


Introduction
AD is a progressive, irreversible neurodegenerative disease that accounts for more than half of the 44 million cases of dementia globally [1]. AD can be either sporadic or familial (inherited). The greatest risk factor for the onset of AD is ageing, and the World Health Organization predicts that by 2050 the number of people over the age of 60 will have increased to 2 billion [2]. With that, the number of people living with AD will increase as well as the economic costs of supporting and treating AD patients.
Symptomatically, AD is initially recognised by mild cognitive impairment (MCI) and problems with short- and long-term memory. As the disease progresses neuropsychiatric symptoms can develop including affective, psychomotor, psychotic and manic syndromes [3]. There are two distinct biomolecular markers within the brain that have long been known to characterise AD, amyloid plaques composed of the amyloid-β (Aβ) peptide and neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau proteins [4]. However, due to their location AD can only be diagnosed using these markers post-mortem.
Mitochondria are also widely observed as dysfunctional in AD, which has resulted in the development of the mitochondrial cascade hypothesis [5, 6]. The dysfunction of mitochondria, and in particular the ETC, has been coupled with the oxidative stress observed in AD [7, 8]. It has been widely debated as to whether amyloid plaques, NFTs or dysfunctional mitochondria play the primary role in the aetiology of AD. We now understand that interactions actually take place between these different biomolecular markers contributing to disease progression [9, 10].
When considering the role of mitochondria in AD, ATP synthase has not been widely discussed. ATPases are present across eukaryotes, prokaryotes and archaea. They can be placed into one of three different classes: F-type, V-type or A-type, similar in structure but differing in function [11, 12]. Mitochondrial ATP synthase is an F-type ATPase and is the final ETC complex of the IMM. It is responsible for the pumping of protons from the inter-membrane space into the matrix while harnessing the chemical energy from this process. The chemical energy is converted into mechanical energy that allows the complex to behave as a molecular motor. Rotation of the motor triggers conformational changes in the catalytic domain of the enzyme that enables the production of ATP, the cellular energy currency of which an estimated 50kg a day is required by the body, from ADP and Pi[13]. In medical research, ATP synthase has been more widely studied in classic mitochondria disorders such as Leigh Syndrome [14, 15].
This review is a synthesis of the data which implicate ATP synthase in the pathology of AD. It then considers ways in which ATP synthase can be therapeutically targeted in order to try and prevent disease onset or to alleviate symptoms.
ATP synthase
What is ATP synthase
F-type ATP synthase is the fifth and final ETC complex of the IMM. It has a large structure with a molecular weight of around 600 kDa and is composed of up to 20 different subunits in mammals [16]. ATP synthase is responsible for the production of the cellular energy carrier ATP from ADP and Pi. This process is driven by the chemiosmotic potential across the IMM first described by Peter Mitchell in the 1960s [17–19]. While F-type ATP synthase is predominantly housed within mitochondria, data have shown that F-type ATP synthase is present at plasma membranes of different cell types both physiologically and pathophysiologically [20, 21].
After ATP synthase’s function was described in vitro, landmark measurements including the kinetic parameters of its three-site cooperative-binding catalytic mechanism and the discovery that protein conformational changes would facilitate the release of tightly bound ATP were reported [22, 23]. The atomic structure of the complex was resolved to 2.8 Å in 1994, revealing a structure which supported the mechanism of rotary catalysis [24]. Since 1994, multiple atomic structures of both eukaryotic and prokaryotic ATP synthase structures have been published, with a recent cryo-EM structure of ATP synthase from S. scrofa shown in Figure 1 [25–28]. ATP synthase consists of two distinct components; a membrane bound FO component and a matrix exposed F1 component. They function cooperatively through a central rotor stalk and a peripheral stator stalk.
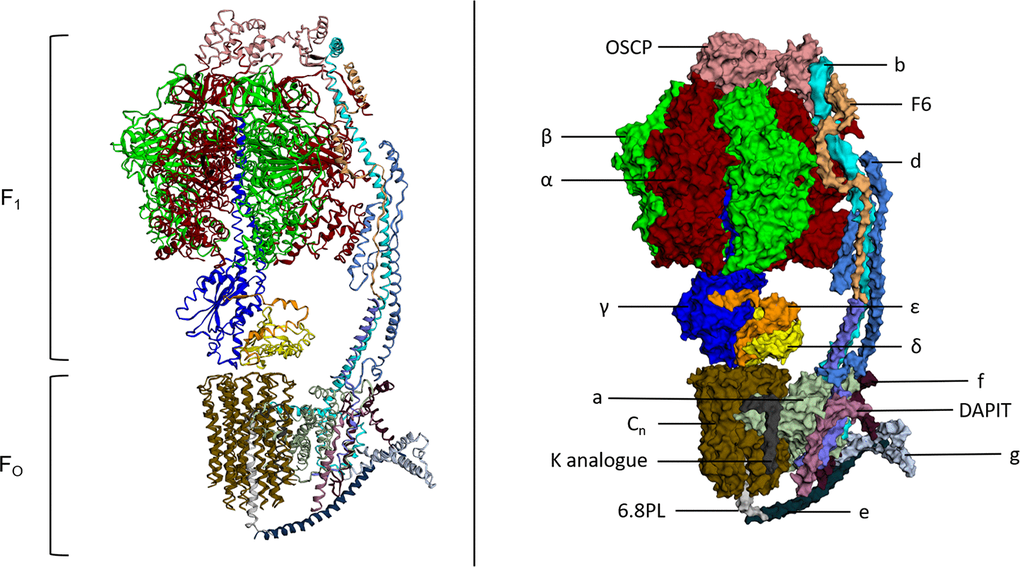
Figure 1. Atomic structure and labelled space fill model of ATP Synthase (S. scrofa). FO and F1 components of the complex both labelled. Individual subunits labelled on the space fill model. This figure was created using image 6J5J from PDB (http://doi.org/10.2210/pdb6J5J/pdbhttps://www.rcsb.org/structure/6J5J) and processed using http://www.sbg.bio.ic.ac.uk/ezmol/.
FO
The FO component of ATP synthase is an insoluble structure that is primarily composed of a ring of varying numbers of c-subunits called the c-ring and has a size that appears to be species dependent [29]. Other FO proteins include subunit a and subunit b as well as others with less well understood roles including subunits d, e, f, g, F6 and 8 (A6L). DAPIT and 6.8PL are present in vertebrates and assist in the assembly of the FO component of ATP synthase [30, 31]. An additional subunit called the oligomycin sensitivity conferring protein (OSCP) is located at the top of the F1 component of ATP synthase. It couples the FO component with the F1 component through its interaction with the peripheral stalk of FO and central stalk of F1 [32]. Protons from the intermembrane space of the mitochondria travel through an aqueous half-channel in subunit a to the c-ring of the FO complex where they bind to conserved acidic c-ring residues, aspartate or glutamate, in the second transmembrane helix of subunit-c [13, 33–35]. These charged proton binding sites are then suggested to be concealed by rotation of α-helices in c subunits which leads to c-ring to rotation along with the central rotary stalk γ-subunit [36, 37]. The rotating FO component transports protons into the matrix through a second aqueous half channel on the matrix side of the membrane and the asymmetric rotor stalk causes conformational changes in F1 which drive the catalytic activity of the β subunits [13].
The role of OSCP
The oligomycin sensitivity conferring protein (OSCP) is part of the peripheral stalk of the FO component of ATP synthase and physically couples the two enzyme components together through its interaction with the central stalk of the F1 component. It is encoded by the ATP5O gene on the long arm of the nuclear chromosome 21. Structurally, OSCP has an N-terminal domain which contains six α-helices and a C-terminal domain consisting of a β-hairpin and two α-helices [32, 38]. While oligomycin does not bind to OSCP, OSCP confers the enzyme’s sensitivity to the antibiotic as it is OSCP that couples the F1 component to the FO component that is bound and inhibited by oligomycin [39].
