



Introduction
Abdominal aortic aneurysm (AAA) is a fatal vascular disease defined as more than 50% arterial dilation of the abdominal aorta [1]. In recent years, the incidence of AAA has increased and it has become one of the top 10 causes of death among people older than 65 years. AAA is a result of the interaction by the environment, heredity and biochemistry and is characterized by chronic inflammation in the vascular wall [2]. Blood vessel wall damage caused by AAA could initiate the immune response, leading recruiting macrophages, T cells and mast cell infiltration to the vascular wall [3]. Then reactive oxygen species production, vascular smooth muscle cell (VSMC) apoptosis and matrix metalloproteinase (MMP) activation increases and leads to extracellular matrix degradation and remodeling, which is the main known pathogenesis factors of AAA [2, 4].
The general treatment of AAA includes classical open surgery and endovascular aneurysm repair. However, the survival rate and quality of life are not significantly improved by early surgical treatment [5–7]. There are still no effective clinical drugs to treat AAA [6] and exploring new pathogenesis factors to find novel therapeutic targets and drug treatment methods for AAA are crucial.
The Notch signal is highly expressed in invertebrates and vertebrates; it mainly consists of the Notch receptor and its ligand, delta-like canonical Notch ligand 4 (DLL4), which are both single transmembrane [8, 9]. When binding to its ligand, the Notch receptor is cut by a disintegrin and metalloproteinase (ADAM) protease and γ-secretase to release the Notch intracellular domain (NICD) with a nuclear positioning signal [10, 11]. The Notch1 signaling pathway plays an important role in regulating the development and transformation of various inflammatory cells such as macrophages and T lymphocytes, and it can modulate the immune response via nuclear factor κB (NF-κB) and phosphatidyl inositol 3-kinase/proteinkinase b (PI3K/Akt) pathways [12–14]. Notch1 signaling also plays a key role in diverse cardiovascular diseases such as valve calcification and left-sided congenital heart disease [8]. The inflammatory response mediated by Notch1 signaling pathway stabilizes the progression of small abdominal aortic aneurysm induced by angiotensin II (AngII) [14]. Furthermore, VSMC-specific Notch1 knockout could prevent AAA by regulating the contraction phenotype of VSMCs and maintaining extracellular matrix homeostasis [15]; therefore, Notch1 may have a pivotal role in AAA formation.
Recently, numerous studies, including our previous work, revealed that paracrine/autocrine factors play a vital role in AAA [16, 17, 27, 28]. For example, angiotensin-converting enzyme 2 [16] and epoxyeicosatrienoic acids [17] are both endogenous vasoactive factors that have benefit effect in suppressing AAA. A crucial vasoactive substance, intermedin (IMD), has been reported to exert important cardiovascular protection effects. IMD is a vasoactive peptide that belongs to the calcitonin gene-related peptide (CGRP) superfamily, discovered by Roh [18] and Takei [19] in 2004. The human IMD gene is located at the end of chromosome 22; it encodes a prepro-IMD consisting of 148 amino acid residues and generates 3 active fragments by proteolysis: IMD1-47, IMD8-47 and IMD1-53. IMD1-53 is likely the main active fragment of IMD [20]. Through the common receptor of CGRP and calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes (CRLR/RAMPs), IMD can increase intracellular cAMP content and participate in various biological effects such as myocardial contraction, vasodilation, regulation of cell proliferation, hypertrophy, migration and apoptosis [21–23]. IMD can also protect the endothelial barrier function and suppress local vascular inflammatory response [21]. In mammals, IMD attributes to protecting against cardiovascular disease such as atherosclerosis, myocardial ischemia-reperfusion injury, cardiac hypertrophy, vascular calcification and AAA [24–28]. Our previous work showed that IMD could restrain the formation of AAA by alleviating oxidative stress and endoplasmic reticulum stress [27, 28]. However, the mechanism of IMD inhibiting AAA has not been systematically studied.
In this study, we explored whether IMD can attenuate AAA by inhibiting Notch1 in AngII- and calcium chloride (CaCl2)-induced mouse models.
Results
Notch1 signaling was activated in murine and human AAA
Notch1 signaling participates in local inflammation in the vessel wall, and inhibition of Notch1 signaling reduced AAA in mice by attenuating macrophage-mediated inflammation [14]. First, we detected Notch1 signaling markers in AngII-induced AAA mouse models. The mRNA levels of Notch1 and its ligand DLL4 were higher by 12.3- and 16.7- fold (both P<0.05) with than without AngII treatment. Meanwhile, Notch1 signaling target genes hes1 and hey1 were increased 30.5- and 5.92-fold (both P<0.05) with AngII treatment to induce AAA (Figure 1A). The protein levels of hes1 and the Notch1 active fragment NICD were increased by 76.4% and 67.1% (both P<0.05) with AAA (Figure 1B, 1C). Immunohistochemistry results were consistent with western blot findings, and hes1 and NICD protein were mainly located in vascular adventitia (Figure 1D, 1E). Furthermore, levels of Notch1 signaling markers NICD, hes1 and inducible nitric oxide synthase (iNOS) were obviously higher in human AAA aortas than in normal aortas (Figure 2A–2C).
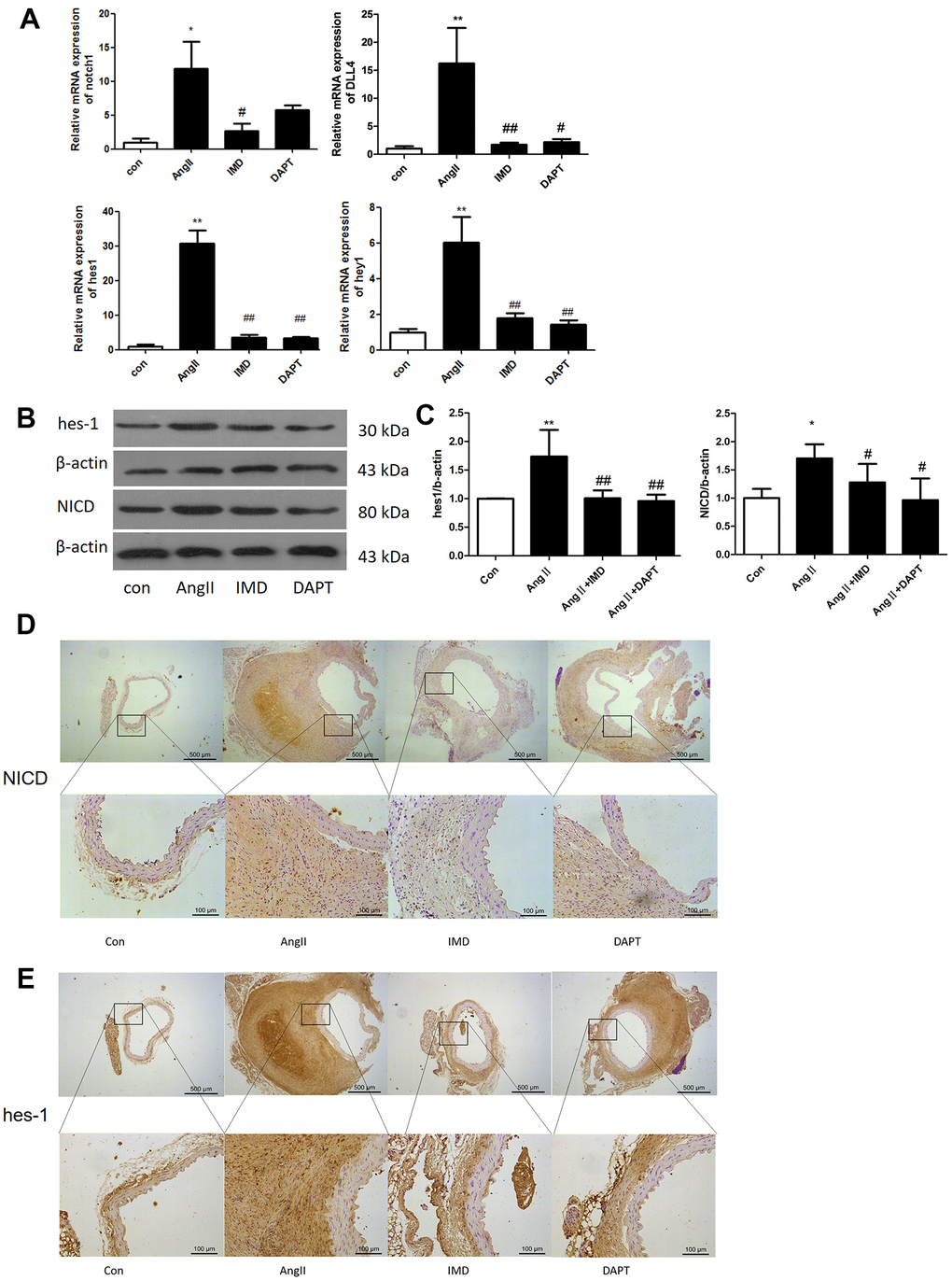
Figure 1. Notch1 signaling was activated in murine AAA. (A) Quantitative real-time PCR of Notch1, DLL4, hes1 and hey1 mRNA expression in aortas of mice with control (saline), AngII, AngII plus IMD and AngII plus DAPT. (B) Western blot analysis of hes1 and Notch1 signaling pathway marker NICD in aortas with Control, AngII, AngII+IMD and AngII+DAPT treatment. β-actin was a control for protein loading. Results are from 1 representative experiment of 3. (C) Quantification is shown as a ratio of β-actin expression. (D, E) Immunohistochemistry of the protein expression of NICD and hes1 in aortas of mice. Scale bar, 500 μm, 100 μm. Boxes and arrows show enlarged areas. Data are mean ± SD. *P<0.05, **P<0.01 vs. Control. #P<0.05, ##P<0.01 vs. AngII.
