



Introduction
The biology of normal wound healing includes sequential yet overlapping inflammatory, proliferative, and remodeling phases that involve complex biological signaling [1–4]. Dysregulation of specific signaling pathways is thought to underlie skin breakdown and poor healing [1–4]. The renin angiotensin system (RAS) is active in connective tissue and skin, and is known to be important in wound healing [5–7]. RAS is involved in the inflammatory response, collagen deposition and in tissue-related growth factor (TGFβ) signaling necessary for wound healing [5–9]. RAS is known to be dysregulated in both aging and in diabetes, with increased AT1R and decreased AT2R expression in diabetic wound healing and in aging [5;10], which may play a role in aging skin vulnerability [5;7;8;10]. Indeed, altered dermal AT1R and AT2R ratio is associated with thinning of epidermis, degeneration of collagen, fracture of dermal layer, and atrophy of subcutaneous fat in diabetic rats [5]. These changes are consistent with those seen clinically in aging skin. AT1R blockers impair fibroblast migration and delay wound healing [11]. The angiotensin subtype 2 receptor is less studied, but its anti-inflammatory, anti-apoptotic and anti-proliferative effects are thought to oppose the effects of AT1R [12]. Virtually nothing is known about the contribution of the AT2R to stages of wound healing. The overarching hypothesis of this study is that a functional balance between skin expression of AT1R and AT2R is required for optimal healing. We further hypothesized that targeted deletion of AT2R would accelerate wound healing rate via un-opposed AT1R activity upregulating skin TGFβ signaling. To dissect the role of AT2R on wound healing we have selected the genetic knockout to avoid the effects of variations in drug delivery to wound bed. Furthermore, given that Angio-tensin II binds with equal affinity to AT1R or AT2R, the knockout of the angiotensin receptors allows for better discrimination of the effects of the receptors by eliminat-ing the possibility of remaining unblocked receptors. In this study we compared C57BL/6J wild-type (WT) mice to age- and gender-matched; AT1R knockout (AT1R−/−) mice and AT2R knockout (AT2R−/−) mice.
Results
To ascertain the influence of angiotensin receptors on wound healing, downstream effectors and healed skin quality, we compared C57BL/6J wild-type (WT) mice to age- and gender-matched; AT1R knockout (AT1R−/−) mice and AT2R knockout (AT2R−/−) mice.
Delayed wound healing in AT1R −/− and accelerated wound healing in AT2R−/− mice
RAS is a key hormonal system whose dysregulation has been linked to aging, inflammation, and impaired wound healing. We show that the AT1R−/− mice were the most delayed in wound healing as compared to WT and the AT2R−/− (Figure 1, panel A, B and C) which is in agreement with previous reports on wound healing in AT1R−/− mice [11]. In contrast, the healing rate in the AT2R−/− mice was accelerated which was unexpected given that AT2R levels in general correlate with positive outcomes [12]. By day 12, 50% of the animals in the AT2R−/− cohort achieved complete wound healing as compared to none in either the AT1R−/− or WT mice(P<0.05). By day 12, only 5% of the size of wounds remained unhealed in the AT2R−/− mice vs. 17% in the AT1R−/− and 11% in WT mouse cohorts (P<0.05; Figure 1, panel C and E). All the AT2R−/− mice were healed completely by day 16 as compared to 10% of WT and 30% of AT1R−/− that remained with open wounds (P<0.05; Figure 1: panel C and E). Further analysis of the fastest healing group, the AT2R−/−, revealed a delayed healing during the inflammatory phase of wound healing (day1-7) and an accelerated healing during proliferative and remodeling phases (day8-16). (Figure 1D) This biphasic pattern of healing observed in the AT2R−/− group, was not seen in either the AT1R−/− or WT mice.
