–16. https://doi.org/10.1016/S2214-109X(13)70145-1 [PubMed]
- 2. Zheng Y, He M, Congdon N. The worldwide epidemic of diabetic retinopathy. Indian J Ophthalmol. 2012; 60:428–31. https://doi.org/10.4103/0301-4738.100542 [PubMed]
- 3. Duh EJ, Sun JK, Stitt AW. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight. 2017; 2:e93751. https://doi.org/10.1172/jci.insight.93751 [PubMed]
- 4. Wubben TJ, Besirli CG, Zacks DN. Pharmacotherapies for retinal detachment. Ophthalmology. 2016; 123:1553–62. https://doi.org/10.1016/j.ophtha.2016.02.040 [PubMed]
- 5. Murakami Y, Notomi S, Hisatomi T, Nakazawa T, Ishibashi T, Miller JW, Vavvas DG. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog Retin Eye Res. 2013; 37:114–40. https://doi.org/10.1016/j.preteyeres.2013.08.001 [PubMed]
- 6. Bressler NM, Beaulieu WT, Bressler SB, Glassman AR, Melia BM, Jampol LM, Jhaveri CD, Salehi-Had H, Velez G, Sun JK; DRCR Retina Network. Anti-vascular endothelial growth factor therapy and risk of traction retinal detachment in eyes with proliferative diabetic retinopathy: Pooled Analysis of Five DRCR Retina Network Randomized Clinical Trials. Retina. 2020; 40:1021–1028. https://doi.org/10.1097/IAE.0000000000002633 [PubMed]
- 7. Stewart MW, Browning DJ, Landers MB. Current management of diabetic tractional retinal detachments. Indian J Ophthalmol. 2018; 66:1751–62. https://doi.org/10.4103/ijo.IJO_1217_18 [PubMed]
- 8. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006; 1:40. https://doi.org/10.1186/1750-1172-1-40 [PubMed]
- 9. Ferrington DA, Fisher CR, Kowluru RA. Mitochondrial defects drive degenerative retinal diseases. Trends Mol Med. 2020; 26:105–18. https://doi.org/10.1016/j.molmed.2019.10.008 [PubMed]
- 10. Wong-Riley MT. Energy metabolism of the visual system. Eye Brain. 2010; 2:99–116. https://doi.org/10.2147/EB.S9078 [PubMed]
- 11. Erickson PA, Fisher SK, Anderson DH, Stern WH, Borgula GA. Retinal detachment in the cat: the outer nuclear and outer plexiform layers. Invest Ophthalmol Vis Sci. 1983; 24:927–42. [PubMed]
- 12. Nakazawa T, Matsubara A, Noda K, Hisatomi T, She H, Skondra D, Miyahara S, Sobrin L, Thomas KL, Chen DF, Grosskreutz CL, Hafezi-Moghadam A, Miller JW. Characterization of cytokine responses to retinal detachment in rats. Mol Vis. 2006; 12:867–78. [PubMed]
- 13. Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA. 2010; 107:21695–700. https://doi.org/10.1073/pnas.1009179107 [PubMed]
- 14. Cook B, Lewis GP, Fisher SK, Adler R. Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest Ophthalmol Vis Sci. 1995; 36:990–96. [PubMed]
- 15. Zacks DN, Hänninen V, Pantcheva M, Ezra E, Grosskreutz C, Miller JW. Caspase activation in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci. 2003; 44:1262–67. https://doi.org/10.1167/iovs.02-0492 [PubMed]
- 16. Roh MI, Murakami Y, Thanos A, Vavvas DG, Miller JW. Edaravone, an ROS scavenger, ameliorates photoreceptor cell death after experimental retinal detachment. Invest Ophthalmol Vis Sci. 2011; 52:3825–31. https://doi.org/10.1167/iovs.10-6797 [PubMed]
- 17. Yoshimura T, Sonoda KH, Sugahara M, Mochizuki Y, Enaida H, Oshima Y, Ueno A, Hata Y, Yoshida H, Ishibashi T. Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS One. 2009; 4:e8158. https://doi.org/10.1371/journal.pone.0008158 [PubMed]
- 18. Nakazawa T, Hisatomi T, Nakazawa C, Noda K, Maruyama K, She H, Matsubara A, Miyahara S, Nakao S, Yin Y, Benowitz L, Hafezi-Moghadam A, Miller JW. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci USA. 2007; 104:2425–30. https://doi.org/10.1073/pnas.0608167104 [PubMed]
- 19. Nakazawa T, Kayama M, Ryu M, Kunikata H, Watanabe R, Yasuda M, Kinugawa J, Vavvas D, Miller JW. Tumor necrosis factor-alpha mediates photoreceptor death in a rodent model of retinal detachment. Invest Ophthalmol Vis Sci. 2011; 52:1384–91. https://doi.org/10.1167/iovs.10-6509 [PubMed]