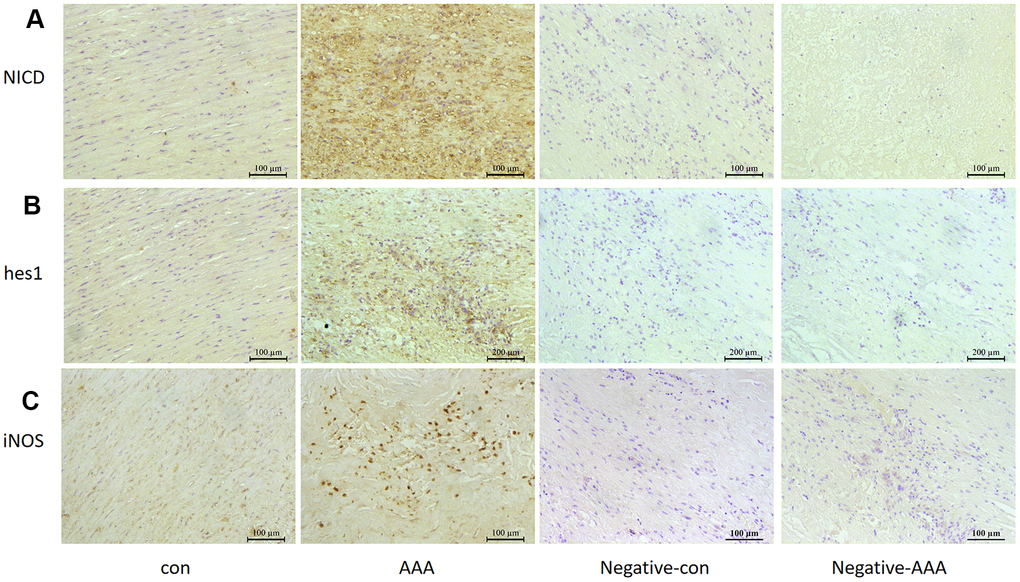
Figure 2. Notch signaling and M1 polarization were activated in human aorta of AAA. Control, Patients with non-abdominal aortic aneurysm; AAA, Patients with abdominal aortic aneurysm; Negative-con, Negative control group of patients with non-abdominal aortic aneurysm; Negative-AAA, Negative control group of patients with abdominal aortic aneurysm. Immunohistochemistry of (A, B) the protein expression of NICD and hes1 in patient aortas and (C) the protein expression of M1-type macrophage marker iNOS in patient aortas.
IMD alleviated AngII-induced AAA by blocking Notch1 signaling
Then, we tested the effects of IMD using AngII-induced AAA model. Negative control animals treated with saline did not show AAA features. The morbidity with AngII treatment was 75%, which was consistent with previous studies [28]. IMD treatment significantly reduced the occurrence of AAA to 46.2% (P<0.05) and N-[N-(3,5-difluorophenacetyl)-Lalany]-S-phenylglycine t-butyl ester (DAPT) (Notch1 pathway inhibitor) had a similar effect as IMD, with 53.3% AAA morbidity (P<0.05) (Figure 3A, 3B). The maximum abdominal aorta diameter of AngII-treated mice was markedly dilated as compared with saline treatment and was reversed with IMD and DAPT treatment (Figure 3B). AAA aortas showed intramural thrombus formation, severe aortic dilatation (Figure 3C), elastin degradation (Figure 3D) and increased MMP activity (Figure 3E), which were all alleviated with IMD infusion. DAPT also had a similar inhibitory effect as IMD.
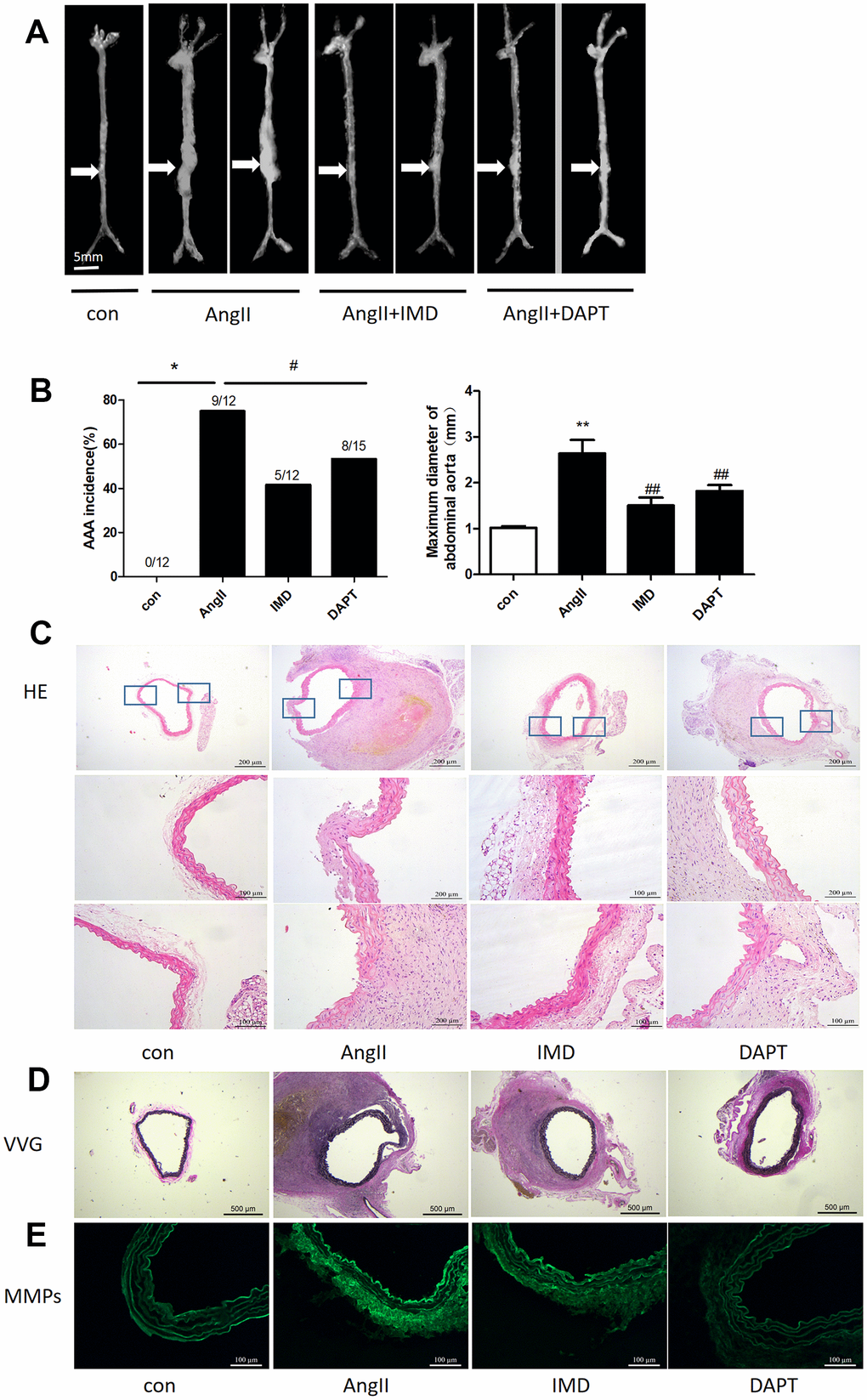
Figure 3. IMD alleviated AngII-induced AAA in mice by blocking Notch1 signaling. (A) Representative photographs of macroscopic features of AAA in male ApoE-/- mice. Control, saline infusion; AngII, AngII (1000 ng/kg/min) infusion; AngII+IMD (300 ng/kg/min) infusion; AngII+DAPT (10 mg/kg) gavage for 4 weeks. The arrows indicate typical AAA in ApoE-/- mice. Scale bars, 5 mm. (B) Incidence of AngII-induced AAA in animals and maximal abdominal aortic diameter of survivor mice at the end of 28 days. (C) Representative HE staining of aortas. Scale bar, 200 μm, 100 μm. (D) Representative VVG staining. Scale bar, 500 μm. (E) Representative in situ zymography of MMP activity in suprarenal aortas from ApoE-/- mice. Scale bar, 100 μm. Data are mean ± SD. *P<0.05, **P<0.01 vs. Control. #P<0.05, ##P<0.01 vs. AngII.
Next we explored whether IMD protected against AAA formation by inhibiting Notch1 signaling activation. Notch1 signaling markers were upregulated during AAA induction (Figure 1). After IMD treatment, the mRNA expression of Notch1, DLL4, hes1 and hey1 were decreased in abdominal aortas 81.3%, 86.8%, 91.2% and 70.1%, respectively (all P<0.05) (Figure 1A), as compared with AngII treatment. Protein levels of hes1 and NICD were reduced 45.6% and 29.6% (both P<0.05) as compared with AngII treatment (Figure 1B, 1C). Immunohistochemistry showed similar results (Figure 1D, 1E). DAPT had similar effects as IMD.
IMD inhibited macrophage-mediated inflammation by Notch1 signaling
Notch1 signaling-mediated macrophage inflammation plays a critical role in AAA pathogenesis [14]. Since activated Notch1 signaling markers were mainly located in vascular adventitia (Figure 1D, 1E), so we supposed that IMD could inhibit AAA by alleviating macrophage-mediated inflammation. First, we detected the expression of representative inflammatory factors in mouse aortas. The mRNA levels of IL-6, monocyte chemotactic protein-1 (MCP-1) and interferon (IFN-γ) were significantly elevated after AngII infusion; IMD and DAPT reversed their expression (Figure 4A). However, the expression of tumor necrosis factor-α (TNF-α) was not changed. It was reported that IL-6 and MCP-1 were the strongest inflammatory factors involved in macrophage migration [3], so we examined the infiltration of macrophages in the vascular wall. Immunofluorescence assay showed that the expression of CD68, a typical macrophage marker, was higher in the vascular wall with AngII than control treatment; IMD and DAPT remarkably reduced the CD68 fluorescence intensity (Figure 4B, 4D). In vitro, using wound-healing assay, lipopolysaccharide (LPS) treatment significantly increased the migration distance of peritoneal macrophages as compared with control treatment; IMD pretreatment could dose-dependently inhibit the migration of macrophages (Figure 4C, 4E).