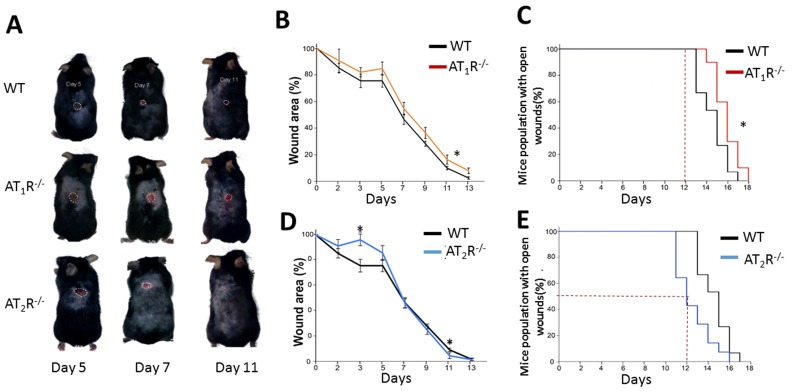
Figure 1. Wound closure measurements in AT1R and AT2R transgenic mice show delayed AT1R−/− and accelerated AT2R−/− healing rate. (A) Representative images from WT, AT1R−/− and AT2R−/− mouse cohorts on day 5, 7 and 11 of wound healing. Plannimetric assessment of wound closure rate in AT1R−/− (B) and AT2R−/− (D). Complete wound closure of AT1R−/− (Panel C) and AT2R−/− (E) mice. Data are means ± SEM *p<0.05.
Increase in wound blood flow in the AT1R−/− mice
Given the prominent role for angiotensin receptors in tissue perfusion and to determine if changes in blood flow to the wounds contributed to the observed healing pattern in AT1R−/− and AT2R−/− mice, wound area blood flow was measured by non-invasive LDPI on days 7 and 11. There were no differences in wound area blood flow among the three groups by day 7, but by day 11 we observed a significantly higher value in the AT1R−/− as compared to the other two groups (P<0.05; Figure 2). Interestingly, the increase in blood flow in the AT1R−/− mice did not correlate with better wound healing. Further, we did not observe differences in wound blood flow the AT2R−/− as compared with WT controls, suggesting that changes in blood flow did not contribute to the accelerated healing observed in the AT2R−/− mice.
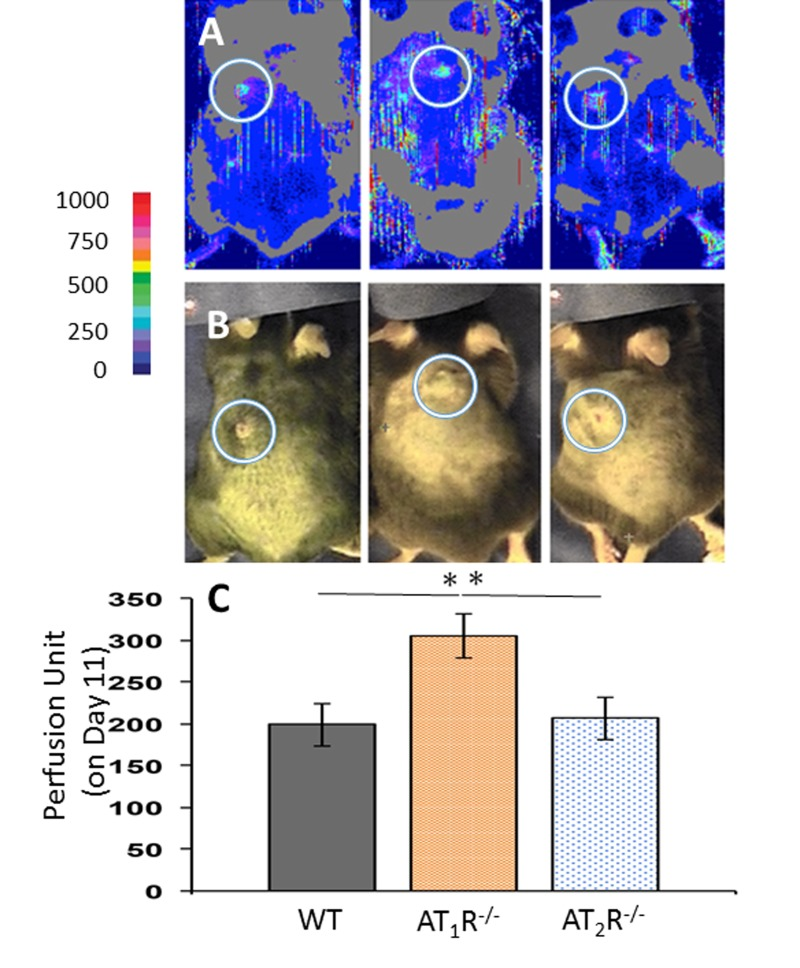
Figure 2. Laser Doppler perfusion imaging of wound area blood flow on day 11 of wound healing shows a higher blood flow in wounds of AT1R−/−. Data are means ± SEM *p<0.05.
