



Introduction
Senescence, also called biological aging, is a functional degradation process of cells and tissues [1], and oxidative stress and free radicals are two critical elements that induce senescence [2]. Accumulation of oxidative stress in cells and tissues causes several age-related diseases that shorten the life span of the organism [3]. Reactive oxygen species (ROS) are a byproduct of normal mitochondrial respiration, specifically the oxidative phosphorylation pathway that activates the production of various superoxides [4]. Superoxide dismutase converts superoxides to hydrogen peroxide (H2O2), which participates in cellular signaling pathways. Maintaining the balance of ROS formation is important for cellular homeostasis [5]. Excessive ROS formation can induce oxidative stress, which plays a critical role in the development of various diseases, including aging [6].
Isocitrate dehydrogenase (IDH) is a highly conserved enzyme that catalyzes isocitrate to α-ketoglutarate (α-KG) [7]. IDH is classified into three isotypes, partly based on cellular localization. IDH1 is found in the cytosol and IDH2 and IDH3 are found in mitochondria. Many studies have focused on IDH2 because of how it functions in mitochondria. IDH2 plays an important role in maintaining mitochondrial redox balance by supplying NAPDH for NADPH-dependent antioxidant enzymes [8]. A recent study suggested that the presence or absence of IDH2 has a critical role in various diseases [9]. Many studies point to IDH2 dysfunction-induced ROS as the main component of disease states. IDH2 deficiency induces a mitochondrial redox imbalance that generates excessive ROS and oxidative stress. As a result, IDH2 deficiency-induced ROS imbalances promote diseases including liver failure [9], cardiac hypertrophy [10], Parkinson’s disease [11], metabolic disorders [12], renal dysfunction [13], hearing loss [14] and kyphosis [15].
Many studies already suggest that ROS are critical elements of accelerated aging [16]. Therefore, regulating excessive ROS and oxidative stress is a compelling target for anti-aging therapies. Previous studies suggested that downregulation of IDH2 is one of the critical factors of ROS generation [17], suggesting that IDH2 could be a potential target for slowing the aging process. In this study, we used mouse embryonic fibroblasts (MEFs) to study senescence. We determined that Idh2 expression is downregulated in senescence-induced MEFs. Furthermore, Idh2 deficiency accelerated senescence-associated phenotypes in MEFs. In conclusion, we suggest that IDH2 gene expression is a crucial regulator of aging and can be a potential target for future anti- aging experiments.
Results
Idh2 expression is decreased in senescent MEFs and tissues
To measure the expression levels of Idh2 in senescence-induced MEFs and aged mouse tissues, we first evaluated the senescence-associated β-gal activity (SA-β-gal), an established hallmark of cellular senescence. Passage 8 (P8) MEFs exhibit higher SA-β-gal staining than Passage 2 (P2) MEFs (Figure 1A and 1B). Levels of well-known senescence marker proteins (p53, p16, and p21) are also increased in P8 MEFs (Figure 1C). Next, we confirmed the expression level of Idh2 in P8 MEFs. Protein and mRNA levels of Idh2 are decreased in P8 MEFs (Figure 1D). Idh2 mRNA expression was also measured using glucose-6-phosphate dehydrogenase (G6PD) as a control to confirm downregulation of Idh2 in P8 MEFs (Supplementary Figure 1). Idh2 levels are also decreased significantly in heart, liver, spleen, and lung tissues from old mice (Figure 1E). From these results, we conclude that expression levels of Idh2 are decreased in p8 MEFs and aged mouse tissues.
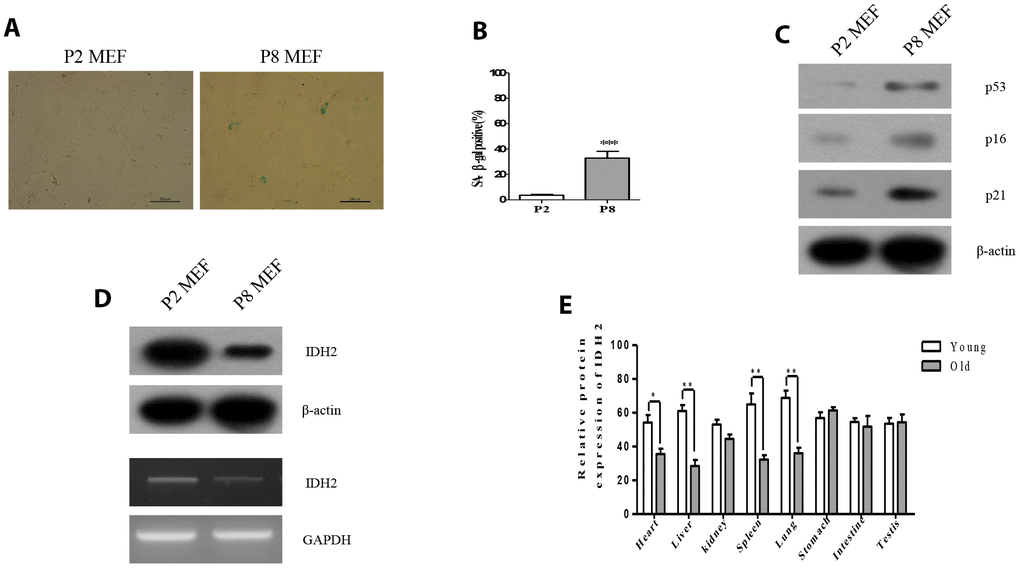
Figure 1. Idh2 was downregulated in senescent mouse embryonic fibroblasts (MEFs) and aging tissues. (A) SA-ß-gal staining of Passage 2 (P2) and Passage 8 (P8) MEFs. Scale bar, 200 µm. (B) Statistical analysis of SA-ß-gal-stained positive cells between P2 and P8 MEFs. (C) Western blot analysis between P2 and P8 MEFs. The following antibodies were used: anti-p53, anti-p16, anti-p21, and anti-ß-actin. (D) Detection of Idh2 expression level in P2 and P8 MEFs. Western blotting and reverse-transcriptase PCR was performed for detecting Idh2. (E)Relative protein expression of Idh2 was detected in mouse tissues using western blot analysis. Ten-week-old and 47-week-old mice were used. Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
Downregulation of Idh2 accelerates senescence in MEFs
Because Idh2 expression is decreased in P8 MEFs, we hypothesized that inhibiting Idh2 expression affects MEF senescence. We decreased Idh2 expression by transfecting small interfering RNA (siRNA) into MEFs and by Idh2 knockout in mice.
Downregulation of Idh2 expression using siRNA (also referred to as “Idh2 knockdown”) results in a marked increase of SA-β-gal staining compared with the level of SA-β-gal staining in response to transfection with scrambled siRNA (Figure 2A and 2B, respectively). Immunofluorescence staining using 5-bromo-2′-deoxyuridine (BrdU) in MEFs revealed that cell proliferation is significantly inhibited in Idh2-knockdown MEFs (Figure 2C and 2D). Next, we measured the level of senescence-related marker proteins in control and Idh2-knockdown MEFs. Interestingly, Idh2-knockdown MEFs show increased levels of p21 and p53, but not p16 (Figure 2E).