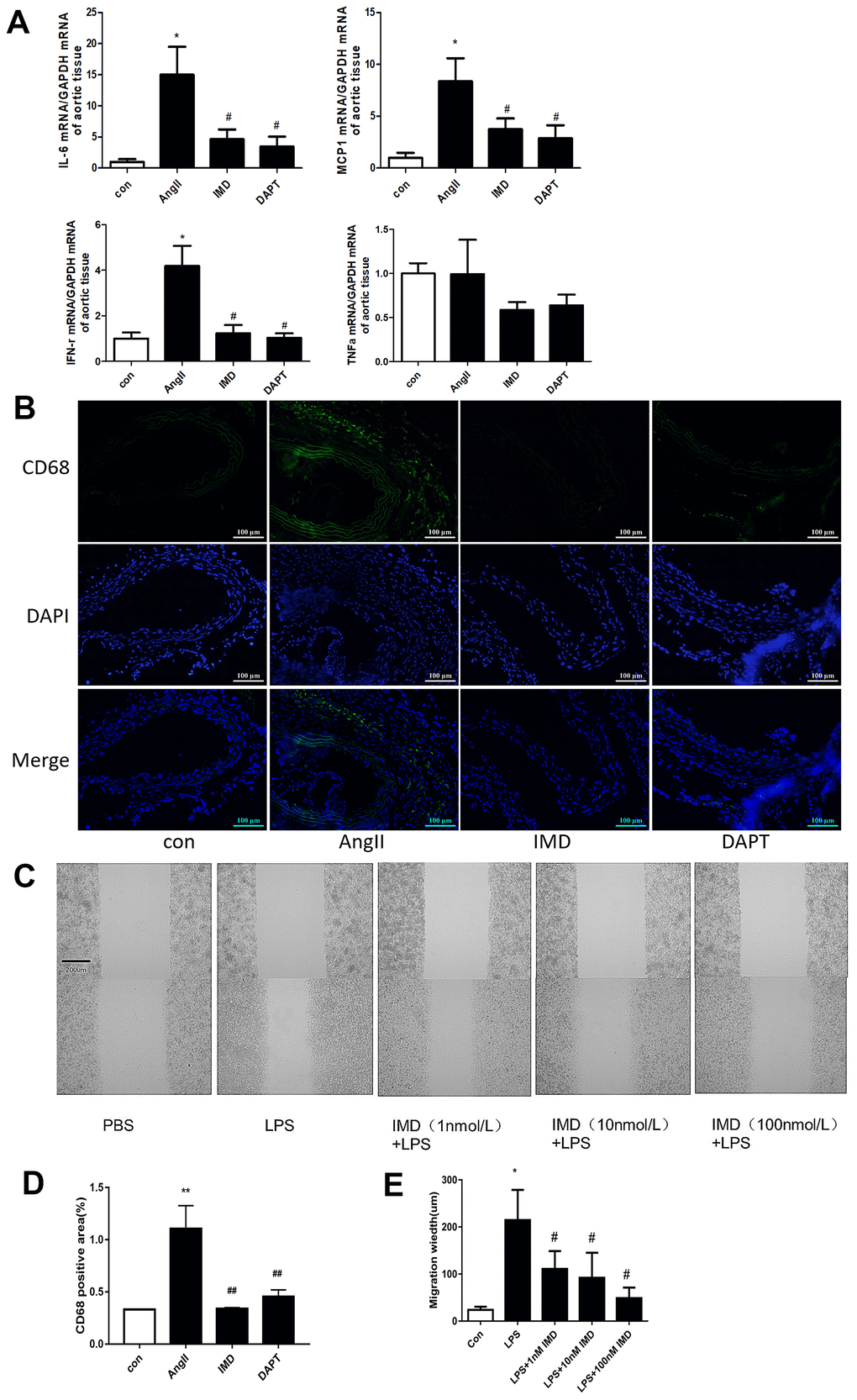
Figure 4. IMD inhibited macrophage-mediated inflammation via Notch1 signaling. (A) Quantitative real-time PCR of mRNA levels of IL-6, MCP-1, IFN-γ and TNF-α in aortas of Control, AngII, AngII+IMD and AngII+DAPT mice. (B) Representative immunofluorescence analysis of the protein expression of CD68 (green) and DAPI (blue) in aortas of mice. Scale bar, 100 μm. (C) Scratch-wound test to detect the transfer ability of mouse peritoneal macrophages in vitro. PBS, control group; LPS, stimulated group (100 ng/mL, 12 h). IMD was pretreated with a concentration gradient (1, 10, 100 nmol/L, 1 h). Cell migration distance was observed at 12 h after scratch wounding. Scale bar, 200 μm. n=3. (D) Quantification of (B). n=3, Data are mean ± SD. **P<0.01 vs. Control. ##P<0.01 vs. AngII. (E) Quantification of scratch-wound test. n=3, Data are mean ± SD. *P<0.05 vs. Control. #P<0.05 vs. LPS.
IMD inhibited NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation and M1 polarization via Notch1 signaling
NLRP3 inflammasome activation leads to production of mature IL-1β and IL-18, then recruitment of a large number of inflammatory cells to the vascular wall, which plays an important role in the initiation of inflammatory responses in the early stages of various diseases [29]. To investigate whether the protective effects of IMD against AAA formation are also mediated by NLRP3 inflammasome activation, we examined the apoptosis-associated speck-like protein containing CARD (ASC) expression. Immunohistochemical results showed that the expression of ASC in abdominal aortic tissues was increased in the AngII aneurysm group and reduced with IMD and DAPT treatment (Figure 5A). Next, we further explored the effect of IMD on NLRP3 inflammasome activation in a macrophage line Raw264.7. The protein level of NLRP3, ASC, caspase-1, IL-18, and IL-1β was upregulated during AAA formation, and IMD and DAPT inhibited NLRP3 inflammasome activation (Figure 5B–5D).
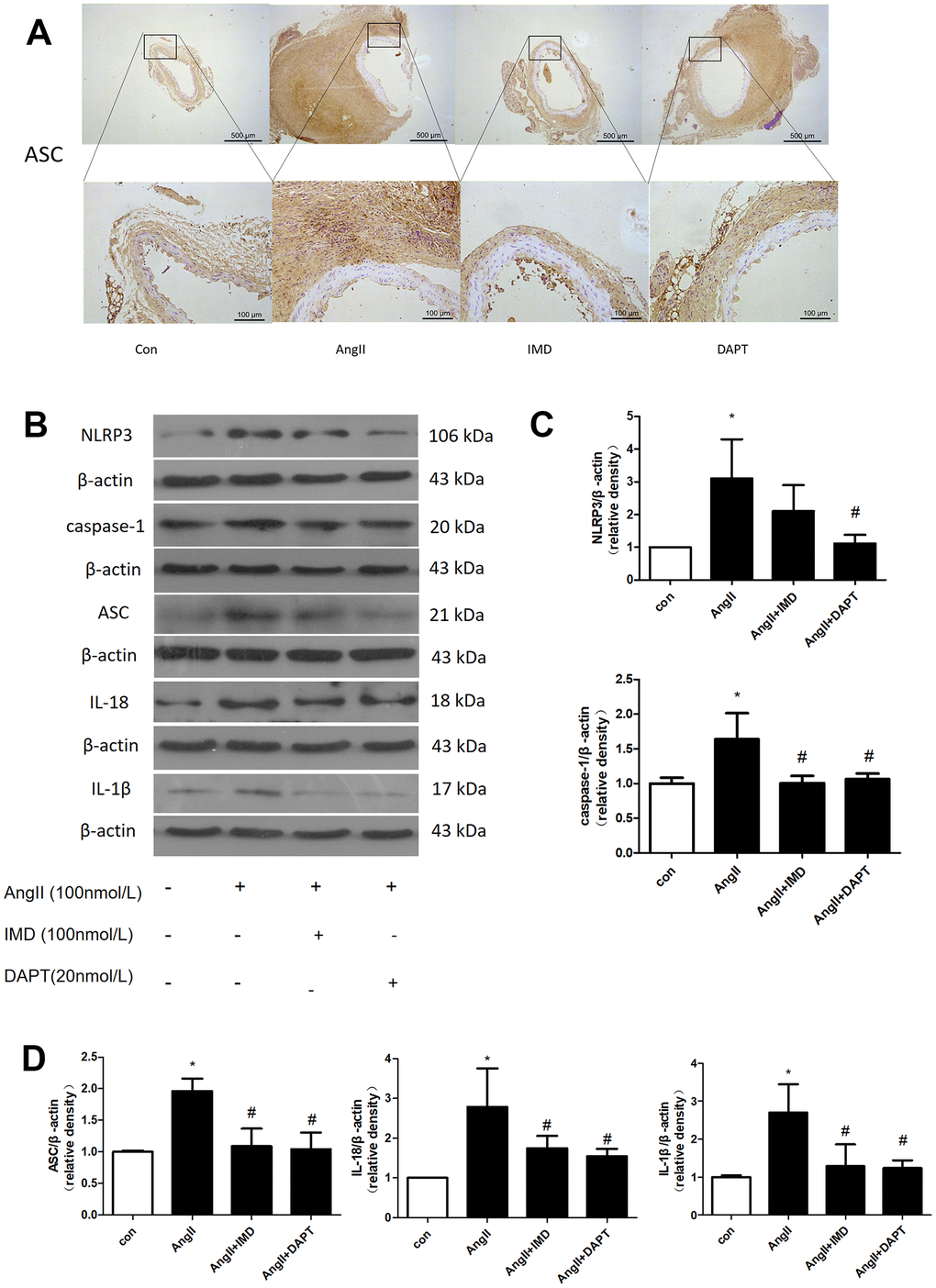
Figure 5. NLRP3 inflammasome activation was inhibited by IMD via Notch1 signaling. (A) Immunohistochemistry analysis of the protein expression of ASC in aortas of Control, AngII, AngII+IMD and AngII+DAPT mice. Control, saline infusion; AngII, AngII (1000 ng/kg/min) infusion; AngII+IMD (300 ng/kg/min) infusion; AngII+DAPT (10 mg/kg) gavage for 4 weeks. Scale bar, 500 μm, 100 μm. Boxes and arrows show enlarged areas. (B) Western blot analysis of protein levels of NLRP3, ASC, caspase-1, IL-1β, IL-18 in Raw264.7 macrophage cell line. The cell line was exposed to PBS, AngII (100 nm, 1 h) and DAPT (20 nm, 1 h). (C, D) Quantification of (B). n=3, Data are mean ± SD. *P<0.05 vs. Control. #P<0.05 vs. AngII.
Under different microenvironments, macrophages can be polarized to assume different phenotypes and show diverse functions. Generally, M1-type macrophages are considered to have pro-inflammatory phenotypes and M2-type macrophages anti-inflammatory phenotypes [30]. M1 and M2 macrophages polarization contribute to AAA formation [2, 3]. Therefore, we detected the effect of IMD on macrophage polarization in bone-marrow-derived macrophages (BMDMs). IFN-γ and interleukin-4 (IL-4) were used to induce BMDM M1 and M2 polarization, respectively. The mRNA expression of the M1 polarization markers IL-12, IL-1β, TNF-α and iNOS was greatly upregulated after IFN-γ treatment; IMD inhibited M1 polarization marker expression (Figure 6A). Similarly, IL-4 promoted the expression of the M2 polarization markers arginase 1 (Arg1), IL-10 and CD206, and IMD further facilitated their expression (Figure 6B). We further explored the protein level of M1 and M2 markers. IFN-γ increased the M1 marker protein levels of iNOS, IL-1β and CD16, and IMD suppressed the protein levels (Figure 6C, 6D). In contrast, IFN-γ suppressed the M2 protein levels of Arg1, IL-10 and CD206, and IMD restored the levels (Figure 6E, 6F). In mice AAA tissue, the M1 macrophage polarization marker iNOS was increased in Ang II group compared with control, IMD and DAPT could decrease the iNOS expression; M2 macrophage polarization marker Arg1 was activated in Ang II group, which increased more in IMD and DAPT group (Supplementary Figure 2A, 2B).
