



Autophagy is probably the best-established anti-aging mechanism. As organisms age, their autophagic capacity declines [1]. Measures to increase autophagy by genetic manipulation (such as overexpression of essential autophagy genes including Atg5 or introduction of gain-of-function mutations in the gene coding for Beclin 1, Becn1) [2,3], caloric restriction or periodic fasting [4], provision of agents that mimic the biochemical consequence of caloric restriction (“caloric restriction mimetics”) [5–7], as well as by other pharmacological inducers of autophagy (such as rapamycin) [8,9], can extend health span and longevity [10,11]. This has been shown for many model organisms (yeast, nematodes, flies, mice) and may apply to non-human primates (in which caloric restriction has beneficial effects) [12] and humans (in thus far that a diet rich in the autophagy inducer spermidine correlates with reduced mortality) [13,14]. These antiaging effects of autophagy may be explained by its contribution to renew (and hence to rejuvenate) the cytoplasm of cells, thereby counteracting many if not most of the molecular and cellular hallmarks of aging [15]. In other words, autophagy may be conceived as (one of) the most important process(es) that antagonize(s) the time-dependent deterioration affecting macromolecules (proteins, ribonucleotides, mitochondrial DNA, membrane lipids etc.) and cytoplasmic organelles (i.e. all organelles except the nucleus), thus acting to decelerate the biological clock [16,17].
The two most frequent lethal monogenetic diseases affecting humans are cystic fibrosis (CF, also called ‘mucoviscidosis’) and Wilson disease (WD). As any other disease, both progress with age, though with rather distinct kinetics. CF kills between childhood and early adulthood, while Wilson disease kills middle-aged adults. Surprisingly, insufficient autophagy appears to be a major determinant of disease pathogenesis for both CF and WD (Figure 1).
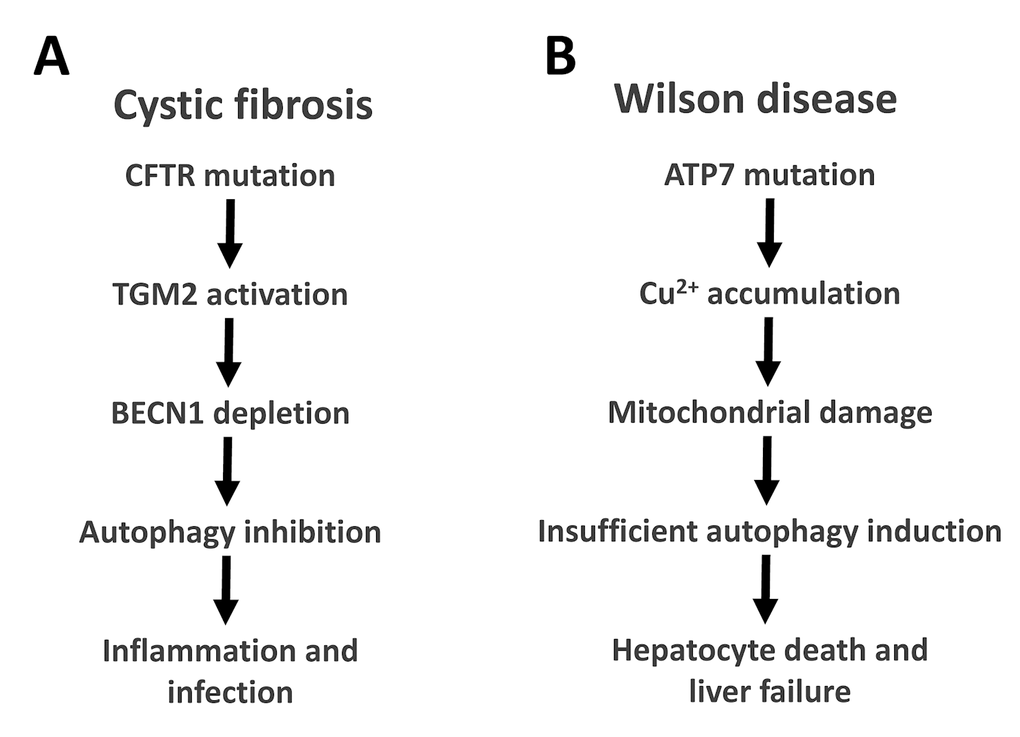
Figure 1. Autophagy in cystic fibrosis and Wilson disease. The relationship between autophagy and CFTR mutations in cystic fibrosis (A) or ATP7B mutations in Wilson disease (B) are depicted. BECN1, Beclin 1; TGM2, transglutaminase 2.