F1
The F1 component is solvent exposed and far more about its activity and structure is understood than its FO counterpart. Its subunit composition is α3β3γδε, with its structure being a six part barrel of alternating α and β subunits, a central asymmetric γ subunit (the aforementioned rotor stalk) protruding through the centre of the barrel while the δ and ε subunits are found at the matrix exposed surface of the FO c-ring [24]. The site of catalysis is located at the interface of the α and β subunits, both of which have nucleotide binding sites and multiple studies of atomic structures have shown nucleotides bound at this interface [40–42]. Interestingly, both α and β subunits possess the same folds despite only sharing around 20% sequence homology [16]. The structural similarities are presented in Figure 2 using the atomic structures of ATP synthase in S. scrofa [27, 43]. Despite the similarity, only the β-subunit possesses catalytic activity due to its ability to form an open conformation as well as possessing a catalytic base for the reverse ATP hydrolysis reaction [44, 45].
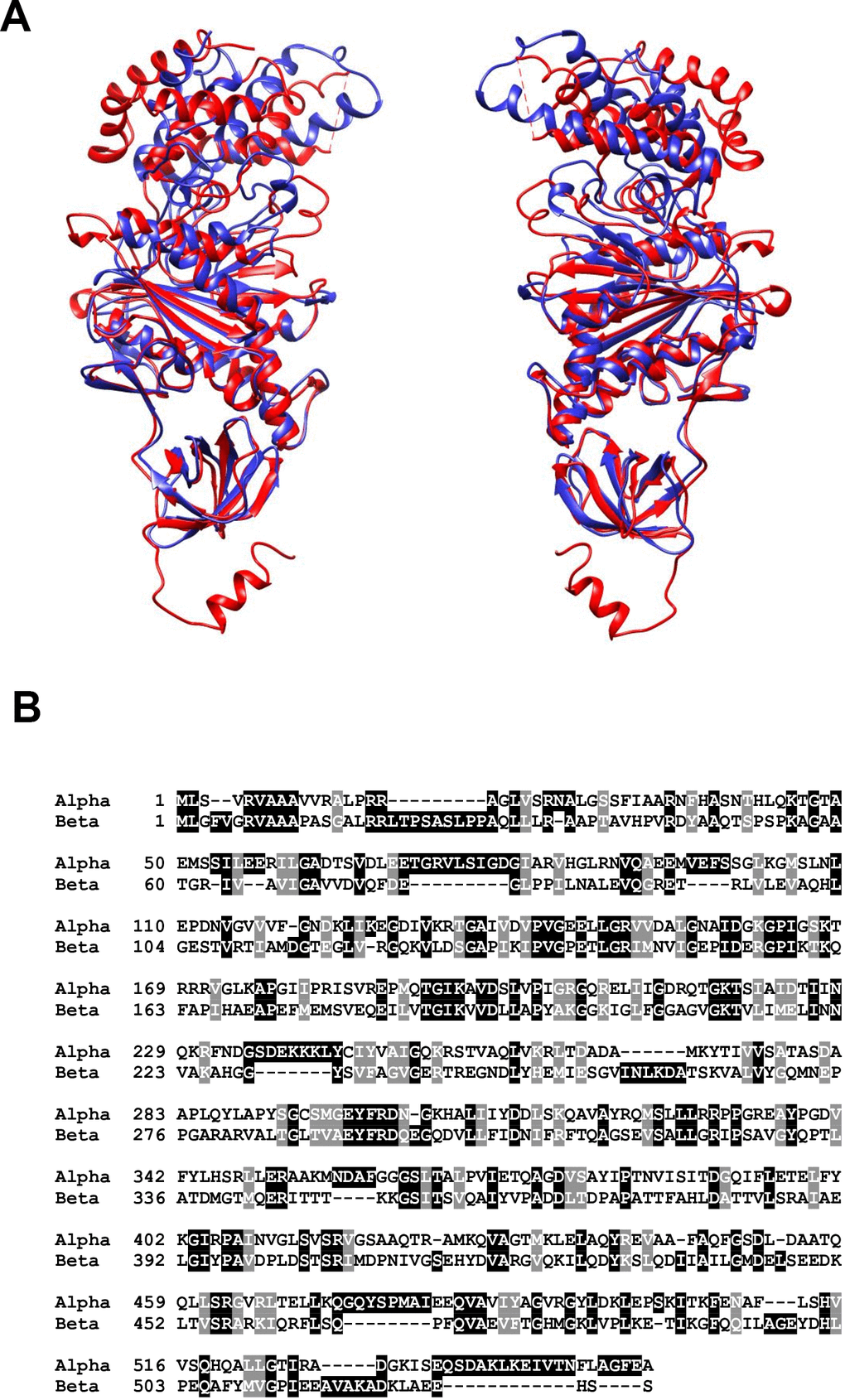
Figure 2. (A) Structural alignment of the alpha (red) and beta (blue) subunits of mitochondrial ATP synthase in S. scrofa. Both subunits are reproduced from image 6J5J in PDB (https://www.rcsb.org/structure/3ZIAhttp://doi.org/10.2210/pdb3ZIA/pdb) and processed using http://www.cgl.ucsf.edu/chimera/. (B) BLAST alignment of the primary amino acid sequences of H. sapiens alpha (UniProt P25705) and beta (UniProt P06576) subunits performed using https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins.
The role of the α-subunit
The α-subunit of the F-type ATP synthase is located in the F1 solvent exposed component of ATP synthase, facing the mitochondrial matrix [24]. It functions as part of a six-part barrel structure (α3β3), and the catalytic nucleotide binding site is located at its interface with the β-subunit [40]. However, the α-subunit displays regulatory activity when compared with the β-subunit which exhibits the catalytic activity of the enzyme [44, 45]. Like its β-subunit counterpart, the α-subunit can be divided into three different domains: a small N-terminal domain, a nucleotide binding domain and a helical C-terminal domain.
The role of the β-subunit
The β-subunit is also located in the solvent exposed F1 component of ATP synthase and it has a largely similar structure to the α-subunit. Its interface with the α-subunit forms the nucleotide binding site and it is the β-subunit that possesses the catalytic activity required for both the synthesis and hydrolysis of ATP. The β-subunit is able to undergo conformational changes to form three distinct conformations in response to the rotation of the γ-subunit in 120° increments [46]. This, coupled with critical arginine, lysine and glutamate residues is what enables β-subunit to catalyse the synthesis and hydrolysis of ATP [47]. For well-illustrated figures of this mechanism, see Feniouk et al., 2008 and Okuno et al., 2011 [48, 49].
ATP synthase in Alzheimer’s Disease (AD)
Mitochondria are known to be dysfunctional in AD patients and this has resulted in the development of the mitochondrial cascade hypothesis [5]. This hypothesis has been developed and revisited several times across the last two decades, serving as a viable alternative to the predominant amyloid hypothesis [6, 50–52]. Much of the focus of the research has been based on the oxidation levels found in the brains of AD patients, and how this observation can be synthesised with the mitochondrial theory of ageing [4, 53]. Despite its physiological relevance to both mitochondrial activity and structure, little attention has been paid to ATP synthase in the formation and development of this theory [54].
The first study implicating ATP synthase in AD aetiology found, through BN-PAGE analysis, decreased expression of the whole complex in the hippocampal tissue of AD patients [55]. Since then, multiple studies have pointed to a decrease in the expression of ATP synthase subunits and they are addressed in this review. There was decreased expression in several of the nuclear encoded ATP synthase genes in the posterior cingulate cortex (11), hippocampal field CA1 (10), middle temporal gyrus (9) and entorhinal cortex (5) [56]. Adult neurogenesis defects are common in AD and it has been suggested that this arises from impaired function of hippocampal neuronal stem cells (NSCs). A study using iPSC-derived NSCs, with familial AD (FAD) associated PS1 mutation M146L, observed a decreased expression of the ATP synthase complex while PS1 expression was kept at physiological levels [57]. In a study with implications for sporadic AD, N2a neuroblastoma cells expressing the ApoE4 allele of the ApoE gene, the major genetic risk factor for sporadic AD, showed a reduction in the levels of all ATP synthase subunits they detected in comparison to ApoE3 controls [58].