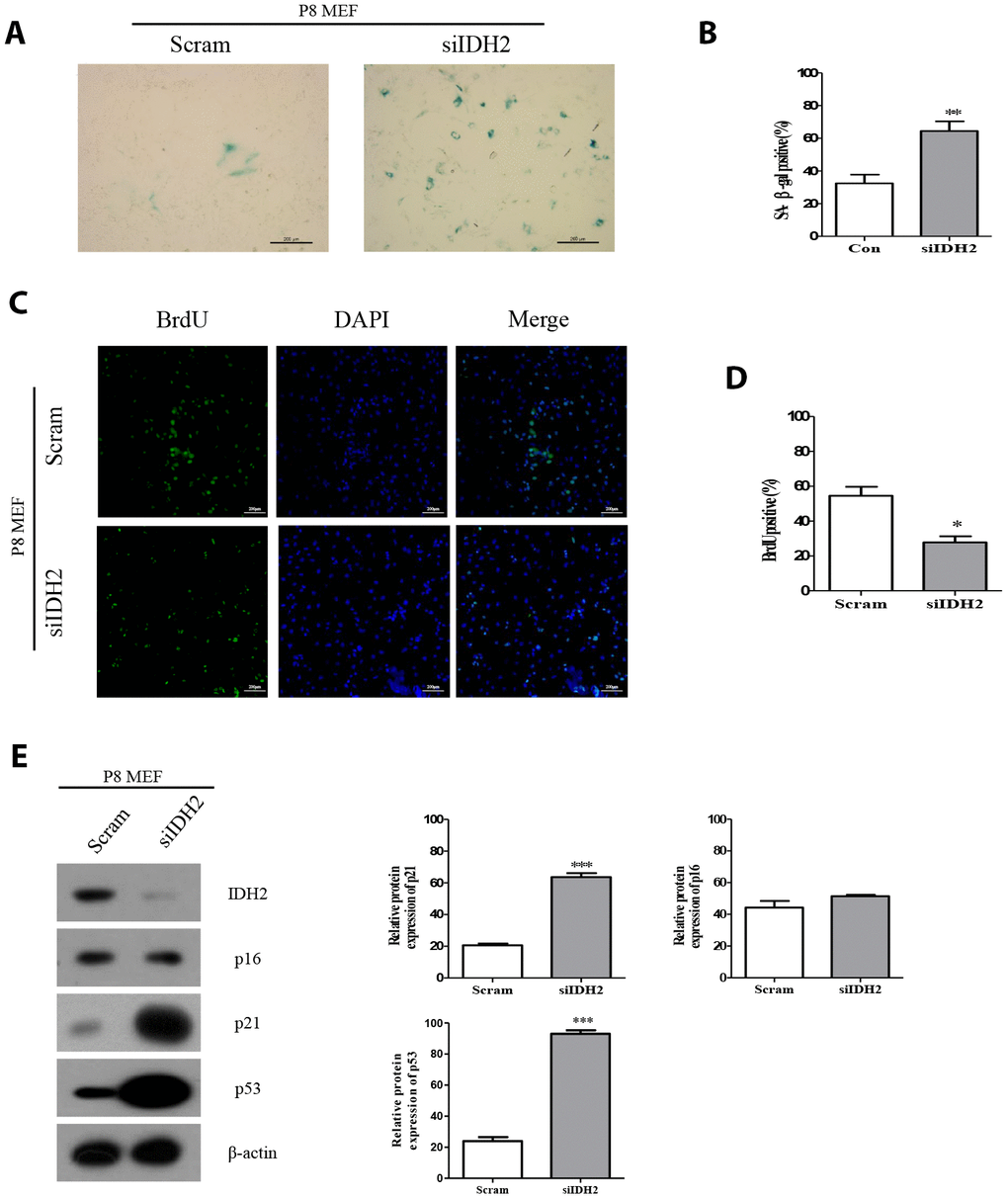
Figure 2. Downregulation of Idh2 in senescent mouse embryonic fibroblasts (MEFs) accelerates the senescence phenotype. (A) SA-β-gal staining of control and Idh2-silenced MEFs. Passage 8 (P8) MEFs were used. Scale bar, 200 μm. (B) Statistical analysis of SA-β-gal-stained positive cells between control and Idh2-knockdown MEFs. (C) BrdU levels between control and Idh2-knockdown MEFs as determined by immunocytochemistry. Nuclei were stained with DAPI, and the merged images show BrdU and DAPI signals. Scale bar, 10 μm. (D) Statistical analysis of BrdU-positive cells between control and Idh2-knockdown MEFs. (E) Western blot analysis between control and Idh2-knockdown MEFs. The following antibodies were used for detection: anti-Idh2, anti-p16, anti-p21, anti-p53, and anti- β-actin. Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
Next, we used Idh2 knockout mice to evaluate senescence in mice that do not express Idh2. MEFs from Idh2 knockout mice (Idh2 knockout MEFs) show a significant increase in SA-β-gal staining compared to that in MEFs from wild type mice (wild type MEFs) (Figure 3A and 3B). BrdU labeling is also decreased in Idh2 knockout MEFs compared to that in wild type MEFs (Figure 3C and 3D). In Idh2 knockout MEFs, levels of senescence marker proteins p21 and p53 are increased, but p16 levels are not (Figure 3E), which is the same result seen with Idh2-knockdown MEFs. In conclusion, downregulation of Idh2 accelerates senescence through p53 and p21 signaling.