- 20. Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018; 27:529–47. https://doi.org/10.1016/j.cmet.2018.02.011 [PubMed]
- 21. Herskovits AZ, Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013; 23:746–58. https://doi.org/10.1038/cr.2013.70 [PubMed]
- 22. Yoshino J, Baur JA, Imai SI. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018; 27:513–28. https://doi.org/10.1016/j.cmet.2017.11.002 [PubMed]
- 23. Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, Nakamura R, Zapata N, Kubota M, Tsubota K, Yoshino J, Imai SI, Apte RS. NAMPT-mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep. 2016; 17:69–85. https://doi.org/10.1016/j.celrep.2016.08.073 [PubMed]
- 24. Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, Schwartzentruber J, Solanki N, Traboulsi EI, et al, and Finding of Rare Disease Genes (FORGE) Canada Consortium. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012; 44:1035–39. https://doi.org/10.1038/ng.2356 [PubMed]
- 25. Perrault I, Hanein S, Zanlonghi X, Serre V, Nicouleau M, Defoort-Delhemmes S, Delphin N, Fares-Taie L, Gerber S, Xerri O, Edelson C, Goldenberg A, Duncombe A, et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat Genet. 2012; 44:975–77. https://doi.org/10.1038/ng.2357 [PubMed]
- 26. Bai S, Sheline CT. NAD(+) maintenance attenuates light induced photoreceptor degeneration. Exp Eye Res. 2013; 108:76–83. https://doi.org/10.1016/j.exer.2012.12.007 [PubMed]
- 27. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010; 5:253–95. https://doi.org/10.1146/annurev.pathol.4.110807.092250 [PubMed]
- 28. Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013 (Suppl 3); 15:26–33. https://doi.org/10.1111/dom.12171 [PubMed]
- 29. Han J, Hubbard BP, Lee J, Montagna C, Lee HW, Sinclair DA, Suh Y. Analysis of 41 cancer cell lines reveals excessive allelic loss and novel mutations in the SIRT1 gene. Cell Cycle. 2013; 12:263–70. https://doi.org/10.4161/cc.23056 [PubMed]
- 30. Mimura T, Kaji Y, Noma H, Funatsu H, Okamoto S. The role of SIRT1 in ocular aging. Exp Eye Res. 2013; 116:17–26. https://doi.org/10.1016/j.exer.2013.07.017 [PubMed]
- 31. Zeng Y, Yang K. Sirtuin 1 participates in the process of age-related retinal degeneration. Biochem Biophys Res Commun. 2015; 468:167–72. https://doi.org/10.1016/j.bbrc.2015.10.139 [PubMed]
- 32. Kowluru RA, Santos JM, Zhong Q. Sirt1, a negative regulator of matrix metalloproteinase-9 in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2014; 55:5653–60. https://doi.org/10.1167/iovs.14-14383 [PubMed]
- 33. Zheng Z, Chen H, Li J, Li T, Zheng B, Zheng Y, Jin H, He Y, Gu Q, Xu X. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes. 2012; 61:217–28. https://doi.org/10.2337/db11-0416 [PubMed]
- 34. Kubota S, Kurihara T, Ebinuma M, Kubota M, Yuki K, Sasaki M, Noda K, Ozawa Y, Oike Y, Ishida S, Tsubota K. Resveratrol prevents light-induced retinal degeneration via suppressing activator protein-1 activation. Am J Pathol. 2010; 177:1725–31. https://doi.org/10.2353/ajpath.2010.100098 [PubMed]
- 35. Chen J, Michan S, Juan AM, Hurst CG, Hatton CJ, Pei DT, Joyal JS, Evans LP, Cui Z, Stahl A, Sapieha P, Sinclair DA, Smith LE. Neuronal sirtuin1 mediates retinal vascular regeneration in oxygen-induced ischemic retinopathy. Angiogenesis. 2013; 16:985–92. https://doi.org/10.1007/s10456-013-9374-5 [PubMed]
- 36. Ke B, Shen XD, Gao F, Ji H, Qiao B, Zhai Y, Farmer DG, Busuttil RW, Kupiec-Weglinski JW. Adoptive transfer of ex vivo HO-1 modified bone marrow-derived macrophages prevents liver ischemia and reperfusion injury. Mol Ther. 2010; 18:1019–25. https://doi.org/10.1038/mt.2009.285 [PubMed]