Considering ATP synthase activity instead of protein expression, an early study investigating AD and ATP synthase found no significant decrease in the enzyme’s catalytic activity when studying the isolated mitochondria from AD patient hippocampal tissue, motor cortex and platelets [59]. However, since this study was published there have been data published that are contradictory to this observation and these are discussed in the following sections of this review.
The α-subunit in AD
ATP synthase subunit α, Amyloid β and NFTs
Transgenic Swedish APP mice (Tg2576) had increased levels of amyloid plaque formation in the brain as they aged, compared with controls. Proteomic analysis of the brains from the Tg2576 mice found that the increase in amyloid plaque deposition with age correlated with an increase in the expression of the α-subunit [60].
An N-glycosylated form of the α-subunit has been shown to act as a binding partner of the extracellular domain of APP and Aβ, with Aβ being the primary component of the AD hallmark amyloid plaques. The α-subunit reaches the membrane via the secretory pathway and it is during this process that it becomes N-glycosylated. Schmidt et al. also demonstrated the localisation of the whole ATP synthase complex at the neuronal membrane and that its extracellular ATPase activity is inhibited by both APP and Aβ. This is especially noteworthy as APP and Aβ share sequence homology with the native ATPase inhibitory factor IF1 [61]. The inhibition was shown to downregulate long-term potentiation (LTP) at the synapses, that Aβ oligomers have since been shown to inhibit alongside the upregulation of long-term depression (LTD) via the NMDA receptors [62].
A study that builds upon the work of Schmidt et al. found that in the cortex and hippocampus of Tg APP PS1 mice, the α-subunit co-localises with insoluble plaques of Aβ – not just the soluble monomeric form of the peptide [63]. Moreover, the authors showed that this interaction occurs at the plasma membrane of neuronal cells, causes inhibition of enzymatic activity and a decrease in the levels of extracellular ATP. These decreases of extracellular ATP may be critical in the cognitive defects which arise in AD due to disruptions in synaptic plasticity, given the important role extracellular ATP plays in LTP [64, 65].
The α-subunit has been observed as part of the NFTs in human AD patient brain samples, one of the characteristic observations in AD patient brains. Monoclonal antibodies that target the insoluble brain lesions in AD found that the α-subunit acted as an antigen to one of the antibodies (AD46). Immunohistochemistry and electron microscopy confirmed the co-localisation of the α-subunit with the NFTs in the cytosol of a degenerating AD neuron [66].
ATP synthase subunit α and oxidative stress
Oxidative stress is a frequently observed phenomenon of AD. C. elegans that over-expressed green fluorescent protein (GFP) as a means of studying the oxidative stress caused by protein aggregation presented carbonylation of the α-subunit [67]. In the hippocampus of AD patients the α-subunit was also shown to be excessively nitrated in comparison to age-matched control brains, as well as having significantly increased protein levels [68].
Another marker of oxidative stress is the level of lipid peroxidation, that arises from the reaction of oxygen radicals with lipids to produce reactive aldehydes. One such example of this is 4-hydroxy-2-nonenal (4-HNE), that covalently attaches to proteins in a Michael addition reaction [69]. The α-subunit of ATP synthase was shown to be HNE modified in the hippocampal tissue of individuals with mild cognitive impairment (MCI), which is symptomatic of early stage AD [70, 71]. The same study also showed that, in the same tissue from MCI patients, ATP synthase had a 35% decrease in activity compared to age-matched controls when measured as a function of ADP production.
A study investigating oxidative stress in the early stages of AD (Braak stages I and II, prior to the onset of MCI) found that the α-subunit of mitochondrial ATP synthase is HNE modified in the entorhinal cortex and that ATP synthase has a decrease in activity of around 30% [72, 73]. The authors chose to use the entorhinal cortex tissue for this study as it is the location of NFTs used to track AD progression during Braak stages I and II. When these data are taken with those from Reed et al. showing 4-HNE modification of the α-subunit and decreased ATP synthase activity, there appears to be correlation between disease progression as measured by the presence of NFTs and the lipoxidation of the α-subunit resulting in reduced ATP synthase activity. Further, the presence of oxidative stress and diminished ATP synthase activity from the earliest stages of AD onset may prove critical to the pathology of the disease. If this oxidative stress precedes the presence of Aβ in the affected tissue, it raises additional questions about the primacy of the amyloid pathology in the aetiology of AD.
Whether or not the α-subunit is oxidised may be dependent upon the tissue that it is found in and the stage of AD pathology in which it is being considered. A line of transgenic mice (J20 Tg) expressing a mutant form of APP that corresponds to the Swedish and Indiana familial forms of AD had a 12.2-fold increase in the expression of the α-subunit in a whole mouse brain homogenate compared with non-Tg mouse brain homogenate [74]. However, there was no indication of oxidation when measured as a function of 3-nitrotyrosine (3-NT) modification of the protein. The authors suggest that this significant increase in the expression of the protein could be a related to cellular stress responses by the brain to maintain energy production. Future studies should look to measure the α-subunit expression of early, middle and late Braak stages of AD in brain tissues shown to have reduced ATP synthase activity as a way to try and validate their suggestion.
Post-translational modification of the α-subunit
Glycosylation of proteins with O-linked β-N-acetylglucosamine (O-GlcNAc) is a widely observed post-translational modification that regulates intracellular events [75]. The α-subunit can be O-GlcNAcylated on the Thr432 residue. However, this modification is reduced in the brains of AD patients, Tg AD mice and in Aβ treated mammalian cell cultures – which resulted in reduced ATP levels [76]. Molecular modelling and co-IP experiments with deletion mutants of the α- and β-subunits with no pocket site showed that Aβ directly blocks the O-GlcNAcylation of the Thr432 residue by mitochondrial O-GlcNAc transferase. Interestingly, the O-GlcNAcylation of Thr432 that had been inhibited by Aβ was rescued by treatment with O-GlcNAcase inhibitor. These findings are particularly noteworthy as they demonstrate a chemical mechanism for the interaction of the Aβ peptide with mitochondrial ATP synthase, and as a result could offer a potential therapeutic target for AD.
The β-subunit in AD
Downregulation of the β-subunit
Several studies have found changes in the expression of the β-subunit of ATP synthase in AD tissue samples and models of AD, and in particular reductions in its expression. Table 1 lists changes in protein expression of ATP synthase subunits, including the β-subunit, that are presented in this review. An early observed instance of reduced expression is the reduction of β-subunit mRNA levels by over 50% in the midtemporal cortex of AD patient brains compared with age-matched controls [77]. In another study, that linked Aβ peptides with ATP synthase in AD, rats that received a bilateral intrahippocampal injection of Aβ showed a significant decrease in the levels of β-subunit compared with controls [78]. Gene expression analysis of the entorhinal cortex of AD patient brains showed reduced expression of ATP5C1 (γ-subunit), ATP5D (δ-subunit), ATP5G1 (subunit c) and ATP5B (β-subunit) [79]. This strengthens the argument that ATP synthase dysfunction plays a role in the disrupted glycometabolism of AD. It must be noted that these studies do not provide a mechanism of how the expression of the β-subunit is downregulated, but early gene mapping studies of the β-subunit reported that ETS domain transcription factors and redox sensitive OXBOX and REBOX transcription factors regulate gene expression [80–82].