Down regulation of TGFβ isoforms and the downstream target proteins (Smads) in AT1R−/− and upregulation of AT1R, TGFβ1 and TGFβ2 during later stages of wound healing in the AT2R−/−
Although not completely characterized, all phases of wound healing appear to be greatly influenced by subtle modulation of TGF-β, which is strongly influenced by RAS [6;7;30–32]. The three isoforms of TGF-β (β1, β2, and β3) signal through the same cell surface receptor, but appear to play distinct functions during wound healing. While TGF β1 and β2 have predominantly pro-scarring roles, TGF β3 have mainly anti-scarring effects [32]. RAS has been tightly linked to TGFβ activity but the specific effects of AT1R and AT2R on the different TGF isoforms are not known. Using qPCR we determined differences in expression of the three different isoforms of TGFβ in the healing skin (day 20) of our mouse cohorts. Our results demonstrate a significant decrease in all the three different isoforms of TGFβ in the wound of the AT1R−/− mice (P<0.05; Figure 3: panel A, B and C). TGF-β1 mRNA levels in AT1R−/− mice were decreased 7.69 fold as compared to WT mice. In contrast the TGFβ1 mRNA levels increased 1.83 fold in AT2R−/− mice as compared to WT mice (P < 0.005; Figure 3: panel A). The expression of TGFβ2 mRNA was also decreased 20 fold in AT1R−/− mice compared with WT mice (P < 0.005; Figure 2: panel B). TGFβ3 mRNA expression was decreased 7.1 fold in AT1R−/− mice in comparison with WT mice (P < 0.005; figure 2: panel C). In contrast, we observed an increase only in TGFβ1 in AT2R−/−, compared to both WT control and AT1R−/− mice (p <0.05; Figure 3: panel A). We quantified changes in AT1R and the three isoforms of TGFβ in days 0, 3, 7 and 9 of wound healing to investigate if the biphasic pattern observed in wound healing of the AT2R−/− corresponded to a stage dependent changes in AT1R and to quantify changes in TGF-β isoforms. Our results suggest that the expression of AT1R was upregulated by day 7 of wound healing (2.18 and 2.56 fold change respectively, p<0.05; Figure 3: panel D). This increase corresponded to an increase in both TGFβ1 and TGFβ2 (p<0.05; Figure 3: panel E and F). No change in TGFβ3 was observed. Changes in AT1R in different stages of wound healing strongly correlated with the changes in TGFβ1 (Pearson r=0.99, p=0.04).
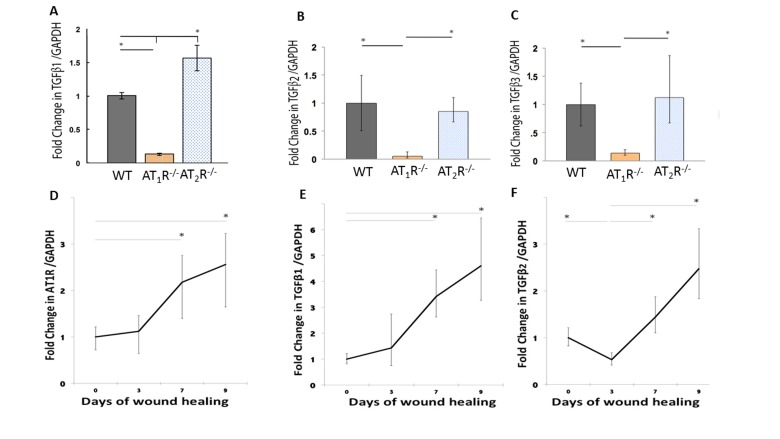
Figure 3. Altered expression of wound TGFβ isoforms in AT1R−/− and AT2R−/− mice. AT1R−/− mice have lower expression of TGFβ1 (A), TGFβ2 (B), and TGFβ3 (C) in healed skin (day20) as compared to WT and AT2R−/− mice. An increase in the expression of AT1R (D), TGFβ1 (E), TGFβ2 (F) correlated with the accelerated healing rate observed in later stages of wound healing in AT2R−/− mice. The length of fold change error bar equal variance 95% confidence interval *p<0.05.