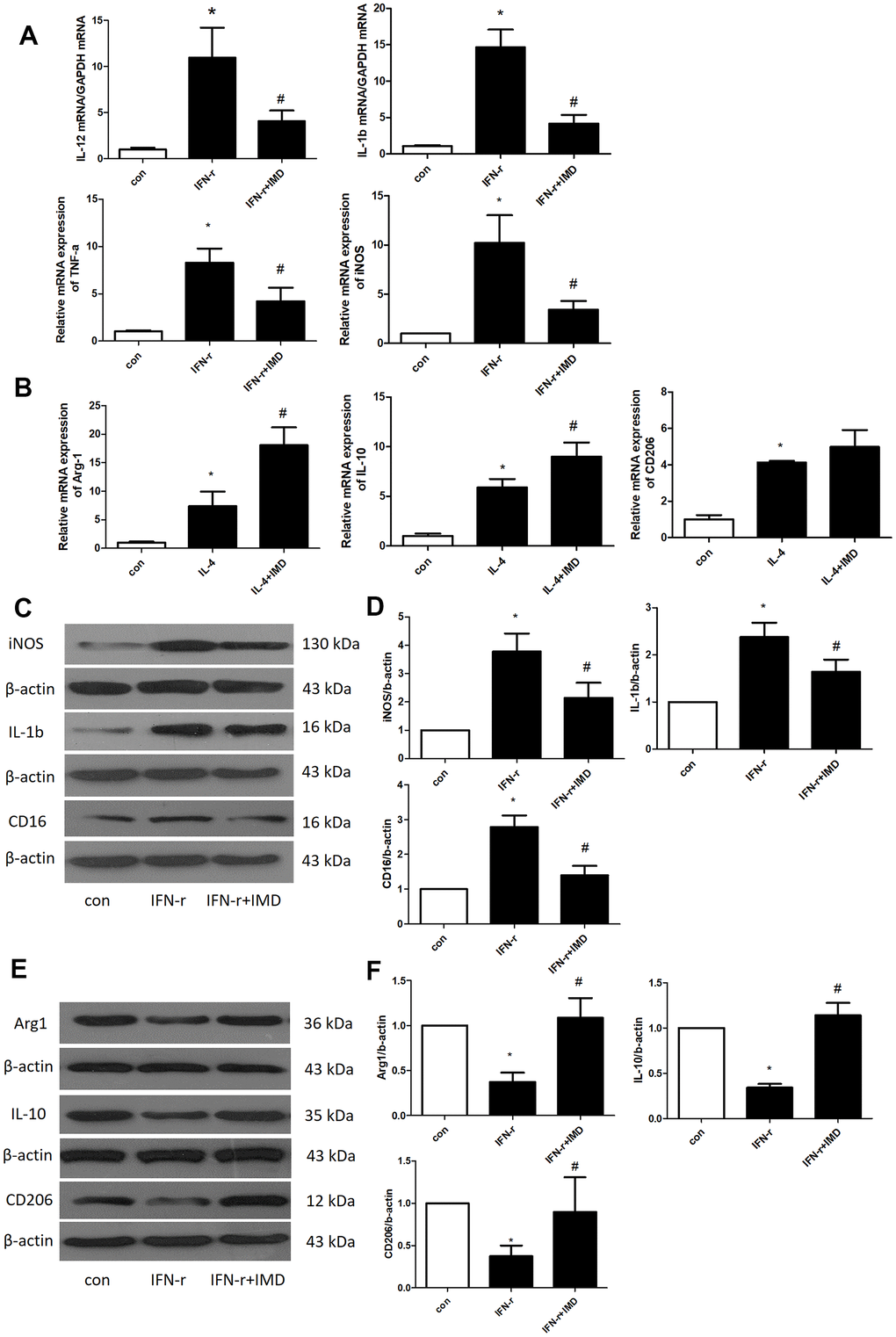
Figure 6. IMD inhibited macrophage M1 polarization. Control, saline treatment group; LPS+ IFN-γ, M1-type macrophage induction group; IFN-γ+IMD, M1-type macrophages pretreated with IMD; IL-4, M2-type macrophage induction group; IL-4+IMD, M2-type macrophages pretreated with IMD. Quantitative real-time PCR analysis of mRNA levels of (A) M1-type macrophage marker IL-12, IL-1β, TNF-α and iNOS in BMDMs and (B) M2-type macrophage marker Arg1, IL-10 and CD206 in BMDMs. Western blot analysis of (C, D) M1-type macrophage markers IL-12, IL-1β and CD16 and (E, F) M2-type macrophage markers Arg1, IL-10 and CD206 in BMDMs. β-actin was a control for protein loading. n=3, Data are mean ± SD. *P<0.05 vs. Control. #P<0.05 vs. IFN-γ.
IMD restrained Notch1 signaling activation by suppressing the expression of ADAM10
Next we explored the molecular mechanism of IMD inhibiting Notch1 signaling activation. ADAM10 and ADAM17 are key proteases during Notch1 signaling pathway activation [11]. In AngII-induced mice abdominal aortas, the mRNA expression of ADAM10 and ADAM17 was increased by 11.9- and 15.1-fold (both P<0.05). IMD decreased ADAM10 and ADAM17 mRNA expression by 68.7% and 70.1% (both P<0.05) (Figure 7A). Results for the protein level of ADAM10 were consistent with mRNA findings (Figure 7B).
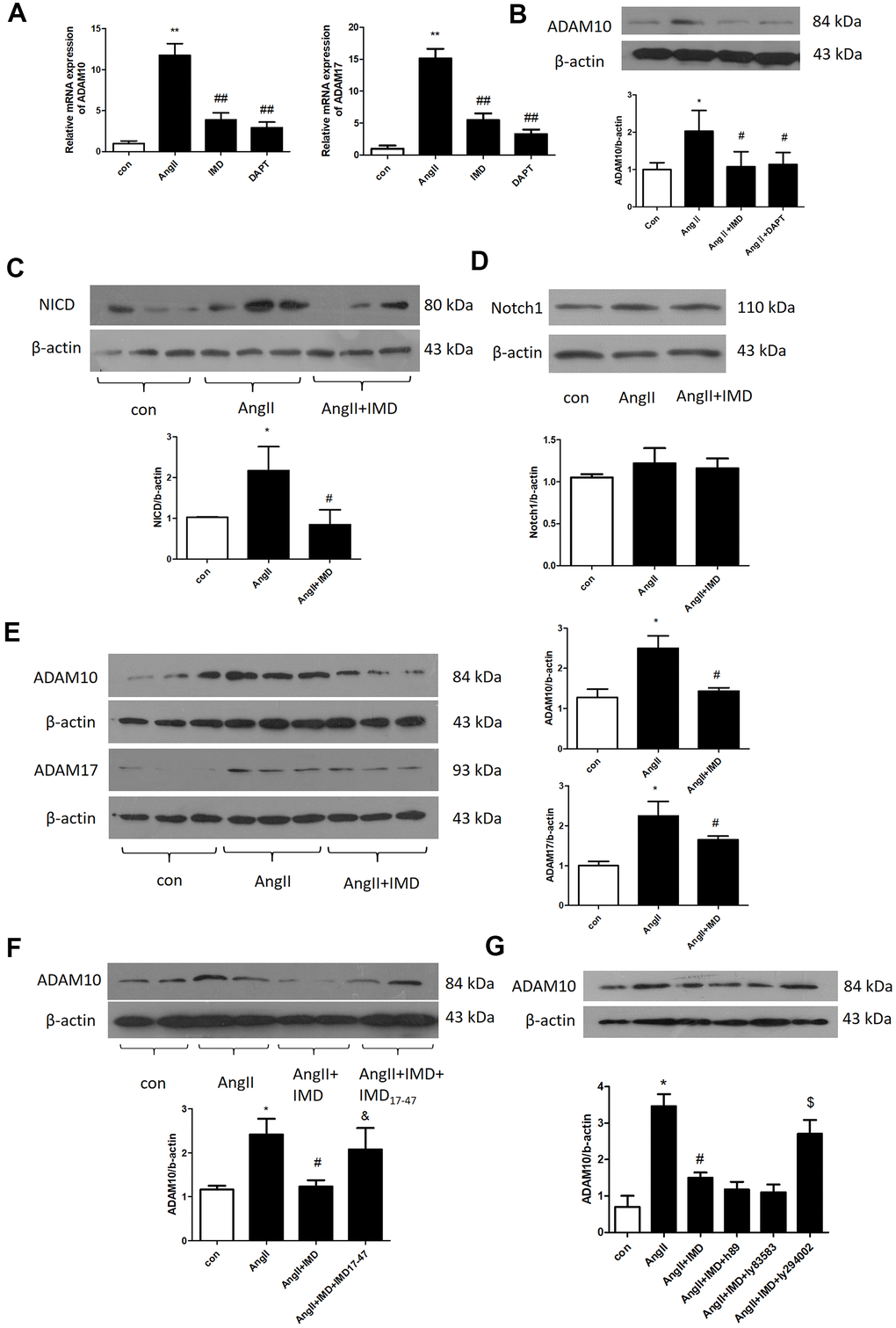
Figure 7. IMD inhibited Notch1 signaling activation by ADAM10. (A) Quantitative real-time PCR analysis of mRNA levels of ADAM10 and ADAM17 in aortas of Control, AngII, AngII+IMD and AngII+DAPT in mice. (B) Western blot analysis of protein level of ADAM10 in aortas of Control, AngII, AngII+IMD and AngII+DAPT mice. Western blot analysis of protein levels of (C, D) NICD, the intracellular fragment of Notch1 and the full length Notch1 receptor and (E) ADAM10 and ADAM17 in AngII-induced macrophage cell line Raw264.7. n=3, Data are mean ± SD. *P<0.05, **P<0.01 vs. Control. #P<0.05, ##P<0.01 vs. AngII. (F) Western blot analysis of protein level of ADAM10 in AngII-induced macrophage cell line Raw264.7. Except for the first 3 groups, IMD receptor blocker IMD17-47 group was added. n=3, Data are mean ± SD. *P<0.05 vs. Control. #P<0.05 vs. AngII. &P<0.05 vs. AngII+IMD. (G) Western blot analysis of protein level of ADAM10 in AngII-induced macrophage cell line Raw264.7. Except for the first 3 groups, cAMP/PKA inhibitor H89, cGMP/PKG inhibitor Ly83583 and PI3K/Akt inhibitor LY294002 were added. n=3, Data are mean ± SD. *P<0.05 vs. Control. #P<0.05 vs. AngII. $P<0.05 vs. AngII+IMD.
We further explored the mechanism in a macrophage lineage Raw264.7. First, we detected the protein levels of NICD and full-length Notch1 receptors in Raw264.7 macrophages by western blot analysis. AngII treatment increased NICD level by 2.19-fold (P<0.05), which was reversed by IMD pre-infusion; Notch1 receptor level was similar in 3 groups (Figure 7C, 7D). Also, ADAM10 and ADAM17 protein levels were elevated by AngII and reversed by IMD in macrophages (Figure 7E). The level of Akt phosphorylation showed the opposite trend as NICD, ADAM10 and ADAM17 (Supplementary Figure 1A). Furthermore, IMD17-47, an IMD receptor blocker, intervened in the effect of IMD on ADAM10 expression (Figure 7F). The PI3K/Akt pathway inhibitor LY294002 also blocked the effect of IMD on ADAM10 expression, but cyclic adenosine monophosphate/proteinkinase A (cAMP/PKA) and cGMP/PKG inhibitors had no effect on ADAM10 expression (Figure 7G).