Table 1. Regulation of individual ATP synthase subunit protein expression levels across different tissue samples from different AD models, summarising data presented in this review.
| Model | Tissue | α-subunit | β-subunit | OSCP | Subunit d | δ-subunit | Reference |
| Aβ injected rat | Hippocampus | Down | Shi, X. et al., 2011 | ||||
| SweAPP Tg mice | Whole brain homogenate | Up | Carrette, O. et al., 2006 | ||||
| 4 months old 5xFAD mice | Synaptic mitochondria | Down | Beck, S. J. et al., 2016 | ||||
| 9 months old 5xFAD mice | Synaptic mitochondria | Down | Beck, S. J. et al., 2016 | ||||
| Non-synaptic mitochondria | Down | Beck, S. J. et al., 2016 | |||||
| 3x Tg AD mouse | Hippocampus | Down | Yu, H. et al., 2018 | ||||
| AD patient | Temporal lobe | Down | Beck, S. J. et al., 2016 | ||||
| Medial frontal gyrus | Down | Adav, S. S. et al., 2019 | |||||
| Temporal cortex | Down | Mukherjee, S. et al., 2017 | |||||
| Frontal cortex | Up | Manczak, M. et al., 2004 |
Autoimmune response to the β-subunit
Autoimmunity is now thought to play a role in the onset of AD [83, 84]. While this hypothesis has not been developed to the same extent as the amyloid, tau and mitochondrial hypotheses, the idea is grounded in the fact that anti-neuronal antibodies have been found in the sera of AD patients. Notably, it was found that the brain of AD patients contains antibodies which target the c-terminal domain of the β-subunit [85]. In neuroblastoma cell lines these antibodies caused a dose dependent decrease in the activity of the ATP synthase complex, and then most strikingly, apoptosis. The apoptotic event was preceded by IMM hyperpolarization and then depolarization.
A study that followed this showed that mice injected into their right cerebral ventricle with anti-β-subunit antibodies isolated from AD patient sera had reduced memory retention [86]. Additionally, an increased rate of apoptosis was detected in the dorsal hippocampal regions of their brains, post-mortem. Taken together, these two studies show a mechanism for the antibodies detected in the brain sera of AD patients to cause neuronal apoptosis and cognitive impairment, both of which are classical symptoms of AD.
Excitotoxicity and cyclin-B1 accumulation
Excitotoxicity is common to neurons in AD and may be mediated by the action of glutamate on the NMDA receptors of excitatory post-synaptic neurons [87]. Interestingly, a mechanism of ATP synthase inhibition via action on the β-subunit has been elucidated in rat cortical neurons and HEK293T cells using glutamate induced excitotoxicity [88]. Cell cultures of rat cortical neurons were treated with glutamate which resulted in an accumulation of cyclin-B1, the cyclin-B1 was shown to form complexes with Cdk1 which accumulated in mitochondria resulting in superoxide production. HEK293T cells were then used in the study to demonstrate that the cyclin-B1-Cdk1 complex phosphorylates Bcl-xL causing its dissociation from the β-subunit of the ATP synthase, a reduction in the enzyme’s catalytic activity and increased oxidative stress. Bcl-xL is a transmembrane mitochondrial protein that acts as a regulator of cell death through its action on proapoptotic factors [89]. Bcl-xL has also been shown to improve the efficiency of neuronal metabolism through its interaction with ATP synthase which decreases membrane-ion leakage [90]. From these data it is clear that a disruption of the interaction between the β-subunit and Bcl-xL could contribute to AD pathology.
OSCP in AD
OSCP downregulation in AD
In 2016 a comprehensive study was published investigating changes in expression of OSCP in the brains of human AD patients, MCI patients and Tg AD mice brains (5xFAD mice) [91]. OSCP was shown to be significantly downregulated in the temporal lobe of AD patients compared to controls. There was also a significant decrease in OSCP expression between the synaptic mitochondria of young and old 5xFAD mice compared with controls as well as in the non-synaptic mitochondria of old 5xFAD mice. Primary cultured mice neurons with downregulated OSCP showed decreased membrane potential, reduced ATP synthesis and elevated levels of superoxide. Beck et al. also demonstrated that there is a physical interaction between Aβ and the OSCP in brain mitochondria which reduced ATP synthase activity, which is supported by evidence of Aβ localising to brain mitochondria [92, 93]. This study is notable as it provides mechanistic detail and also presents another case of Aβ peptides interacting with ATP synthase subunits in a detrimental fashion, as is the case with the α-subunit.
Interaction with Cyclophilin D (Ppif)
Cyclophilin D (CypD) is one of the only proteins which appears to be essential to the elusive molecular make-up of the mitochondrial permeability transition pore (mPTP) [94]. CypD has also been demonstrated to interact with ATP synthase, regulating the formation of the respiratory efficiency enhancing synthasome [95]. Of note, it was also shown that synthasome assembly and mPTP formation are inversely proportional. A study of mice in 2017 found that CypD levels increased with ageing, as did the physical interaction between OSCP and CypD, despite a decrease in the expression levels of OSCP [96]. These changes resulted in decreased ATP synthase activity and an increase in mitochondrial dysfunction, including a decreased ATP:oxygen ratio. A follow up study found that the temporal lobe of AD patient brains and 5xFAD mice had increased formation of CypD-OSCP complexes, and that the presence of Aβ substantially decreased the KD of this interaction [97]. The authors also showed that in 5xFAD mice CypD promotes the OSCP-Aβ interaction as well as the ubiquitin mediated degradation of OSCP. However, CypD deficient 5xFAD mice had improved cognitive function and attenuated ATP synthase deregulation compared to their 5xFAD littermates.
Subunit d in AD
Decreased expression and gene locus risk factor
Subunit d of mitochondrial ATP synthase is a component of the FO peripheral stalk which is encoded by the ATP5H (ATP5PD) gene, located on the long arm of nuclear chromosome 17. A genome wide association study (GWAS) found that the shared locus of ATP5H and KCTD2 could be a genetic risk factor for AD, where until a few years ago APOE4 was thought to be the only instance of this [98]. A study of 3x Tg AD mice found significantly decreased expression of ATP5H in hippocampal tissue [99]. An LC-MS/MS-based iTRAQ quantitative proteomics study also demonstrated that multiple proteins from the mitochondrial proteome are under-expressed in the medial frontal gyrus of AD human patients including ATP5H, ATP5B, ATP5I and ATP5J compared with age-matched controls [100]. Perhaps most interestingly, another GWAS found the ATP5H gene to be a candidate gene of interest in late-onset AD (LOAD) and that its expression was decreased in the temporal cortex of AD patients [101]. An RNAi knockdown of C. elegans Tg for Aβ peptide proved to be protective against Aβ toxicity. From these data we can see that the ATP5H gene appears to be associated with LOAD, but any kind of molecular mechanism for this association is yet to be elucidated.
The δ-subunit in AD
Upregulation in AD
The δ-subunit of mitochondrial ATP synthase is part of the F1 component and associates with the γ-subunit of the rotary stalk, in proximity of the F0 c-ring. It is encoded by the ATP5D gene located on the short arm of nuclear chromosome 19. In 2004 a study from Manczak et al. showed increased mRNA levels for ATP6 and ATP8 genes in AD patient brains, while immunofluorescence analysis of the frontal cortex of AD patients found increased levels of the δ-subunit of ATP synthase [102]. While isolated, these data show yet another example of altered patterns of subunit expression across different tissues of the human AD brain.
ATP synthase therapeutics in AD
J147
J147 was identified in 2011 through a drug discovery scheme that sought to target age associated pathologies, as opposed to amyloid plaques, due to age being the greatest risk-factor in AD onset [103]. J147 is a neurotrophic compound that has proven safe to use in animal studies and has been shown to rescue cognitive defects in aged mouse models of AD [104]. The cognitive rescue effects seen in this study are shown to correlate with the induction of the neurotrophic factors NGF (nerve growth factor) and BDNF (brain derived neurotrophic factor). A later study from the same group demonstrated that the α-subunit is a molecular target of J147 and that J147 modulates ATP synthase activity [105]. The mild inhibition of ATP synthase by J147 may be neuroprotective. J147 also activates the canonical longevity pathway of AMPK/mTOR via CamKK2 and its administration was shown to extend the lifespan of Drosophila. This is noteworthy due to the fact that ageing is the biggest risk factor for the onset of AD.