CF is due to loss-of-function mutations of the cystic fibrosis transmembrane conductance regulator (CFTR), reducing the expression or function of this chloride channel at the plasma membrane. Among 2000 different CFTR mutations, the most frequent one is CFTRdel506, accounting for the pathogenesis of 70-90% of all CF cases [18]. As a consequence of CFTR mutations, the function of epithelia (mostly in the lung but also in the gastrointestinal tract) and macrophages are compromised, ultimately causing defective clearance of mucus and infectious microorganisms [19,20]. This then triggers chronic pulmonary infection and inflammation as the primary cause of morbidity and mortality. The treatment of cystic fibrosis remains largely symptomatic, although drugs that increase the expression or improve the function of CFTR are being developed. Importantly, the CFTR defect (be it a lack of expression or a lack of function) compromises autophagy by several mechanisms including the depletion of the essential pro-autophagic protein Beclin 1 (BECN1), locking the cell in a state of deficient proteostasis [18,21]. Successful pharmacological treatment of patients bearing the CFTRdel506 mutation with a combination of epigallocatechin gallate (EGCG, an inhibitor of the autophagy-inhibitory acetyl transferase EP300, an important regulator of autophagy) [22–25] and cysteamine (an inhibitor of transglutaminase-2) can induce autophagy in vivo, and enhance the expression of the mutated CFTRdel506 protein at the cell surface, of nasal respiratory epithelial cells from CF patients [26–28]. Of note, mice that bear the CFTRdel506 mutation respond to this combination therapy (EGCG plus cysteamine) only if they are autophagy-competent, yet fail to do so, if they are autophagy-deficient due to the heterozygous knockout of Becn1. These experiments confirm that autophagy is required for the treatment to work [18,21].
WD results from the loss-of-function mutation of the gene coding for ATPase copper transporting beta (ATP7B), a plasma member protein that pumps out copper from cells [29]. Depending on the nutritional copper uptake and modulatory factors (such as obesity, which accelerates disease pathogenesis), copper then accumulates in particular cell types (mostly in hepatocytes, but also in cardiomyocytes and neurons) beyond a critical threshold that causes cell death, hepatic inflammation and insufficiency (and more rarely cardiomyopathy and neurodegeneration). Excessive cytosolic copper electrophoretically enriches in mitochondria, causing the crosslinking of proteins from the inner and the outer membrane of these organelles, ultimately resulting in mitochondrial destruction and cell death [30]. The only known treatments of WD aim at reducing the copper content in the diet and at chelating copper by suitable molecules such as penicillamine [31]. Recent evidence suggests that WD pathogenesis is also linked to autophagy. Indeed, the livers of WD patients and those of ATP7B-/- rats manifest an increase in autophagic flux [32]. In vitro experiments demonstrate that excessive incorporation of copper into cells triggers autophagy, which acts a cellular defense mechanism to reduce the probability of cell death. Hence, autophagy has a cytoprotective function that is, however, insufficient to avoid the pathogenesis of WD [32]. That said, it remains to be determined whether pharmacological stimulation of autophagy would reduce copper toxicity in WD patients.
