



Introduction
Distal cholangiocarcinoma (dCCA) originates from epithelial cells of the common bile duct and accounts for approximately 20%-30% of all cholangiocarcinoma (CCA) cases [1]. The five-year overall survival of dCCA patients remains dismal, ranging from 11%-48% [2–4]. Most patients are diagnosed in the progressive stage and lost the opportunity for surgical resection. This delayed diagnosis is mainly due to a lack of specific biomarkers and a limited understanding of the molecular mechanisms regulating the oncogenesis of dCCA.
Recently, circular (circ)RNA, a type of non-coding RNA, has become recognized as a functional molecule in human diseases [5]. Unlike linear RNA, circRNA contains covalently closed loops without 3′ and 5′ ends, making it more stable and resistant to degradation. CircRNA has been shown to play essential regulatory roles in multiple biological processes, including transcriptional splicing, protein-protein interactions (PPIs), ribosomal RNA processing, and micro (mi)RNAs sponging [6]. Accumulating studies have demonstrated that circRNAs are differentially expressed (DE) in human cancers. For instance, hsa_circ_0007142 is upregulated and promotes cell proliferation in colorectal cancer [7]; in non-small cell lung cancer cell lines, hsa_circ_100146 is highly expressed [8]; in CCA cells, hsa_circ_0001649 is downregulated and exhibits tumor-suppressive activity [9]; and circRNA Cdr1as is upregulated in CCA cells and associated with poor prognosis [10].
A growing number of studies have revealed that circRNAs function as a sponge of miRNAs and inhibit their activities. Examples include the upregulation of hsa_circ_0005230, which facilitates cell growth and metastasis in CCA via sponging of miR-1238 and miR-1299 [11]. In hepatocellular carcinoma (HCC), hsa_circ_100338 activates the mammalian target of rapamycin signaling pathway by absorbing miR-141-3p [12]. In gastric cancer, hsa_circ_100269 inhibits cell proliferation by sponging miR-630 [13]. Therefore, at the post-transcriptional level, circRNAs regulate protein-coding mRNAs by competing for miRNAs; they are known as competing endogenous RNAs (ceRNAs) in this context [14]. Early studies demonstrated that circRNAs could be detected in body fluids, serum, plasma, and tissue samples [15, 16]. Consequently, they have the real potential of being promising molecular biomarkers and therapeutic targets in human diseases.
However, there has been little systematic evaluation of circRNAs in dCCA, and the function of dysregulated circRNAs remains to be further investigated. In this study, we constructed a comprehensive circRNA expression profile by microarray, performed DE analyses, identified a novel circRNA biomarker, and established a circRNA-miRNA-mRNA network to understand the possible functions of circRNAs in dCCA (Figure 1).
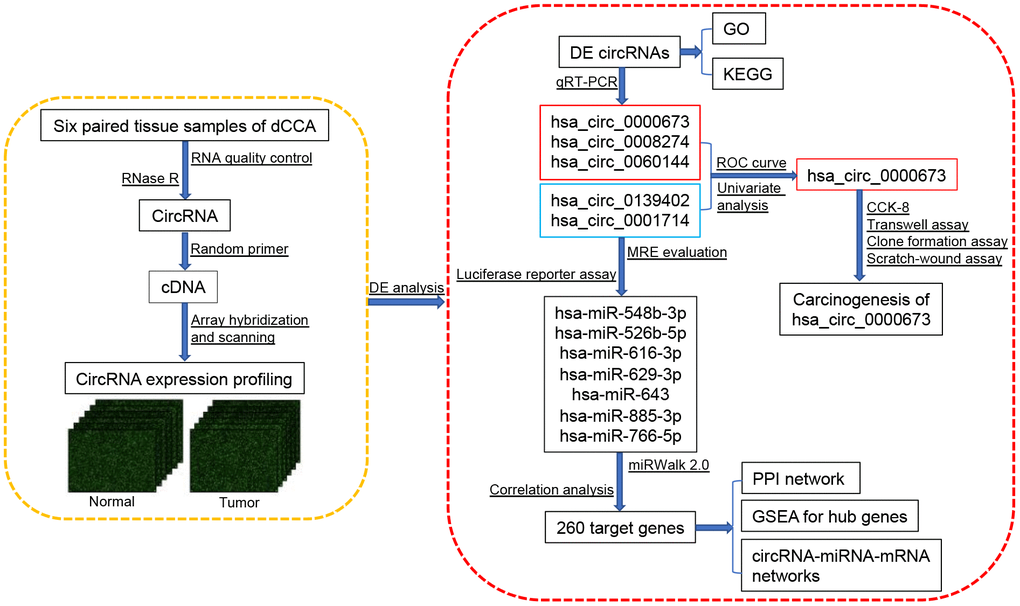
Figure 1. Flowchart of the current study. Abbreviations: dCCA: distal cholangiocarcinoma; circRNA: circular RNA; DE circRNAs: differentially expressed circular RNAs; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Gene and Genomes; qRT-PCR: quantitative real-time polymerase chain reaction; ROC curve: receiver operating characteristic curve; CCK-8: cell counting kit-8; MRE: microRNA response element; miRNA: microRNA; PPI: protein-protein interaction; GSEA: gene set enrichment analysis.
Results
Identification of dysregulated circular RNAs in distal cholangiocarcinoma
Six dCCA patients were enrolled in this study. The clinicopathological characteristics of the patients are summarized in Table 1. Microarray profiling analyses detected a total of 12,934 circRNAs. The distribution of the current dataset was appropriate (Figure 2A). A total of 132 up- and 39 downregulated circRNAs (|log2(fold change) (log2FC)| > 1 and p < 0.05) were identified and visualized with scatter and volcano plots (Figure 2B, 2C). The results indicated that more than 80% of the circRNAs originated from exons of host genes (Figure 2D). Upregulated circRNAs were mainly distributed in chromosomes 1 and 16, whereas downregulated circRNAs were enriched in chromosomes 17 and 19 (Figure 2E). In hierarchical clustering analyses, 171 DE circRNAs were distinguishable between dCCA and adjacent normal samples (Figure 3). The majority of DE circRNAs were also derived from exons. The characteristics of the top 10 dysregulated circRNAs are listed in Supplementary Table 1. All the microarray datasets were deposited in the Gene Expression Omnibus (GEO) site (GEO accession number: GSE148561).
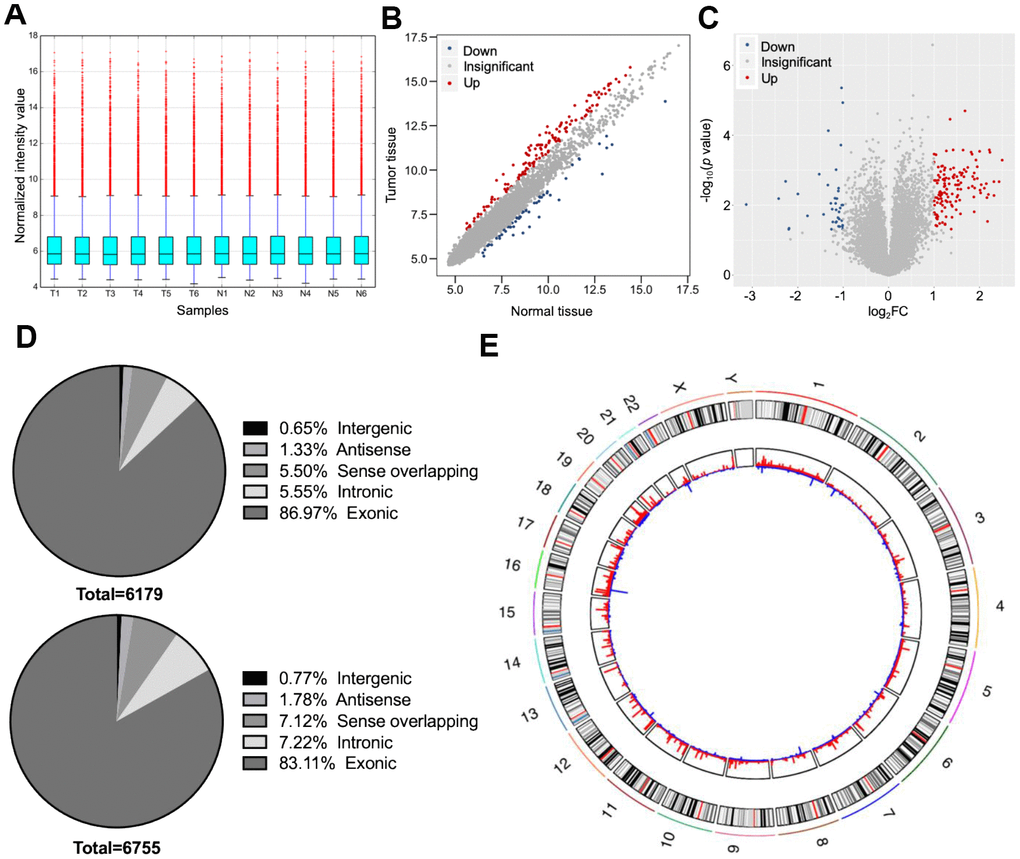
Figure 2. Overall characteristics of circular (circ)RNA expression profiling of distal cholangiocarcinoma (dCCA). (A) The distributions of circRNA expression values after quantile normalization and log2 processing. (T: dCCA tissues; N: paired adjacent normal tissues). (B) The scatter plot describing the difference of circRNA levels between dCCA and normal tissues. The red and blue dots indicate more than 1.5-fold change (FC) in dCCA and normal tissues, respectively. (C) The volcano plot of differentially expressed circRNAs (|log2FC| > 1 and p < 0.05). The red and blue dots represent up- and downregulated circRNAs in tumor. (D) The classifications of circRNAs (up: upregulated circRNAs; down: downregulated circRNAs). (E) The circle diagram describes the location of circRNAs on chromosomes. The red and blue lines represent the up- and downregulated circRNAs in tumor tissues. The segment length indicates the FC value.
