



Introduction
There is a strong association between Alzheimer's disease (AD) and type-2 diabetes [1-3]. Patients with type 2-diabetes have twice the risk of controls to develop AD [4-9], and the percentage of type 2-diabetes among AD patients is significantly higher than among age-matched non-AD controls [4, 10].
Insulin receptor (IR) and insulin-like growth factor-1 receptor (IGF-1R) are abundant in the normal human brain and are significantly decreased in AD. Insulin receptor substrate-1 (IRS-1) binds not only to IR and IGF-1R but also to the amyloid precursor protein (APP). Moreover, insulin-degrading enzyme (IDE) not only degrades insulin, IDE also degrades APP, amyloid beta (Aβ) [11] and amylin [12]. Insulin and insulin-like growth factor (IGF-1) prevent amyloid formation by decreasing APP in AD [13].
Amyloidosis is a group of conditions of diverse etiologies characterized by the accumulation of insoluble fibrillar proteins in various organs. Amyloidosis is a key pathological feature of both AD and type 2-diabetes, and chronic bacterial infections are frequently associated with amyloidosis. Bacteria and their toxic components, the bacterial endotoxin lipopolysaccharide (LPS) and bacterial cell wall peptidoglycan (BPG) are amyloidogenic and have been successfully used for almost a century in models of experimental inflammation and amyloidosis.
Because of the pioneering work of Warren and Marshall [14] infection with Helicobacter pylori (H. pylori) is accepted as a cause of gastro-duodenal ulcers. Recent observations show that infectious agents are factors in various chronic inflammatory disorders, including atherosclerosis, cardiovascular and cerebrovascular disorders [15-23], chronic lung diseases, [24-26], inflammatory bowel diseases, diabetes [27] and neuropsychiatric disorders, including AD [28, 29]. Polymorphisms in inflammatory genes are also implicated as risk factors in these age-related chronic disorders [30-32].
Increasing evidence supports an association between periodontal and systemic disorders [33, 34]. Periodontal pathogens are predominantly Gram-negative bacteria, and include Porphyromonas gingivalis, various oral Treponema (T) spirochetes e.g. T. denticola, T. pectinovorum, T. amylovorum, T. maltophilum, T. medium and T. socranskii, as well as various herpes viruses [21, 35-38]. Periodontitis is a risk factor for several chronic inflammatory disorders including atherosclerosis, stroke, diabetes and AD [33]. Aβ binds and disrupts lipid bilayers of bacterial membranes causing Ca(2+) influx and bacteriolysis [39]. Islet amyloid amylin is also able to form ion channels on lipid bilayers, causing Ca(2+) influx and cell destruction [40] and recent observations demonstrate antimicrobial activity of amylin [41, 42]. Thus, both Aβ and amylin accumulation are a response of the innate immune system to invading pathogens. This review considers recent information related to the role of local inflammation and involvement of pathogens in type 2 diabetes and AD.
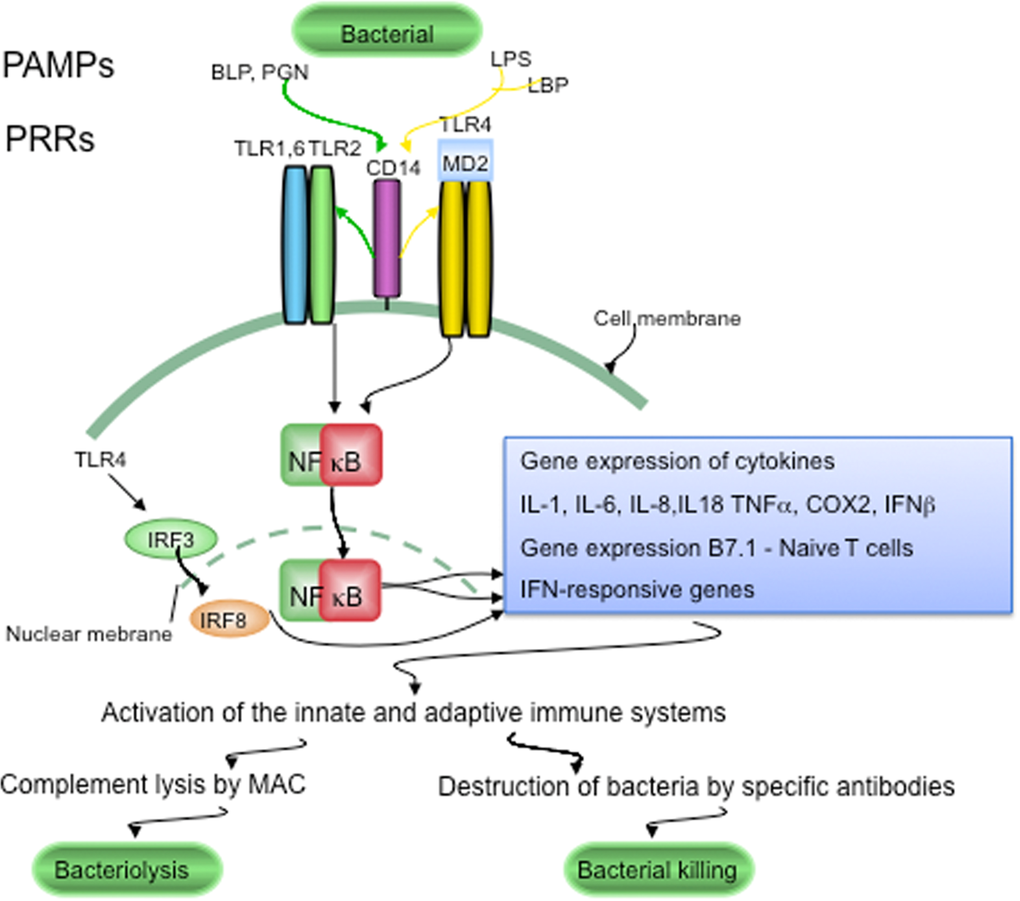
Figure 1. LTR signaling in host defenses against pathogens Conserved components unique to microorganisms, like bacterial lipopolysaccharide (LPS), bacterial peptidoglycan (PGN) and bacterial lipoproteins (BLP), are called pathogen-associated molecular patterns (PAMPs). PAMPs are sensed by pattern recognition receptors (PRRs), which include CD14 and various Toll-like receptors (TLRs). LPS is recognized following its binding to lipoprotein binding protein (LBP). CD14, is part of the LPS receptor complex, and together with the functionally linked TLR2 and TLR4, and the associated molecule MD-2 (lymphocyte antigen 96) are expressed in the endocrine cells of human pancreatic islets. PRRs and TLRs signaling pathways play a major role in maintaining pathogen-free host tissues. When TLRs are activated by PAMPs, through NFκB signaling the innate and adaptive immune systems are activated. Invading bacteria are killed by the terminal attack complex (MAC) of the classical complement pathway and by specific anti-bacterial antibodies provided by the adaptive immune system.
AD is characterized by a slowly progressive dementia and brain atrophy predominantly localized to the cerebral cortex and hippocampus. Senile plaques, and neurofibrillary tangles accumulate in parallel with disease progression. Senile plaques and tangles, argyrophilic filamentous structures, named curly fibers, neuropil threads or dystrophic neurites accumulate in abundance in the cerebral cortex [43, 44]. These slender curly structures are independent filaments, which do not form continuous networks. Their main length as determined by morphometric analysis is about 22 μm [44]. Amyloid deposits in AD mainly consist of Aβ, which include peptides of 40, 42 and 43 amino acid length, deriving from the larger 120 kDa APP by β- and γ-secretase cleavages [46-50]. APP is implicated in cellcell interactions, in regulation of immune system responses, and in T-cell differentiation [51-53]. Neurofibrillary tangles contain paired helical filaments composed mainly of the microtubule-associated protein tau, which is in a hyperphosphorylated state. The role of inflammation as a factor in the pathogenesis of AD is well established [32, 54-57]. McGeer, Rogers and Griffin documented first the importance of local inflammation and IL-1 signaling in AD [55, 58, 59]. Historic and recent observations indicate that pathogens are able to initiate and sustain chronic infection and inflammation and to reproduce the pathological and biological hallmarks of AD [29]. An analysis of the available historic and recent data following Koch's and Hill's criteria favors a causal relationship between spirochetal infection and AD [28]. Co-infection with several spirochetes also occurs in AD [28, 29, 60]. Chlamydophyla (Chlamydia) pneumoniae (C. pneumoniae), Porphyromonas gingivalis, H. Pylori and Herpes simplex virus type 1 (HSV-1) are other microorganisms, which are demonstrated in AD [61-64].