Following this, a computational modelling paper of mitochondrial αβγ was published which demonstrated a mechanism of how J147 could bind to the α-subunit and modulate enzymatic activity [106]. Soliman et al. then used their per-residue energy decomposition (PRED) protocol to identify three compounds from a molecular library which could modulate ATP synthase activity in a similar manner to J147 [107]. The compounds which they identified had a higher binding propensity for the α-subunit than J147 and specifically targeted Arg1112 and Gln426 for binding.
While J147 may have potential as an effective treatment for AD, it is noteworthy that the mechanism of action is through an inhibition of ATP synthase activity given that ATP synthase activity inhibition has been observed as part of the pathophysiology of AD. It may be that there are subtle but significant differences in the decreased levels of enzymatic activity between those induced by J147 and those observed in studies of AD. We suggest that the stage of disease progression should also be considered with J147 administration, as it may be the case that the positive outcomes of J147 treatment may not be observed once pathological ATP synthase activity inhibition crosses a certain threshold.
Taken together, these data present J147 as a potentially suitable AD drug which alleviates cognitive symptoms after they have presented with a known mechanism of action. Currently J147 is undergoing clinical trials to assess its safety and efficacy as a treatment for AD.
Salvianolic acid B (SalB)
Salvianolic acid B (SalB) is a polyphenolic compound which possesses therapeutic potential as a treatment of AD. SalB has been suggested to act on multiple different pathologies present in various neurodegenerative diseases, and in particular mitochondrial dysfunction [108]. In both cellular and mouse models of AD it has been reported that SalB can inhibit Aβ generation and may help to prevent neuroinflammation [109–111]. Alongside mitochondrial dysfunction, these two phenomena are pathologies classically associated with AD.
With regards to ATP synthase and SalB, a study in 2018 showed that in mouse neuronal cell cultures treated with Aβ SalB was able suppress superoxide production, preserve mitochondrial dynamics and mitigate the decrease in ATP synthase activity [112]. While no mechanism is offered by the authors of the paper, this is a line of investigation that we believe should be further pursued.
Conclusion
AD is widely studied due to the hugely debilitating effects it exhibits on the individual, as well as its prevalence in countries with ageing populations. The dysfunction of mitochondria is heavily implicated in the aetiology of the sporadic and familial forms of AD. There is some debate about whether mitochondrial dysfunction is the primary lesion in the disease onset. Likely mitochondrial dysfunction is a convergence point for several concurrent lesions resulting in disease pathology and progression. However, little attention has so far been paid to the role that ATP synthase may play in AD. The data presented in this review suggest that this is an oversight and that the dysfunction of ATP synthase and its constituent components not only leads to disease onset, but that the enzyme complex can be targeted pharmacologically to treat the disease. In J147 there is a candidate drug currently undergoing clinical trials, and we follow these developments with cautious optimism. While only one study so far has investigated the efficacy of SalB as a potential therapeutic agent for AD, the data produced is encouraging and we hope to see this investigated further. Due to both the structural and functional complexity of ATP synthase, we see that its contribution to both disease pathology and its potential therapeutic targeting should be considered with enthusiasm and studied with intellectual nuance.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council [grant number BB/J014508/1], via awards to BE and TLI.
References
- 1. GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019; 18:88–106. https://doi.org/10.1016/S1474-4422(18)30403-4 [PubMed]
- 2. Mitchell E, Walker R. Global ageing: successes, challenges and opportunities. Br J Hosp Med (Lond). 2020; 81:1–9. https://doi.org/10.12968/hmed.2019.0377 [PubMed]
- 3. Neugroschl J, Wang S. Alzheimer’s disease: diagnosis and treatment across the spectrum of disease severity. Mt Sinai J Med. 2011; 78:596–612. https://doi.org/10.1002/msj.20279 [PubMed]
- 4. Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010; 362:329–44. https://doi.org/10.1056/NEJMra0909142 [PubMed]
- 5. Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004; 63:8–20. https://doi.org/10.1016/j.mehy.2003.12.045 [PubMed]
- 6. Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis. 2018; 62:1403–16. https://doi.org/10.3233/JAD-170585 [PubMed]
- 7. Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013; 2013:316523. https://doi.org/10.1155/2013/316523 [PubMed]
- 8. Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014; 1842:1240–47. https://doi.org/10.1016/j.bbadis.2013.10.015 [PubMed]
- 9. Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer’s disease. J Alzheimers Dis. 2010 (Suppl 2); 20:S569–78. https://doi.org/10.3233/JAD-2010-100357 [PubMed]
- 10. Cheng Y, Bai F. The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front Neurosci. 2018; 12:163. https://doi.org/10.3389/fnins.2018.00163 [PubMed]
- 11. Grüber G, Wieczorek H, Harvey WR, Müller V. Structure-function relationships of A-, F- and V-ATPases. J Exp Biol. 2001; 204:2597–605. [PubMed]
- 12. Kühlbrandt W, Davies KM. Rotary ATPases: a new twist to an ancient machine. Trends Biochem Sci. 2016; 41:106–16. https://doi.org/10.1016/j.tibs.2015.10.006 [PubMed]
- 13. von Ballmoos C, Wiedenmann A, Dimroth P. Essentials for ATP synthesis by F1F0 ATP synthases. Annu Rev Biochem. 2009; 78:649–72. https://doi.org/10.1146/annurev.biochem.78.081307.104803 [PubMed]
- 14. Ruhoy IS, Saneto RP. The genetics of leigh syndrome and its implications for clinical practice and risk management. Appl Clin Genet. 2014; 7:221–34. https://doi.org/10.2147/TACG.S46176 [PubMed]
- 15. Dautant A, Meier T, Hahn A, Tribouillard-Tanvier D, di Rago JP, Kucharczyk R. ATP synthase diseases of mitochondrial genetic origin. Front Physiol. 2018; 9:329. https://doi.org/10.3389/fphys.2018.00329 [PubMed]
- 16. Xu T, Pagadala V, Mueller DM. Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microb Cell. 2015; 2:105–25. https://doi.org/10.15698/mic2015.04.197 [PubMed]
- 17. Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961; 191:144–48. https://doi.org/10.1038/191144a0 [PubMed]
- 18. Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev Camb Philos Soc. 1966; 41:445–502. https://doi.org/10.1111/j.1469-185x.1966.tb01501.x [PubMed]
- 19. Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans. 2013; 41:1–16. https://doi.org/10.1042/BST20110773 [PubMed]