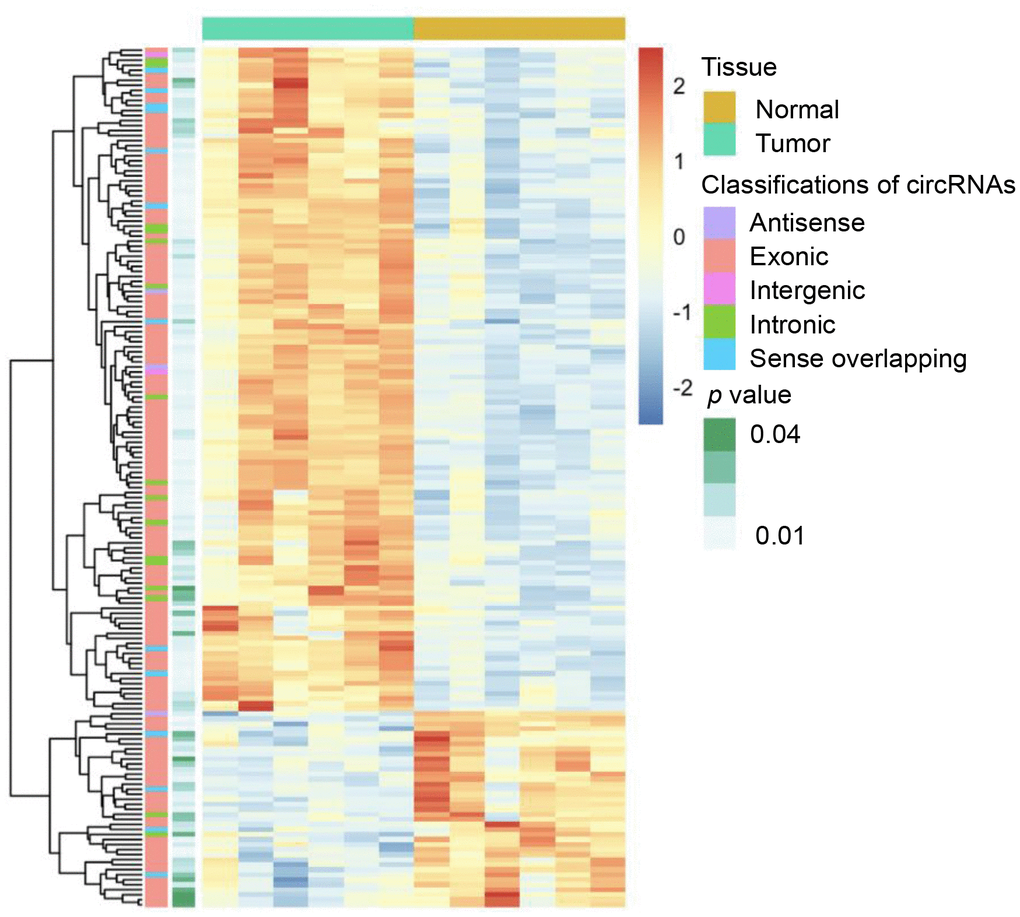
Figure 3. Heatmap of differentially expressed circular (circ)RNAs in distal cholangiocarcinoma. In total, 132 up- and 39 downregulated circRNAs were identified with microarray (|log2(fold change)|> 1 and p < 0.05). The green and yellow bars on the top represent tumor and normal tissues. In the middle, the color of boxes changes from red to blue, indicating high to low-expression of circRNAs. The first and second columns on the left represent classifications of circRNAs and the p-value of the differential analysis.
Table 1. Clinicopathological characteristics of six patients with distal cholangiocarcinoma.
| No. | Gender | Age | Histological differentiation | T | N | M | TNM stagea | CA19-9 (U/mL) | Positive lymph nodes | Total lymph nodes | Perineuronal invasion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Male | 70 | Moderate | T2 | N0 | M0 | IIA | 40.30 | 0 | 37 | No | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Male | 62 | Moderate-poor | T2 | N0 | M0 | IIA | 26.47 | 0 | 14 | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Male | 64 | Moderate | T3 | N1 | M1 | IV | 643.47 | 1 | 15 | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Male | 69 | Moderate | T3 | N0 | M0 | IIB | 74.04 | 0 | 21 | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Male | 53 | Moderate | T3 | N0 | M0 | IIB | 9.16 | 0 | 14 | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Female | 56 | Poor | T3 | N1 | M0 | IIB | 177.21 | 1 | 29 | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| aTNM stage is based on the 8th edition of the American Joint Committee on Cancer. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CA 19-9, carbohydrate antigen 19-9. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Functions of differentially expressed circular RNAs
The host genes of the 171 DE circRNAs were selected for Gene Ontology (GO) analyses. The results indicated that the term “response to nutrient” was most enriched in the biological process category (Figure 4A), and “adherens junction” was the top enriched term in the cellular component category (Figure 4B). The most significant number of genes in the molecular functions category were in “cell adhesion molecule binding” (Figure 4C). In Kyoto Encyclopedia of Gene and Genomes (KEGG) pathway enrichment analysis, five genes were associated with regulation of “actin cytoskeleton,” “focal adhesion,” “carbon metabolism,” and “calcium signaling pathway” (Figure 4D).
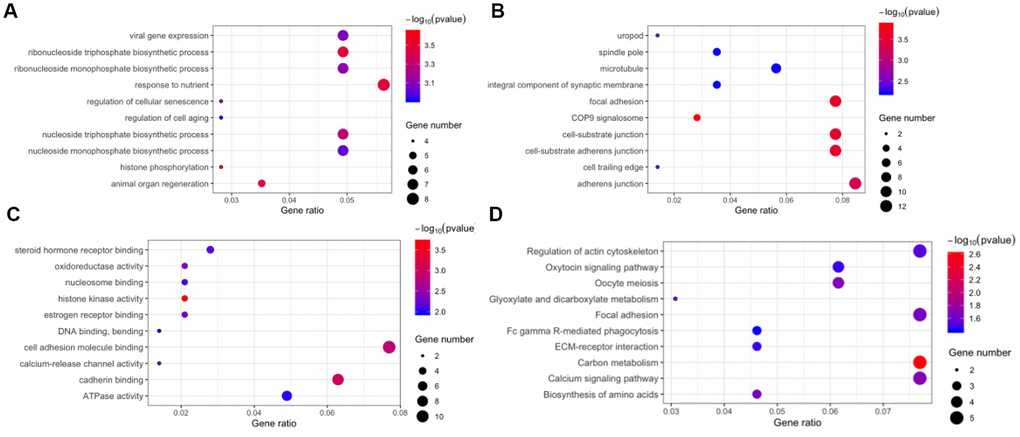
Figure 4. Function annotation and pathway enrichment for the host genes of differentially expressed circular RNAs. Gene Ontology, including biological process (A), cellular component (B), and molecular function (C). (D) Kyoto Encyclopedia of Gene and Genomes pathway enrichment.