Type 2 diabetes
Type 2 diabetes is a major health problem worldwide, and the number of AD cases is predicted to double during the next decades [65]. In order to solve this urgent problem, prompt action is recommended for both diseases [28, 66]. Type 2-diabetes, previously termed non-insulin dependent diabetes mellitus (NIDDM) or adult-onset diabetes, is characterized by progressive destruction of islet β-cells, resulting in decreased insulin production and decreased action of insulin on peripheral tissues. Type 2 diabetes is the most common form of diabetes, comprising 90-95% of all diabetic cases [67]. Local amyloid deposits are present in more than 95% of type 2 diabetic patients [68] and are mainly composed of amylin [69-71]. This islet amyloid polypeptide of 37 amino acids is derived by proteolytic cleavage of the 89-amino acid islet amyloid precursor protein or proamylin [69, 70, 72]. Amylin is related to the calcitonin/calcitonin gene-related peptide family [73]. Along with insulin, amylin is produced by β-cells in the Langerhans islets of the pancreas. The extent of amyloid deposition correlates with the clinical severity of diabetes, with the impairment in insulin secretion and glucose metabolism, and with the severity of beta-cell loss [69, 70, 74]. The cause of type 2 diabetes and the pathophysiological processes involved in amyloid formation are not well understood.
Aβ and insulin share a common sequence recognition motif [75], and Aβ is a direct competitive inhibitor of insulin binding and action, which reduces insulin receptor autophosphorylation [75]. Aβ and insulin are both substrates for the same insulin degrading enzyme (IDE). High-affinity interaction between Aβ and proamylin result in cross-suppression of cytotoxic selfassembly of both peptides, further suggesting a molecular association between AD and type 2 diabetes [76]. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c- Jun N-terminal kinase signaling [77]. It is noteworthy that insulin dysfunction can lead to tau phosphorylation in vivo [78].
Aβ and tau in the pancreas in type 2 diabetes
Tau mRNA and protein expression are observed in normal and tumoral pancreatic and β-cell-derived cell lines [79-82]. Six predominant tau isoforms are identified by immunoblotting, which form tau deposits detectable by immunofluorescence and sarkosylinsoluble pellets [83]. The expression of APP and tau mRNAs by RT-PCR in normal and type 2 diabetes pancreas and in insulinoma beta cells (INS), as well as the detection of APP and tau immunoreactive bands by Western blot, indicates that APP and tau are present in the pancreatic tissue and in islet beta cells [82]. Slight upregulation of tau expression is also defined at the gene level in pancreatic islets in type 2 diabetic patients compared to normal age matched controls [84].
Aggregated Aβ, hyperphosphorylated tau, ubiquitin, apolipoprotein E, apolipoprotein(a), IB1/JIP-1 and JNK1 are immuno-expressed in the affected Langerhans islets in patients with type 2 diabetes [82, 85, 86] and Aβ is colocalized with amylin [82]. The secondary structure of islet amyloid deposits as analyzed in situ by Synchrotron InfraRed MicroSpectroscopy (SIMRS) reveals a protein (Amide I) absorbance maximum near 1630 cm-1, which is representative of beta-sheet protein structure and identical with the in situ infrared microspectra of amyloid deposits of senile plaques [82, 87, 88].
In transgenic mice carrying the carboxyl-terminus of the beta APP gene, Aβ is demonstrated not only in the brain, but also in the kidney and pancreas [89-91]. Formation of Aβ plaques is also observed in the pancreas in transgenic NORBA mice overexpressing human APP [92]. Remarkably, accumulation of Aβ and hyperphosphorylated tau also occur in brains of rat models of spontaneous diabetes, particularly of type 2 diabetes [93]. All these results indicate that Aβ formation and tau phosphorylation are also features of type 2 diabetes.
It is noteworthy that islet amyloid amylin is also observed in other organs than the pancreas, including the gut and kidney. Seventy-two of 149 patients with diabetic nephropathy showed amylin deposition in the kidney [94]. Amylin deposition in human brain and high affinity amylin binding sites in rat brains were also documented [95, 96].
Local inflammation in type 2 diabetes
Systemic, rather than pancreatic inflammation, has been associated with type 2 diabetes since 1997 [97, 98]. Circulating markers of inflammation, acute-phase reactants and interleukin (IL)-6, the major cytokine mediator of acute-phase responses, are strong predictors of the development of type 2 diabetes [99-102]. Recent prospective studies have strengthened the association between type 2 diabetes and markers of systemic inflammation [103-110].
Only recently have studies focused on the possibility that, similarly to AD, local inflammatory processes might play an important role in type 2 diabetes. On haematoxylin and eosin-stained sections, discrete or moderate lymphocytic infiltrates were present in 2 of 8 diabetic cases analysed. Despite the apparent lack of lymphocytic infiltration in the remaining 6 diabetic cases, use of more sensitive markers revealed significant numbers of CD4 and CD 8 positive T lymphocytes in affected Langerhans islets [27]. Clumps of HLA-DR positive reactive macrophages accumulate around the islet amyloid deposits. Inflammatory cells expressing complement 3 receptor are also present in association with islet amyloid deposits and in the wall of some affected blood vessels [27]. This finding, together with abundant immunoreactivity to complement proteins C3d, C4d and C5b9 in association with islet lesions, indicates that the classical complement pathway is strongly activated in type 2 diabetes [27]. Recent observations demonstrate that both Aβ and amylin activate complement pathways [76]. These results show that, as in AD, local immune responses play an important role in the pathogenesis of type 2 diabetes [27]. In support of the importance of local inflammation in type 2 diabetes, an elevated number of immune cells are detected in the pancreatic islets in conjunction with increased levels of cytokines, chemokines, and IL-1 [111-114]. Animal models of type 2 diabetes investigated also display islet immune cell infiltration [114, 115].
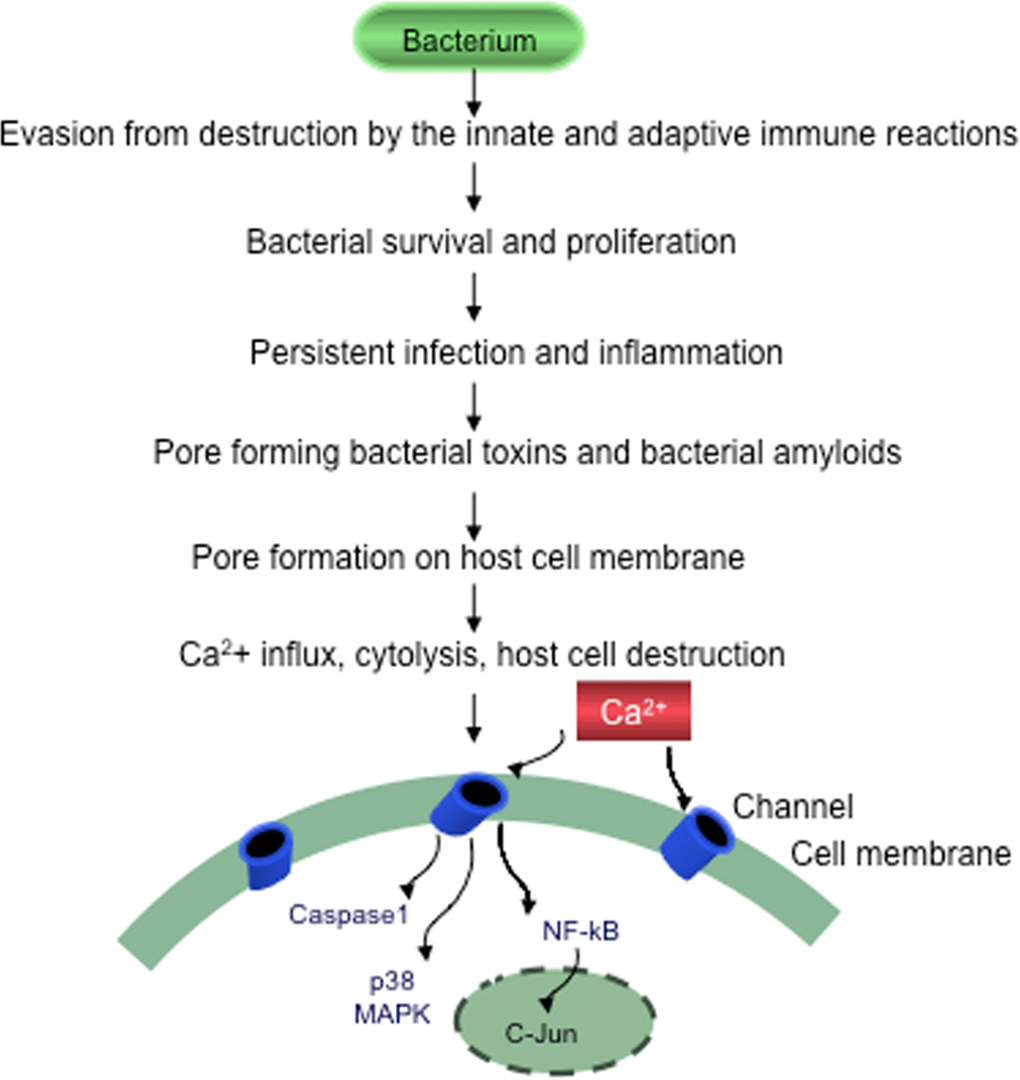
Figure 2. Evasion of bacteria from destruction by the host immune systems Bacteria by suppressing, subverting or escaping host defenses will survive, proliferate and cause persistent chronic infection, sustained inflammation and slowly progressive host cell destruction.