Endogenous IMD alleviated CaCl2-induced AAA formation
Our previous studies reported that exogenous IMD protected against AAA formation by inhibiting oxidative stress and endoplasmic reticulum stress [27, 28]. To further explore the role of endogenous IMD in AAA, we created IMD transgenic and knockout mice and induced aneurysm by incubation of CaCl2 in the abdominal aorta. With CaCl2 incubation, AAA was induced in 73% of mice in a C57 background (Figure 8A, 8B), the maximum abdominal aorta diameter was increased and the protein levels of NICD, hes1 and M1 macrophage marker iNOS were upregulated (Figure 8A, 8B, 8D, 8E, 8F). Moreover, 90% of IMD-knockout mice (P<0.05) developed AAA and showed more increased maximum abdominal aorta diameter (Figure 8A, 8B). However, AAA was induced in only 50% of IMD transgenic mice (P<0.05) and the maximum abdominal aorta diameter was decreased (Figure 8A, 8B). HE staining showed that CaCl2 induction caused dilated abdominal aorta, vascular wall thickening and damaged vascular elastic plates, which was alleviated by IMD overexpression and aggravated by IMD deficiency (Figure 8C). Moreover, Notch1 pathway markers were upregulated in aortas of IMD-knockout mice and were downregulated in IMD transgenic mice (Figure 8D–8F).
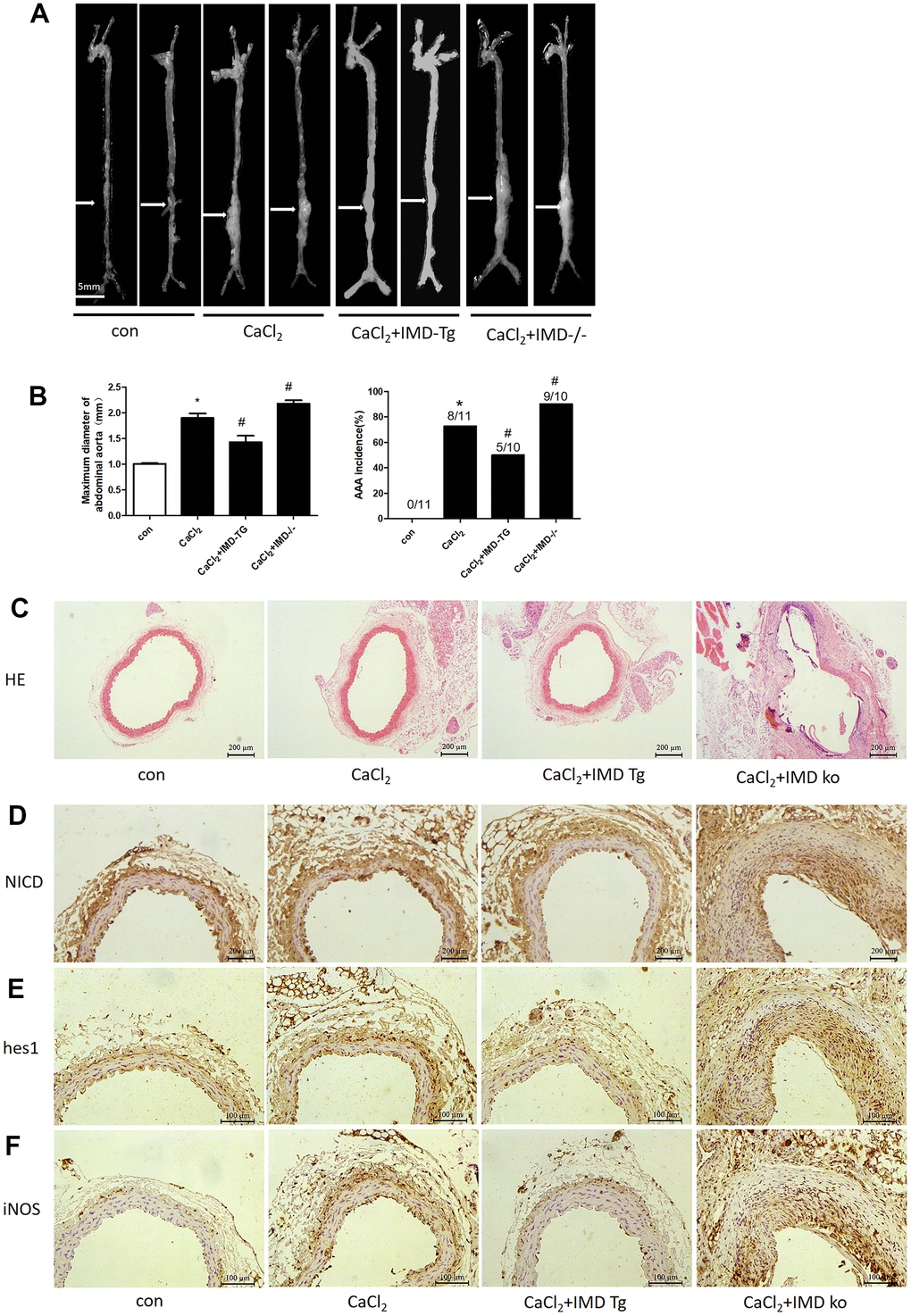
Figure 8. Endogenous IMD alleviated CaCl2-induced AAA and Notch1 signaling and M1 polarization. Control, wild type C57 mice with saline incubation; CaCl2, wild-type C57 mice with CaCl2 incubation; CaCl2+IMD-Tg, IMD transgenic mice with CaCl2 incubation to induce AAA; CaCl2+IMD ko, IMD-knockout mice with CaCl2 incubation to induce AAA. (A) Representative photographs of macroscopic features of aneurysms in male C57 mice at the end of 28 days. Scale bar, 5 mm. (B) Incidence of CaCl2-induced AAA in animals and maximal abdominal aortic diameter of surviving mice at the end of 28 days. The vascular diameter > 50% of the average diameter of the control group was considered AAA. *P<0.05 vs. Control. #P<0.05 vs. CaCl2. (C) Representative HE staining. Scale bar, 200 μm. (D–F) Immunohistochemistry of the protein expression of NICD, hes1 and iNOS in aortas of mice.
Discussion
In the present study, the Notch1 signaling pathway was significantly activated during the pathogenesis of AAA. As an important vasoactive factor, exogenous and endogenous IMD could significantly decrease the occurrence and development of AAA by suppressing the Notch1 signaling pathway. IMD inhibited the expression of the protease ADAM10 through the PI3K/Akt signaling pathway via a CRLR/RAMP receptor and thereby inhibited activation of the Notch1 signaling pathway. Suppression of Notch1 signaling pathway by IMD further prevented NLRP3 activation and M1 polarization of macrophages, promoted M2 macrophage polarization, thus reducing the inflammatory response in local aortas to prevent AAA formation. Thus, IMD might be a vital target for treating AAA in clinical practice.
First, we investigated the regulatory role of exogenous IMD in an AngII-induced AAA model in ApoE-/- mouse background. AngII can affect endothelial cell function, elevate blood pressure, and promote macrophage activation and pro-angiogenesis to accelerate AAA formation [28, 31]. As a classical model of AAA, the AngII-induced mice model is widely used in studying the pathogenesis of AAA, simulating most of the pathological features in human AAA, such as atherosclerosis plaque and thrombosis, infiltration of inflammatory cells in the local vasculature, and severe destruction of vascular elastin [32, 33]. Our results were consistent with those in the literature. Thus, IMD has a protective effect in the development of AAA.
Then we explored how IMD inhibited AAA. As shown in previous studies, Notch signal plays an important role in the pathogenesis of various diseases, such as tumor and cardiovascular diseases [34]. Notch1 affects vascular calcification and participates in the phenotypic changes of vascular smooth muscle cells via multiple signaling pathways [35, 36]. Here, we found that the levels of the Notch1 pathway marker NICD and hes1 were increased in the vascular tissues of patients with AAA, suggesting the Notch1 signaling pathway activation. We also found increased expression of NICD and hes1 in aortas of AngII-induced mice; IMD infusion could significantly inhibit this protein activation. We also used DAPT, a Notch1 inhibitor, which had similar inhibitory effect as IMD. So IMD might prevent AAA via the Notch1 signaling pathway.
AAA is a chronic vascular disease characterized by a local invasive inflammatory response. Inflammatory cells infiltrate the vessel wall and release a large number of inflammatory factors and produce a large amount of reactive oxygen species, which is a critical process in the pathogenesis of aneurysm [37, 38]. Inflammatory cells, especially T lymphocytes and macrophages, are primarily located in the media and adventitia of the aorta [2] and produce a large amount of MMPs, inflammatory cytokines and chemokines, such as IL-1β, IL-6 and MCP-1, affecting extracellular matrix homeostasis and promoting the apoptosis of smooth muscle cells [39, 40]. We found that IMD could reverse the increased levels of inflammatory factors. Immunofluorescence assay showed increased expression of the macrophage marker CD68 in the vascular wall of mice with AAA and IMD reversed the CD68 level, which might be related to the impaired migration capacity of macrophages after IMD infusion, as testified by the scratch-wound assay. Therefore, the anti-inflammatory effect of IMD might be mediated by inhibiting Notch1.
NLRP3 is the most studied inflammasome and has extensive functions. Activation of NLRP3 needs two signals: Toll-like receptors as the first signal activate NF-κB [41], which increases the precursor expression of NLRP3, IL-1β and IL-18. When receiving the second signal, NLRP3 proceeds with oligomerization and recruits molecular ASC. ASC assembles filamentous structure recruitment and enables pro-caspase-1 to shear and activate, then produces the mature IL-1β and IL-18 from their precursors [29, 42]. Mitochondrial oxidative stress in macrophages can activate NLPR3, which plays a role in initiating the inflammatory response in early AAA formation [7]. In this study, AngII promoted NLRP3 activation both in AAA mouse model and cultured macrophages; both IMD and DAPT reversed NLRP3 expression, so IMD may inhibit the activation of NLRP3 in macrophages by blocking the Notch1 signaling pathway, thereby preventing the initiation of the early inflammatory response of AAA.
We also investigated the effect of IMD on macrophage polarization in mouse BMDMs. IMD pre-treatment significantly inhibited macrophage M1 polarization and promoted macrophage M2 polarization. Macrophages have diverse functional characteristics in different microenvironments, showing obvious heterogeneity. M1 macrophages, as the pro-inflammatory phenotype, have a strong promoting effect on inflammatory diseases such as AAA, whereas M2 macrophages can alleviate the local inflammatory response of vessels and have a certain protective effect on AAA [43–45]. IMD also inhibited AAA by regulating macrophage polarization in mice aortas. Overall, IMD may inhibit the formation of AAA by inhibiting M1-type macrophage polarization and promoting M2-type macrophage polarization.