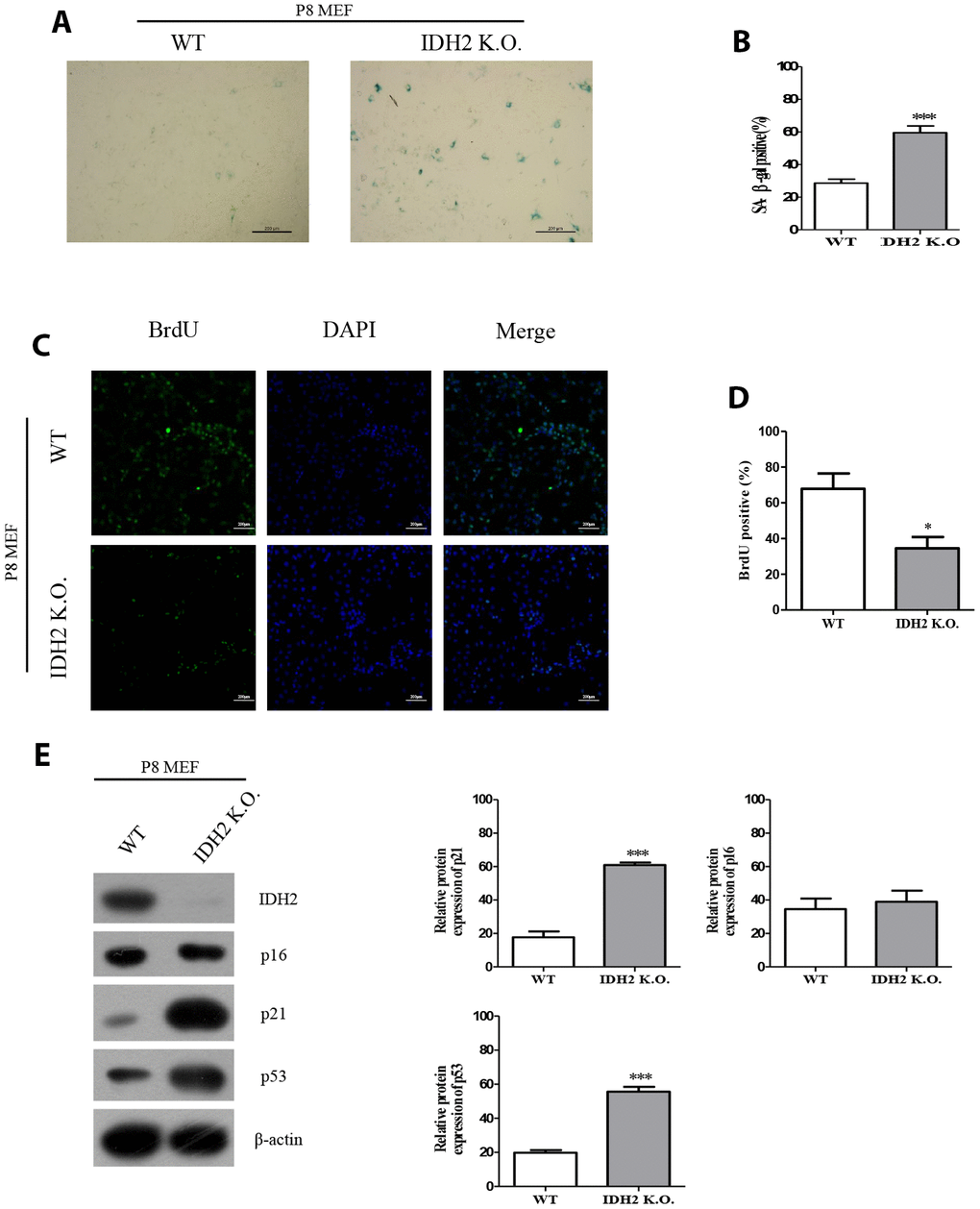
Figure 3. The senescence phenotype is promoted in Idh2 knockout mouse embryonic fibroblasts (MEFs). (A) SA-β-gal staining of wild type and Idh2 knockout MEFs. Passage 8 (P8) MEFs were used. Scale bar, 200 μm. (B) Statistical analysis of SA-β-gal-stained positive cells between wild type and Idh2 knockout MEFs. (C) BrdU level between wild type and Idh2 knockout MEFs as determined by immunocytochemistry. Nuclei were stained with DAPI, and the merged images show BrdU and DAPI signals. Scale bar, 10 μm. (D) Statistical analysis of BrdU-positive cells between wild type and Idh2 knockout MEFs. (E) Western blot analysis between wild type and Idh2 knockout MEFs. The following antibodies were used for detection: anti-Idh2, anti-p16, anti-p21, anti-p53, and anti-β-actin. Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
Idh2 knockout mice show unusual histopathology in lung and spleen tissue
In addition to accelerating senescence via p53 and p21 signaling, Idh2 knockout causes histopathology differences in mice. We first examined p21 levels in Idh2 knockout and wild type mice by immunohistochemistry. Among the tissues examined, lung and spleen exhibit significantly upregulated p21 and p53 expression in Idh2 knockout mice (Figure 4A and 4B). Hematoxylin and eosin staining (H&E staining) was used to detect histopathological changes. Lungs of Idh2 knockout mice exhibit significantly more alveolar wall destruction than lungs from wild type mice (Figure 4C). Spleens of Idh2 knockout mice also exhibit a reduction in white pulp, which consists of lymphoid tissue (Figure 4D). Therefore, Idh2 knockout accelerates senescence in lung and spleen tissues through upregulation of p21 expression.
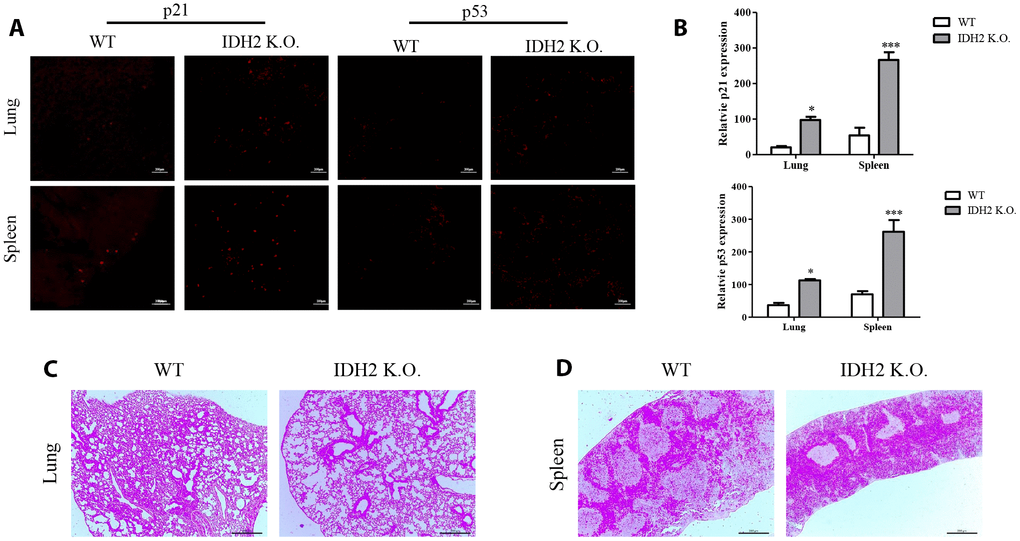
Figure 4. Acceleration of senescence in several tissues is observed in Idh2 knockout mice. (A) P21 and p53 levels in lung and spleen tissues from wild type and Idh2 knockout mice as determined by immunohistochemistry. Images show p21 and p53 signals. Scale bar, 10 μm (n = 6/group). (B) Statistical analysis of p21 and p53-positive signals between lung and spleen tissues from wild type and Idh2 knockout mice. (C) Representative H&E-stained sections from wild type and Idh2 knockout mouse lungs (n = 6/group). (D) Representative H&E-stained sections from wild type and Idh2 knockout mouse spleens (n = 6/group).