Validation of differentially expressed circular RNAs
We selected four significantly upregulated circRNAs, including hsa_circ_0060144, hsa_circ_0008274, hsa_circ_0000673, and hsa_circ_0072088, and two significantly downregulated circRNAs, including hsa_circ_0139402 and hsa_circ_0001714 for quantitative real-time polymerase chain reaction (qRT-PCR) validation. The relative intensity values measured with microarray are shown in Supplementary Figure 1, and the structures of the six circRNAs were visualized using the Cancer-Specific CircRNA Database (CSCD) in Figure 5. The qRT-PCR results revealed that the expression levels of hsa_circ_0060144, hsa_circ_0008274, and hsa_circ_0000673 were significantly upregulated (Figure 6A–6C), and the expression levels of hsa_circ_0139402 and hsa_circ_0001714 were significantly downregulated (Figure 6E, 6F). However, the expression of hsa_circ_0072088 did not differ between tumor and healthy tissues (Figure 6D). Thus, the five verified DE circRNAs were selected to perform further bioinformatics analyses.
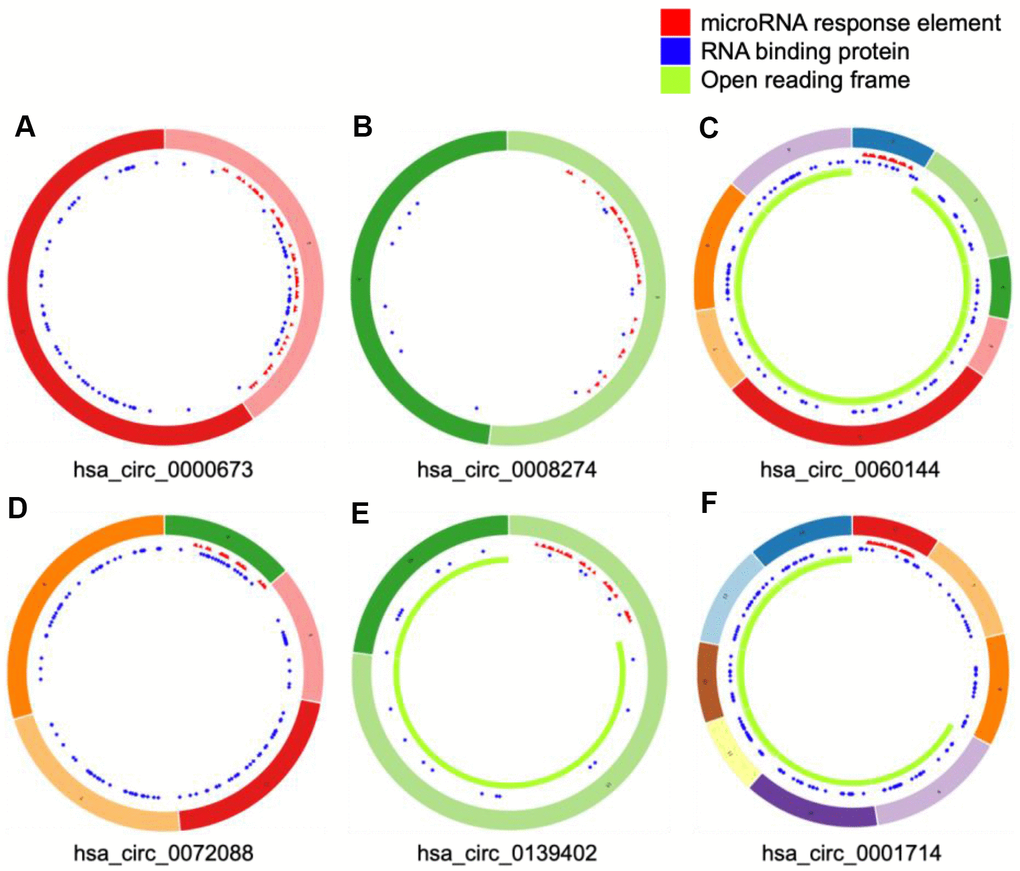
Figure 5. Structural patterns of six differentially expressed circular (circ)RNAs. The features of four upregulated circRNAs, including hsa_circ_0000673, hsa_circ_0008274, hsa_circ_0060144, and hsa_circ_0072088 (A–D), and two downregulated circRNAs, including hsa_circ_0139402 and hsa_circ_0001714 (E and F), were visualized by Cancer-Specific CircRNAs Database.
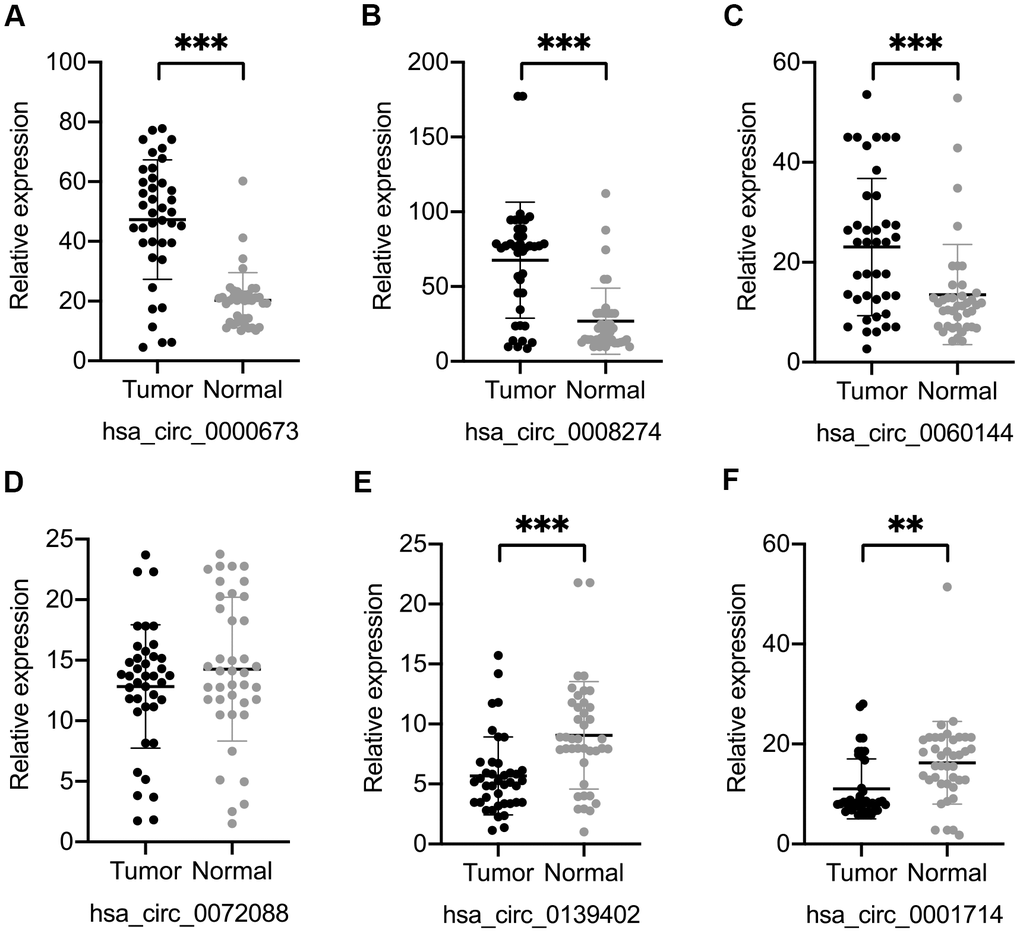
Figure 6. Validation of six differentially expressed circular (circ)RNAs by qRT-PCR. Three circRNAs, including hsa_circ_0000673 (A), hsa_circ_0008274 (B), and hsa_circ_0060144 (C) were significantly upregulated, and two circRNAs, including hsa_circ_0139402 (E) and hsa_circ_0001714 (F) were significantly downregulated in tumor tissues. (D) The level of hsa_circ_0072088 was not different between tumor and normal tissues. **p < 0.01, ***p < 0.001.
A circRNA-miRNA-mRNA interacting network
Prior studies have shown that circRNAs act as miRNA sponge molecules and increase the expression of downstream genes. Hence, we predicted nine target miRNAs of the five verified DE circRNAs using CircInteractome and CSCD databases. The results of luciferase reporter assay validated the bindings between five circRNAs and seven miRNAs, including hsa_circ_0000673/miR-548b-3p, hsa_circ_0008274/miR-526b-5p, hsa_circ_0060144/ (miR-616-3p, miR-629-3p, and miR-643), hsa_circ_0139402/miR-885-3p, and hsa_circ_0001714/miR-766-5p (Supplementary Figure 2).