Next we sought to determine the impact of the disruption of angiotensin receptors on the downstream target proteins of the TGFβ signaling pathway. TGFβ signals through Smad2 and Smad3 that are phosphorylated by TGFβ receptors and translocate to the nucleus with the common-mediator (co-Smad) Smad4 [33;34]. Our results showed no significant difference in Smad2 in wounds of AT1R−/− or AT2R−/− mice as compared to WT mice (Figure 4). Consistent with the reduction in the three isoforms of TGFβ, we have observed a significant reduction in both Smad3 and the common-mediator Smad4 in wounds of AT1R−/− mice as compared with WT mice (P<0.001, Figure 4). Our results also demonstrate reduction in phosphorylation of Smad2 and Smad3 only in wounds of AT1R−/− mice as compared with WT mice (P<0.01, Figure 5). Interestingly, we have observed a similar decrease in Smad3 in AT2R−/− mice as compared to WT mice (P<0.0001, Figure 4). There were no differences in Smad4, phospho-Smad2 or phospho-Smad3 in wounds of AT2R−/− mice.
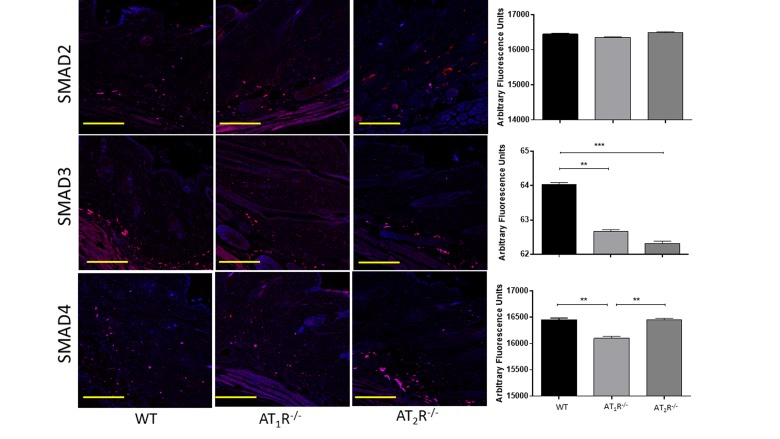
Figure 4. Changes in the TGFβ downstream signaling proteins in wounds of AT1R−/− and AT2R−/− mice. AT1R−/− mice have lower expression of Smad3 and the common mediator Smad4 in healed skin (day20) as compared to WT. A decrease in the expression of Smad3 was also observed in the AT2R−/− mice. The photomicrographs presented in red fluorescent staining with a blue DAPI counter stain for nuclei at 10x magnification. Quantification of mean fluorescence intensity of Smads in wild-type and mutant mice is shown. Scale bar 200 μm. **p<0.005, ***p<0.0005.
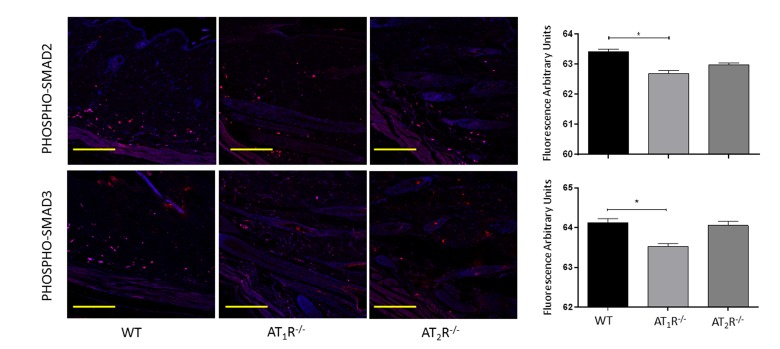
Figure 5. Changes in levels of phosphorylated TGFβ downstream signaling proteins in wounds of AT1R−/− and AT2R−/− mice. AT1R−/− mice have lower expression of Phospho-Smad2 and Phospho-Smad3 in healed skin (day20) as compared to WT. The photomicrographs presented in red fluorescent staining with a blue DAPI counter stain for nuclei at 10x magnification. Quantification of mean fluorescence intensity of phospho-Smads in wild-type and mutant mice is shown. Scale bar 200 μm. *p<0.05.
Reduced repair and proliferation activity in wounds of AT1R−/− mice
The angiotensin receptors have been linked to changes in cell differentiation and proliferation. While AT1R have been shown to increase cell proliferation, AT2R have been shown critical for cell differentiation [12].