Idh2 deficiency induces ROS generation and increases p21 expression
Previous studies demonstrated that excessive ROS production accelerates the aging process and IDH2 deficiency elevates ROS generation. However, there have been no studies on the relationship between IDH2 deficiency-induced ROS generation and aging. Our studies suggest that Idh2 deficiency accelerates aging through the p21 signaling pathway. Therefore, we used siRNA and Idh2 knockout MEFs to downregulate Idh2 expression. Before these experiments, we evaluated off-target effects for the Idh2 siRNA sequence by testing another predesigned siRNA (Supplementary Figure 2). All siRNAs show similar expression patterns of p21, p53, and p16 in MEFs. Idh2 knockdown in MEFs results in increased levels of pro-inflammatory mediators, iNOS and Cox-2, and a well-known ROS marker protein, Prx-SO3 (Figure 5A). Idh2 knockout MEFs also show increased levels of iNOS and Cox-2 and Prx-SO3 (Figure 5B). Furthermore, increased levels of total ROS are seen in Idh2-knockdown and Idh2 knockout MEFs using flow cytometry (Figure 5C and 5D, respectively). Next, we used H2O2 to detect changes in ROS levels in P8 MEFs (Figure 5E). H2O2 is a well-known ROS-generating material that induces premature senescence. H2O2 treatment increases levels of senescence marker proteins p21 and p53. Furthermore, an ROS scavenger, N-acetyl cysteine (NAC), prevents the upregulation of senescence and ROS marker proteins. Idh2 knockdown also results in a similar pattern as H2O2 treatment in MEFs. NAC treatment also prevents Idh2 knockdown-induced upregulation of senescence marker proteins. We conclude that Idh2 downregulation induced ROS generation, which promoted an increase in p21 expression.
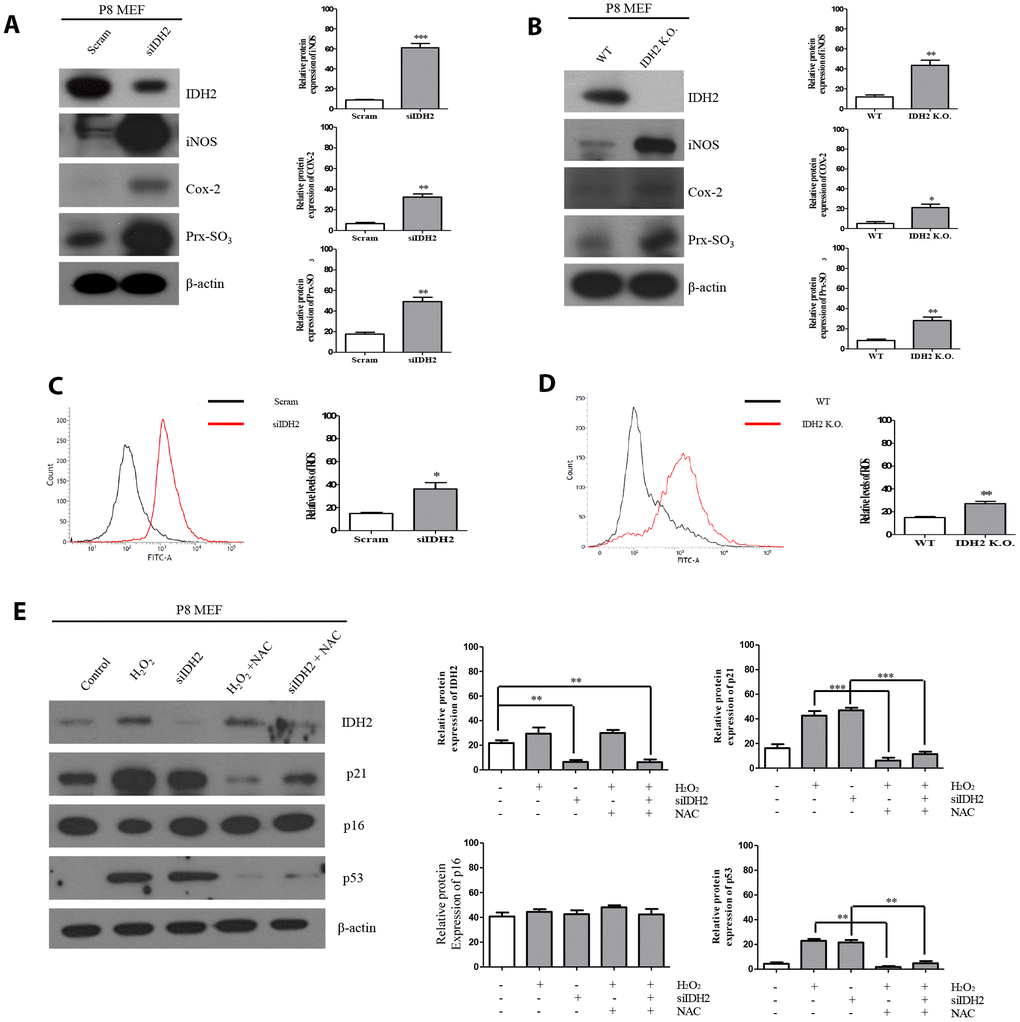
Figure 5. Idh2 deficiency-mediated reactive oxygen species (ROS) generation activates p21 expression. (A) Western blot analysis of pro-inflammatory mediators and oxidative stress marker proteins in control and Idh2-knockdown mouse embryonic fibroblasts (MEFs). The following antibodies were used for detection: anti-Idh2, anti-iNOS, anti-Cox-2, anti-Prx-SO3, and anti-β-actin. (B) Western blot analysis of pro-inflammatory mediators and oxidative stress marker proteins between wild type and Idh2 knockout MEFs. The following antibodies were used for detection: anti-Idh2, anti-iNOS, anti-Cox-2, anti-Prx-SO3, and anti-β-actin. (C) Control and Idh2-knockdown MEFs were incubated with DCF-DA for 15 min at 37°C and intracellular ROS levels were analyzed by flow cytometry. (D) Wild type and Idh2 knockout MEFs were incubated with DCF-DA for 15 min at 37°C and intracellular ROS levels were analyzed by flow cytometry. (E) After transfecting MEFs with siIdh2, H2O2 was added for 3 days. NAC was added 4 h before H2O2 treatment. Western blot analysis was detected with the following antibodies. Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
The absence of Idh2 promotes senescence by inhibiting cyclin-dependent kinase 2
Generally, the induction of senescence is associated with cell cycle regulation. Cyclin-dependent kinase proteins participate in regulating the cell cycle, including Cdk1, Cdk2, Cdk4, and Cdk6. Multiple studies have suggested that dysfunction of cyclin-dependent kinase proteins induces cell cycle arrest, which can promote cellular senescence [18]. Therefore, we examined cell cycle arrest and dysfunction of cyclin-dependent kinase proteins in Idh2 knockout mice by propidium iodide staining to evaluate cell cycle by flow cytometry. Compared with wild type MEFs, Idh2-knockout MEFs show a low percentage of cell cycle distribution in G0 and G1 phases (Figure 6A). Furthermore, cyclin-dependent kinase protein expression levels show that only Cdk2 is decreased in Idh2 knockout MEFs (Figure 6B). In addition, we measured the Cdk2 mRNA level in Idh2 knockout P8 MEFs (Supplementary Figure 3). In contrast with the protein level, Cdk2 mRNA levels are unaffected by Idh2 knockout. Therefore, Idh2 knockout induces cell cycle arrest through inhibiting Cdk2 at the protein level.