Using the DIANA-miRPath platform, we explored and visualized the KEGG pathways associated with the above seven miRNAs. The results showed that these miRNAs were mostly enriched in cancer-related pathways, basal cell carcinoma, TGF-beta, and Wnt signaling pathways (Supplementary Figure 3).
With miRWalk 2.0, we predicted the mRNAs with binding sites corresponding to the seven miRNAs. After that, we performed correlation analyses between miRNA and mRNA with the CCA cohort of The Cancer Genome Atlas (TCGA). A total of 260 significantly negatively correlated mRNAs were regarded as the potential target genes of the seven miRNAs. Thus, a circRNA-miRNA-mRNA interacting network was constructed, including five circRNAs, seven miRNAs, and 260 mRNAs (Figure 7).
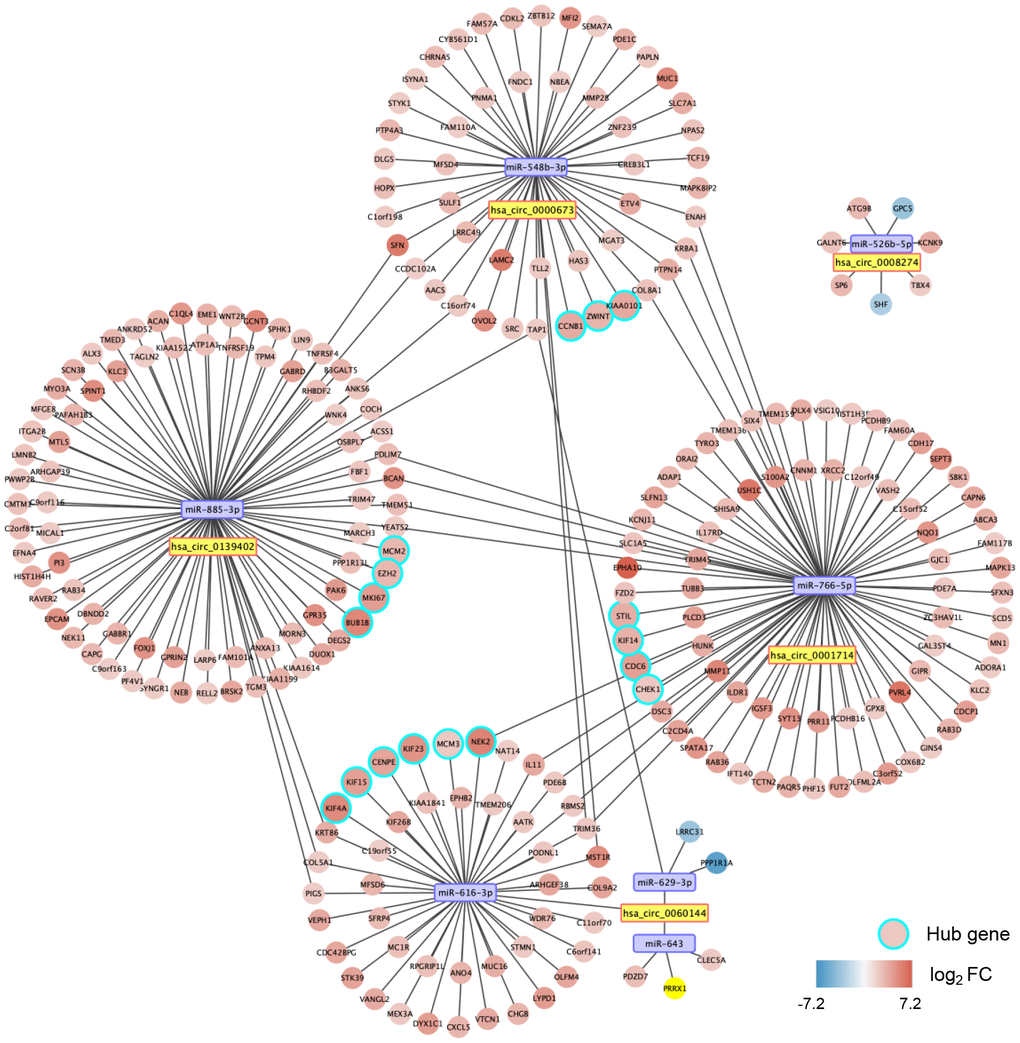
Figure 7. A circular (circ)RNA-micro(mi)RNA-mRNA interacting network in dCCA. The network contains five dysregulated circRNAs, seven miRNAs, 260 mRNAs, and 17 hub genes. The node color changes from blue to red, representing the fold change of mRNA expression value in the cholangiocarcinoma cohort of The Cancer Genome Atlas (tumor versus normal).
Gene set enrichment analysis of hub genes
A PPI network for the 260 target genes was constructed based on the Search Tool for the Retrieval of Interacting Genes (STRING) database and visualized with Cytoscape software (Supplementary Figure 4). Using the Molecular Complex Detection (MCODE) approach, we screened out 17 hub genes (Supplementary Figure 4). Thus, we established a potential critical ceRNA network for dCCA, including four DE circRNAs, four miRNAs, and 17 hub genes (Figure 7).
We validated the expression of the 17 hub genes with GSE32879 containing 23 CCA and seven normal samples. The results showed that 15 hub genes, including CCNB1, KIF23, KIF4A, ZWINT, CENPE, KIF15, MCM3, NEK2, BUB1B, EZH2, MCM2, MIKI67, CHEK1, KIF14, and PCLAF were significantly upregulated in tumor tissues (Supplementary Figure 5). Gene set enrichment analysis (GSEA) was performed based on the mRNA expression of the CCA cohort of the TCGA. As shown in Figure 8, compared with healthy tissue samples, BUB1B, CCNB1, CDC6, CHEK1, and MCM2 were enriched in “negative regulation of cell cycle transition” in GO term of biological process and associated with “cell cycle” pathways in KEGG analyses. In summary, these results elucidate that the central circRNA-miRNA-mRNA network possibly regulates cell proliferation in dCCA.
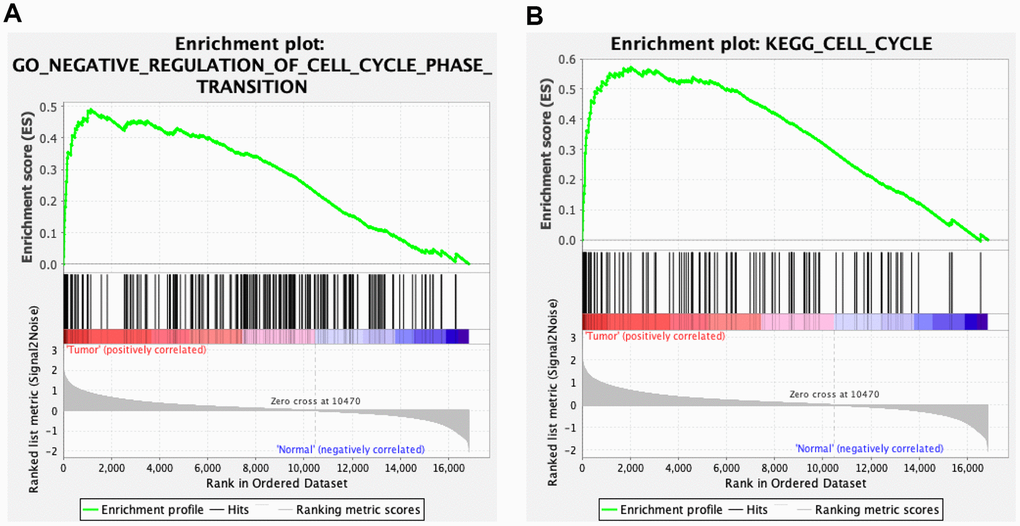
Figure 8. Gene set enrichment analysis (GSEA) for cholangiocarcinoma (CCA). Five hub genes of CCA, including BUB1B, CCNB1, CDC6, CHEK1, and MCM2 were enriched in the cell cycle based on biological process (A) and Kyoto Encyclopedia of Genes and Genomes analyses (B).
Association between hsa_circ_0000673 and dCCA progression
The results of the receiver operating characteristic (ROC) curve indicated that, in distinguishing tumor from healthy tissue samples, hsa_circ_0000673 had the highest area under the curve (AUC) value among the five circRNAs (Supplementary Figure 6). As shown in Table 2, overexpression of hsa_circ_0000673 was significantly associated with tumor invasion (p = 0.001), poor differentiation (p = 0.041), and residual tumor (p = 0.044). These results indicated that hsa_circ_0000673 was associated with malignant phenotypes of dCCA.