Pathogens and type 2 diabetes
The onset of diabetes often occurs during or after an acute infection. Infections that lead to pancreatitis may produce diabetes [116]. Circulating LPS is higher in diabetic subjects compared to non-diabetics [117-119]. The association of diabetes with tuberculosis, syphilis and leprosy is well known [89, 90, 120-127]. It is noteworthy that various bacterial and viral infections, including H. pylori, enteroviruses, rubella, mumps, rotavirus, parvovirus and cytomegalovirus (CMV) have been proposed as potential etiological factors in type 1 diabetes [128-130].
An increased prevalence of C. pneumoniae IgA was observed in type 2 diabetes [131] and C. pneumoniae reinfection accelerated the development of insulin resistance and diabetes in obese C57BL/6 mice [132]. KKAy diabetic mice showed a significant increase in blood glucose, serum tumor necrosis factor-alpha (TNFα) and IL-6 levels after inoculation with the periodontal pathogen Porphyromonas gingivalis [133]. A high prevalence of H. pylori infection is observed in type 2 diabetes by many authors [134-145], with some exceptions [146-148]. A significantly higher percentage of positive fluorescent treponemal antibody reaction is observed among diabetic patients compared with nondiabetics [78, 149]. Diabetes mellitus is more prevalent among Borrelia-infected patients [150].
Periodontal disorders are risk factors for type 2 diabetes [151-157] and are highly prevalent, affecting up to 90% of the worldwide population [153]. Both type 1 and type 2 diabetic patients show a three- to four-fold increased risk of periodontitis [158-161]. Type 2 diabetic patients have significantly more severe periodontitis, a higher plaque index and a higher prevalence and magnitude of root surface caries than non-diabetic subjects. Periodontitis is predominantly caused by Gram-negative anaerobic bacteria [151-154] and several spirochetes are also demonstrated to be periodontal pathogens. Checkerboard DNA-DNA hybridization showed that significantly more diabetic subjects have higher levels of T. denticola compared to controls [162]. Treatment of periodontal infections improves glycemic control in diabetic patients [151-154, 163].
The association of periodontitis and diabetes suggests that microorganisms are pathogenic in type 2 diabetes [27, 152-154]. The presence of various bacteria associated with islet lesions in patients with type 2 diabetes is demonstrated pathologically [27]. Anti-core- LPS J5, anti-Lipid A and anti-BPG antibodies reveal positive LPS and BPG immune-reactions in association with islet amyloid deposits in the pancreas of 8 patients with clinically and pathologically confirmed type 2 diabetes [27]. On doubly immune-stained sections, LPS and BPG were immuno-co-localized with amylin, and LPS-positive and BPG-positive helically shaped, round, and fusiform bacteria are observed in the affected islets. Intracellularly located immune-reactive C. pneumoniae was observed in the affected Langerhans islets in 5 of 6 diabetic cases tested, and extracellularly located, slightly coiled H. pylori bacteria are demonstrated in 3 of these same diabetic patients. Spirochetes are also observed in the affected islets, explaining the high frequency of fluorescent Treponemal antibodies found in diabetes [27]. These bacterial structures are immunoreactive for C5b-9, the complement membrane attack complex (MAC) intended to lyse bacteria. As MAC assembles on cell membranes in vivo, the possibility is eliminated that C5b-9 immuno-reactive bacterial structures may correspond to post mortem bacterial growth. Moderate LPS and BPG immuno-reactivity was also observed in the pancreas of three controls in association with mild islet amyloid deposition, corresponding to pre-clinical stages of type 2 diabetes in these cases. The remaining 9 controls were negative [27].
In agreement with epidemiological studies, the presence of bacteria, including C. pneumoniae, H. pylori and various types spirochetes in pancreatic islets supports a pathogenic role of bacterial infection in type 2 diabetes. Simultaneous occurrence of various types of bacteria in the pancreatic islets of the same diabetic patient suggests that concurrent infection by several pathogens occurs in type 2 diabetes, as observed in atherosclerosis [21, 22] and AD [28, 29, 60-62, 64]. C. pneumoniae [15, 16], H. pylori [17, 18], several periodontal pathogens, including invasive oral spirochetes [19, 20] and herpes viruses, have also been demonstrated in human atherosclerotic lesions. Some microorganisms were shown to enhance atherosclerosis in experimental animals [20, 21].
Improvement of glycaemic control was observed after eradication of H. pylori infection in diabetic patients, suggesting that early antibiotic and anti-inflammatory treatment may be an effective way to prevent or slow down the disease process [164].
Host pathogen interaction through toll-like receptors
Whole bacteria and specific microbial components (LPS, BPG) are demonstrated in islet lesions in type 2 diabetes [27, 118, 165]. Such conserved microbial motifs (LPS, BPG, various bacterial lipoproteins, bacterial DNA etc.) are called pathogen-associated molecular patterns (PAMPs). PAMPs are sensed by pattern recognition receptors (PRRs) [166], which trigger an immediate response against invading pathogens. The major forms of PRRs are Toll-like receptors (TLRs) and some nucleotide-binding oligomerization domain (NOD) receptors called Nodlike receptors (NLRs) [167]. Once TLRs and NLRs are activated by PAMPs through signaling pathways, they induce innate and adaptive immune responses [167, 168]. Thus signaling by TLRs and NLRs is a key component of immune responses to microbial infection [169].
CD14, which is part of the LPS receptor complex, together with the functionally linked TLR2 and TLR4, and the associated molecule MD-2, are all expressed in the endocrine cells of human pancreatic islets. SV40- transformed islet cells (HP62) synthesize and secrete CD14 in response to LPS in a time- and dose-dependent manner. In vitro experiments using rat islets, which also express CD14, as well as HP62 cells, showed that LPS influences glucose-dependent insulin secretion and induces formation of inflammatory cytokines such as IL-1α, IL-6 and TNFα [170]. LPS also induces increased APP and tau levels in neuronal and nonneuronal cells in vitro [82].
TLRs are involved in a variety of diseases including atherosclerosis, type 2 diabetes, liver disease, inflammatory bowel diseases and AD [165]. NLRs together with TLRs induce IL-1β and IL-18, which are important mediators in most inflammatory disorders [168]. Expression of various PAMPs and PRRs, including CD14 and TLRs, together with local immune responses in association with the lesions in both AD and type 2 diabetes, indicates that microorganisms and PAMPs induce and sustain chronic infection and inflammation in these chronic amyloidogenic disorders. The role of bacteria as sources of PAMPs via TLR2 and TLR4 stimulation, in atherosclerosis, type 2 diabetes, AD suggests a common pathogenic process in these diseases.
Antimicrobial peptides (AMPs) are another important group of molecules of the innate immune system, which combat invading microorganisms. Recent observations reveal that Aβ, the most important biological marker of AD, is an innate immune molecule, and shares properties with AMPs [39]. Soluble Aβ 1-42 oligomers form channels on lipid cell membranes and cause Ca(2+) influx and cell destruction [171]. Aβ at high doses exerts antimicrobial activity in vivo against eight common and clinically relevant microorganisms of the 12 tested. Antimicrobial activity of brain homogenates was attenuated by immune-depletion of Aβ [38]. CT105 peptide, a C terminal fragment of APP also forms ion channels or pores [172] and the microtubule binding sites of tau have been shown to harbor somewhat similar properties [173].
The level of human serum amyloid A (SAA), an acute phase protein, which rises during infection, also forms channels on lipid bilayer membranes with resulting Ca(2+) influx. Expression of a recombinant acute phase isoform variant of human SAA 1.1 (SAAp) induces bacteriolysis, suggesting an important role in host defenses [174]. It is noteworthy that amylin also forms ion channels on lipid bilayers with consequent Ca(2+) influx and cell lysis [40, 175]. Recent observations show that amylin is an antimicrobial peptide, which can augment host defenses against on-going infection, as also observed for Aβ [39] and SAA.
Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in various neurodegenerative disorders [176]. Blockade of an amyloid peptide channel by zinc, and inhibition by Congo red, has been recently reported [177].
It has been hypothesized that amyloid pores may in fact be beta-sheet barrels similar to pore forming bacterial toxins [178]. Pore-forming toxins are the most common class of bacterial protein toxins and are often important virulence factors. These toxins are typically oligomers of soluble, monomeric proteins or peptides, which form transmembrane channels. Channel formation in the membrane of targeted cells triggering cellular ion imbalance is a frequent form of bacterial attack [179, 180]. These pore-forming bacterial toxins generate calcium-dependent and lipid-mediated signaling on host cell surfaces, leading to a variety of events such as tyrosine phosphorylation [181], actin rearrangement [182], NF-κB activation [183] and regulation of gene expression through histone modification [184].
It is noteworthy that amyloid proteins constitute a previously overlooked integral part of the cellular envelope of many bacteria [185-189]. Amyloid fibril formation not only results in toxic aggregates, but also provides biologically functional molecules [186, 187, 190], which play a role in virulence, invasion and host cell destruction. Recent observations show that the amyloid oligomers associated with human diseases, perforin from cytotoxic T lymphocytes and poreforming bacterial toxins, share structural homology and the same mechanism of membrane permeabilization [191].
The amyloidogenic properties of perforin and the bacterial pore-forming toxin alpha-hemolysin were demonstrated spectroscopically and morphologically [191].
The observations on amyloidogenesis suggest that host cells and bacteria, during host-pathogen interactions, use similar molecular mechanisms to induce host cell lysis and bacteriolysis. Further studies will be required to determine whether host cell destruction predominates over bacteriolysis in chronic sustained infections and inflammations.
In addition, genetic predisposition of the host, the virulence and biology of the invading pathogens, and environmental factors, including nutrition and demographic conditions, are also key determinants of disease expression.
Establishment of chronic infection, inflammation and progressive cell damage
During infection, pathogens employ a broad range of strategies to overcome antigenic recognition, phagocytosis and complement lysis. Blockade of the complement cascade allows their survival even in immune competent hosts. If pathogens are not recognized by the immune system, and in the absence of cell-mediated immune responses, the microorganism can spread freely and accumulate in affected host tissues [192]. Under such conditions, microorganisms will establish chronic infection, inflammation and progressive tissue damage. Host responses to bacterial infections are genetically controlled. Polymorphisms of proinflammatory cytokine genes are associated with susceptibility to infection [193]. TNF-α and Class II major histocompatibility genes are critical mediators of host defences against infection by influencing host immune responses to bacterial and viral infections. Polymorphisms in the gene encoding TNF-α may determine a strong cell-mediated immune response or a weak or absent cellular response [193, 194]. Human leukocyte antigen (HLA) also controls cell-mediated responses [195]. HLA-DR isotypes are associated with a protective response, whereas HLA-DQ isotypes have a more limited cellular response but with a higher number of microorganisms. Accordingly, a polarity in host reactions can be observed in various infections. In tuberculoid or paucibacillary leprosy, inflammatory cell infiltration is strong and the number of microorganisms is low. However in lepromatous or bacillary leprosy, the poor or absent inflammatory cell infiltrates are accompanied by a high number of Mycobacterium leprae bacilli. A similar polarity in host reactions also occurs in neurosyphilis. Strong cell-mediated immune responses and a low number of spirochetes characterize the infiltrative form of general paresis. In the atrophic form of general paresis, the lymphoplasmocytic infiltrates are poor or absent, but the number of spirochetes is high [196-199].
If infectious agents are involved in diabetes, one may expect that such polarities in host reactions might also be present. According to this view, type 1 diabetes is characterized by strong cell mediated immune response with a low number of microorganisms and type 2 diabetes is characterized by poor or absent lymphoplasmocytic infiltrates with high number of microorganisms. Further studies are needed to support this suggestion.
Discussion
Common cellular and molecular mechanisms are implicated in the pathogenesis of AD and type 2 diabetes. In addition to amylin, Aβ and hyperphosphorylated tau accumulations are also features of islet lesions in type 2 diabetes. Ubiquitin, Apo-E, Apo(a), IB1/JIP-1 and JNK-1 are all associated with brain lesions in AD and islet lesions in type 2 diabetes. As in AD, both cellular and humoral components of local immune responses are involved in the pathogenesis of type 2 diabetes, as indicated by the presence of PAMPs and PRRs in the affected Langerhans islets. The classical complement pathway is activated in both AD and type 2 diabetes. Using sensitive immuno-markers of B and T lymphocytes, increased numbers of CD4, and particularly of CD 8 positive T cells, are demonstrated in both AD and type 2 diabetes, indicating a minimal involvement of the adaptive immune system.
Epidemiological and pathological evidence demonstrate the prevalence of pathogens in several chronic inflammatory diseases, including atherosclerosis, AD and type 2 diabetes. An increased prevalence of C. pneumoniae IgA, H. pylori infection, and a significantly higher percentage of positive fluorescent treponemal antibody levels are observed among diabetic patients compared to controls, indicating the involvement of spirochetes in diabetes. These observations indicate that various types of spirochetes are also involved in diabetes. As AD, atherosclerosis and diabetes are all associated with periodontal disorders, to consider periodontal pathogens, including Porphyromonas gingivalis and various periodontal pathogen spirochetes and various intestinal bacteria, including intestinal spirochetes, in the etiology of these chronic inflammatory disorders is important. Borrelia burgdorferi and other Borrelias might also be candidate spirochetes in some cases with type 2 diabetes. The observation of several of these bacteria in association with islet lesions suggests that chronic bacterial infection could be directly involved in the pathogenesis of type 2 diabetes. Similarly to periodontitis, simultaneous infection with multiple pathogens also occurs in type 2 diabetes. Invading pathogens, and their persisting toxic PAMPs sensed by PRRs through TLRs signaling, induce innate and adaptive immune responses. In the affected pancreas, pathogenic bacteria and their toxic components can be observed, along with a host immunological reaction, which is characteristic of a localized inflammatory process associated with the sites of tissue damage, similar to the inflammatory process observed in AD [58, 59, 200, 201].
Conclusion
Increasing evidence supports the hypothesis that bacteria or their slowly degradable remnants, may initiate a cascade of events leading to persistent chronic infection, β-cell loss and amyloid deposition in type 2 diabetes. Increased Aβ and amylin accumulation, and the resulting AD and islet pathology, may be mediated by a response of the innate immune system to infection. An infectious origin may give an explanation of the common pathogenic mechanisms and inflammatory gene polymorphisms involved in both AD and type 2 diabetes. This interpretation may have important implications for current and future treatment strategies, opening the possibility of preventing infection, inflammation and amyloidosis in both AD and type 2 diabetes.
Acknowledgments
We would like to thank all those who contributed in a direct or an indirect way in the realization of this review.
Funding
This work is supported by the Prevention Alzheimer International Foundation, Switzerland and the Pacific Alzheimer Research Foundation, British Columbia University, Vancouver, Canada.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1. Voisin T, Lugardon S, Balardy L, Vellas B. Vascular risk factors and Alzheimer's disease.Rev Med Interne.2003;24:Suppl 3288s-291s..
- 2. Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study.Diabetologia.1996;39:1392-1397..
- 3. Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease.Diabetes.2004;53:474-481..
- 4. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study.Neurology.1999;53:1937-1942..
- 5. Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, et al. The risk of dementia among persons with diabetes mellitus: a population-based cohort study.Ann N Y Acad Sci.1997;826:422-427..
- 6. Grodstein F, Chen J, Wilson RS, Manson JE. Type 2 diabetes and cognitive function in community-dwelling elderly women.Diabetes Care.2001;24:1060-1065..
- 7. Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study.Diabetes.2002;51:1256-1262..
- 8. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function.Arch Neurol.2004;61:661-666..
- 9. Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study.Neurology.2004;63:1181-1186..
- 10. Kuusisto J, Koivisto K, Mykkanen L, Helkala EL, Vanhanen M, Hanninen T, et al. Association between features of the insulin resistance syndrome and Alzheimer's disease independently of apolipoprotein E4 phenotype: cross sectional population based study.BMJ.1997;315:1045-1049..
- 11. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo.Proc Natl Acad Sci U S A.2003;100:4162-4167..