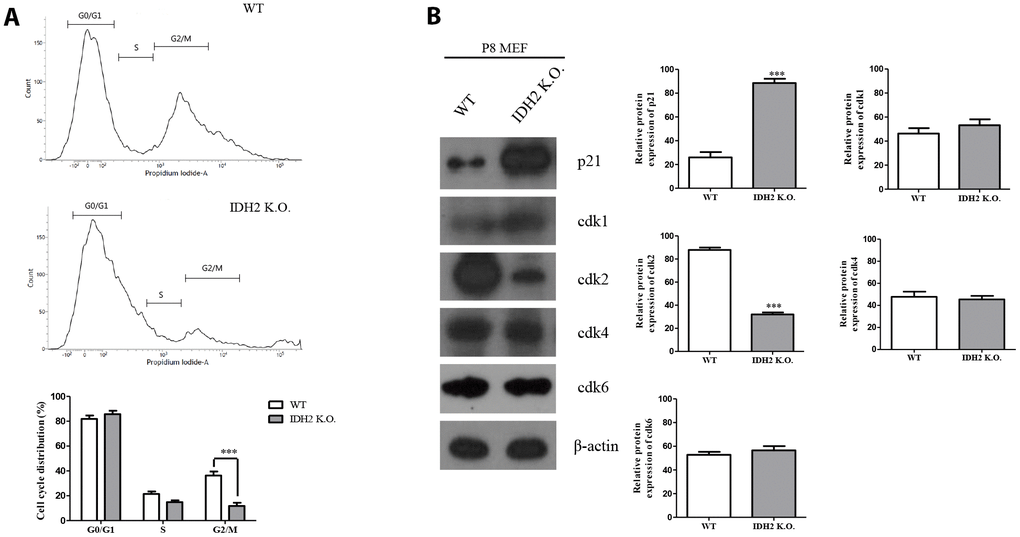
Figure 6. Downregulation of Idh2 promotes senescence in mouse embryonic fibroblasts (MEFs) through suppressing Cdk2. (A) Wild type and Idh2 knockout MEFs were incubated with propidium iodide for 15 min at 37°C. Cell cycle was detected with flow cytometry. (B) Cyclin-dependent kinase protein levels were detected with western blot analysis in wild type and Idh2 knockout MEFs. The following antibodies were used for detection: anti-p21, anti-Cdk1, anti-Cdk2, anti-Cdk4, anti-Cdk6, and anti-β-actin. Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
Upregulation of Idh2 protects against senescence
Our results suggested that downregulation of Idh2 accelerates the development of senescence through the p21 signaling pathway. We next examined whether overexpression of Idh2 could protect against senescence. Idh2-overexpressing P8 MEFs show fewer SA-β-gal-positive cells than normal P8 MEFs (Figure 7A and 7B). Furthermore, the increased p21 and p53 levels seen in P8 MEFs are downregulated by overexpressing Idh2 (Figure 7C). Senescence-related ROS generation was also inhibited in Idh2-overexpressing P8 MEFs. The increased levels of iNOS, COX-2, and Prx-SO3 seen in P8 MEFs were diminished by Idh2 overexpression. Furthermore, Cdk2 levels were higher in P8 MEFs that overexpress Idh2 than in control P8 MEFs (Figure 7D). Cellular ROS levels were also decreased in Idh2-overexpressing P8 MEFs (Figure 7E). We conclude that upregulation of Idh2 expression can decelerate cellular senescence.
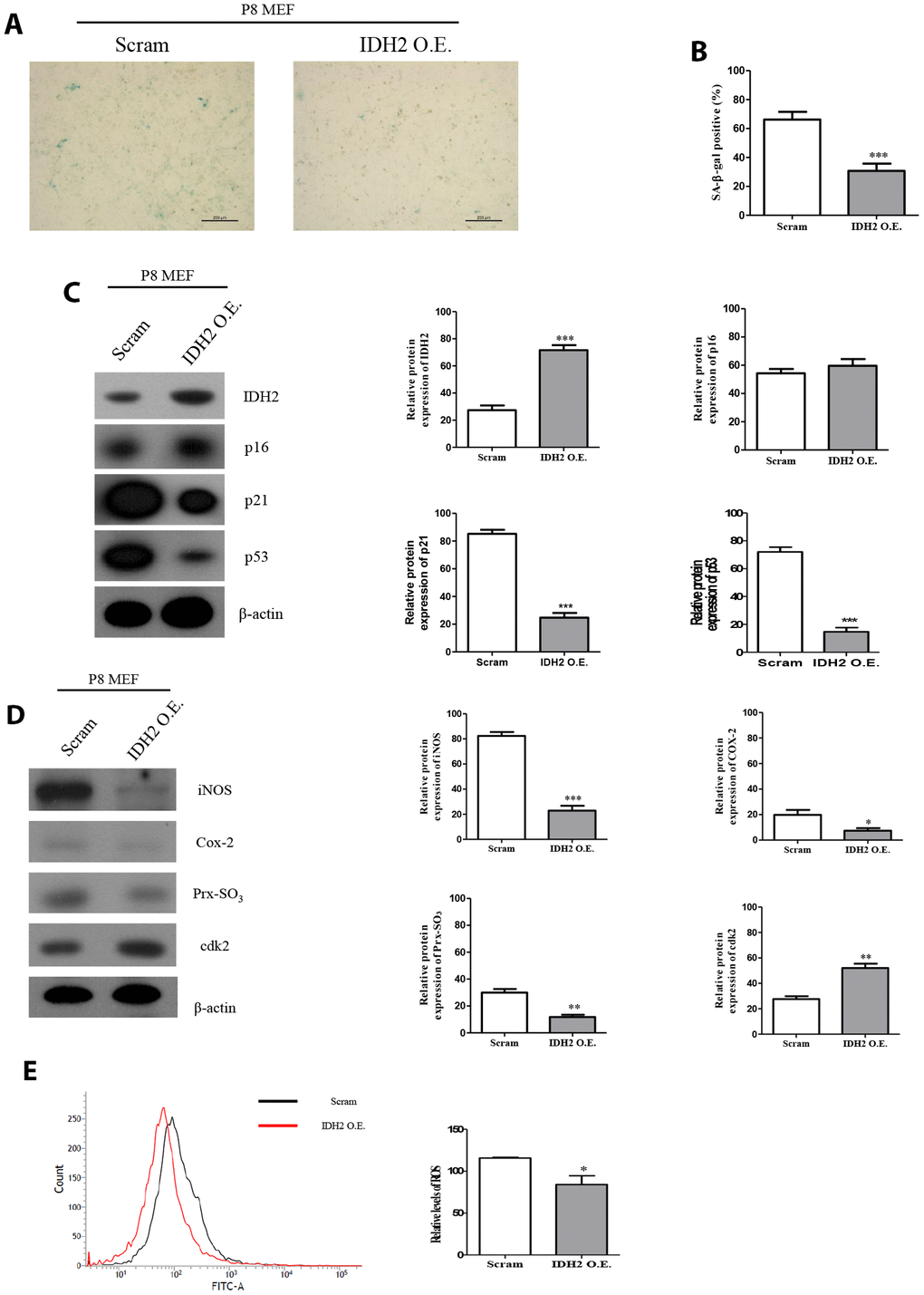
Figure 7. Overexpression of Idh2 prevents the acceleration of senescence in mouse embryonic fibroblasts (MEFs). (A) SA-β-gal staining of control and Idh2-overexpressing MEFs. pLenti 6.3 Idh2 plasmid was transfected into Passage 8 (P8) MEFs. (B) Statistical analysis of SA-β-gal-stained positive cells between control and Idh2-overexpressing MEFs. (C) Senescence-associated marker proteins were detected using western blot analysis in control and Idh2-overexpressing MEFs. The following antibodies were used for detection: anti-Idh2, anti-p16, anti-p21, anti-p53, and anti-β-actin. (D) Pro-inflammatory mediators, reactive oxygen species (ROS) marker proteins, and cyclin-dependent kinase 2 were detected using western blot analysis. The following antibodies were used for detection: anti-iNOS, anti-Cox-2, anti-Prx-SO3, and anti-β-actin. (E) Relative intracellular ROS levels were detected in control and Idh2-overexpressing MEFs. Intracellular ROS were detected by flow cytometry. Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
In this study, we examined the effects of Idh2 deficiency in cellular senescence, which accelerates aging. Idh2 levels are decreased in heart, liver, spleen, and lung tissues in aged mice. Using wild type and Idh2 knockout MEFs, SA-β-gal staining and BrdU assay data suggested that Idh2 deficiency promotes cellular senescence. Passage 8 Idh2 knockout MEFs exhibited senescence-associated phenotypes. We confirmed that Idh2 deficiency increased ROS generation, one of the major factors that promote cellular senescence. Furthermore, we demonstrated that Idh2 deficiency-induced ROS promote senescence signaling through p53 and p21, thereby inhibiting Cdk2, a well-known cyclin-dependent kinase that functions in the cell cycle.