- 37. Huang J, Shen XD, Yue S, Zhu J, Gao F, Zhai Y, Busuttil RW, Ke B, Kupiec-Weglinski JW. Adoptive transfer of heme oxygenase-1 (HO-1)-modified macrophages rescues the nuclear factor erythroid 2-related factor (Nrf2) antiinflammatory phenotype in liver ischemia/reperfusion injury. Mol Med. 2014; 20:448–55. https://doi.org/10.2119/molmed.2014.00103 [PubMed]
- 38. Meng X, Yuan Y, Shen F, Li C. Heme oxygenase-1 ameliorates hypoxia/reoxygenation via suppressing apoptosis and enhancing autophagy and cell proliferation though Sirt3 signaling pathway in H9c2 cells. Naunyn Schmiedebergs Arch Pharmacol. 2019; 392:189–98. https://doi.org/10.1007/s00210-018-1575-4 [PubMed]
- 39. Nakamura K, Zhang M, Kageyama S, Ke B, Fujii T, Sosa RA, Reed EF, Datta N, Zarrinpar A, Busuttil RW, Araujo JA, Kupiec-Weglinski JW. Macrophage heme oxygenase-1-SIRT1-p53 axis regulates sterile inflammation in liver ischemia-reperfusion injury. J Hepatol. 2017; 67:1232–42. https://doi.org/10.1016/j.jhep.2017.08.010 [PubMed]
- 40. Shyong MP, Lee FL, Hen WH, Kuo PC, Wu AC, Cheng HC, Chen SL, Tung TH, Tsao YP. Viral delivery of heme oxygenase-1 attenuates photoreceptor apoptosis in an experimental model of retinal detachment. Vision Res. 2008; 48:2394–402. https://doi.org/10.1016/j.visres.2008.07.017 [PubMed]
- 41. Narayanan SV, Dave KR, Saul I, Perez-Pinzon MA. Resveratrol preconditioning protects against cerebral ischemic injury via nuclear erythroid 2-related factor 2. Stroke. 2015; 46:1626–32. https://doi.org/10.1161/STROKEAHA.115.008921 [PubMed]
- 42. Xue F, Huang JW, Ding PY, Zang HG, Kou ZJ, Li T, Fan J, Peng ZW, Yan WJ. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav Brain Res. 2016; 309:1–8. https://doi.org/10.1016/j.bbr.2016.04.045 [PubMed]
- 43. Harlan BA, Pehar M, Killoy KM, Vargas MR. Enhanced SIRT6 activity abrogates the neurotoxic phenotype of astrocytes expressing ALS-linked mutant SOD1. FASEB J. 2019; 33:7084–91. https://doi.org/10.1096/fj.201802752R [PubMed]
- 44. Jaliffa C, Ameqrane I, Dansault A, Leemput J, Vieira V, Lacassagne E, Provost A, Bigot K, Masson C, Menasche M, Abitbol M. Sirt1 involvement in rd10 mouse retinal degeneration. Invest Ophthalmol Vis Sci. 2009; 50:3562–72. https://doi.org/10.1167/iovs.08-2817 [PubMed]
- 45. Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. 2003; 100:10794–99. https://doi.org/10.1073/pnas.1934713100 [PubMed]
- 46. Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014; 33:1321–40. https://doi.org/10.1002/embj.201386917 [PubMed]
- 47. Song W, Song Y, Kincaid B, Bossy B, Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1α. Neurobiol Dis. 2013; 51:72–81. https://doi.org/10.1016/j.nbd.2012.07.004 [PubMed]
- 48. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, Chang HY, Bohr VA, Ried T, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008; 452:492–96. https://doi.org/10.1038/nature06736 [PubMed]
- 49. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006; 124:315–29. https://doi.org/10.1016/j.cell.2005.11.044 [PubMed]
- 50. Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008; 102:703–10. https://doi.org/10.1161/CIRCRESAHA.107.164558 [PubMed]
- 51. Yang X, Park SH, Chang HC, Shapiro JS, Vassilopoulos A, Sawicki KT, Chen C, Shang M, Burridge PW, Epting CL, Wilsbacher LD, Jenkitkasemwong S, Knutson M, et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J Clin Invest. 2017; 127:1505–16. https://doi.org/10.1172/JCI88574 [PubMed]
- 52. Cao W, Hong Y, Chen H, Wu F, Wei X, Ying W. SIRT2 mediates NADH-induced increases in Nrf2, GCL, and glutathione by modulating Akt phosphorylation in PC12 cells. FEBS Lett. 2016; 590:2241–55. https://doi.org/10.1002/1873-3468.12236 [PubMed]