Table 2. Association between hsa_circ_0000673 level and clinicopathological parameters of distal cholangiocarcinoma patients.
| Clinicopathological parameters | hsa_circ_0000673a | p value | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Low(n = 20) | High(n = 20) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age (years) | 66 | 69 | 0.436 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender | 0.731 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Male | 15 | 13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Female | 5 | 7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pathological differentiation | 0.041* | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Moderate and high | 17 | 10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Poor | 3 | 10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Numbers of positive lymph node | 13.5±11.3 | 12.8±10.7 | 0.842 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vascular invasion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 17 | 18 | 1.000 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 3 | 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNM stageb | 1.000 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I and II | 8 | 9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| III and IV | 12 | 11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Primary tumor | 0.001** | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T1 and T2 | 15 | 4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T3 and T4 | 5 | 16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reginal lymph nodes invasion | 1.000 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N0 and N1 | 8 | 9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | 12 | 11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Distant metastases | 0.4872 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | 20 | 18 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Positive | 0 | 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Resection | 0.044* | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R0 | 19 | 13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R1 and R2 | 1 | 7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *p < 0.05; **p < 0.01. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| aUsing median expression value of hsa_circ_0000673 as a cut-off. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bTNM stage is based on the 8th edition of the American Joint Committee on Cancer. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R0: a negative resection margin; R1: a resection margin with microscopic residual tumor; R2: a resection margin with macroscopic residual tumor. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Inhibition of hsa_circ_0000673 suppresses CCA cell proliferation, migration, and invasion in vitro
In order to investigate the biological functions of hsa_circ_0000673, we first measured its expression level in four CCA cell lines. As shown in Figure 9A, RBE showed the highest expression, and HuH28 exhibited the lowest expression. Therefore, we chose RBE and KMBC for the silencing of hsa_circ_0000673. The small interfering RNA (siRNA) targeting the back-splice junction sites of hsa_circ_0000673 was designed and shown to decrease the expression level significantly (Figure 9B). A cell counting kit-8 (CCK-8) assay showed that inhibition of hsa_circ_0000673 significantly suppressed cell proliferation in both RBE and KMBC cells (Figure 9C). In transwell assays, the silencing of hsa_circ_0000673 significantly inhibited the migration and invasion abilities of RBE and KMBC cells (Figure 9D). A scratch-wound assay revealed that the migration abilities of RBE and KMBC cells were significantly inhibited following treatment with siRNA (Figure 9E). Moreover, in colony formation assays, siRNA significantly decreased the colony-forming ability of RBE and KMBC cells (Figure 9F). These in vitro experiments collectively suggested that inhibition of hsa_circ_0000673 could suppress the proliferation, migration, and invasion of CCA cells.
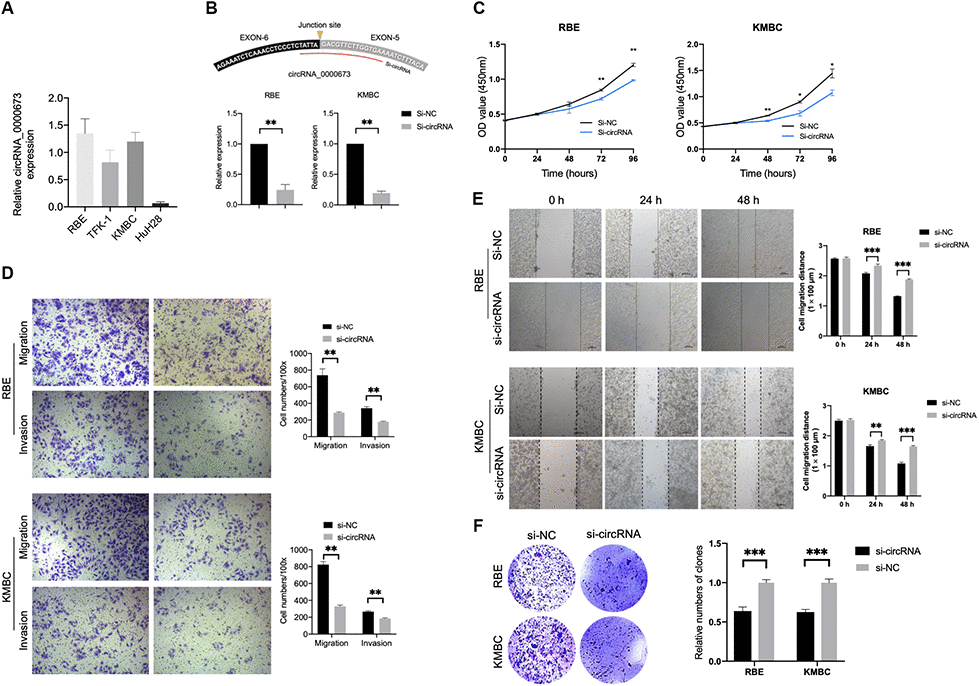
Figure 9. Inhibition of hsa_circ_0000673 suppresses cholangiocarcinoma (CCA) cell proliferation, migration, and invasion. (A) Relative expression of hsa_circ_0000673 in four CCA cell lines. (B) Specific small interfering RNA (siRNA) inhibits the expression of hsa_circ_000067 in CCA cells. Cell counting kit-8 assays (C), transwell migration and invasion assays (D), scratch-wound assays (E), and colony formation assays (F) of CCA cells transfected with negative control and si-hsa_circ_0000673. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Cholangiocarcinoma, an aggressive tumor, is classified as intrahepatic CCA, perihilar CCA, or distal CCA based on anatomical location. Each subtype has distinct clinicopathological characteristics and therapeutic strategies [17]. To our knowledge, this is the first study conducting circRNA expression profiling of dCCA and revealing 132 up- and 39 downregulated circRNAs. The functions of DE circRNAs were illustrated with GO and KEGG analyses. Three up- and two downregulated circRNAs were validated with qRT-PCR. A circRNA-miRNA-mRNA network was constructed, including five circRNAs, seven miRNAs, and 260 mRNAs in dCCA. Finally, hsa_circ_0000673 was identified as a potential biomarker for dCCA, and inhibition of this circRNA suppressed proliferation, migration, and invasion of CCA cells in vitro.
In the GO term of biological process, “response to nutrient” and “ribonucleoside triphosphate biosynthetic process” were most enriched for DE circRNAs, which indicated that dysfunction of nutrient and energy metabolism might promote oncogenesis of dCCA. In cellular component and molecular functions, the term “adherens junction” was mainly enriched, which was in agreement with a previous study [18]. In KEGG analysis, “regulations of cytoskeleton” were significantly enriched, supporting the correlation between DE circRNAs and cellular migration [11].
In the current study, six circRNAs were selected for qRT-PCR according to the following criteria: first, the mature sequence length of circRNA was between 200–4000 bp; second, circRNAs better contained many binding sites with miRNA and RNA binding proteins; third, the top 20 dysregulated circRNAs were preferred. By doing this, the candidate circRNAs were more likely to be functional and suitable for subsequent experiments. Among the five verified circRNAs, hsa_circ_0000673 was identified as a biomarker for dCCA diagnosis with optimal efficiency (AUC = 0.85). It was associated with features of malignancy such as tumor invasion, poor histological stage, and residual tumor. The results of in vitro studies indicated that inhibition of hsa_circ_0000673 suppressed the migration and invasion abilities of CCA cells. Therefore, our results demonstrated that hsa_circ_0000673 could be an oncogene in dCCA and could serve as a potential biomarker and therapeutic target for dCCA diagnosis and treatment.
Hsa_circ_0000673 is derived from the fifth and sixth exons of RSL1D1 containing 251 bp. According to our study, hsa_circ_0000673 was verified to bind to miR-548b-3p, which may target CCNB1. In the CCA cohort of TCGA, CCNB1 was significantly overexpressed in tumor tissues. Hence, hsa_circ_0000673 possibly promotes dCCA progression via miR-548b-3p/CCNB1. However, the potential regulatory mechanism is needed for further verification.
Our results identified five miRNAs that bind with the three upregulated circRNAs, and among them, miR-548b-3p, miR-526b-5p, and miR-616-3p are linked to pathways in cancer according to miRPath analyses. Studies revealed the antitumor activities of these miRNAs. For example, in glioma, miR-526b-5p and miR-548b-3p acted as tumor suppressors by targeting WEE1 [19] and MTA2 [20], respectively. In lung cancer, miR-548b-3p was downregulated in tumor specimens and induced cell apoptosis via the PI3K/AKT signaling pathway [21]. Moreover, results of KEGG analyses indicated that miR-629-3p and miR-643 were associated with the cell cycle pathway that promoted tumor progression. In pancreatic and gastric cancers, miR-629 acts as an oncogene, promoting cell proliferation and inhibiting apoptosis by targeting FOXO3 [22, 23]. In osteosarcoma, miR-643 was revealed to suppress tumorigenesis by ZEB1 [24].