The aforementioned results suggest that autophagy plays a prominent disease-decelerating function in both CF and WD (Figure 1). Of note, these finding may have broader implications. Indeed, recent evidence suggests that CFTR function of enterocytes is inhibited in celiac disease (also called ‘gluten enteropathy’), due to its direct inhibition by gluten-derived peptides [33]. As in CF, CFTR inhibition results in reduced expression of Beclin 1 protein, thus compromising autophagy. Of note, potentiation of CFTR function by suitable drugs (‘CFTR potentiators’) can reverse the pro-inflammatory effect of gluten-derived peptides and restore Beclin 1 expression [33]. Decreased CFTR expression has also been observed in several mouse models of autoimmune disease that respond to pharmacological treatment with CFTR potentiators, suggesting that defective CFTR function (and presumably its downstream consequence, autophagy inhibition) might play a rather general role in the pathophysiology of distinct disease entities [34,35]. Similarly, the accumulation of toxic heavy metals (such as cadmium, copper, lead and mercury) may play a general role in accelerating age-related diseases [36–38]. Circumstantial evidence suggests that the toxicity of heavy metals such as cadmium is counteracted by autophagy as well [39].
Based on the aforementioned examples, it is tempting to speculate that autophagy has a general role in slowing down time-dependent processes that ultimately lead to age-related diseases. Indeed, genetic defects in different autophagy-relevant genes cause a broad range of distinct pathologies across a wide spectrum of cardiovascular, infectious, inflammatory, metabolic, neoplastic, neurodegenerative diseases (Levine and Kroemer, in press). It remains to be determined, however, which would be the optimal strategy to increase autophagy for extending the health span in the general population without such gene defects.
Conflicts of Interest
A patent application by LM is pending (filing date, July 26, 2017. N° 102017000085714). GK is a scientific co-founder of Samsara Therapeutics.
Funding
This project was supported by the Italian Institute of Health, under the frame of E-Rare-2, the ERANet for Research on Rare Diseases "RescueCFTRpreclinic” (to LM and GK). GK is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche M.dicale (FRM); a donation by Elior; the European Commission (ArtForce); the European Research Council (ERC); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Searave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE) and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
References
- 1. Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018; 19:579–93. https://doi.org/10.1038/s41580-018-0033-y [PubMed]
- 2. Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013; 4:2300. https://doi.org/10.1038/ncomms3300 [PubMed]
- 3. Fernández AF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, He C, Ting T, Liu Y, Chiang WC, Marciano DK, Schiattarella GG, Bhagat G, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018; 558:136–40. https://doi.org/10.1038/s41586-018-0162-7 [PubMed]
- 4. Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, Michaud M, Madeo F, Tavernarakis N, Kroemer G. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010; 1:e10. https://doi.org/10.1038/cddis.2009.8 [PubMed]
- 5. Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009; 11:1305–14. https://doi.org/10.1038/ncb1975 [PubMed]
- 6. Madeo F, Pietrocola F, Eisenberg T, Kroemer G. Caloric restriction mimetics: towards a molecular definition. Nat Rev Drug Discov. 2014; 13:727–40. https://doi.org/10.1038/nrd4391 [PubMed]