We found that IMD could suppress AAA formation via the Notch1 signaling pathway and therefore restrain NLRP3 activation and macrophage M1 polarization. However, how IMD inhibited Notch1 needs elucidation. The activation of Notch1 is regulated by multiple signaling pathways. The binding of the ligand to the receptor leads to two protein cleavages of the receptor, which is the main mechanism of Notch signaling pathway activation. The first cleavage is mediated by members of the ADAM metalloproteinase family (mainly ADAM10 and ADAM17) and the second is completed by a protein-secreting enzyme complex. Proteases ADAM10 and ADAM17 have key regulatory effects on the activation of the Notch1 pathway [11]. In human patients with non-small cell lung cancer, ADAM10 overexpression activated the Notch1 signaling pathway, leading to increased cell migration and invasion [46]. ADAM17 could regulate the chemical sensitivity of colorectal cancer stem cells by activating the Notch1 signaling pathway [47]. Therefore, we hypothesized that regulation of the Notch1 signaling pathway by IMD was achieved via the proteases ADAM10 and ADAM17. The expression of ADAM10 and ADAM17 was significantly higher in the abdominal aortas of mice with AAA than controls and was significantly reduced after IMD treatment. In Raw264.7 macrophages, IMD significantly inhibited the protein level of NICD but had no significant effect on the full-length Notch1 receptor. These results indicate that IMD blocks the activation of the Notch1 signaling pathway by inhibiting the expression of the protease ADAM10.
We further explored the mechanism of IMD regulating the expression of ADAM10. IMD exerts its biological effects via nonselective interaction with the CRLR/RAMP receptor complex, and the ligand-binding selectivity of CRLR is regulated by RAMP1, 2, and 3 [20]. We found that IMD14-47, a CRLR/RAMP receptor blocker, and PI3K/Akt inhibitor LY294002 blocked the regulation of IMD on ADAM10 expression. Therefore, IMD could inhibit ADAM10 protein expression via its CRLR/RAMP receptor and the PI3K/Akt signaling pathway, thereby inhibiting the activation of Notch1.
In the present work we revealed the protective role of exogenous IMD in an AAA mouse model. To further explore the role of endogenous IMD in AAA, we constructed IMD transgenic and knockout mice in a C57/BL background. We used an AAA mouse model induced by CaCl2, characterized by a severe inflammatory response in local vessels [29]. As compared with wild-type mice, IMD transgenic mice showed significantly reduced incidence of AAA and average abdominal aorta maximum diameter, and IMD-knockout mice showed the reverse results. With CaCl2 incubation, the abdominal aorta was dilated, the vascular wall was thickened and vascular elastic plates were damaged; all were alleviated in IMD transgenic mice and aggravated in IMD-knockout mice. These results indicate that endogenous IMD plays a key role in inhibiting AAA and maintaining the homeostasis of vascular structure and function. Our previous study reported that IMD transgenic mice showed alleviated the neointima formation in a carotid-artery-ligation model [22]. In this study, we first confirmed the protective effect of endogenous IMD in AAA formation. We also detected the Notch1 pathway and M1 macrophage markers in IMD transgenic and knockout mice with CaCl2 incubation. These proteins were higher in aortas with IMD deficiency and downregulated with IMD overexpression, which suggests that endogenous IMD suppressed Notch1 signaling pathway activation and M1-type macrophage polarization in an AAA model induced by calcium chloride.
Two different AAA models were used in this study, Ang II induction for 4 weeks in ApoE-/- background and CaCl2 incubation in C57 background mice. Ang II induced model is a classic AAA model and it has been widely used to study the pathogenesis of AAA and can simulate most of the pathological features of human AAA, such as atherosclerotic plaques and thrombosis, local vascular inflammatory cell infiltration and severe elastin damage [32, 33]. However, the formation of CaCl2 induced AAA is mainly dependent on the destruction of elastic fibers by calcium deposition in the medium and the activation of inflammatory response, which is mainly manifested as calcification, vascular smooth muscle cell apoptosis, infiltration of inflammatory cells in the adventitia and media and more serious degradation of elastins [48]. Both models showed significant inflammatory cell infiltration and elastin degradation. In this study, although the models were inconsistent, they both proved that the inflammation mediated by Notch1 signaling pathway was involved in the occurrence and development of AAA and IMD could protect AAA by inhibiting Notch1 mediated inflammation.
Numerous studies in animal models have shown significant therapeutic potential targets for treating AAA; however, there is still no effective target for clinical application [6]. So it is essential to find new targets. In our previous and present work, IMD has shown some benefits for inhibiting AAA. First, as an endogenous bioactive peptide, IMD is widely distributed in the cardiovascular system. Also, IMD could suppress early inflammatory response via the Notch1 signaling pathway to prevent AAA to achieve the purpose of early treatment. Nevertheless, the mechanism by which IMD inhibits Notch1 needs further exploration.
Taken together, our results provide evidence that exogenous and endogenous IMD can reduce the occurrence of AAA by inhibiting Notch1 signaling pathway-mediated inflammation. IMD binds to its receptor non-selectively to suppress ADAM10 protein level via the PI3K/Akt signaling pathway, thereby restraining the activation of Notch1. Inactivation of Notch1 further inhibits the activation of NLRP3 inflammasome and M1 macrophage polarization, and reduces MMP activity, thus slowing the genesis and development of AAA. This study also uncovered a new inflammatory mechanism of endogenous IMD inhibiting the occurrence of AAA, which provides new directions for therapeutic targets of AAA.
Materials and Methods
Materials
Synthetic human IMD1–53, rat IMD17–47 and AngII were from Phoenix Pharmaceuticals (Belmont, CA, USA). Alzet Mini-osmotic pumps (Alzet model 1004) were from DURECT Corp. (Cupertino, CA, USA). DAPT, LPS, IFN-γ, IL-4, PI3K/Akt inhibitor LY294002, cAMP/PKA inhibitor H89 and cGMP/PKG inhibitor Ly83583 were from Sigma (MO, USA). Primary antibodies for Notch1 (ab52627), hes1 (ab71559), NICD (ab8925), iNOS (ab3523), NLRP3 (ab214185), caspase-1 (ab1872), CD68 (ab955), CD16 (ab203883), Arg1 (ab91279), CD206 (ab64693, ADAM10 (ab1997), ADAM17 (ab2051), IL-1β (ab2105), IL-10 (ab9969) and IL-18 (ab191860) were from Abcam (Cambridge, UK). Primary antibodies for p-Akt (Ser473) (4060) and Akt (9272) were from Cell Signaling Technology(CST, USA). Primary antibodies for β-actin (sc-1616), GAPDH (sc-25778), ASC (sc-514414), and all secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Other chemicals and reagents were of analytical grade. Mouse macrophage line Raw264.7 were bought from American Type Culture Collection (ATCC, USA).
IMD transgenic and knockout mice
IMD transgenic and knockout mice were generated by Model Animal Research Center of Nanjing University. All animals were maintained at C57BL/6 genetic background, kept under specific pathogen-free conditions, approved by the Animal Care and Use Committee of Peking University Health Science Center. Male mice 8-12 weeks old were used for experiments.
AngII-induced AAA model
ApoE-/- mice in a C57BL/6 background were from the Animal Center, Peking University Health Science Center (Beijing). All animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011). ApoE-/- mice (5 months old, male) were randomly assigned to 4 groups for treatment: infusion of saline; AngII (1000 ng/kg/min) [7]; AngII plus IMD1-53 (300 ng/kg/h) [28]; and AngII plus DAPT. AngII, saline and IMD1-53 were infused for 28 days. DAPT was administered 3 days before AngII infusion. AngII and IMD1-53 were administered subcutaneously via an Alzet Mini-osmotic pump. Four weeks later, surviving mice were intraperitoneally injected with pentobarbital (50 mg/kg) for anesthetization.
CaCl2-induced AAA model
Briefly, as described previously [48], C57BL/6 and IMD transgenic and knockout mice were anesthetized with an intraperitoneal injection of pentobarbital (40 mg/kg) before undergoing laparotomy. The abdominal aorta between the renal arteries and bifurcation of the iliac arteries was isolated from the surrounding retroperitoneal structures. The AAA model was induced with extraluminal CaCl2. After baseline measurements, 0.5 mol/L CaCl2 was applied to the external surface of the aorta for 15 min. The aorta was rinsed with 0.9% sterile saline and the incision was closed. NaCl (0.9%) was substituted for CaCl2 in sham control mice.
Cell culture
BM macrophages were extracted and cultured as previously described [7] with minor modification. Mouse tibias were collected and the BM was flushed with Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) and differentiated into primary BMDMs. Cells were cultured in DMEM containing 10% fetal bovine serum (FBS; Hyclone, USA) and incubated at 37° C in a humidified atmosphere containing 5% CO2. Macrophages were stimulated with IFN-γ and IL-4 to induce M1 and M2 macrophage polarization, respectively [30].
The mouse peritoneal macrophages were collected and cultured as previously described [49] with minor modification. The mouse were injected intraperitoneally with 2 ml of brewer modified thioglycollate medium (BD, USA). Three days later, cells were harvested by peritoneal lavage with 5 ml RPMI-1640 medium (Gibco, USA). Then cells were washed with cold RPMI-1640 medium and seeded on plastic plates. After two hours incubation, the non-adherent cells were washed with RPMI-1640 medium and the adherent cells were monolayer macrophages.
Wound healing assay
The mouse peritoneal macrophages was used in wound healing assay. Macrophages were cultured in 6-well plates and starved for 12 hours in medium supplemented with 0.5%FBS. Then macrophages were scraped, washed with PBS and incubated under 37° C in the presence of LPS or IMD1-53 for 24 hours in an atmosphere containing 5% CO2. Images were captured through a Olympus microscope.
Histology and immunofluorescence of mice
Mice were killed and the abdominal aortas were harvested, fixed for 12 hr and embedded in paraffin, and cross-sections (6 μm) were prepared. Paraffin sections were stained with hematoxylin and eosin (HE) and Verhoeff-van Gieson (VVG).
For immunofluorescence assay, freshly cut OCT-embedded aorta sections were incubated with the antibodies anti-CD68 (1:100 dilution) for 1 hr at 37° C, then at 4° C overnight, washed with PBS, then incubated with secondary TRITC-conjugated goat anti-rabbit IgG (1:300 dilution). IgG was a negative control. DAPI was used to stain the nuclei.
Immunohistochemistry of human and mice
Human and mouse AAA aortas were used in immunohistochemistry assay. Surgical specimens were obtained from AAA patients undergoing open surgical repair at Beijing An Zhen Hospital. The control aortic samples were obtained from heart transplantation donors (without AAA) at Beijing An Zhen Hospital. Informed consent was obtained from all patients according to protocols approved by the Medical Ethical Committee of Beijing An Zhen Hospital that comply with the principles outlined in the Declaration of Helsinki.