On the other hand, miR-766-5p and miR-885-3p bind to the two downregulated circRNAs, and they were enriched in multiple cancer-related pathways. For example, in HCC, miR-885-3p and miR-766 played a role in tumor progression by targeting TIGAR and NR3C2, respectively [25, 26]. Likewise, these two miRNAs increased cell proliferation and migration by targeting SOCS5 and SCAI in colorectal cancer [27, 28].
In the PPI network, 17 hub genes corresponding to four DE circRNAs and four miRNAs were identified. Interestingly, most of the hub genes were related to carcinogenesis. For example, the protein encoded by CCNB1 plays a crucial role in regulating the G2/M transition. In gastric, breast, and colorectal cancer cells, inhibition of CCNB1 induced G2/M phase cell cycle arrest and promoted apoptosis [29, 30]. Furthermore, CHEK1 [31], BUB1B [32], and MCM2 [33] could promote cancer cell proliferation, and which were enriched in cell cycle pathways in GSEA analysis for CCA. Therefore, these circRNA-miRNA-mRNA networks probably play significant roles in regulating the cell cycle.
Notably, ZWINT was overexpressed in multiple tumors such as HCC [34], prostate, and lung cancer [35], and it was associated with poor survival through upregulating cell-cycle proteins. A high level of NEK2 was also associated with a poor prognosis in pancreatic cancer [36]. In HCC, NEK2 activated the PP1/Akt, Wnt, and mitogen-activated protein kinase pathways [37], thereby exerting a carcinogenic function. Thus, our results outline the potential circRNAs-related signaling pathways such as hsa_circRNA_0000673/miR-548b-3p/CCNB1 and hsa_circRNA_0060144/miR-616-3p/NEK2.
There are several limitations to this study. First, mechanistic studies are needed to verify the molecular functions of the five DE circRNAs. Second, due to a lack of following-up, survival analyses were not performed on hsa_circ_0000673. Third, a high false-positive was inevitable due to the sample size of the microarray and qRT-PCR. These limitations indicate that more clinical data and dCCA cases are needed to complete analyses for more convincing conclusions. Systematic molecular biology experiments should be carried out to verify the potential signaling pathways presented in the current study.
Conclusions
In summary, we first obtained a comprehensive circRNA profile in dCCA and identified 171 dysregulated circRNAs. Three up- and two downregulated circRNAs were validated with qRT-PCR. A circRNA-miRNA-mRNA network was constructed, and which may regulate the cell cycle in dCCA. The expression of hsa_circRNA_0000673 was upregulated in dCCA tissues and associated with tumor progression. In vitro experiments indicate that hsa_circRNA_0000673 may function as an oncogene and serve as a potential biomarker and a therapeutic target for dCCA treatment.
Materials and Methods
Patients and samples
From January to December 2019, 40 patients diagnosed with dCCA who subsequently underwent pancreaticoduodenectomy were enrolled in the current study, at the Department of Hepatobiliary Surgery in Beijing Chao-Yang Hospital, Capital Medical University (Beijing, China). The diagnosis was confirmed with pathology results. Adjuvant radiotherapy and chemotherapy were not performed preoperatively. Paired tumor and adjacent healthy fresh tissues were immediately frozen in liquid nitrogen for two hours, then stored at -80° C. Among the 40 pairs of dCCA tissues, six pairs were selected for circRNA microarray analysis, and all the tissue samples (including the samples for microarray) were used for qRT-PCR validation. All the patients signed the informed consent form, and the study was approved by the hospital ethics committee.
Cell culture and transfection
The human CCA cell lines RBE, TFK-1, KMBC, and HuH28 were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured with RPMI-1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% FBS and penicillin/streptomycin at 37° C with 5% CO2.
SiRNA against hsa_circ_0000673 (si-circRNA: 5′-TATTAGACGTTCTTGGTGAAAATCTTTACA-3′) was designed and synthesized by GenePharma (Shanghai, China). Following the manufacturer’s instructions, 50 nM of siRNA was transfected into RBE and KMBC cells using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA).
Circular RNA microarray detection and differential expression analyses
The paired tumor and adjacent normal tissue samples from six dCCA patients were used for microarray detection. Total RNA from each sample was extracted with TRIzol reagent (Invitrogen) and quantified using the NanoDrop ND-1000 (Thermo Fisher, USA). The products were digested with Rnase R (Epicentre, Madison, WI, USA), and the linear RNAs were removed. The enriched circRNAs were amplified and transcribed into fluorescent cRNA utilizing a random primer method (Super RNA Labeling Kit, Arraystar, Rockville, MD, USA). Labeled cRNAs were hybridized onto the Arraystar Human circRNA Array V2 (8x15K) and incubated for 17 h at 65° C in a hybridization oven (Agilent, Santa Clara, CA, USA). After washing the slides, the arrays were scanned using an Agilent G2505C scanner.
Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Quantile normalization of the raw data was performed using R software. CircRNA samples with at least three out of six having flags in “P” or “M” (“All Targets Value”) were retained for further analyses. The limma R package was applied to identify DE circRNAs between tumor and normal tissues (|log2FC| > 1 and p < 0.05). We used pheatmap and Rcircos R packages to visualize the DE circRNAs and their distributions in human chromosomes.
GO and KEGG analyses for differentially expressed circular RNAs
To further understand the function of DE circRNAs, the clusterProfiler R package was used to perform GO and KEGG pathway enrichment analyses for host genes of DE circRNAs [38]. The results were visualized with the ggplot2 R package [39].
Quantitative real-time polymerase chain reaction
Total RNAs from cells and tissue samples were isolated with TRIzol reagent (Invitrogen) and transcribed into the first-strand cDNA using rtStar™ First-Strand cDNA Synthesis Kit (Arraystar Inc.). Specific primers were designed with Primer 5.0 and synthesized by Yingjun Biotechnology Co., Ltd (Shanghai, China). Forward and reverse primers were located at the 3’ and 5’ ends of circRNA, respectively, which ensured that the amplified sequence included the back-splice junction site of circRNAs (the sequences are shown in Supplementary Table 2). Following the manufacturer’s instructions, qRT-PCR was performed with Arraystar SYBR® Green Real-time qPCR Master Mix (Arraystar Inc.). The expression of circRNA was analyzed using the 2-ΔΔCt method and normalized to β-actin expression levels.
Identification of a prognostic biomarker for distal cholangiocarcinoma
To evaluate whether the expression values of circRNAs could distinguish the tumor from normal tissue samples, ROC curves were applied, and AUCs were calculated. Meanwhile, clinicopathological variables of dCCA patients including tumor size, regional lymph node metastasis, distant metastasis, TNM stage grouping, histological grade, and resection status were extracted and classified according to the 8th edition of the AJCC. To assess the relationship between hsa_circ_0000673 level and these variables, the patients were divided into high and low-level groups according to the median value of this circRNA. Then the percentages of T1-T2 versus T3-T4, N0-N1 versus N2, negative versus positive metastasis, TNM stage I-II versus stage III-IV, well to moderate versus poor histologic grades, and R0 versus R1 were calculated and compared between the two groups with Fisher’s exact tests.
Competing miRNA prediction
The potential target miRNAs of the five DE circRNAs were screened out with CSCD (http://gb.whu.edu.cn/CSCD/) and CircInteractome database (https://circinteractome.nia.nih.gov/index.html). An intersection analysis between the results of the two datasets was then performed to identify the possible competing miRNAs.
Luciferase reporter assay
The wild-type (WT) and mutant (MUT) sequences of five DE circRNAs containing miRNA binding sites were cloned and inserted into the pmirGLO Dual-Luciferase vectors (Promega, Madison, WI, USA), respectively. HEK 293T cells were co-transfected with 50 nM miRNA mimics or negative control and 500 ng/ml wild-type or mutated pmirGLO-circRNA with Lipofectamine 2000 (Invitrogen). After 48 hours, the luciferase activity of each group was measured by the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) following the manufacture’s instruction. All assays were performed in triplicate.
KEGG pathway enrichment for miRNAs
To illustrate the functions of the target miRNAs of the five DE circRNAs, DIANA-miRPath V2.0 was used to find and visualize the enriched KEGG pathways.