- 20. Taurino F, Giannoccaro C, Sardanelli AM, Cavallo A, De Luca E, Santacroce S, Papa S, Zanotti F, Gnoni A. Function and expression study uncovered hepatocyte plasma membrane ecto-ATP synthase as a novel player in liver regeneration. Biochem J. 2016; 473:2519–30. https://doi.org/10.1042/BCJ20160065 [PubMed]
- 21. Chang HY, Huang TC, Chen NN, Huang HC, Juan HF. Combination therapy targeting ectopic ATP synthase and 26S proteasome induces ER stress in breast cancer cells. Cell Death Dis. 2014; 5:e1540. https://doi.org/10.1038/cddis.2014.504 [PubMed]
- 22. Boyer PD, Cross RL, Momsen W. A new concept for energy coupling in oxidative phosphorylation based on a molecular explanation of the oxygen exchange reactions. Proc Natl Acad Sci USA. 1973; 70:2837–39. https://doi.org/10.1073/pnas.70.10.2837 [PubMed]
- 23. Gresser MJ, Myers JA, Boyer PD. Catalytic site cooperativity of beef heart mitochondrial F1 adenosine triphosphatase. Correlations of initial velocity, bound intermediate, and oxygen exchange measurements with an alternating three-site model. J Biol Chem. 1982; 257:12030–8. [PubMed]
- 24. Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 a resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994; 370:621–28. https://doi.org/10.1038/370621a0 [PubMed]
- 25. Sobti M, Smits C, Wong AS, Ishmukhametov R, Stock D, Sandin S, Stewart AG. cryo-EM structures of the autoinhibited E. Coli ATP synthase in three rotational states. Elife. 2016; 5:e21598. https://doi.org/10.7554/eLife.21598 [PubMed]
- 26. Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science. 1999; 286:1700–05. https://doi.org/10.1126/science.286.5445.1700 [PubMed]
- 27. Gu J, Zhang L, Zong S, Guo R, Liu T, Yi J, Wang P, Zhuo W, Yang M. cryo-EM structure of the mammalian ATP synthase tetramer bound with inhibitory protein IF1. Science. 2019; 364:1068–75. https://doi.org/10.1126/science.aaw4852 [PubMed]
- 28. Reynolds CR, Islam SA, Sternberg MJ. EzMol: a web server wizard for the rapid visualization and image production of protein and nucleic acid structures. J Mol Biol. 2018; 430:2244–48. https://doi.org/10.1016/j.jmb.2018.01.013 [PubMed]
- 29. Nesci S, Trombetti F, Ventrella V, Pagliarani A. The c-ring of the F1FO-ATP synthase: facts and perspectives. J Membr Biol. 2016; 249:11–21. https://doi.org/10.1007/s00232-015-9860-3 [PubMed]
- 30. Ohsakaya S, Fujikawa M, Hisabori T, Yoshida M. Knockdown of DAPIT (diabetes-associated protein in insulin-sensitive tissue) results in loss of ATP synthase in mitochondria. J Biol Chem. 2011; 286:20292–96. https://doi.org/10.1074/jbc.M110.198523 [PubMed]
- 31. He J, Ford HC, Carroll J, Douglas C, Gonzales E, Ding S, Fearnley IM, Walker JE. Assembly of the membrane domain of ATP synthase in human mitochondria. Proc Natl Acad Sci USA. 2018; 115:2988–93. https://doi.org/10.1073/pnas.1722086115 [PubMed]
- 32. Antoniel M, Giorgio V, Fogolari F, Glick GD, Bernardi P, Lippe G. The oligomycin-sensitivity conferring protein of mitochondrial ATP synthase: emerging new roles in mitochondrial pathophysiology. Int J Mol Sci. 2014; 15:7513–36. https://doi.org/10.3390/ijms15057513 [PubMed]
- 33. Angevine CM, Fillingame RH. Aqueous access channels in subunit a of rotary ATP synthase. J Biol Chem. 2003; 278:6066–74. https://doi.org/10.1074/jbc.M210199200 [PubMed]
- 34. Symersky J, Pagadala V, Osowski D, Krah A, Meier T, Faraldo-Gómez JD, Mueller DM. Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformation. Nat Struct Mol Biol. 2012; 19:485–91. https://doi.org/10.1038/nsmb.2284 [PubMed]
- 35. Allegretti M, Klusch N, Mills DJ, Vonck J, Kühlbrandt W, Davies KM. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an f-type ATP synthase. Nature. 2015; 521:237–40. https://doi.org/10.1038/nature14185 [PubMed]
- 36. Noji H, Yasuda R, Yoshida M, Kinosita K
Jr . Direct observation of the rotation of F1-ATPase. Nature. 1997; 386:299–302. https://doi.org/10.1038/386299a0 [PubMed] - 37. Fillingame RH, Angevine CM, Dmitriev OY. Mechanics of coupling proton movements to c-ring rotation in ATP synthase. FEBS Lett. 2003; 555:29–34. https://doi.org/10.1016/s0014-5793(03)01101-3 [PubMed]
- 38. Rees DM, Leslie AG, Walker JE. The structure of the membrane extrinsic region of bovine ATP synthase. Proc Natl Acad Sci USA. 2009; 106:21597–601. https://doi.org/10.1073/pnas.0910365106 [PubMed]
- 39. Giorgio V, Fogolari F, Lippe G, Bernardi P. OSCP subunit of mitochondrial ATP synthase: role in regulation of enzyme function and of its transition to a pore. Br J Pharmacol. 2019; 176:4247–57. https://doi.org/10.1111/bph.14513 [PubMed]
- 40. Menz RI, Walker JE, Leslie AG. Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell. 2001; 106:331–41. https://doi.org/10.1016/s0092-8674(01)00452-4 [PubMed]
- 41. Braig K, Menz RI, Montgomery MG, Leslie AG, Walker JE. Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ADP and aluminium fluoride. Structure. 2000; 8:567–73. https://doi.org/10.1016/s0969-2126(00)00145-3 [PubMed]
- 42. Kagawa R, Montgomery MG, Braig K, Leslie AG, Walker JE. The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J. 2004; 23:2734–44. https://doi.org/10.1038/sj.emboj.7600293 [PubMed]
- 43. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004; 25:1605–12. https://doi.org/10.1002/jcc.20084 [PubMed]
- 44. Dittrich M, Hayashi S, Schulten K. On the mechanism of ATP hydrolysis in F1-ATPase. Biophys J. 2003; 85:2253–66. https://doi.org/10.1016/S0006-3495(03)74650-5 [PubMed]
- 45. Sakamoto J. Identification of the nucleotide-binding site for ATP synthesis and hydrolysis in mitochondrial soluble F1-ATPase. J Biochem. 1984; 96:475–81. https://doi.org/10.1093/oxfordjournals.jbchem.a134859 [PubMed]
- 46. Ma J, Flynn TC, Cui Q, Leslie AG, Walker JE, Karplus M. A dynamic analysis of the rotation mechanism for conformational change in F1-ATPase. Structure. 2002; 10:921–31. https://doi.org/10.1016/s0969-2126(02)00789-x [PubMed]
- 47. Senior AE, Nadanaciva S, Weber J. The molecular mechanism of ATP synthesis by F1F0-ATP synthase. Biochim Biophys Acta. 2002; 1553:188–211. https://doi.org/10.1016/s0005-2728(02)00185-8 [PubMed]
- 48. Feniouk BA, Yoshida M. Regulatory mechanisms of proton-translocating FOF1-ATP synthase. Results Probl Cell Differ. 2008; 45:279–308. https://doi.org/10.1007/400_2007_043 [PubMed]
- 49. Okuno D, Iino R, Noji H. Rotation and structure of FoF1-ATP synthase. J Biochem. 2011; 149:655–64. https://doi.org/10.1093/jb/mvr049 [PubMed]
- 50. Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010 (Suppl 2); 20:S265–79. https://doi.org/10.3233/JAD-2010-100339 [PubMed]
- 51. Makin S. The amyloid hypothesis on trial. Nature. 2018; 559:S4–S7. https://doi.org/10.1038/d41586-018-05719-4 [PubMed]
- 52. Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front Neurosci. 2018; 12:25. https://doi.org/10.3389/fnins.2018.00025 [PubMed]
- 53. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016; 61:654–666. https://doi.org/10.1016/j.molcel.2016.01.028 [PubMed]