- 7. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016; 22:1428–38. https://doi.org/10.1038/nm.4222 [PubMed]
- 8. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460:392–95. https://doi.org/10.1038/nature08221 [PubMed]
- 9. Blagosklonny MV. Rapamycin extends life- and health span because it slows aging. Aging (Albany NY). 2013; 5:592–98. https://doi.org/10.18632/aging.100591 [PubMed]
- 10. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011; 146:682–95. https://doi.org/10.1016/j.cell.2011.07.030 [PubMed]
- 11. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011; 333:1109–12. https://doi.org/10.1126/science.1201940 [PubMed]
- 12. Longo VD, Panda S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016; 23:1048–59. https://doi.org/10.1016/j.cmet.2016.06.001 [PubMed]
- 13. Kiechl S, Pechlaner R, Willeit P, Notdurfter M, Paulweber B, Willeit K, Werner P, Ruckenstuhl C, Iglseder B, Weger S, Mairhofer B, Gartner M, Kedenko L, et al. Higher spermidine intake is linked to lower mortality: a prospective population-based study. Am J Clin Nutr. 2018; 108:371–80. https://doi.org/10.1093/ajcn/nqy102 [PubMed]
- 14. Madeo F, Carmona-Gutierrez D, Kepp O, Kroemer G. Spermidine delays aging in humans. Aging (Albany NY). 2018; 10:2209–11. https://doi.org/10.18632/aging.101517 [PubMed]
- 15. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 16. Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015; 125:85–93. https://doi.org/10.1172/JCI73946 [PubMed]
- 17. López-Otín C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic Control of Longevity. Cell. 2016; 166:802–21. https://doi.org/10.1016/j.cell.2016.07.031 [PubMed]
- 18. Maiuri L, Raia V, Kroemer G. Strategies for the etiological therapy of cystic fibrosis. Cell Death Differ. 2017; 24:1825–44. https://doi.org/10.1038/cdd.2017.126 [PubMed]
- 19. Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, D’Eletto M, Tosco A, De Gregorio F, Izzo V, Maiuri MC, Kroemer G, Raia V, Maiuri L. Cysteamine re-establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis-relevant F508del-CFTR mutation. Cell Death Dis. 2017; 8:e2544. https://doi.org/10.1038/cddis.2016.476 [PubMed]
- 20. Zhang S, Stoll G, Pedro JM, Sica V, Sauvat A, Obrist F, Kepp O, Li Y, Maiuri L, Zamzami N, Kroemer G. Evaluation of autophagy inducers in epithelial cells carrying the ΔF508 mutation of the cystic fibrosis transmembrane conductance regulator CFTR. Cell Death Dis. 2018; 9:191. https://doi.org/10.1038/s41419-017-0235-9 [PubMed]
- 21. Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, Sepe A, Salvadori L, Buonpensiero P, Di Pasqua A, Grassia R, Leone CA, Guido S, et al. A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016; 23:1380–93. https://doi.org/10.1038/cdd.2016.22 [PubMed]
- 22. Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014; 53:710–25. https://doi.org/10.1016/j.molcel.2014.01.016 [PubMed]
- 23. Pietrocola F, Castoldi F, Markaki M, Lachkar S, Chen G, Enot DP, Durand S, Bossut N, Tong M, Malik SA, Loos F, Dupont N, Mariño G, et al. Aspirin Recapitulates Features of Caloric Restriction. Cell Reports. 2018; 22:2395–407. https://doi.org/10.1016/j.celrep.2018.02.024 [PubMed]
- 24. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015; 21:805–21. https://doi.org/10.1016/j.cmet.2015.05.014 [PubMed]
- 25. Pietrocola F, Lachkar S, Enot DP, Niso-Santano M, Bravo-San Pedro JM, Sica V, Izzo V, Maiuri MC, Madeo F, Mariño G, Kroemer G. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015; 22:509–16. https://doi.org/10.1038/cdd.2014.215 [PubMed]
- 26. De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, Salvadori L, Grassia R, Leone CA, De Rosa G, Maiuri MC, Pettoello-Mantovani M, Guido S, et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy. 2014; 10:2053–74. https://doi.org/10.4161/15548627.2014.973737 [PubMed]