Previous studies demonstrated that ROS act on various cellular organelles and signaling pathways [19]. Excessive cellular ROS production affects mitochondria by damaging mitochondrial DNA [20]. As seen in Figure 1, we confirmed that Idh2 expression was diminished in senescence-induced conditions. Because IDH2 functions in metabolism, mitochondrial function, and ROS generation, we expected that IDH2 would be a senescence-inducing factor. ROS are related to IDH2 and are known to play a major role in accelerating senescence and accompany changes in cellular function and senescence-related diseases.
Multiple studies have already suggested that excessive ROS generation affects Ras, p53, p21, and p16 signaling pathways, which are closely related to cell senescence [21]. P53 activation triggered by excessive ROS can inhibit autophagic functions, resulting in mitochondrial dysfunction, which in turn, promotes cell senescence [22]. Therefore, maintenance of a proper balance of ROS is important for cellular homeostasis. In Figures 2 and 3, our data also supported that activation of p53 by downregulation of Idh2 expression upregulates p21, but not p16, which promotes cellular senescence. Increased p21 expression was also detected in tissues from Idh2 knockout mice, such as lung and spleen (Figure 4). H&E staining data also supported that Idh2 knockout mice show age-related features such as alveolar wall destruction in lung, necrotic debris accumulation in liver and a diminished number of inflammatory cells in spleen. Furthermore, we confirmed that excessive ROS generation triggered by Idh2 knockdown induced p53 activation, which in turn triggered the upregulation of p21 (Figure 5). Finally, upregulation of p21 expression inhibited Cdk2, which has a critical role in cell cycle regulation (Figure 6).
Additionally, we confirmed the apoptotic signaling pathway in Idh2-deficient conditions. Because IDH2 is one of the most critical enzymes in the tricarboxylic acid (TCA) cycle, we predicted that generating Idh2-deficient conditions by siRNA or Idh2 knockout would affect apoptosis. We confirmed that neither caspase 3 nor PARP, which are well known apoptotic marker proteins in mitochondria-mediated apoptosis, were induced by Idh2 knockdown (Supplementary Figure 4). Furthermore, non-mitochondrial apoptosis was also confirmed through Annexin V in flow cytometry. Contrary to our prediction, no significant changes in marker proteins and Annexin V were detected in Idh2 knockdown MEFs.
In the present study, Idh2 deficiency results in excessive ROS generation. Recent studies suggest that IDH2, a critical enzyme in the TCA cycle in mitochondria, is one of the major factors correlated with ROS-induced cellular dysfunction. Many studies support that IDH2 deficiency promotes several diseases, including cancer [23], ischemia-reperfusion injury [9], inflammation [24], and age-related spinal deformities [17]. However, the effect of IDH2 deficiency on cellular senescence and senescence-related diseases has yet to be elucidated. Through this study, we demonstrate that Idh2 deficiency-induced ROS generation activates p53 and p21 expression, thereby inhibiting Cdk2. However, more studies are needed to examine the relationship between Idh2 deficiency and age-related diseases further.
In Figure 7, we examined whether overexpression of Idh2 could slow the development of cellular senescence. Upregulation of Idh2 inhibited the senescence-associated phenotype in MEFs. Furthermore, Idh2 overexpression decreased levels of senescence-associated marker proteins, pro-inflammatory mediators, and ROS markers. Furthermore, Cdk2 expression was also elevated upon Idh2 overexpression. However, we cannot conclusively say that upregulation of Idh2 has an anti-senescence effect; we can only suggest the possibility that Idh2 is potentially a useful protein for studying senescence. Therefore, more studies are needed on the relationship between Idh2 and cellular senescence.
In conclusion, we demonstrated that Idh2 expression decreased cellular senescence, and downregulation of Idh2 promoted it. Furthermore, Idh2 deficiency-induced cell cycle arrest by inhibiting Cdk2 through the p53 and p21 signaling pathways (Figure 8). Interestingly, overexpression of Idh2 alleviated senescence-associated phenotypes as shown by decreased SA-β-gal staining and expression of p21 and p53. Furthermore, Idh2 overexpression increased levels of pro-inflammatory mediators and ROS. Further studies are needed to investigate which factors decrease Idh2 levels in age-dependent conditions. In addition, the anti-senescence effect of Idh2 should also be studied further. Although anti-aging studies have long been a popular focus for many studies, as life expectancy increases, aging studies are receiving more attention. As mentioned above, although more studies are needed, IDH2 represents a fascinating target of future cellular senescence studies.