- 54. Blum TB, Hahn A, Meier T, Davies KM, Kühlbrandt W. Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc Natl Acad Sci USA. 2019; 116:4250–55. https://doi.org/10.1073/pnas.1816556116 [PubMed]
- 55. Schägger H, Ohm TG. Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur J Biochem. 1995; 227:916–21. https://doi.org/10.1111/j.1432-1033.1995.tb20219.x [PubMed]
- 56. Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, Kukull W, Morris JC, Hulette CM, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci USA. 2008; 105:4441–46. https://doi.org/10.1073/pnas.0709259105 [PubMed]
- 57. Martín-Maestro P, Sproul A, Martinez H, Paquet D, Gerges M, Noggle S, Starkov AA. Autophagy induction by bexarotene promotes mitophagy in presenilin 1 familial Alzheimer’s disease iPSC-derived neural stem cells. Mol Neurobiol. 2019; 56:8220–36. https://doi.org/10.1007/s12035-019-01665-y [PubMed]
- 58. Orr AL, Kim C, Jimenez-Morales D, Newton BW, Johnson JR, Krogan NJ, Swaney DL, Mahley RW. Neuronal apolipoprotein E4 expression results in proteome-wide alterations and compromises bioenergetic capacity by disrupting mitochondrial function. J Alzheimers Dis. 2019; 68:991–1011. https://doi.org/10.3233/JAD-181184 [PubMed]
- 59. Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI, Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol Aging. 2002; 23:371–76. https://doi.org/10.1016/s0197-4580(01)00314-1 [PubMed]
- 60. Carrette O, Burgess JA, Burkhard PR, Lang C, Côte M, Rodrigo N, Hochstrasser DF, Sanchez JC. Changes of the cortex proteome and apolipoprotein E in transgenic mouse models of Alzheimer’s disease. J Chromatogr B Analyt Technol Biomed Life Sci. 2006; 840:1–9. https://doi.org/10.1016/j.jchromb.2006.05.019 [PubMed]
- 61. Schmidt C, Lepsverdize E, Chi SL, Das AM, Pizzo SV, Dityatev A, Schachner M. Amyloid precursor protein and amyloid beta-peptide bind to ATP synthase and regulate its activity at the surface of neural cells. Mol Psychiatry. 2008; 13:953–69. https://doi.org/10.1038/sj.mp.4002077 [PubMed]
- 62. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008; 14:837–42. https://doi.org/10.1038/nm1782 [PubMed]
- 63. Xing SL, Chen B, Shen DZ, Zhu CQ. Β-amyloid peptide binds and regulates ectopic ATP synthase α-chain on neural surface. Int J Neurosci. 2012; 122:290–97. https://doi.org/10.3109/00207454.2011.649867 [PubMed]
- 64. Fujii S. ATP- and adenosine-mediated signaling in the central nervous system: the role of extracellular ATP in hippocampal long-term potentiation. J Pharmacol Sci. 2004; 94:103–06. https://doi.org/10.1254/jphs.94.103 [PubMed]
- 65. Yamazaki Y, Fujii S. Extracellular ATP modulates synaptic plasticity induced by activation of metabotropic glutamate receptors in the hippocampus. Biomed Res. 2015; 36:1–9. https://doi.org/10.2220/biomedres.36.1 [PubMed]
- 66. Sergeant N, Wattez A, Galván-valencia M, Ghestem A, David JP, Lemoine J, Sautiére PE, Dachary J, Mazat JP, Michalski JC, Velours J, Mena-López R, Delacourte A. Association of ATP synthase alpha-chain with neurofibrillary degeneration in Alzheimer’s disease. Neuroscience. 2003; 117:293–303. https://doi.org/10.1016/s0306-4522(02)00747-9 [PubMed]
- 67. Boyd-Kimball D, Poon HF, Lynn BC, Cai J, Pierce WM
Jr , Klein JB, Ferguson J, Link CD, Butterfield DA. Proteomic identification of proteins specifically oxidized in caenorhabditis elegans expressing human Aβ(1-42): implications for Alzheimer’s disease. Neurobiol Aging. 2006; 27:1239–49. https://doi.org/10.1016/j.neurobiolaging.2005.07.001 [PubMed] - 68. Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis. 2006; 22:76–87. https://doi.org/10.1016/j.nbd.2005.10.004 [PubMed]
- 69. Reed TT. Lipid peroxidation and neurodegenerative disease. Free Radic Biol Med. 2011; 51:1302–19. https://doi.org/10.1016/j.freeradbiomed.2011.06.027 [PubMed]
- 70. Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2008; 30:107–20. https://doi.org/10.1016/j.nbd.2007.12.007 [PubMed]
- 71. Lindeboom J, Weinstein H. Neuropsychology of cognitive ageing, minimal cognitive impairment, Alzheimer’s disease, and vascular cognitive impairment. Eur J Pharmacol. 2004; 490:83–86. https://doi.org/10.1016/j.ejphar.2004.02.046 [PubMed]
- 72. Terni B, Boada J, Portero-Otin M, Pamplona R, Ferrer I. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol. 2010; 20:222–33. https://doi.org/10.1111/j.1750-3639.2009.00266.x [PubMed]
- 73. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991; 82:239–59. https://doi.org/10.1007/BF00308809 [PubMed]
- 74. Robinson RA, Lange MB, Sultana R, Galvan V, Fombonne J, Gorostiza O, Zhang J, Warrier G, Cai J, Pierce WM, Bredesen DE, Butterfield DA. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: effects of the amyloid-β peptide of amyloid precursor protein. Neuroscience. 2011; 177:207–22. https://doi.org/10.1016/j.neuroscience.2011.01.005 [PubMed]
- 75. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017; 18:452–65. https://doi.org/10.1038/nrm.2017.22 [PubMed]
- 76. Cha MY, Cho HJ, Kim C, Jung YO, Kang MJ, Murray ME, Hong HS, Choi YJ, Choi H, Kim DK, Choi H, Kim J, Dickson DW, et al. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum Mol Genet. 2015; 24:6492–504. https://doi.org/10.1093/hmg/ddv358 [PubMed]
- 77. Chandrasekaran K, Hatanpää K, Rapoport SI, Brady DR. Decreased expression of nuclear and mitochondrial DNA-encoded genes of oxidative phosphorylation in association neocortex in Alzheimer disease. Brain Res Mol Brain Res. 1997; 44:99–104. https://doi.org/10.1016/s0169-328x(96)00191-x [PubMed]
- 78. Shi X, Lu X, Zhan L, Liu L, Sun M, Gong X, Sui H, Niu X, Liu S, Zheng L, Chen J, Zhou Y. Rat hippocampal proteomic alterations following intrahippocampal injection of amyloid beta peptide (1-40). Neurosci Lett. 2011; 500:87–91. https://doi.org/10.1016/j.neulet.2011.06.009 [PubMed]
- 79. Ding B, Xi Y, Gao M, Li Z, Xu C, Fan S, He W. Gene expression profiles of entorhinal cortex in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2014; 29:526–32. https://doi.org/10.1177/1533317514523487 [PubMed]
- 80. Chung AB, Stepien G, Haraguchi Y, Li K, Wallace DC. Transcriptional control of nuclear genes for the mitochondrial muscle ADP/ATP translocator and the ATP synthase beta subunit. Multiple factors interact with the OXBOX/REBOX promoter sequences. J Biol Chem. 1992; 267:21154–61. [PubMed]
- 81. Haraguchi Y, Chung AB, Neill S, Wallace DC. OXBOX and REBOX, overlapping promoter elements of the mitochondrial F0F1-ATP synthase beta subunit gene. Oxbox/REBOX in the ATPsyn beta promoter. J Biol Chem. 1994; 269:9330–34. [PubMed]
- 82. Villena JA, Martin I, Viñas O, Cormand B, Iglesias R, Mampel T, Giralt M, Villarroya F. ETS transcription factors regulate the expression of the gene for the human mitochondrial ATP synthase beta-subunit. J Biol Chem. 1994; 269:32649–54. [PubMed]
- 83. D’Andrea MR. Add Alzheimer’s disease to the list of autoimmune diseases. Med Hypotheses. 2005; 64:458–63. https://doi.org/10.1016/j.mehy.2004.08.024 [PubMed]