- 12. Bennett RG, Duckworth WC, Hamel FG. Degradation of amylin by insulin-degrading enzyme.J Biol Chem.2000;275:36621-36625..
- 13. Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, et al. Clearance mechanisms of Alzheimer's amyloid-beta peptide: implications for therapeutic design and diagnostic tests.Mol Psychiatry.2009;14:469-486..
- 14. Marshall BJ and Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration.Lancet.1984;1:1311-1315..
- 15. Laitinen K, Laurila A, Pyhala L, Leinonen M, Saikku P. Chlamydia pneumoniae infection induces inflammatory changes in the aortas of rabbits.Infect Immun.1997;65:4832-4835..
- 16. Saikku P. Epidemiology of Chlamydia pneumoniae in atherosclerosis.Am Heart J.1999;138:S500-503..
- 17. Mendall MA, Goggin PM, Molineaux N, Levy J, Toosy T, Strachan D, et al. Relation of Helicobacter pylori infection and coronary heart disease.Br Heart J.1994;71:437-439..
- 18. Martin-de-Argila C, Boixeda D, Canton R, Gisbert JP, Fuertes A. High seroprevalence of Helicobacter pylori infection in coronary heart disease.Lancet.1995;346:310.
- 19. Renvert S, Pettersson T, Ohlsson O, Persson GR. Bacterial profile and burden of periodontal infection in subjects with a diagnosis of acute coronary syndrome.J Periodontol.2006;77:1110-1119..
- 20. Zaremba M, Gorska R, Suwalski P, Kowalski J. Evaluation of the incidence of periodontitis-associated bacteria in the atherosclerotic plaque of coronary blood vessels.J Periodontol.2007;78:322-327..
- 21. Chiu B. Multiple infections in carotid atherosclerotic plaques.Am Heart J.1999;138:S534-536..
- 22. Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques.J Periodontol.2000;71:1554-1560..
- 23. Rassu M, Cazzavillan S, Scagnelli M, Peron A, Bevilacqua PA, Facco M, et al. Demonstration of Chlamydia pneumoniae in atherosclerotic arteries from various vascular regions.Atherosclerosis.2001;158:73-79..
- 24. Martin RJ. Infections and asthma.Clin Chest Med.2006;27:87-98..
- 25. MacDowell AL and Bacharier LB. Infectious triggers of asthma.Immunol Allergy Clin North Am.2005;25:45-66..
- 26. Micillo E, Bianco A, D'Auria D, Mazzarella G, Abbate GF. Respiratory infections and asthma.Allergy.2000;55:Suppl 6142-45..
- 27. Miklossy J, Martins R, Darbinian N, Khalili K, McGeer P. Type 2 diabetes: Local Inflammation and Direct Effect of Bacterial Toxic Components.Open Pathol J.2008;2:86-95..
- 28. Miklossy J. Alzheimer's disease -a neurospirochetosis. Analysis of the evidence following Koch's and Hill's criteria.J Neuroinflammation.2011;8:190.
- 29. Miklossy J. Emerging roles of pathogens in Alzheimer disease.Expert Rev Mol Med.2011;13:e30.
- 30. DeGraba TJ. Immunogenetic susceptibility of atherosclerotic stroke: implications on current and future treatment of vascular inflammation.Stroke.2004;35S:2712-2719..
- 31. Kolb H and Mandrup-Poulsen T. An immune origin of type 2 diabetes?Diabetologia.2005;48:1038-1050..
- 32. McGeer PL and McGeer EG. Polymorphisms in inflammatory genes and the risk of Alzheimer disease.Arch Neurol.2001;58:1790-1792..
- 33. Kamer AR, Dasanayake AP, Craig RG, Glodzik-Sobanska L, Bry M, de Leon MJ. Alzheimer's disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis.J Alzheimer's Dis.2008;13:437-449..
- 34. Santacroce L, Carlaio RG, Bottalico L. Does it make sense that diabetes is reciprocally associated with periodontal disease?Endocr Metab Immune Disord Drug Targets.2010;10:57-70..
- 35. Seppaa B and Ainamo J. Dark field microscopy of the subgingival microflora in insulin-dependent diabetics.J Clin Periodontol.1996;23:63-67..
- 36. Ling LJ, Ho CC, Wu CY, Chen YT, Hung SL. Association between human herpesviruses and the severity of periodontitis.J Periodontol.2004;75:1479-1485..
- 37. Heydenrijk K, Meijer HJ, van der Reijden WA, Raghoebar GM, Vissink A, Stegenga B. Microbiota around root-form endosseous implants: a review of the literature.Int J Oral Maxillofac Implants.2002;17:829-838..
- 38. O'Brien-Simpson NM, Veith PD, Dashper SG, Reynolds EC. Antigens of bacteria associated with periodontitis.Periodontol 2000.2004;35:101-134..
- 39. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid betaprotein is an antimicrobial peptide.PLoS One.2010;5:e9505.
- 40. Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer's betaamyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line.J Biol Chem.2000;275:14077-14083..
- 41. Wang L, Liu Q, Chen JC, Cui YX, Zhou B, Chen YX, et al. Antimicrobial activity of human islet amyloid polypeptides: an insight into amyloid peptides' connection with antimicrobial peptides.Biol Chem.2012;393:641-646..
- 42. Wang G. Human antimicrobial peptides and proteins.Pharmaceuticals (Basel).2014;7:545-594..
- 43. Gallyas F. Silver staining of Alzheimer's neurofibrillary changes by means of physical development.Acta Morphol Acad Sci Hung.1971;19:1-8..
- 44. Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer's disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques.Neurosci Lett.1986;65:351-355..
- 45. Markesbery WR, Wang HZ, Kowall NW, Kosik KS, McKee AC. Morphometric image analysis of neuropil threads in Alzheimer's disease.Neurobiol Aging.1993;14:303-307..
- 46. Glenner GG and Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein.Biochem Biophys Res Commun.1984;120:885-890..
- 47. Glenner GG, Wong CW, Quaranta V, Eanes ED. The amyloid deposits in Alzheimer's disease: their nature and pathogenesis.Appl Pathol.1984;2:357-369..
- 48. Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor.Nature.1987;325:733-736..
- 49. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome.Proc Natl Acad Sci U S A.1985;82:4245-4249..
- 50. Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease.Trends Cell Biol.1998;8:447-453..
- 51. Monning U, Konig G, Prior R, Mechler H, Schreiter-Gasser U, Masters CL, et al. Synthesis and secretion of Alzheimer amyloid beta A4 precursor protein by stimulated human peripheral blood leucocytes.FEBS Lett.1990;277:261-266..
- 52. Allen JS, Murphy GM Jr., Eng LF, Stultz KE, Davies HD, Pickford LB, et al. Alzheimer's disease: beta-amyloid precursor protein mRNA expression in mononuclear blood cells.Neurosci Lett.1991;132:109-112..
- 53. Ledoux S, Rebai N, Dagenais A, Shaw IT, Nalbantoglu J, Sekaly RP, et al. Amyloid precursor protein in peripheral mononuclear cells is up-regulated with cell activation.J Immunol.1993;150:5566-5575..
- 54. McGeer PL and Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer's disease.Neurology.1992;42:447-449..
- 55. Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, et al. Inflammation and Alzheimer's disease pathogenesis.Neurobiol Aging.1996;17:681-686..
- 56. McGeer PL and McGeer EG. Local neuroinflammation and the progression of Alzheimer's disease.J Neurovirol.2002;8:529-538..
- 57. McGeer PL and McGeer EG. The possible role of complement activation in Alzheimer disease.Trends Mol Med.2002;8:519-523..
- 58. McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR.Neurosci Lett.1987;79:195-200..
- 59. Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease.Proc Natl Acad Sci U S A.1989;86:7611-7615..
- 60. Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer's disease.Oral Microbiol Immunol.2002;17:113-118..
- 61. Balin BJ, Little CS, Hammond CJ, Appelt DM, Whittum-Hudson JA, Gerard HC, et al. Chlamydophila pneumoniae and the etiology of late-onset Alzheimer's disease.J Alzheimers Dis.2008;13:371-380..
- 62. Itzhaki RF and Wozniak MA. Herpes simplex virus type 1 in Alzheimer's disease: the enemy within.J Alzheimer's Dis.2008;13:393-405..
- 63. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue.J Alzheimer's Dis.2013;36:665-677..
- 64. Kountouras J, Tsolaki M, Gavalas E, Boziki M, Zavos C, Karatzoglou P, et al. Relationship between Helicobacter pylori infection and Alzheimer disease.Neurology.2006;66:938-940..
- 65. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030.Diabetes Care.2004;27:1047-1053..