- 53. Nguyen GT, Gertz M, Steegborn C. Crystal structures of Sirt3 complexes with 4'-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem Biol. 2013; 20:1375–85. https://doi.org/10.1016/j.chembiol.2013.09.019 [PubMed]
- 54. Sanz A, Soikkeli M, Portero-Otín M, Wilson A, Kemppainen E, McIlroy G, Ellilä S, Kemppainen KK, Tuomela T, Lakanmaa M, Kiviranta E, Stefanatos R, Dufour E, et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc Natl Acad Sci USA. 2010; 107:9105–10. https://doi.org/10.1073/pnas.0911539107 [PubMed]
- 55. Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 2002; 368:545–53. https://doi.org/10.1042/BJ20021121 [PubMed]
- 56. Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003; 86:1101–07. https://doi.org/10.1046/j.1471-4159.2003.01908.x [PubMed]
- 57. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014; 94:909–50. https://doi.org/10.1152/physrev.00026.2013 [PubMed]
- 58. Nimmagadda VK, Bever CT, Vattikunta NR, Talat S, Ahmad V, Nagalla NK, Trisler D, Judge SI, Royal W
3rd , Chandrasekaran K, Russell JW, Makar TK. Overexpression of SIRT1 protein in neurons protects against experimental autoimmune encephalomyelitis through activation of multiple SIRT1 targets. J Immunol. 2013; 190:4595–607. https://doi.org/10.4049/jimmunol.1202584 [PubMed] - 59. Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011; 14:528–36. https://doi.org/10.1016/j.cmet.2011.08.014 [PubMed]
- 60. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013; 25:1939–48. https://doi.org/10.1016/j.cellsig.2013.06.007 [PubMed]
- 61. Cameron AM, Castoldi A, Sanin DE, Flachsmann LJ, Field CS, Puleston DJ, Kyle RL, Patterson AE, Hässler F, Buescher JM, Kelly B, Pearce EL, Pearce EJ. Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat Immunol. 2019; 20:420–32. https://doi.org/10.1038/s41590-019-0336-y [PubMed]
- 62. Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, Contrepois K, Wang Q, Lee BA, Coronado M, Bernstein D, Snyder MP, Migaud M, et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat Immunol. 2019; 20:50–63. https://doi.org/10.1038/s41590-018-0255-3 [PubMed]
- 63. Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J, Imai SI. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016; 24:795–806. https://doi.org/10.1016/j.cmet.2016.09.013 [PubMed]
- 64. Matsumoto H, Miller JW, Vavvas DG. Retinal detachment model in rodents by subretinal injection of sodium hyaluronate. J Vis Exp. 2013; 50660. https://doi.org/10.3791/50660 [PubMed]
- 65. Park JH, Long A, Owens K, Kristian T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol Dis. 2016; 95:102–10. https://doi.org/10.1016/j.nbd.2016.07.018 [PubMed]
- 66. Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, Schuh RA. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015; 15:19. https://doi.org/10.1186/s12883-015-0272-x [PubMed]
- 67. Uddin GM, Youngson NA, Doyle BM, Sinclair DA, Morris MJ. Nicotinamide mononucleotide (NMN) supplementation ameliorates the impact of maternal obesity in mice: comparison with exercise. Sci Rep. 2017; 7:15063. https://doi.org/10.1038/s41598-017-14866-z [PubMed]
- 68. Zhao TT, Tian CY, Yin ZQ. Activation of Müller cells occurs during retinal degeneration in RCS rats. Adv Exp Med Biol. 2010; 664:575–83. https://doi.org/10.1007/978-1-4419-1399-9_66 [PubMed]
- 69. Grosche J, Härtig W, Reichenbach A. Expression of glial fibrillary acidic protein (GFAP), glutamine synthetase (GS), and Bcl-2 protooncogene protein by Müller (glial) cells in retinal light damage of rats. Neurosci Lett. 1995; 185:119–22. https://doi.org/10.1016/0304-3940(94)11239-f [PubMed]
- 70. Tan E, Ding XQ, Saadi A, Agarwal N, Naash MI, Al-Ubaidi MR. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Invest Ophthalmol Vis Sci. 2004; 45:764–68. https://doi.org/10.1167/iovs.03-1114 [PubMed]