Construction of a circRNA-miRNA-mRNA network
The putative target mRNAs of miRNAs were obtained by using the miRWalk2.0 database (http://mirwalk.umm.uni-heidelberg.de). We extracted the mRNA expression profile of the CCA cohort from Broad TCGA GDAC (http://gdac.broadinstitute.org), which included 36 tumorous and nine normal samples. A Pearson correlation analysis between miRNA and mRNA were performed, and the significantly negatively correlated (p < 0.05) mRNAs were regarded as the target genes of miRNAs.
A PPI network was obtained using the STRING database (https://string-db.org) [40] and visualized with Cytoscape software. A combined score ranging from 0 (low) to 1 (high) represents the correlation of each PPI relationship pair. In the current study, we used an interaction score > 0.4 (moderate) as the cut-off criterion. The MCODE plug-in in Cytoscape software was used to identify the hub genes of the PPI network [41]. The screening conditions were set as degree cut-off = 2, K-Core = 2, and Node Score Cutoff = 0.2. Finally, a circRNA-miRNA-mRNA regulating network in dCCA was constructed. Besides, we verified the expression of hub genes using the mRNA data of GSE32879 [42].
Cell viability assay
Cell proliferation ability was measured using a CCK-8 assay (Sigma-Aldrich) according to the manufacturer’s instructions. Approximately 2 × 103 cells per well were incubated in 96-well plates. At 0, 24, 48, 72, and 96 h, CCK-8 solution (10μl) was added to each well, and the cells were incubated at 37° C for two h. The optical density (OD) at 450 nm was determined with a SpectraMax microtiter plate reader (Molecular Devices, Carlsbad, CA, USA). Cells in each group were tested in three replicates.
Transwell assay
Cell migration and invasion assays were performed using a transwell chamber (8 μ pore size; Corning Incorporated, Corning, NY, USA) following the manufacturer’s instructions. Polycarbonate (PC) membrane (Corning) was pre-coated with or without matrigel (Corning) for invasion or migration assays. At 24 h after transfection, 2 × 104 cells in 200 μl serum-free DMEM were seeded into the upper compartment. The lower chamber was filled with 700 μl 20% FBS. After 24 h incubation, the cells located in the upper chamber were erased, and the cells in the lower chamber were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 15 min (Beyotime Institute of Biotechnology, Jiangsu, China). The stained cells were photographed and counted in five fields with a light microscope.
Colony formation assay
After 24 h transfection, cells were seeded into 6-well plates and incubated for 14 days. After that, the cells were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 15 min. The cell colonies were photographed and counted with a light microscope.
Cell scratch wound assay
In brief, 5 × 107 cells per well were seeded in a 24-well plate and cultured overnight. After that, these cells were scratched with a 200-μl pipette tip to create a wound and washed twice with phosphate-buffered saline (PBS). The cells were photographed at 0, 24, and 48 h, and the width of the wounds were subsequently measured. The wound width in each group was tested in three replicates.
Statistical analyses
R software version 3.6.0 was used to integrate and analyze the data. A paired sample t-test was used to compare continuous variables between two groups. Continuous variables were shown as the mean ± standard deviation. Fisher’s exact test was applied to compare the categorical variables among groups. The figures were produced using GraphPad Prism 8.0 (GraphPad Software, Inc. La Jolla, CA, USA). A p-value < 0.05 was considered statistically significant.
Author Contributions
The conception and design of the study, as well as manuscript writing were performed by Xin Zhao and Xinxue Zhang; administrative support was provided by Qiang He and Lixin Li; the provision of study materials was by Ren Lang, Zhe Liu, and Jiqiao Zhu; the collection and assembly of data were by Zhigang Zhang; the experiments were performed by Shaocheng Lyu. All authors read and approved the final manuscript.
Acknowledgments
We thank Drs. Hua Fan, Xianliang Li, Yu Liu, and Jiantao Kou for general support and providing samples.
Conflicts of Interest
The authors disclose no potential conflicts of interest.
Funding
This work was supported by the Beijing Chao-Yang Hospital Talent Development Program (17-1351).
References
- 1. Hoyos S, Navas MC, Restrepo JC, Botero RC. Current controversies in cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis. 2018; 1864:1461–67. https://doi.org/10.1016/j.bbadis.2017.07.027 [PubMed]
- 2. Strijker M, Belkouz A, van der Geest LG, van Gulik TM, van Hooft JE, de Meijer VE, Haj Mohammad N, de Reuver PR, Verheij J, de Vos-Geelen J, Wilmink JW, Groot Koerkamp B, Klümpen HJ, Besselink MG, and Dutch Pancreatic Cancer Group. Treatment and survival of resected and unresected distal cholangiocarcinoma: a nationwide study. Acta Oncol. 2019; 58:1048–55. https://doi.org/10.1080/0284186X.2019.1590634 [PubMed]
- 3. Courtin-Tanguy L, Turrini O, Bergeat D, Truant S, Darnis B, Delpero JR, Mabrut JY, Regenet N, Sulpice L. Multicentre study of the impact of factors that may affect long-term survival following pancreaticoduodenectomy for distal cholangiocarcinoma. HPB (Oxford). 2018; 20:405–10. https://doi.org/10.1016/j.hpb.2017.10.016 [PubMed]
- 4. Kim BH, Kim K, Chie EK, Kwon J, Jang JY, Kim SW, Oh DY, Bang YJ. Long-term outcome of distal cholangiocarcinoma after pancreaticoduodenectomy followed by adjuvant chemoradiotherapy: a 15-year experience in a single institution. Cancer Res Treat. 2017; 49:473–83. https://doi.org/10.4143/crt.2016.166 [PubMed]
- 5. Yuan X, Yuan Y, He Z, Li D, Zeng B, Ni Q, Yang M, Yang D. The regulatory functions of circular RNAs in digestive system cancers. Cancers (Basel). 2020; 12:770. https://doi.org/10.3390/cancers12030770 [PubMed]
- 6. Hsiao KY, Sun HS, Tsai SJ. Circular RNA - new member of noncoding RNA with novel functions. Exp Biol Med (Maywood). 2017; 242:1136–41. https://doi.org/10.1177/1535370217708978 [PubMed]
- 7. Zhu CL, Sha X, Wang Y, Li J, Zhang MY, Guo ZY, Sun SA, He JD. Circular RNA hsa_circ_0007142 is upregulated and targets miR-103a-2-5p in colorectal cancer. J Oncol. 2019; 2019:9836819. https://doi.org/10.1155/2019/9836819 [PubMed]
- 8. Chen L, Nan A, Zhang N, Jia Y, Li X, Ling Y, Dai J, Zhang S, Yang Q, Yi Y, Jiang Y. Circular RNA 100146 functions as an oncogene through direct binding to miR-361-3p and miR-615-5p in non-small cell lung cancer. Mol Cancer. 2019; 18:13. https://doi.org/10.1186/s12943-019-0943-0 [PubMed]
- 9. Xu Y, Yao Y, Zhong X, Leng K, Qin W, Qu L, Cui Y, Jiang X. Downregulated circular RNA hsa_circ_0001649 regulates proliferation, migration and invasion in cholangiocarcinoma cells. Biochem Biophys Res Commun. 2018; 496:455–61. https://doi.org/10.1016/j.bbrc.2018.01.077 [PubMed]
- 10. Jiang XM, Li ZL, Li JL, Xu Y, Leng KM, Cui YF, Sun DJ. A novel prognostic biomarker for cholangiocarcinoma: circRNA Cdr1as. Eur Rev Med Pharmacol Sci. 2018; 22:365–71. https://doi.org/10.26355/eurrev_201801_14182 [PubMed]