- 84. Wotton CJ, Goldacre MJ. Associations between specific autoimmune diseases and subsequent dementia: retrospective record-linkage cohort study, UK. J Epidemiol Community Health. 2017; 71:576–83. https://doi.org/10.1136/jech-2016-207809 [PubMed]
- 85. Vacirca D, Delunardo F, Matarrese P, Colasanti T, Margutti P, Siracusano A, Pontecorvo S, Capozzi A, Sorice M, Francia A, Malorni W, Ortona E. Autoantibodies to the adenosine triphosphate synthase play a pathogenetic role in Alzheimer’s disease. Neurobiol Aging. 2012; 33:753–66. https://doi.org/10.1016/j.neurobiolaging.2010.05.013 [PubMed]
- 86. Berry A, Vacirca D, Capoccia S, Bellisario V, Malorni W, Ortona E, Cirulli F. anti-ATP synthase autoantibodies induce neuronal death by apoptosis and impair cognitive performance in C57BL/6J mice. J Alzheimers Dis. 2013; 33:317–21. https://doi.org/10.3233/JAD-2012-121312 [PubMed]
- 87. Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int. 2004; 45:583–95. https://doi.org/10.1016/j.neuint.2004.03.007 [PubMed]
- 88. Veas-Pérez de Tudela M, Delgado-Esteban M, Maestre C, Bobo-Jiménez V, Jiménez-Blasco D, Vecino R, Bolaños JP, Almeida A. Regulation of bcl-xL-ATP synthase interaction by mitochondrial cyclin B1-cyclin-dependent kinase-1 determines neuronal survival. J Neurosci. 2015; 35:9287–301. https://doi.org/10.1523/JNEUROSCI.4712-14.2015 [PubMed]
- 89. Michels J, Kepp O, Senovilla L, Lissa D, Castedo M, Kroemer G, Galluzzi L. Functions of BCL-X L at the interface between cell death and metabolism. Int J Cell Biol. 2013; 2013:705294. https://doi.org/10.1155/2013/705294 [PubMed]
- 90. Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, et al. bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011; 13:1224–33. https://doi.org/10.1038/ncb2330 [PubMed]
- 91. Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, Gauba E, Lu L, Pascual JM, Kroener S, Du H. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun. 2016; 7:11483. https://doi.org/10.1038/ncomms11483 [PubMed]
- 92. Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005; 19:2040–41. https://doi.org/10.1096/fj.05-3735fje [PubMed]
- 93. Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of a beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006; 15:1437–49. https://doi.org/10.1093/hmg/ddl066 [PubMed]
- 94. Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. 2016; 26:655–67. https://doi.org/10.1016/j.tcb.2016.04.006 [PubMed]
- 95. Beutner G, Alanzalon RE, Porter GA
Jr . Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci Rep. 2017; 7:14488. https://doi.org/10.1038/s41598-017-14795-x [PubMed] - 96. Gauba E, Guo L, Du H. Cyclophilin D promotes brain mitochondrial F1FO ATP synthase dysfunction in aging mice. J Alzheimers Dis. 2017; 55:1351–62. https://doi.org/10.3233/JAD-160822 [PubMed]
- 97. Gauba E, Chen H, Guo L, Du H. Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol Dis. 2019; 121:138–47. https://doi.org/10.1016/j.nbd.2018.09.020 [PubMed]
- 98. Boada M, Antúnez C, Ramírez-Lorca R, DeStefano AL, González-Pérez A, Gayán J, López-Arrieta J, Ikram MA, Hernández I, Marín J, Galán JJ, Bis JC, Mauleón A, et al, and Alzheimer’s Disease Neuroimaging Initiative. ATP5H/KCTD2 locus is associated with Alzheimer’s disease risk. Mol Psychiatry. 2014; 19:682–87. https://doi.org/10.1038/mp.2013.86 [PubMed]
- 99. Yu H, Lin X, Wang D, Zhang Z, Guo Y, Ren X, Xu B, Yuan J, Liu J, Spencer PS, Wang JZ, Yang X. Mitochondrial molecular abnormalities revealed by proteomic analysis of hippocampal organelles of mice triple transgenic for Alzheimer disease. Front Mol Neurosci. 2018; 11:74. https://doi.org/10.3389/fnmol.2018.00074 [PubMed]
- 100. Adav SS, Park JE, Sze SK. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol Brain. 2019; 12:8. https://doi.org/10.1186/s13041-019-0430-y [PubMed]
- 101. Mukherjee S, Russell JC, Carr DT, Burgess JD, Allen M, Serie DJ, Boehme KL, Kauwe JS, Naj AC, Fardo DW, Dickson DW, Montine TJ, Ertekin-Taner N, et al. Systems biology approach to late-onset Alzheimer’s disease genome-wide association study identifies novel candidate genes validated using brain expression data and caenorhabditis elegans experiments. Alzheimers Dement. 2017; 13:1133–42. https://doi.org/10.1016/j.jalz.2017.01.016 [PubMed]
- 102. Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004; 5:147–62. https://doi.org/10.1385/NMM:5:2:147 [PubMed]
- 103. Chen Q, Prior M, Dargusch R, Roberts A, Riek R, Eichmann C, Chiruta C, Akaishi T, Abe K, Maher P, Schubert D. A novel neurotrophic drug for cognitive enhancement and Alzheimer’s disease. PLoS One. 2011; 6:e27865. https://doi.org/10.1371/journal.pone.0027865 [PubMed]
- 104. Prior M, Dargusch R, Ehren JL, Chiruta C, Schubert D. The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice. Alzheimers Res Ther. 2013; 5:25. https://doi.org/10.1186/alzrt179 [PubMed]
- 105. Goldberg J, Currais A, Prior M, Fischer W, Chiruta C, Ratliff E, Daugherty D, Dargusch R, Finley K, Esparza-Moltó PB, Cuezva JM, Maher P, Petrascheck M, Schubert D. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell. 2018; 17:e12715. https://doi.org/10.1111/acel.12715 [PubMed]
- 106. Emmanuel IA, Olotu FA, Agoni C, Soliman ME. Deciphering the ‘Elixir of life’: dynamic perspectives into the allosteric modulation of mitochondrial ATP synthase by J147, a novel drug in the treatment of Alzheimer’s disease. Chem Biodivers. 2019; 16:e1900085. https://doi.org/10.1002/cbdv.201900085 [PubMed]
- 107. Emmanuel IA, Olotu F, Agoni C, Soliman ME. Broadening the horizon: integrative pharmacophore-based and cheminformatics screening of novel chemical modulators of mitochondria ATP synthase towards interventive Alzheimer’s disease therapy. Med Hypotheses. 2019; 130:109277. https://doi.org/10.1016/j.mehy.2019.109277 [PubMed]
- 108. Zhao R, Liu X, Zhang L, Yang H, Zhang Q. Current progress of research on neurodegenerative diseases of salvianolic acid B. Oxid Med Cell Longev. 2019; 2019:3281260. https://doi.org/10.1155/2019/3281260 [PubMed]
- 109. Lee YW, Kim DH, Jeon SJ, Park SJ, Kim JM, Jung JM, Lee HE, Bae SG, Oh HK, Son KH, Ryu JH. Neuroprotective effects of salvianolic acid B on an Aβ25-35 peptide-induced mouse model of Alzheimer’s disease. Eur J Pharmacol. 2013; 704:70–77. https://doi.org/10.1016/j.ejphar.2013.02.015 [PubMed]
- 110. Tang Y, Huang D, Zhang MH, Zhang WS, Tang YX, Shi ZX, Deng L, Zhou DH, Lu XY. Salvianolic acid B inhibits Aβ generation by modulating BACE1 activity in SH-SY5Y-APPsw cells. Nutrients. 2016; 8:333. https://doi.org/10.3390/nu8060333 [PubMed]
- 111. Liu J, Wang Y, Guo J, Sun J, Sun Q. Salvianolic acid B improves cognitive impairment by inhibiting neuroinflammation and decreasing Aβ level in Porphyromonas gingivalis-infected mice. Aging (Albany NY). 2020; 12:10117–28. https://doi.org/10.18632/aging.103306 [PubMed]
- 112. He Y, Jia K, Li L, Wang Q, Zhang S, Du J, Du H. Salvianolic acid B attenuates mitochondrial stress against Aβ toxicity in primary cultured mouse neurons. Biochem Biophys Res Commun. 2018; 498:1066–72. https://doi.org/10.1016/j.bbrc.2018.03.119 [PubMed]