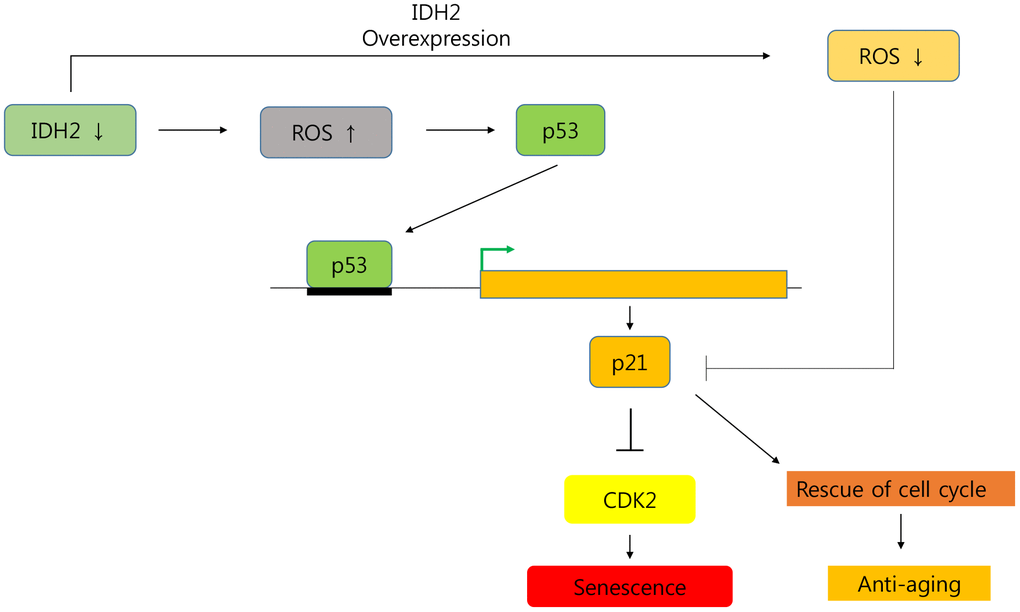
Figure 8. Graphical abstract of the senescence pathway in Idh2-deficient conditions.Idh2 deficiency-induced ROS generation accelerates p53 and p21 signaling pathways which inhibit Cdk2 expression. Furthermore, overexpression of Idh2 prevented senescence-associated phenotypes by decreasing the levels of senescence signaling pathway-associated proteins p21 and p53.
Materials and Methods
Cell culture and treatment
MEFs and NIH 3T3 cells were cultured in Dulbecco’s modified Eagle medium (Welgene, Daegu, Korea) containing 10% fetal bovine serum (Thermo Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin (Welgene) and incubated at 37°C in a 5% CO2 incubator (SANYO, Osaka, Japan). MEFs and NIH 3T3 cells were grown to 60% confluency and then transfected with 10 pmol of siRNA against Idh2 (siIdh2; Bioneer, Daejeon, Korea) or scrambled siRNA control.
Preparation of MEFs
All animal experiments were approved and conducted per the guidelines of the Animal Care Committee of Kyungpook National University. MEFs were generated from 10 to 12.5-day-old embryos derived from WT and Idh2 knockout mice. Embryos were minced, dispersed in 0.05% trypsin/EDTA, and incubated in 5% CO2 at 37°C for 30 min. After incubation, the mass was pipetted for dispersion and then allowed to settle for 1 min, after which the pellet was removed. The cell suspension was plated and incubated at 37°C until confluence. Then, confluent MEFs were stocked using stock solution (Sigma-Aldrich, St. Louis, MO, USA).
SA-β-gal staining
SA-β-gal staining was carried out using a commercial kit (Cell signaling, Danvers, MA, USA). Before staining, the medium was removed from the cells, and cells were washed three times with phosphate-buffered saline (PBS). Then, cells were fixed for 15 min at room temperature. After fixation, cells were washed with PBS, β-galactosidase staining solution was added, and cells were incubated at 37°C overnight in a dry incubator without CO2. Stained cells were observed with a light microscope.
BrdU assay
BrdU (Thermo Scientific) was dissolved as a 10 mM solution in DMSO (Sigma). BrdU solution was then diluted to 10 μM in cell culture medium. Culture medium was removed and replaced with BrdU labeling solution. Cells were incubated at 37°C for 2 hours. After incubation, cells were fixed using 3.7% paraformaldehyde. BrdU was labeled by an anti-BrdU primary antibody (SantaCruz, Dallas, TX, USA) at room temperature overnight. After labeling with an appropriate secondary antibody, BrdU was detected using a confocal microscope (Carl Zeiss, Oberkochen, Germany).
Western blot analysis
Whole-cell lysates were obtained using PRO-PREP protein extraction solution (Intron Biotechnology, Gyeonggi-do, Korea). Proteins were separated by SDS-PAGE, and bands were transferred to nitrocellulose membranes (Pall Corporation, Pensacola, FL, USA). The membranes were then incubated with the following primary antibodies at 4°C overnight: anti-p53, anti-Idh2, anti-B-actin, anti-Cdk1, anti-Cdk2, anti-Cdk4, anti-Cdk6, anti-iNOS, anti-Caspase3, and anti-PARP (Cell signaling); anti-p16 and anti-p21 (Abcam, Cambridge, UK); anti-COX-2 (SantaCruz); and anti-Prx-SO3 (Ab Frontier, Seoul, Korea). After washing with buffer, the membranes were incubated with the appropriate secondary antibody (Thermo Scientific) at room temperature for 6 hours.
RNA isolation and RT-PCR
Total RNA was isolated using TRI-Reagent (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized using Reverse Transcription Premix (Bioneer). PCR was performed using gene-specific primers and PCR premix (Bioneer). The following primers were used: Idh2, 5′-ATCAAGCAGAAGCTCATCCTGC-3′ (forward) and 5′-TCTGTGGCCTTGTACTGGTCG-3′ (reverse); Idh1, 5′-GTCGTCATGCTTATGGGGAT-3′ (forward) and 5′-CAACACCACCACCTTCTTCA-3′ (reverse); Idh3, 5′-ACCTTATAGCAAACACGGCG-3′ (forward) and 3′-TCTTATCATTCCCCACATTCCC-3′ (reverse); Cdk2, 5′-GCTTTCTGCCATTCTCATCG-3′ (forward) and 5′-GTCCCCAGAGTCCGAAAGAT-3′ (reverse); G6PD, 5′-TGAGGGTCGTGGGGGCTATTTTGA-3′ (forward) and 5′-GCATCAGGGAGCTTCACATTCTTG-3′ (reverse); GAPDH, 5′-ACCACAGTCCATGCCATCAC-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse). Multi-Gauge software (Fujifilm, Japan) was used to analyze band intensity.
Immunohistochemistry
Before tissue collection from mice, animals were perfused. After tissue collection, all tissues were fixed in 10% formalin at 4°C overnight. Tissues were cryosectioned using an HM525 NX Cryostat (Thermo Scientific). Sectioned tissues were incubated with an anti-p21 and anti-p53 antibody (Abcam) at 4°C overnight. Tissues were then incubated with Alexa 549 goat anti-rabbit secondary antibody (Thermo Scientific) at 4°C overnight. Images were obtained using an LSM-710 confocal microscope (Carl Zeiss).
Measurement of ROS and cell cycle
Total ROS levels were assessed using DCF-DA (Invitrogen, Carlsbad, CA, USA), and cell cycle determination was performed using propidium iodide (PI; BD Bioscience, Franklin Lakes, NJ, USA). Trypsinized cells were incubated with 5 μM DCF-DA and PI at 37°C for 15 min, washed with PBS, and then analyzed with a flow cytometer (BD Bioscience, San Jose, CA, USA).