- 11. Xu Y, Yao Y, Liu Y, Wang Z, Hu Z, Su Z, Li C, Wang H, Jiang X, Kang P, Sun D, Zhong X, Cui Y. Elevation of circular RNA circ_0005230 facilitates cell growth and metastasis via sponging miR-1238 and miR-1299 in cholangiocarcinoma. Aging (Albany NY). 2019; 11:1907–17. https://doi.org/10.18632/aging.101872 [PubMed]
- 12. Huang XY, Huang ZL, Zhang PB, Huang XY, Huang J, Wang HC, Xu B, Zhou J, Tang ZY. CircRNA-100338 is associated with mTOR signaling pathway and poor prognosis in hepatocellular carcinoma. Front Oncol. 2019; 9:392. https://doi.org/10.3389/fonc.2019.00392 [PubMed]
- 13. Zhang Y, Liu H, Li W, Yu J, Li J, Shen Z, Ye G, Qi X, Li G. CircRNA_100269 is downregulated in gastric cancer and suppresses tumor cell growth by targeting miR-630. Aging (Albany NY). 2017; 9:1585–94. https://doi.org/10.18632/aging.101254 [PubMed]
- 14. Chan JJ, Tay Y. Noncoding RNA:RNA regulatory networks in cancer. Int J Mol Sci. 2018; 19:1310. https://doi.org/10.3390/ijms19051310 [PubMed]
- 15. Ojha R, Nandani R, Chatterjee N, Prajapati VK. Emerging role of circular RNAs as potential biomarkers for the diagnosis of human diseases. Adv Exp Med Biol. 2018; 1087:141–57. https://doi.org/10.1007/978-981-13-1426-1_12 [PubMed]
- 16. Zhang Z, Yang T, Xiao J. Circular RNAs: promising biomarkers for human diseases. EBioMedicine. 2018; 34:267–74. https://doi.org/10.1016/j.ebiom.2018.07.036 [PubMed]
- 17. Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018; 15:95–111. https://doi.org/10.1038/nrclinonc.2017.157 [PubMed]
- 18. Li H, Long J, Xie F, Kang K, Shi Y, Xu W, Wu X, Lin J, Xu H, Du S, Xu Y, Zhao H, Zheng Y, Gu J. Transcriptomic analysis and identification of prognostic biomarkers in cholangiocarcinoma. Oncol Rep. 2019; 42:1833–42. https://doi.org/10.3892/or.2019.7318 [PubMed]
- 19. Wu M, Li X, Liu Q, Xie Y, Yuan J, Wanggou S. miR-526b-3p serves as a prognostic factor and regulates the proliferation, invasion, and migration of glioma through targeting WEE1. Cancer Manag Res. 2019; 11:3099–110. https://doi.org/10.2147/CMAR.S192361 [PubMed]
- 20. Pan Y, Liang W, Zhao X, Liu L, Qing Y, Li Y. miR-548b inhibits the proliferation and invasion of Malignant gliomas by targeting metastasis tumor-associated protein-2. Neuroreport. 2016; 27:1266–73. https://doi.org/10.1097/WNR.0000000000000690 [PubMed]
- 21. Wang Z, Wu X, Hou X, Zhao W, Yang C, Wan W, Chen L. miR-548b-3p functions as a tumor suppressor in lung cancer. Lasers Med Sci. 2020; 35:833–39. https://doi.org/10.1007/s10103-019-02865-7 [PubMed]
- 22. Li M, Wang Y, Liu X, Zhang Z, Wang L, Li Y. miR-629 targets FOXO3 to promote cell apoptosis in gastric cancer. Exp Ther Med. 2020; 19:294–300. https://doi.org/10.3892/etm.2019.8168 [PubMed]
- 23. Yan H, Li Q, Wu J, Hu W, Jiang J, Shi L, Yang X, Zhu D, Ji M, Wu C. MiR-629 promotes human pancreatic cancer progression by targeting FOXO3. Cell Death Dis. 2017; 8:e3154. https://doi.org/10.1038/cddis.2017.525 [PubMed]
- 24. Wang H, Xing D, Ren D, Feng W, Chen Y, Zhao Z, Xiao Z, Peng Z. MicroRNA-643 regulates the expression of ZEB1 and inhibits tumorigenesis in osteosarcoma. Mol Med Rep. 2017; 16:5157–64. https://doi.org/10.3892/mmr.2017.7273 [PubMed]
- 25. Yang C, Ma X, Guan G, Liu H, Yang Y, Niu Q, Wu Z, Jiang Y, Bian C, Zang Y, Zhuang L. MicroRNA-766 promotes cancer progression by targeting NR3C2 in hepatocellular carcinoma. FASEB J. 2019; 33:1456–67. https://doi.org/10.1096/fj.201801151R [PubMed]
- 26. Zou S, Rao Y, Chen W. miR-885-5p plays an accomplice role in liver cancer by instigating TIGAR expression via targeting its promoter. Biotechnol Appl Biochem. 2019; 66:763–71. https://doi.org/10.1002/bab.1767 [PubMed]
- 27. Jia B, Xia L, Cao F. The role of miR-766-5p in cell migration and invasion in colorectal cancer. Exp Ther Med. 2018; 15:2569–74. https://doi.org/10.3892/etm.2018.5716 [PubMed]
- 28. Su M, Qin B, Liu F, Chen Y, Zhang R. miR-885-5p upregulation promotes colorectal cancer cell proliferation and migration by targeting suppressor of cytokine signaling. Oncol Lett. 2018; 16:65–72. https://doi.org/10.3892/ol.2018.8645 [PubMed]
- 29. Kim SM, Vetrivel P, Ha SE, Kim HH, Kim JA, Kim GS. Apigetrin induces extrinsic apoptosis, autophagy and G2/M phase cell cycle arrest through PI3K/AKT/mTOR pathway in AGS human gastric cancer cell. J Nutr Biochem. 2020; 83:108427. https://doi.org/10.1016/j.jnutbio.2020.108427 [PubMed]
- 30. El-Huneidi W, Shehab NG, Bajbouj K, Vinod A, El-Serafi A, Shafarin J, Bou Malhab LJ, Abdel-Rahman WM, Abu-Gharbieh E. Micromeria fruticosa induces cell cycle arrest and apoptosis in breast and colorectal cancer cells. Pharmaceuticals (Basel). 2020; 13:115. https://doi.org/10.3390/ph13060115 [PubMed]
- 31. Choi C, Cho WK, Park S, Shin SW, Park W, Kim H, Choi DH. Checkpoint kinase 1 (CHK1) inhibition enhances the sensitivity of triple-negative breast cancer cells to proton irradiation via Rad51 downregulation. Int J Mol Sci. 2020; 21:2691. https://doi.org/10.3390/ijms21082691 [PubMed]
- 32. Ma Q, Liu Y, Shang L, Yu J, Qu Q. The FOXM1/BUB1B signaling pathway is essential for the tumorigenicity and radioresistance of glioblastoma. Oncol Rep. 2017; 38:3367–75. https://doi.org/10.3892/or.2017.6032 [PubMed]
- 33. Cheung CH, Hsu CL, Chen KP, Chong ST, Wu CH, Huang HC, Juan HF. MCM2-regulated functional networks in lung cancer by multi-dimensional proteomic approach. Sci Rep. 2017; 7:13302. https://doi.org/10.1038/s41598-017-13440-x [PubMed]
- 34. Ying H, Xu Z, Chen M, Zhou S, Liang X, Cai X. Overexpression of zwint predicts poor prognosis and promotes the proliferation of hepatocellular carcinoma by regulating cell-cycle-related proteins. Onco Targets Ther. 2018; 11:689–702. https://doi.org/10.2147/OTT.S152138 [PubMed]
- 35. Song ZY, Chao F, Zhuo Z, Ma Z, Li W, Chen G. Identification of hub genes in prostate cancer using robust rank aggregation and weighted gene co-expression network analysis. Aging (Albany NY). 2019; 11:4736–56. https://doi.org/10.18632/aging.102087 [PubMed]
- 36. Ning Z, Wang A, Liang J, Liu J, Zhou T, Yan Q, Wang Z. Abnormal expression of Nek2 in pancreatic ductal adenocarcinoma: a novel marker for prognosis. Int J Clin Exp Pathol. 2014; 7:2462–69. [PubMed]
- 37. Zhang MX, Xu XM, Zhang P, Han NN, Deng JJ, Yu TT, Gan YY, He XQ, Long ZX. Effect of silencing NEK2 on biological behaviors of HepG2 in human hepatoma cells and MAPK signal pathway. Tumour Biol. 2016; 37:2023–35. https://doi.org/10.1007/s13277-015-3993-y [PubMed]
- 38. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–87. https://doi.org/10.1089/omi.2011.0118 [PubMed]
- 39. Maag JL. Gganatogram: an R package for modular visualisation of anatograms and tissues based on ggplot2. F1000Res. 2018; 7:1576. https://doi.org/10.12688/f1000research.16409.2 [PubMed]
- 40. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–52. https://doi.org/10.1093/nar/gku1003 [PubMed]
- 41. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003; 4:2. https://doi.org/10.1186/1471-2105-4-2 [PubMed]
- 42. Oishi N, Kumar MR, Roessler S, Ji J, Forgues M, Budhu A, Zhao X, Andersen JB, Ye QH, Jia HL, Qin LX, Yamashita T, Woo HG, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology. 2012; 56:1792–803. https://doi.org/10.1002/hep.25890 [PubMed]