First, abdominal aortas were fixed in paraformaldehyde for 12 hr and embedded in paraffin, and cross-sections (6 μm) were prepared. For immunohistochemistry, paraffin sections from human or mouse aortas were incubated at 37° C for 1 hr, then at 4° C overnight with primary antibodies for hes1 (1:300 dilution), NICD (1:100 dilution), iNOS (1:200 dilution), Arg1 (1:300 dilution) or ASC (1:100 dilution). On day 2, arterial sections were rinsed with 0.01 mol/L phosphate buffered saline (PBS), then incubated with horseradish peroxidase-conjugated secondary antibodies diluted in 0.1 mol/L PBS for 1 hr at room temperature and redyeing the nucleus with hematoxylin. Normal serum was a negative control.
In situ zymography
Freshly OCT-embedded mouse aorta sections (7.00 um) were used to detect MMP activity. Sections were incubated in a dark and humid chamber at 37° C for 24 hr with MMP fluorogenic substrate DQ–gelatin–FITC (Invitrogen), then washed with PBS. Negative controls were incubated with PBS.
Western blot analysis
Aortic tissue or cells were homogenized in lysis buffer. Then equal amounts of total protein were resolved by 10% or 15% SDS-PAGE (15% for small proteins). Proteins were transferred to a nitrocellulose membrane and blocked with 5% nonfat dried milk for 1 hr, then incubated with the primary antibodies β-actin (1:4000 dilution), GAPDH (1:2000), NLRP3 (1:1000), caspase-1 (1:500), ASC (1:1000), Arg1 (1:1000), IL-1β (1:1000), IL-18 (1:1000), IL-10 (1:200), CD16 (1:300), CD206 (1:1000), Notch1 (1:500), hes1 (1:1000), NICD (1:500), iNOS (1:500), ADAM10 (1:500), ADAM17 (1:500), p-Akt (1:1000) or Akt (1:1000) overnight at 4° C, then with horseradish peroxidase-conjugated secondary antibody for 1 hr. Protein expression was analyzed by using NIH ImageJ and normalized to β-actin expression. All experiments were repeated at least 3 times.
Quantitative real-time PCR
Total RNA from aortic tissue and BMDMs was extracted with Trizol (Applygen, Beijing) and reverse transcribed by using a reverse transcription system (Applygen, Beijing). Real-time PCR amplification included Applied Biosystems 7500 fast PCR System (Life Technologies, USA) and SYBR Green I reagent (Tiangen Biotech, Beijing). The cycle threshold (Ct) was determined as the number of PCR cycles required for a given reaction to reach an arbitrary fluorescence value within the linear amplification range. Relative quantification was performed according to the 2-ΔΔCt method, with GAPDH as a reference. The primers for real-time PCR were in Table 1.
Table 1. Primer sequence for RT-PCR.
| Target | Sequence | |
| Mouse Notch1 | Forward | 5’-ACCCACTCTGTCTCCCACAC-3’ |
| Reverse | 5’-GCTTCCTTGCTACCACAAGC-3’ | |
| Mouse Dll4 | Forward | 5’-TTAAGCACTTCCAGGCAACC-3’ |
| Reverse | 5’-ACCACTGCCGCTATTCTTGT-3’ | |
| Mouse hes1 | Forward | 5’-GGCGAAGGGCAAGAATAAAT-3’ |
| Reverse | 5’-TGCTTCACAGTCATTTCCAGA-3’ | |
| Mouse hey1 | Forward | 5’-GGAGGGTCAGCAAAGCATTA-3’ |
| Reverse | 5’-CTCCCTTCACCTCACTGCTC-3’ | |
| Mouse Adam10 | Forward | 5’-CCAGCTCTGATGGCAAAGAT-3’ |
| Reverse | 5’-CACTGAACTGCTTGCTCCAC-3’ | |
| Mouse Adam17 | Forward | 5’-TGGCAAATGTGAGAAACGAG-3’ |
| Reverse | 5’-AAACCAGAACAGACCCAACG-3’ | |
| Mouse Il6 | Forward | 5’-GCCTTCTTGGGACTGATGCT-3’ |
| Reverse | 5’-TGCCATTGCACAACTCTTTTC-3’ | |
| Mouse Mcp1 | Forward | 5’-AGGGACTGAGGCACTCCAGA-3’ |
| Reverse | 5’-TGACGACGAGACTTCCAGACTACA-3’ | |
| Mouse Ifng | Forward | 5’-TGAGACAATGAACGCTACACACT-3’ |
| Reverse | 5’-GTCACCATCCTTTTACCAGT-3’ | |
| Mouse Il12 | Forward | 5’-CTGTGCCTTGGTAGCATCTATG-3’ |
| Reverse | 5’-GCAGAGTCTCGCCATTATGATTC-3’ | |
| Mouse Il10 | Forward | 5’-GCTCTTACTGACTGGCATGAG-3’ |
| Reverse | 5’-CGCAGCTCTAGGAGCATGTG-3’ | |
| Mouse iNOS | Forward | 5’-GGAGTGACGGCAAACATGACT-3’ |
| Reverse | 5’-TCGATGCACAACTGGGTGAAC-3’ | |
| Mouse Arg1 | Forward | 5’-TTTTTCCAGCAGACCAGCTT-3’ |
| Reverse | 5’-AGAGATTATCGGAGCGCCTT-3’ | |
| Mouse Il1b | Forward | 5’-GCAACTGTTCCTGAACTCAACT-3 |
| Reverse | 5’-ATCTTTTGGGGTCCGTCAACT-3 | |
| Mouse Tnf | Forward | 5’-CCCTCACACTCAGATCATCTTCT-3’ |
| Reverse | 5’-GCTACGACGTGGGCTACAG-3’ | |
| Mouse Gapdh | Forward | 5’-ACTTTGTCAAGCTCATTTCC-3’ |
| Reverse | 5’-TGCAGCGAACTTTATTGATG-3’ |
Statistical analysis
GraphPad Prism v5.00 for Windows (GraphPad Software Inc, San Diego, CA, USA) was used for analysis; data are expressed as mean ± SD. First, Kolmogorov-Smirnov test was used to evaluate the normality of data distribution (P>0.1) and F-test was used to compare variances (P>0.1 take for equal variance). Then Student t test was used to compare 2 groups. One-way ANOVA was used to compare more than 2 groups, and when significant (P<0.05), the Tukey HSD test was used to test for differences between groups. P<0.05 was considered statistically significant. Fisher’s exact test was used for the incidence of aneurysm. P<0.05 was considered statistically significant.
Supplementary Materials
Author Contributions
Yong-Fen Qi and Xian-Qiang Ni conceived and designed the study. Xian-Qiang Ni, Ya-Rong Zhang, Wei-Wei Lu, Qing Zhu, Jin-Ling Ren, Yao Chen, Lin-Shuang Zhang and Xin Liu performed the experiments. Li-Xin Jia provided abdominal aortas of healthy controls and AAA patients. Xian-Qiang Ni and Ya-Rong Zhang wrote the paper. Ya-Rong Zhang, Li-Xin Jia, Jie Du, Zhong-Ping Ning, Mo-Zhi Jia, Yan-Rong Yu, Chao-Shu Tang and Yong-Fen Qi reviewed and edited the manuscript. All authors have read and approved the submitted manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 81670434, 31872790, 32071113 to YFQ), the Major Research Plan of the National Natural Science Foundation of China (grant no. 91339203 to YFQ), the Outstanding Clinical Discipline Project of Shanghai Pudong, Shanghai Municipal Health Commission (ZK2019B25) and Peking University Clinical Scientist Program (BMU2019LCKXJ001).
References
- 1. Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011; 8:92–102. https://doi.org/10.1038/nrcardio.2010.180 [PubMed]
- 2. Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. 2019; 16:225–42. https://doi.org/10.1038/s41569-018-0114-9 [PubMed]
- 3. Li H, Bai S, Ao Q, Wang X, Tian X, Li X, Tong H, Hou W, Fan J. Modulation of immune-inflammatory responses in abdominal aortic aneurysm: emerging molecular targets. J Immunol Res. 2018; 2018:7213760. https://doi.org/10.1155/2018/7213760 [PubMed]
- 4. Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006; 26:987–94. https://doi.org/10.1161/01.ATV.0000214999.12921.4f [PubMed]
- 5. Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. 2005; 111:816–28. https://doi.org/10.1161/01.CIR.0000154569.08857.7A [PubMed]
- 6. Robertson L, Atallah E, Stansby G. Pharmacological treatment of vascular risk factors for reducing mortality and cardiovascular events in patients with abdominal aortic aneurysm. Cochrane Database Syst Rev. 2017; 1:CD010447. https://doi.org/10.1002/14651858.CD010447.pub3 [PubMed]
- 7. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, Sagara J, Taniguchi S, Takahashi M. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015; 35:127–36. https://doi.org/10.1161/ATVBAHA.114.303763 [PubMed]
- 8. Kerstjens-Frederikse WS, van de Laar IM, Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, Klein Wassink-Ruiter JS, van der Zwaag PA, du Marchie Sarvaas GJ, Bergman KA, Bilardo CM, Roos-Hesselink JW, Janssen JH, et al. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet Med. 2016; 18:914–23. https://doi.org/10.1038/gim.2015.193 [PubMed]
- 9. Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC. Structural biology. Structural basis for Notch1 engagement of delta-like 4. Science. 2015; 347:847–53. https://doi.org/10.1126/science.1261093 [PubMed]
- 10. Pagie S, Gérard N, Charreau B. Notch signaling triggered via the ligand DLL4 impedes M2 macrophage differentiation and promotes their apoptosis. Cell Commun Signal. 2018; 16:4. https://doi.org/10.1186/s12964-017-0214-x [PubMed]
- 11. Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009; 29:5679–95. https://doi.org/10.1128/MCB.00406-09 [PubMed]
- 12. Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, Thompson B, Spaulding C, Macaroun S, Alegre ML, Kee BL, Ferrando A, Miele L, Aifantis I. Targeting the NF-kappaB signaling pathway in Notch1-induced t-cell leukemia. Nat Med. 2007; 13:70–77. https://doi.org/10.1038/nm1524 [PubMed]
- 13. Dail M, Wong J, Lawrence J, O’Connor D, Nakitandwe J, Chen SC, Xu J, Lee LB, Akagi K, Li Q, Aster JC, Pear WS, Downing JR, et al. Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature. 2014; 513:512–16. https://doi.org/10.1038/nature13495 [PubMed]
- 14. Cheng J, Koenig SN, Kuivaniemi HS, Garg V, Hans CP. Pharmacological inhibitor of notch signaling stabilizes the progression of small abdominal aortic aneurysm in a mouse model. J Am Heart Assoc. 2014; 3:e001064. https://doi.org/10.1161/JAHA.114.001064 [PubMed]