Statistical analysis
Data were shown as the mean and standard deviation (SD) of three independent experiments (n = 3). Differences observed between experimental groups were tested for statistical significance using one-way and two-way ANOVA with GraphPad Prism software. A p-value of < 0.05 was deemed statistically significant and is shown in the figures by an asterisk. P-values of < 0.01 and < 0.001 are shown by two and three asterisks, respectively.
Supplementary Materials
Author Contributions
J.W. Park, S.R. Lee, HJ. Lee, H.S Lee, and D.S. Lee designed the study; U.B. Chae performed the experiments and data analysis. D.S. Lee and U.B. Chae wrote the original manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors have declared that no competing interests exist.
Funding
This work was primarily supported by National Research Foundation of Korea grants funded by the Korea government (NRF-2017R1A5A2015391, NRF-2015R1A4A1042271, 2017R1A2B4008176) and the Korea Research Institute of Bioscience & Biotechnology (KRIBB) Research Initiative Program (KGM4621922)
References
- 1. LeBrasseur NK, Tchkonia T, Kirkland JL. Cellular Senescence and the Biology of Aging, Disease, and Frailty. Nestle Nutr Inst Workshop Ser. 2015; 83:11–18. https://doi.org/10.1159/000382054 [PubMed]
- 2. Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. 2012; 2012:646354. https://doi.org/10.1155/2012/646354 [PubMed]
- 3. Shaw PX, Werstuck G, Chen Y. Oxidative stress and aging diseases. Oxid Med Cell Longev. 2014; 2014:569146. https://doi.org/10.1155/2014/569146 [PubMed]
- 4. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014; 94:909–50. https://doi.org/10.1152/physrev.00026.2013 [PubMed]
- 5. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015; 163:560–69. https://doi.org/10.1016/j.cell.2015.10.001 [PubMed]
- 6. Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid Med Cell Longev. 2016; 2016:3565127. https://doi.org/10.1155/2016/3565127 [PubMed]
- 7. Smolková K, Ježek P. The Role of Mitochondrial NADPH-Dependent Isocitrate Dehydrogenase in Cancer Cells. Int J Cell Biol. 2012; 2012:273947. https://doi.org/10.1155/2012/273947 [PubMed]
- 8. Kong MJ, Han SJ, Kim JI, Park JW, Park KM. Mitochondrial NADP+-dependent isocitrate dehydrogenase deficiency increases cisplatin-induced oxidative damage in the kidney tubule cells. Cell Death Dis. 2018; 9:488. https://doi.org/10.1038/s41419-018-0537-6 [PubMed]
- 9. Han SJ, Choi HS, Kim JI, Park JW, Park KM. IDH2 deficiency increases the liver susceptibility to ischemia-reperfusion injury via increased mitochondrial oxidative injury. Redox Biol. 2018; 14:142–53. https://doi.org/10.1016/j.redox.2017.09.003 [PubMed]
- 10. Ku HJ, Ahn Y, Lee JH, Park KM, Park JW. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic Biol Med. 2015; 80:84–92. https://doi.org/10.1016/j.freeradbiomed.2014.12.018 [PubMed]
- 11. Kim H, Kim SH, Cha H, Kim SR, Lee JH, Park JW. IDH2 deficiency promotes mitochondrial dysfunction and dopaminergic neurotoxicity: implications for Parkinson’s disease. Free Radic Res. 2016; 50:853–60. https://doi.org/10.1080/10715762.2016.1185519 [PubMed]
- 12. Lee SJ, Kim SH, Park KM, Lee JH, Park JW. Increased obesity resistance and insulin sensitivity in mice lacking the isocitrate dehydrogenase 2 gene. Free Radic Biol Med. 2016; 99:179–88. https://doi.org/10.1016/j.freeradbiomed.2016.08.011 [PubMed]
- 13. Lee SJ, Cha H, Lee S, Kim H, Ku HJ, Kim SH, Park JH, Lee JH, Park KM, Park JW. Idh2 deficiency accelerates renal dysfunction in aged mice. Biochem Biophys Res Commun. 2017; 493:34–39. https://doi.org/10.1016/j.bbrc.2017.09.082 [PubMed]
- 14. White K, Kim MJ, Han C, Park HJ, Ding D, Boyd K, Walker L, Linser P, Meneses Z, Slade C, Hirst J, Santostefano K, Terada N, et al. Loss of IDH2 Accelerates Age-related Hearing Loss in Male Mice. Sci Rep. 2018; 8:5039. https://doi.org/10.1038/s41598-018-23436-w [PubMed]
- 15. Chae U, Park NR, Kim ES, Choi JY, Yim M, Lee HS, Lee SR, Lee S, Park JW, Lee DS. IDH2-deficient mice develop spinal deformities with aging. Physiol Res. 2018; 67:487–94. https://doi.org/10.33549/physiolres.933711 [PubMed]
- 16. Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013; 60:1–4. https://doi.org/10.1016/j.freeradbiomed.2013.02.011 [PubMed]
- 17. Oh IU, Inazawa J, Kim YO, Song BJ, Huh TL. Assignment of the human mitochondrial NADP(+)-specific isocitrate dehydrogenase (IDH2) gene to 15q26.1 by in situ hybridization. Genomics. 1996; 38:104–06. https://doi.org/10.1006/geno.1996.0602 [PubMed]
- 18. Schwartz GK. CDK inhibitors: cell cycle arrest versus apoptosis. Cell Cycle. 2002; 1:122–23. https://doi.org/10.4161/cc.1.2.112 [PubMed]
- 19. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev. 2016; 2016:4350965. https://doi.org/10.1155/2016/4350965 [PubMed]
- 20. Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009; 37:2539–48. https://doi.org/10.1093/nar/gkp100 [PubMed]
- 21. Lawless C, Jurk D, Gillespie CS, Shanley D, Saretzki G, von Zglinicki T, Passos JF. A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS One. 2012; 7:e32117. https://doi.org/10.1371/journal.pone.0032117 [PubMed]
- 22. Kongara S, Karantza V. The interplay between autophagy and ROS in tumorigenesis. Front Oncol. 2012; 2:171. https://doi.org/10.3389/fonc.2012.00171 [PubMed]
- 23. Li J, He Y, Tan Z, Lu J, Li L, Song X, Shi F, Xie L, You S, Luo X, Li N, Li Y, Liu X, et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1α in lung cancer. Theranostics. 2018; 8:4050–61. https://doi.org/10.7150/thno.21524 [PubMed]
- 24. Choi SJ, Piao S, Nagar H, Jung SB, Kim S, Lee I, Kim SM, Song HJ, Shin N, Kim DW, Irani K, Jeon BH, Park JW, Kim CS. Isocitrate dehydrogenase 2 deficiency induces endothelial inflammation via p66sh-mediated mitochondrial oxidative stress. Biochem Biophys Res Commun. 2018; 503:1805–11. https://doi.org/10.1016/j.bbrc.2018.07.117 [PubMed]