- 66. Matthews DR and Matthews PC. Banting Memorial Lecture 2010. Type 2 diabetes as an ‘infectious’ disease: is this the Black Death of the 21st century?Diabet Med.2011;28:2-9..
- 67. O'Brien JA, Patrick AR, Caro J. Estimates of direct medical costs for microvascular and macrovascular complications resulting from type 2 diabetes mellitus in the United States in 2000.Clin Ther.2003;25:1017-1038..
- 68. Lopes DH, Colin C, Degaki TL, de Sousa AC, Vieira MN, Sebollela A, Martinez AM, Bloch C Jr., Ferreira ST, Sogayar MC. Amyloidogenicity and cytotoxicity of recombinant mature human islet amyloid polypeptide (rhIAPP).J Biol Chem.2004;279:42803-42810..
- 69. Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients.Proc Natl Acad Sci U S A.1987;84:8628-8632..
- 70. Cooper GJ, Willis AC, Reid KB, Clark A, Baker CA, Turner RC, et al. Diabetes-associated peptide.Lancet.1987;2:966.
- 71. Westermark P, Wernstedt C, O'Brien TD, Hayden DW, Johnson KH. Islet amyloid in type 2 human diabetes mellitus and adult diabetic cats contains a novel putative polypeptide hormone.Am J Pathol.1987;127:414-417..
- 72. Sanke T, Bell GI, Sample C, Rubenstein AH, Steiner DF. An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing.J Biol Chem.1988;263:17243-17246..
- 73. Cooper GJ, Leighton B, Dimitriadis GD, Parry-Billings M, Kowalchuk JM, Howland K, et al. Amylin found in amyloid deposits in human type 2 diabetes mellitus may be a hormone that regulates glycogen metabolism in skeletal muscle.Proc Natl Acad Sci U S A.1988;85:7763-7766..
- 74. Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes.J Clin Endocrinol Metab.2004;89:3629-3643..
- 75. Miragliotta G, Del Prete R, Mosca A. Helicobacter pylori infection and coronary heart disease.Lancet.1994;344:751.
- 76. Klegeris A and McGeer PL. Complement activation by islet amyloid polypeptide (IAPP) and alpha-synuclein 112.Biochem Biophys Res Commun.2007;357:1096-1099..
- 77. Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin.J. Neurosci.2009;29:9078-9089..
- 78. Brauner A, Carlsson B, Sundkvist G, Ostenson CG. Falsepositive treponemal serology in patients with diabetes mellitus.J Diabetes. Complications.1994;8:57-62..
- 79. Vanier MT, Neuville P, Michalik L, Launay JF. Expression of specific tau exons in normal and tumoral pancreatic acinar cells.J Cell Sci.111:1998;1419-1432..
- 80. Jimeno A, Hallur G, Chan A, Zhang X, Cusatis G, Chan F, et al. Development of two novel benzoylphenylurea sulfur analogues and evidence that the microtubule-associated protein tau is predictive of their activity in pancreatic cancer.Mol Cancer Ther.2007;6:1509-1516..
- 81. Michalik L, Neuville P, Vanier MT, Launay JF. Pancreatic tau related maps: biochemical and immunofluorescence analysis in a tumoral cell line.Mol Cell Biochem.1995;143:107-112..
- 82. Miklossy J, Qing H, Radenovic A, Kis A, Vileno B, Laszlo F, et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes.Neurobiol Aging.2010;31:1503-1515..
- 83. Maj M, Gartner W, Ilhan A, Neziri D, Attems J, Wagner L. Expression of TAU in insulin-secreting cells and its interaction with the calcium-binding protein secretagogin.J Endocrinol.2010;205:25-36..
- 84. Gunton JE, Kulkarni RN, Yim S, Okada T, Hawthorne WJ, Tseng YH, et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes.Cell.2005;122:337-349..
- 85. Charge SB, Esiri MM, Bethune CA, Hansen BC, Clark A. Apolipoprotein E is associated with islet amyloid and other amyloidoses: implications for Alzheimer's disease.J Pathol.1996;179:443-447..
- 86. Miklossy J, Taddei K, Martins R, Escher G, Kraftsik R, Pillevuit O, et al. Alzheimer disease: curly fibers and tangles in organs other than brain.J Neuropathol Exp Neurol.1999;58:803-814..
- 87. Choo LP, Wetzel DL, Halliday WC, Jackson M, LeVine SM, Mantsch HH. In situ characterization of beta-amyloid in Alzheimer's diseased tissue by synchrotron Fourier transform infrared microspectroscopy.Biophys J.1996;71:1672-1679..
- 88. Miklossy J, Kis A, Radenovic A, Miller L, Forro L, Martins R, et al. Beta-amyloid deposition and Alzheimer's type changes induced by Borrelia spirochetes.Neurobiol Aging.2006;27:228-236..
- 89. Rosenbloom J. The relation between diabetes mellitus and clinical syphilis.JAMA.1917;68:1232-1234..
- 90. Warthin A and Wilson F. The Coincidence of Latent Syphilis and Diabetes.Am J Med Sci.1916;152:157-164..
- 91. Fukuchi K, Hart M, Yan Z, Hassell JR, Li L. Transgenic mice overexpressing both amyloid beta-protein and perlecan in pancreatic acinar cells.Histol Histopathol.2004;19:845-852..
- 92. Nicolau C, Greferath R, Balaban TS, Lazarte JE, Hopkins RJ. A liposome-based therapeutic vaccine against beta -amyloid plaques on the pancreas of transgenic NORBA mice.Proc Natl Acad Sci U S A.2002;99:2332-2337..
- 93. Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes.Diabetes.2007;56:1817-1824..
- 94. Gong W, Liu ZH, Zeng CH, Peng A, Chen HP, Zhou H, et al. Amylin deposition in the kidney of patients with diabetic nephropathy.Kidney Int.2007;72:213-218..
- 95. Jackson K, Barisone GA, Diaz E, Jin LW, DeCarli C, Despa F. Amylin deposition in the brain: A second amyloid in Alzheimer disease?Annals of neurology.2013;74:517-526..
- 96. Beaumont K, Kenney MA, Young AA, Rink TJ. High affinity amylin binding sites in rat brain.Mol Pharmacol.1993;44:493-497..
- 97. Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acutephase reactants and interleukin-6 with metabolic syndrome X.Diabetologia.1997;40:1286-1292..
- 98. Pickup JC and Crook MA. Is type II diabetes mellitus a disease of the innate immune system?Diabetologia.1998;41:1241-1248..
- 99. Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, et al. Markers of inflammation and prediction of diabetes mellitus in adults : a cohort study.Lancet.1999;353:1649-1652..
- 100. Duncan BB, Schmidt MI, Offenbacher S, Wu KK, Savage PJ, Heiss G. Factor VIII and other hemostasis variables are related to incident diabetes in adults. The Atherosclerosis Risk in Communities (ARIC) Study.Diabetes Care.1999;22:767-772..
- 101. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. Creactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus.JAMA.2001;286:327-334..
- 102. Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes.Diabetes Care.2004;27:813-823..
- 103. Hu FB, Meigs JB, Li TY, Rifai N, Manson JE. Inflammatory markers and risk of developing type 2 diabetes in women.Diabetes.2004;53:693-700..
- 104. Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study.Diabetes.2003;52:812-817..
- 105. Thorand B, Löwel H, Schneider A, Kolb H, Meisinger C, Fröhlich M, et al. C-reactive protein as a predictor for incident diabetes mellitus among middle-aged men: results from the MONICA Augsburg cohort study, 1984-1998.Arch Intern Med.2003;163:93-99..
- 106. Thorand B, Kolb H, Baumert J, Koenig W, Chambless L, Meisinger C, et al. Elevated levels of interleukin-18 predict the development of type 2 diabetes: results from the MONICA/KORA Augsburg Study, 1984-2002.Diabetes.2005;54:2932-2938..
- 107. Doi Y, Kiyohara Y, Kubo M, Ninomiya T, Wakugawa Y, Yonemoto K, et al. Elevated C-reactive protein is a predictor of the development of diabetes in a general Japanese population: the Hisayama Study.Diabetes Care.2005;28:2497-2500..
- 108. Nakanishi S, Yamane K, Kamei N, Okubo M, Kohno N. Elevated C-reactive protein is a risk factor for the development of type 2 diabetes in Japanese Americans.Diabetes Care.2003;26:2754-2757..
- 109. Rutter MK, Meigs JB, Sullivan LM, D'Agostino RB Sr., Wilson PW. C-reactive protein, the metabolic syndrome, and prediction of cardiovascular events in the Framingham Offspring Study.Circulation.2004;110:380-385..