- 27. Izzo V, Pietrocola F, Sica V, Durand S, Lachkar S, Enot D, Bravo-San Pedro JM, Chery A, Esposito S, Raia V, Maiuri L, Maiuri MC, Kroemer G. Metabolic interactions between cysteamine and epigallocatechin gallate. Cell Cycle. 2017; 16:271–79. https://doi.org/10.1080/15384101.2016.1249550 [PubMed]
- 28. Romani L, Oikonomou V, Moretti S, Iannitti RG, D’Adamo MC, Villella VR, Pariano M, Sforna L, Borghi M, Bellet MM, Fallarino F, Pallotta MT, Servillo G, et al. Thymosin α1 represents a potential potent single-molecule-based therapy for cystic fibrosis. Nat Med. 2017; 23:590–600. https://doi.org/10.1038/nm.4305 [PubMed]
- 29. Lalioti V, Sandoval I, Cassio D, Duclos-Vallée JC. Molecular pathology of Wilson’s disease: a brief. J Hepatol. 2010; 53:1151–53. https://doi.org/10.1016/j.jhep.2010.07.008 [PubMed]
- 30. Zischka H, Lichtmannegger J, Schmitt S, Jägemann N, Schulz S, Wartini D, Jennen L, Rust C, Larochette N, Galluzzi L, Chajes V, Bandow N, Gilles VS, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest. 2011; 121:1508–18. https://doi.org/10.1172/JCI45401 [PubMed]
- 31. Lichtmannegger J, Leitzinger C, Wimmer R, Schmitt S, Schulz S, Kabiri Y, Eberhagen C, Rieder T, Janik D, Neff F, Straub BK, Schirmacher P, DiSpirito AA, et al. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J Clin Invest. 2016; 126:2721–35. https://doi.org/10.1172/JCI85226 [PubMed]
- 32. Polishchuk EV, Merolla A, Lichtmannegger J, Romano A, Indrieri A, Ilyechova EY, Concilli M, De Cegli R, Crispino R, Mariniello M, Petruzzelli R, Ranucci G, Iorio R, et al. Activation of autophagy, observed in liver tissues from patients with Wilson disease and from Atp7b-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology. 2018. Epub ahead of print. https://doi.org/10.1053/j.gastro.2018.11.032 [PubMed]
- 33. Villella VR, Venerando A, Cozza G, Esposito S, Ferrari E, Monzani R, Spinella MC, Oikonomou V, Renga G, Tosco A, Rossin F, Guido S, Silano M, et al. A pathogenic role for cystic fibrosis transmembrane conductance regulator in celiac disease. EMBO J. 2018e100101. https://doi.org/10.15252/embj.2018100101 [PubMed]
- 34. Ko SB, Mizuno N, Yatabe Y, Yoshikawa T, Ishiguro H, Yamamoto A, Azuma S, Naruse S, Yamao K, Muallem S, Goto H. Corticosteroids correct aberrant CFTR localization in the duct and regenerate acinar cells in autoimmune pancreatitis. Gastroenterology. 2010; 138:1988–96. https://doi.org/10.1053/j.gastro.2010.01.001 [PubMed]
- 35. Zeng M, Szymczak M, Ahuja M, Zheng C, Yin H, Swaim W, Chiorini JA, Bridges RJ, Muallem S. Restoration of CFTR Activity in Ducts Rescues Acinar Cell Function and Reduces Inflammation in Pancreatic and Salivary Glands of Mice. Gastroenterology. 2017; 153:1148–59. https://doi.org/10.1053/j.gastro.2017.06.011 [PubMed]
- 36. Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014; 13:1045–60. https://doi.org/10.1016/S1474-4422(14)70117-6 [PubMed]
- 37. Eid A, Zawia N. Consequences of lead exposure, and it’s emerging role as an epigenetic modifier in the aging brain. Neurotoxicology. 2016; 56:254–61. https://doi.org/10.1016/j.neuro.2016.04.006 [PubMed]
- 38. Sensi SL, Granzotto A, Siotto M, Squitti R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol Sci. 2018; 39:1049–63. https://doi.org/10.1016/j.tips.2018.10.001 [PubMed]
- 39. Pi H, Li M, Tian L, Yang Z, Yu Z, Zhou Z. Enhancing lysosomal biogenesis and autophagic flux by activating the transcription factor EB protects against cadmium-induced neurotoxicity. Sci Rep. 2017; 7:43466. https://doi.org/10.1038/srep43466 [PubMed]