- 15. Sachdeva J, Mahajan A, Cheng J, Baeten JT, Lilly B, Kuivaniemi H, Hans CP. Smooth muscle cell-specific Notch1 haploinsufficiency restricts the progression of abdominal aortic aneurysm by modulating CTGF expression. PLoS One. 2017; 12:e0178538. https://doi.org/10.1371/journal.pone.0178538 [PubMed]
- 16. Moran CS, Biros E, Krishna SM, Wang Y, Tikellis C, Morton SK, Moxon JV, Cooper ME, Norman PE, Burrell LM, Thomas MC, Golledge J. Resveratrol inhibits growth of experimental abdominal aortic aneurysm associated with upregulation of angiotensin-converting enzyme 2. Arterioscler Thromb Vasc Biol. 2017; 37:2195–203. https://doi.org/10.1161/ATVBAHA.117.310129 [PubMed]
- 17. Zhang LN, Vincelette J, Cheng Y, Mehra U, Chen D, Anandan SK, Gless R, Webb HK, Wang YX. Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb Vasc Biol. 2009; 29:1265–70. https://doi.org/10.1161/ATVBAHA.109.186064 [PubMed]
- 18. Roh J, Chang CL, Bhalla A, Klein C, Hsu SY. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem. 2004; 279:7264–74. https://doi.org/10.1074/jbc.M305332200 [PubMed]
- 19. Takei Y, Inoue K, Ogoshi M, Kawahara T, Bannai H, Miyano S. Identification of novel adrenomedullin in mammals: a potent cardiovascular and renal regulator. FEBS Lett. 2004; 556:53–58. https://doi.org/10.1016/s0014-5793(03)01368-1 [PubMed]
- 20. Yang JH, Jia YX, Pan CS, Zhao J, Ouyang M, Yang J, Chang JK, Tang CS, Qi YF. Effects of intermedin(1-53) on cardiac function and ischemia/reperfusion injury in isolated rat hearts. Biochem Biophys Res Commun. 2005; 327:713–19. https://doi.org/10.1016/j.bbrc.2004.12.071 [PubMed]
- 21. Aslam M, Pfeil U, Gündüz D, Rafiq A, Kummer W, Piper HM, Noll T. Intermedin (adrenomedullin2) stabilizes the endothelial barrier and antagonizes thrombin-induced barrier failure in endothelial cell monolayers. Br J Pharmacol. 2012; 165:208–22. https://doi.org/10.1111/j.1476-5381.2011.01540.x [PubMed]
- 22. Zhu Q, Ni XQ, Lu WW, Zhang JS, Ren JL, Wu D, Chen Y, Zhang LS, Yu YR, Tang CS, Qi YF. Intermedin reduces neointima formation by regulating vascular smooth muscle cell phenotype via cAMP/PKA pathway. Atherosclerosis. 2017; 266:212–22. https://doi.org/10.1016/j.atherosclerosis.2017.10.011 [PubMed]
- 23. Li P, Sun HJ, Han Y, Wang JJ, Zhang F, Tang CS, Zhou YB. Intermedin enhances sympathetic outflow via receptor-mediated cAMP/PKA signaling pathway in nucleus tractus solitarii of rats. Peptides. 2013; 47:1–6. https://doi.org/10.1016/j.peptides.2013.05.002 [PubMed]
- 24. Zhang X, Gu L, Chen X, Wang S, Deng X, Liu K, Lv Z, Yang R, He S, Peng Y, Huang D, Jiang W, Wu K. Intermedin ameliorates atherosclerosis in ApoE null mice by modifying lipid profiles. Peptides. 2012; 37:189–93. https://doi.org/10.1016/j.peptides.2012.07.011 [PubMed]
- 25. Teng X, Song J, Zhang G, Cai Y, Yuan F, Du J, Tang C, Qi Y. Inhibition of endoplasmic reticulum stress by intermedin(1-53) protects against myocardial injury through a PI3 kinase-Akt signaling pathway. J Mol Med (Berl). 2011; 89:1195–205. https://doi.org/10.1007/s00109-011-0808-5 [PubMed]
- 26. Chang JR, Guo J, Wang Y, Hou YL, Lu WW, Zhang JS, Yu YR, Xu MJ, Liu XY, Wang XJ, Guan YF, Zhu Y, Du J, et al. Intermedin1-53 attenuates vascular calcification in rats with chronic kidney disease by upregulation of α-Klotho. Kidney Int. 2016; 89:586–600. https://doi.org/10.1016/j.kint.2015.12.029 [PubMed]
- 27. Lu WW, Jia LX, Ni XQ, Zhao L, Chang JR, Zhang JS, Hou YL, Zhu Y, Guan YF, Yu YR, Du J, Tang CS, Qi YF. Intermedin1-53 attenuates abdominal aortic aneurysm by inhibiting oxidative stress. Arterioscler Thromb Vasc Biol. 2016; 36:2176–90. https://doi.org/10.1161/ATVBAHA.116.307825 [PubMed]
- 28. Ni XQ, Lu WW, Zhang JS, Zhu Q, Ren JL, Yu YR, Liu XY, Wang XJ, Han M, Jing Q, Du J, Tang CS, Qi YF. Inhibition of endoplasmic reticulum stress by intermedin1-53 attenuates angiotensin II-induced abdominal aortic aneurysm in ApoE KO mice. Endocrine. 2018; 62:90–106. https://doi.org/10.1007/s12020-018-1657-6 [PubMed]
- 29. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018; 17:688. https://doi.org/10.1038/nrd.2018.149 [PubMed]
- 30. Koh YC, Yang G, Lai CS, Weerawatanakorn M, Pan MH. Chemopreventive effects of phytochemicals and medicines on M1/M2 polarized macrophage role in inflammation-related diseases. Int J Mol Sci. 2018; 19:2208. https://doi.org/10.3390/ijms19082208 [PubMed]
- 31. Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, Sadybekov A, Zamlynny B, Rudd MT, Hollenstein K, Tolstikova A, White TA, Hunter MS, et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature. 2017; 544:327–32. https://doi.org/10.1038/nature22035 [PubMed]
- 32. Remus EW, O’Donnell RE
Jr , Rafferty K, Weiss D, Joseph G, Csiszar K, Fong SF, Taylor WR. The role of lysyl oxidase family members in the stabilization of abdominal aortic aneurysms. Am J Physiol Heart Circ Physiol. 2012; 303:H1067–75. https://doi.org/10.1152/ajpheart.00217.2012 [PubMed] - 33. Obama T, Tsuji T, Kobayashi T, Fukuda Y, Takayanagi T, Taro Y, Kawai T, Forrester SJ, Elliott KJ, Choi E, Daugherty A, Rizzo V, Eguchi S. Epidermal growth factor receptor inhibitor protects against abdominal aortic aneurysm in a mouse model. Clin Sci (Lond). 2015; 128:559–65. https://doi.org/10.1042/CS20140696 [PubMed]
- 34. Pei H, Song X, Peng C, Tan Y, Li Y, Li X, Ma S, Wang Q, Huang R, Yang D, Li D, Gao E, Yang Y. TNF-α inhibitor protects against myocardial ischemia/reperfusion injury via Notch1-mediated suppression of oxidative/nitrative stress. Free Radic Biol Med. 2015; 82:114–21. https://doi.org/10.1016/j.freeradbiomed.2015.02.002 [PubMed]
- 35. Clark CR, Bowler MA, Snider JC, Merryman WD. Targeting cadherin-11 prevents Notch1-mediated calcific aortic valve disease. Circulation. 2017; 135:2448–50. https://doi.org/10.1161/CIRCULATIONAHA.117.027771 [PubMed]
- 36. Zhou S, Fang X, Xin H, Li W, Qiu H, Guan S. Osteoprotegerin inhibits calcification of vascular smooth muscle cell via down regulation of the Notch1-RBP-Jκ/Msx2 signaling pathway. PLoS One. 2013; 8:e68987. https://doi.org/10.1371/journal.pone.0068987 [PubMed]
- 37. Davis FM, Rateri DL, Daugherty A. Mechanisms of aortic aneurysm formation: translating preclinical studies into clinical therapies. Heart. 2014; 100:1498–505. https://doi.org/10.1136/heartjnl-2014-305648 [PubMed]
- 38. Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017; 14:457–71. https://doi.org/10.1038/nrcardio.2017.52 [PubMed]
- 39. Johnston WF, Salmon M, Pope NH, Meher A, Su G, Stone ML, Lu G, Owens GK, Upchurch GR
Jr , Ailawadi G. Inhibition of interleukin-1β decreases aneurysm formation and progression in a novel model of thoracic aortic aneurysms. Circulation. 2014; 130:S51–9. https://doi.org/10.1161/CIRCULATIONAHA.113.006800 [PubMed] - 40. Tieu BC, Lee C, Sun H, Lejeune W, Recinos A
3rd , Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009; 119:3637–51. https://doi.org/10.1172/JCI38308 [PubMed] - 41. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009; 183:787–91. https://doi.org/10.4049/jimmunol.0901363 [PubMed]
- 42. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010; 10:210–15. https://doi.org/10.1038/nri2725 [PubMed]
- 43. Rubio-Navarro A, Amaro Villalobos JM, Lindholt JS, Buendía I, Egido J, Blanco-Colio LM, Samaniego R, Meilhac O, Michel JB, Martín-Ventura JL, Moreno JA. Hemoglobin induces monocyte recruitment and CD163-macrophage polarization in abdominal aortic aneurysm. Int J Cardiol. 2015; 201:66–78. https://doi.org/10.1016/j.ijcard.2015.08.053 [PubMed]
- 44. Dale MA, Xiong W, Carson JS, Suh MK, Karpisek AD, Meisinger TM, Casale GP, Baxter BT. Elastin-derived peptides promote abdominal aortic aneurysm formation by modulating M1/M2 macrophage polarization. J Immunol. 2016; 196:4536–43. https://doi.org/10.4049/jimmunol.1502454 [PubMed]
- 45. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013; 496:445–55. https://doi.org/10.1038/nature12034 [PubMed]
- 46. Guo J, He L, Yuan P, Wang P, Lu Y, Tong F, Wang Y, Yin Y, Tian J, Sun J. ADAM10 overexpression in human non-small cell lung cancer correlates with cell migration and invasion through the activation of the Notch1 signaling pathway. Oncol Rep. 2012; 28:1709–18. https://doi.org/10.3892/or.2012.2003 [PubMed]
- 47. Wang R, Ye X, Bhattacharya R, Boulbes DR, Fan F, Xia L, Ellis LM. A disintegrin and metalloproteinase domain 17 regulates colorectal cancer stem cells and chemosensitivity via Notch1 signaling. Stem Cells Transl Med. 2016; 5:331–38. https://doi.org/10.5966/sctm.2015-0168 [PubMed]
- 48. Wang Y, Krishna S, Golledge J. The calcium chloride-induced rodent model of abdominal aortic aneurysm. Atherosclerosis. 2013; 226:29–39. https://doi.org/10.1016/j.atherosclerosis.2012.09.010 [PubMed]
- 49. Harada T, Yoshimura K, Yamashita O, Ueda K, Morikage N, Sawada Y, Hamano K. Focal adhesion kinase promotes the progression of aortic aneurysm by modulating macrophage behavior. Arterioscler Thromb Vasc Biol. 2017; 37:156–65. https://doi.org/10.1161/ATVBAHA.116.308542 [PubMed]