- 110. Festa A, D'Agostino R Jr., Tracy RP, Haffner SM. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study.Diabetes.2002;51:1131-1137..
- 111. Donath MY, Boni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes.Physiology (Bethesda).2009;24:325-331..
- 112. Boni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation.J Clin Endocrinol Metab.2008;93:4065-4074..
- 113. Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. Islet-associated macrophages in type 2 diabetes.Diabetologia.2009;52:1686-1688..
- 114. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased number of islet-associated macrophages in type 2 diabetes.Diabetes.2007;56:2356-2370..
- 115. Ehses JA, Ellingsgaard H, Boni-Schnetzler M, Donath MY. Pancreatic islet inflammation in type 2 diabetes: from alpha and beta cell compensation to dysfunction.Arch Physiol Biochem.2009;115:240-247..
- 116. Cecil RL. Text-book of Medicine.Saunders companyPhiladelphia and London1938;1614.
- 117. Devaraj S, Dasu MR, Park SH, Jialal I. Increased levels of ligands of Toll-like receptors 2 and 4 in type 1 diabetes.Diabetologia.2009;52:1665-1668..
- 118. Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, Khanolkar M, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes.Am J Physiol Endocrinol Metab.2007;292:E740-747..
- 119. Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes.Diabetes Care.2011;34:392-397..
- 120. Nigam P, Dayal SG, Srivastava P, Joshi LD, Goyal BM, Dutt B, et al. Diabetic status in leprosy.Hansenol Int.1979;4:7-14..
- 121. Restrepo BI. Convergence of the tuberculosis and diabetes epidemics: renewal of old acquaintances.Clin Infect Dis.2007;45:436-438..
- 122. Perez A, Brown HS 3rd, Restrepo BI. Association between tuberculosis and diabetes in the Mexican border and non-border regions of Texas.Am J Trop Med Hyg.2006;74:604-611..
- 123. Maalej S, Belhaoui N, Bourguiba M, Mahouachi R, Chtourou A, Taktak S, et al. [Pulmonary tuberculosis and diabetes. A retrospective study of 60 patients in Tunisia].Presse medicale.2009;38:20-24..
- 124. Fisher-Hoch SP, Whitney E, McCormick JB, Crespo G, Smith B, Rahbar MH, et al. Type 2 diabetes and multidrug-resistant tuberculosis.Scand J Inf Dis.2008;40:888-893..
- 125. Price FW. Textbook of the Practice of Medicine.Oxford Medical Publication, Humphrey Milford Publisher, Oxford University PressLondon and Edinburgh1941;2032.
- 126. Stevenson CR, Critchley JA, Forouhi NG, Roglic G, Williams BG, Dye C, et al. Diabetes and the risk of tuberculosis: a neglected threat to public health?Chronic Illn.2007;3:228-245..
- 127. Dixon B. Diabetes and tuberculosis: an unhealthy partnership.Lancet Infect Dis.2007;7:444.
- 128. van der Werf N, Kroese FG, Rozing J, Hillebrands JL. Viral infections as potential triggers of type 1 diabetes.Diabetes Metab Res Rev.2007;23:169-183..
- 129. Goldberg E and Krause I. Infection and type 1 diabetes mellitus -a two edged sword?Autoimmun Rev.2009;8:682-686..
- 130. Avasthi TS and Ahmed N. Helicobacter pylori and type 1 diabetes mellitus: possibility of modifying chronic disease susceptibility with vaccinomics at the anvil.Omics.2011;15:589-596..
- 131. Toplak H, Haller EM, Lauermann T, Weber K, Bahadori B, Reisinger EC, et al. Increased prevalence of IgA-Chlamydia antibodies in NIDDM patients.Diabetes Res Clin Pract.1996;32:97-101..
- 132. Wang C, Gao D, Kaltenboeck B. Acute Chlamydia pneumoniae reinfection accelerates the development of insulin resistance and diabetes in obese C57BL/6 mice.J Inf Dis.2009;200:279-287..
- 133. Nishihara R, Sugano N, Takano M, Shimada T, Tanaka H, Oka S, et al. The effect of Porphyromonas gingivalis infection on cytokine levels in type 2 diabetic mice.J Periodontal Res.2009;44:305-310..
- 134. Kozak R, Juhasz E, Horvat G, Harcsa E, Lovei L, Sike R, Szele K. Helicobacter pylori infection in diabetic patients].Orv Hetil.1999;14:993-995..
- 135. Quatrini M, Boarino V, Ghidoni A, Baldassarri AR, Bianchi PA, Bardella MT. Helicobacter pylori prevalence in patients with diabetes and its relationship to dyspeptic symptoms.J Clin Gastroenterol.2001;32:215-217..
- 136. Gulcelik NE, Kaya E, Demirbas B, Culha C, Koc G, et al. Helicobacter pylori prevalence in diabetic patients and its relationship with dyspepsia and autonomic neuropathy.J Endocrinol Invest.2005;28:214-217..
- 137. Papamichael KX, Papaioannou G, Karga H, Roussos A, Mantzaris GJ. Helicobacter pylori infection and endocrine disorders: is there a link?World J Gastroenterol. WJG.2009;15:2701-2707..
- 138. Quadri R, Rossi C, Catalfamo E, Masoero G, Lombardo L, Della Monica P, et al. Helicobacter pylori infection in type 2 diabetic patients.Nutr Metab Cardiovasc Dis.2000;10:263-266..
- 139. Ohnishi M, Fukui M, Ishikawa T, Ohnishi N, Ishigami N, Yoshioka K, et al. Helicobacter pylori infection and arterial stiffness in patients with type 2 diabetes mellitus.Metabolism.2008;57:1760-1764..
- 140. Hamed SA, Amine NF, Galal GM, Helal SR, Tag El-Din LM, Shawky OA, et al. Vascular risks and complications in diabetes mellitus: the role of helicobacter pylori infection.J Stroke Cerebrovasc Dis.2008;17:86-94..
- 141. Bener A, Micallef R, Afifi M, Derbala M, Al-Mulla HM, Usmani MA. Association between type 2 diabetes mellitus and Helicobacter pylori infection.Turk J Gastroenterol.2007;18:225-229..
- 142. Perdichizzi G, Bottari M, Pallio S, Fera MT, Carbone M, Barresi G. Gastric infection by Helicobacter pylori and antral gastritis in hyperglycemic obese and in diabetic subjects.New Microbiol.1996;19:149-154..
- 143. Oldenburg B, Diepersloot RJ, Hoekstra JB. High seroprevalence of Helicobacter pylori in diabetes mellitus patients.Dig Dis Sci.1996;41:458-461..
- 144. Han X, Li Y, Wang J, Liu B, Hu H, Li X, et al. Helicobacter pylori infection is associated with type 2 diabetes among a middle-and old-age Chinese population.Diabetes Metab Res Rev.2015;.
- 145. Roubaud Baudron C, Franceschi F, Salles N, Gasbarrini A. Extragastric diseases and Helicobacter pylori.Helicobacter.2013;18:Suppl 144-51..
- 146. Pellicano R, Astegiano M, Rizzetto M. Helicobacter pylori and type 2 diabetes mellitus. Negative results and goals of future studies.Saudi Med J.2008;29:1213.
- 147. Demir M, Gokturk HS, Ozturk NA, Kulaksizoglu M, Serin E, Yilmaz U. Helicobacter pylori prevalence in diabetes mellitus patients with dyspeptic symptoms and its relationship to glycemic control and late complications.Dig Dis Sci.2008;53:2646-2649..
- 148. Ko GT, Chan FK, Chan WB, Sung JJ, Tsoi CL, To KF, et al. Helicobacter pylori infection in Chinese subjects with type 2 diabetes.Endocr Res.2001;27:171-177..
- 149. Hughes MK, Fusillo MH, Roberson BS. Positive fluorescent treponemal antibody reactions in diabetes.Appl Microbiol.1970;19:425-428..
- 150. Bjerkhoel A, Carlsson M, Ohlsson J. Peripheral facial palsy caused by the Borrelia spirochete.Acta Otolaryngol.1989;108:424-430..
- 151. Fenesy KE. Periodontal disease: an overview for physicians.Mt Sinai J Med.1998;65:362-369..
- 152. Grossi SG and Genco RJ. Periodontal disease and diabetes mellitus: a two-way relationship.Ann Periodontol.1998;3:51-61..
- 153. Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases.Lancet.2005;366:1809-1820..
- 154. Taylor GW. Bidirectional interrelationships between diabetes and periodontal diseases: an epidemiologic perspective.Ann Periodontol.2001;6:99-112..
- 155. Gurav A and Jadhav V. Periodontitis and risk of diabetes mellitus.J Diabetes.2011;3:21-28..
- 156. Mealey BL and Rose LF. Diabetes mellitus and inflammatory periodontal diseases.Curr Opin Endocrinol Diabetes Obes.2008;15:135-141..
- 157. Meurman JH and Hamalainen P. Oral health and morbidity--implications of oral infections on the elderly.Gerodontology.2006;23:3-16..
- 158. Loe H. Periodontal disease. The sixth complication of diabetes mellitus.Diabetes Care.1993;16:329-334..
- 159. Nelson RG, Shlossman M, Budding LM, Pettitt DJ, Saad MF, Genco RJ, et al. Periodontal disease and NIDDM in Pima Indians.Diabetes Care.1990;13:836-840..
- 160. Emrich LJ, Shlossman M, Genco RJ. Periodontal disease in non-insulin-dependent diabetes mellitus.J periodontology.1991;62:123-131..
- 161. Taylor GW, Burt BA, Becker MP, Genco RJ, Shlossman M, Knowler WC, et al. Non-insulin dependent diabetes mellitus and alveolar bone loss progression over 2 years.Journal of periodontology.1998;69:76-83..
- 162. Hintao J, Teanpaisan R, Chongsuvivatwong V, Ratarasan C, Dahlen G. The microbiological profiles of saliva, supragingival and subgingival plaque and dental caries in adults with and without type 2 diabetes mellitus.Oral Microbiol Immunol.2007;22:175-181..
- 163. Grossi SG, Skrepcinski FB, DeCaro T, Robertson DC, Ho AW, Dunford RG, et al. Treatment of periodontal disease in diabetics reduces glycated hemoglobin.J Periodontol.1997;68:713-719..
- 164. de Luis DA, Garcia Avello A, Lasuncion MA, Aller R, Martin de Argila C, Boixeda de Miquel D, et al. Improvement in lipid and haemostasis patterns after Helicobacter pylori infection eradication in type 1 diabetic patients.Clin Nutr.1999;18:227-231..
- 165. Erridge C. Diet, commensals and the intestine as sources of pathogen-associated molecular patterns in atherosclerosis, type 2 diabetes and non-alcoholic fatty liver disease.Atherosclerosis.2011;216:1-6..
- 166. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity.Cell.2006;124:783-801..
- 167. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body.Ann Rev Immunol.2009;27:229-265..
- 168. Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders.Semin Immunol.2009;21:242-253..
- 169. Lien E and Ingalls RR. Toll-like receptors.Crit Care Med.2002;30S:S1-11..
- 170. Vives-Pi M, Somoza N, Fernandez-Alvarez J, Vargas F, Caro P, Alba A, et al. Evidence of expression of endotoxin receptors CD14, toll-like receptors TLR4 and TLR2 and associated molecule MD-2 and of sensitivity to endotoxin (LPS) in islet beta cells.Clin Exp Immunology.2003;133:208-218..
- 171. Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT Jr. Neurodegenerative disease: amyloid pores from pathogenic mutations.Nature.2002;418:291.
- 172. Suh YH, Chong YH, Kim SH, Choi W, Min K, Jeong SJ, et al. Molecular physiology, biochemistry, and pharmacology of Alzheimer's amyloid precursor protein (APP).Ann N Y Acad Sci.1996;786:169-183..
- 173. Kobayashi N, Masuda J, Kudoh J, Shimizu N, Yoshida T. Binding sites on tau proteins as components for antimicrobial peptides.Biocontrol Sci.2008;13:49-56..
- 174. Hirakura Y, Carreras I, Sipe JD, Kagan BL. Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense.Amyloid.2002;9:13-23..
- 175. Anguiano M, Nowak RJ, Lansbury PT Jr. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a porelike mechanism that may be relevant to type II diabetes.Biochemistry.2002;41:11338-11343..
- 176. Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, et al. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases.J Biol Chem.2004;279:46363-46366..
- 177. Hirakura Y, Yiu WW, Yamamoto A, Kagan BL. Amyloid peptide channels: blockade by zinc and inhibition by Congo red (amyloid channel block).Amyloid.2000;7:194-199..
- 178. Kagan BL and Thundimadathil J. Amyloid peptide pores and the beta sheet conformation.Adv Exp Med Biol.2010;677:150-167..
- 179. Gekara NO, Westphal K, Ma B, Rohde M, Groebe L, Weiss S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes.Cell Microbiol.2007;9:2008-2021..
- 180. Gonzalez MR, Bischofberger M, Pernot L, van der Goot FG, Freche B. Bacterial pore-forming toxins: the (w)hole story?Cell Mol Life Sci.2008;65:493-507..
- 181. Gekara NO and Weiss S. Lipid rafts clustering and signalling by listeriolysin O.Biochem Soc Trans.2004;32:712-714..
- 182. Cossart P and Lecuit M. Interactions of Listeria monocytogenes with mammalian cells during entry and actin-based movement: bacterial factors, cellular ligands and signaling.EMBO J.1998;17:3797-3806..
- 183. Kayal S, Lilienbaum A, Poyart C, Memet S, Israel A, Berche P. Listeriolysin O-dependent activation of endothelial cells during infection with Listeria monocytogenes: activation of NFkappa B and upregulation of adhesion molecules and chemokines.Mol Microbiol.1999;31:1709-1722..
- 184. Hamon MA, Batsche E, Regnault B, Tham TN, Seveau S, Muchardt C, Cossart P. Histone modifications induced by a family of bacterial toxins.Proc Natl Acad Sci U S A.2007;104:13467-13472..
- 185. Kim W and Hecht MH. Sequence determinants of enhanced amyloidogenicity of Alzheimer A{beta}42 peptide relative to A{beta}40.J Biol Chem.2005;280:35069-35076..
- 186. Otzen D and Nielsen PH. We find them here, we find them there: functional bacterial amyloid.Cell Mol Life Sci.2008;65:910-927..
- 187. Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, et al. Role of Escherichia coli curli operons in directing amyloid fiber formation.Science.2002;295:851-855..
- 188. Larsen P, Nielsen JL, Dueholm MS, Wetzel R, Otzen D, Nielsen PH. Amyloid adhesins are abundant in natural biofilms.Environ Microbiol.2007;9:3077-3090..
- 189. Jordal PB, Dueholm MS, Larsen P, Petersen SV, Enghild JJ, Christiansen G, et al. Widespread abundance of functional bacterial amyloid in mycolata and other gram-positive bacteria.Appl Environ Microbiol.2009;75:4101-4110..
- 190. Wang X, Hammer ND, Chapman MR. The molecular basis of functional bacterial amyloid polymerization and nucleation.J Biol Chem.2008;283:21530-21539..
- 191. Yoshiike Y, Kayed R, Milton SC, Takashima A, Glabe CG. Pore-forming proteins share structural and functional homology with amyloid oligomers.Neuromolecular Med.2007;9:270-275..
- 192. Roy S, McGuire W, Mascie-Taylor CG, Saha B, Hazra SK, Hill AV, et al. Tumor necrosis factor promoter polymorphism and susceptibility to lepromatous leprosy.J Infect Dis.1997;176:530-532..
- 193. Knight JC and Kwiatkowski D. Inherited variability of tumor necrosis factor production and susceptibility to infectious disease.Proc Assoc Am Physicians.1999;111:290-298..
- 194. Shaw MA, Donaldson IJ, Collins A, Peacock CS, Lins-Lainson Z, Shaw JJ, et al. Association and linkage of leprosy phenotypes with HLA class II and tumour necrosis factor genes.Genes Immun.2001;2:196-204..
- 195. Lagrange PH and Abel L. The genetic susceptibility to leprosy in humans.Acta Leprol.1996;10:11-27..
- 196. Pacheco e Silva AC. Localisation du Treponema Pallidum dans le cerveau des paralytiques généraux.Revista de Neurologia.1926;2:558-565..
- 197. Miklossy J. Biology and neuropathology of dementia in syphilis and Lyme disease.Handb Clin Neurol.2008;89:825-844..
- 198. Miklossy J, Khalili K, Gern L, Ericson RL, Darekar P, Bolle L, et al. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease.J Alzheimers Dis.2004;6:639-649.discussion 673-681.
- 199. Pacheco e Silva AC. Espirochetose dos centros nervos. Memorias do hospicio de Juquery, anno III-IV 3-4, 1926-27:1-27.
- 200. Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease.Neurobiol Aging.1988;9:339-349..
- 201. McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients.Neurosci Lett.1989;107:341-346..