Given the observed delayed wound healing noted in AT1R−/− mice, we wanted to determine if this delay was driven by changes in cellular proliferative activity. We quantified changes in Proliferating cell nuclear antigen (PCNA), a nuclear protein essential for DNA replication and repair and is a marker for cellular growth and proliferation. To further investigate the mitotic activity at the wound site, we studied expression levels of the phosphorylated form of the histone protein H3. Histone H3 phosphorylation is linked to cells that are actively dividing. Consistent with the delayed healing rate in AT1R−/− mice, we have observed a significant reduction in PCNA (P<0.005, Figure 6) and in mitotic histone H3 phosphorylation at several residues, including serines 28 (P<0.005) as well as threonines 3 (0.005) and 11(0.05) (Figure 7). Surprisingly, we have observed a similar decrease in PCNA and phosphorylation of Histone 3 Threonine 3 residue in AT2R−/− mice. The impact of the differential phosphorylation of certain histone H3 residues in AT2R−/− mice on wound healing activity and scar quality is currently unclear.
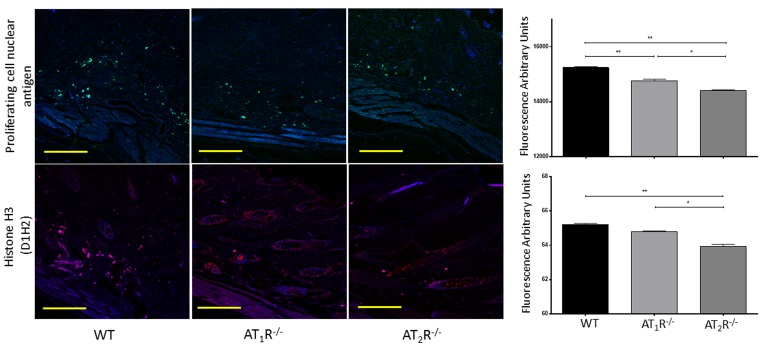
Figure 6. Down regulation of Proliferating Cell Nuclear Antigen in wounds of AT1R−/− and AT2R−/− mice. AT2 R−/− mice have lower expression of total Histone H3 in healed skin (day20) as compared to WT. The photomicrographs presented in green (PCNA) or red (Total Histone H3) fluorescent staining with a blue DAPI counter stain for nuclei at 10x magnification. Quantification of mean fluorescence intensity of PCNA and Histone H3 in wild-type and mutant mice is shown. Scale bar 200 μm. *p<0.05, **p<0.005 .
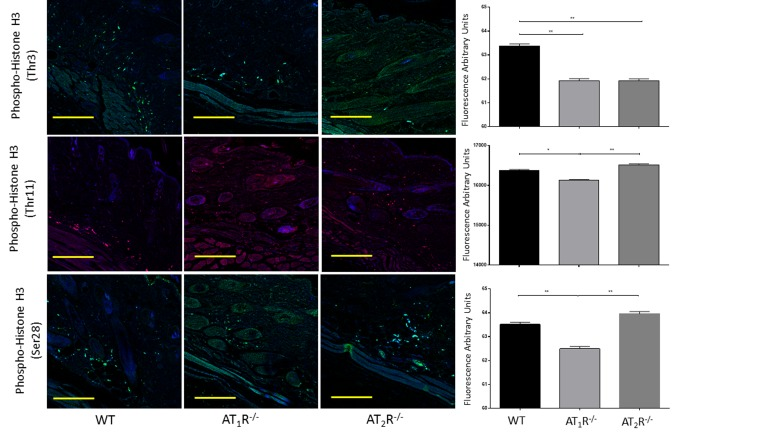
Figure 7. Down regulation of mitotic histone H3 phosphorylation in wounds of AT1R−/− at several residues, including serine 28 as well as threonine 3 and 11. AT2 R−/− mice have lower expression of Phospho-Histone H3 (Thr3) in healed skin (day20) as compared to WT. The photomicrographs presented in green (Thr3 and S28) or red (Thr11) fluorescent staining with a blue DAPI counter stain for nuclei at 10x magnification. Quantification of mean fluorescence intensity of Phospho-Histone H3 in wild-type and mutant mice is shown. Scale bar 200 μm. *p<0.05, **p<0.005.
Tensiometry shows wound fragility in AT2R−/− mice
Given the pro-scarring and fibrotic effects of TGFβ1, we next sought to determine if there was a difference in the healed skin's physical characteristics (Peak force, total work and compliance). Our results show that despite the accelerated healing rate observed in the AT2R−/− mice, the healed skin in the AT2R−/− mice was more fragile, fracturing more easily (Panel 8B), being more compliant (Panel 8C), and breaking with less work (Panel 8D) than wounds from AT1R−/− or WT mice. (P<0.05). Masson's trichrome staining of healing skin shows increase of subcutaneous fat in both AT1R−/− and AT2R−/−. A reduction in dermal collagen zone in AT1R−/− was also observed (Figure 9).
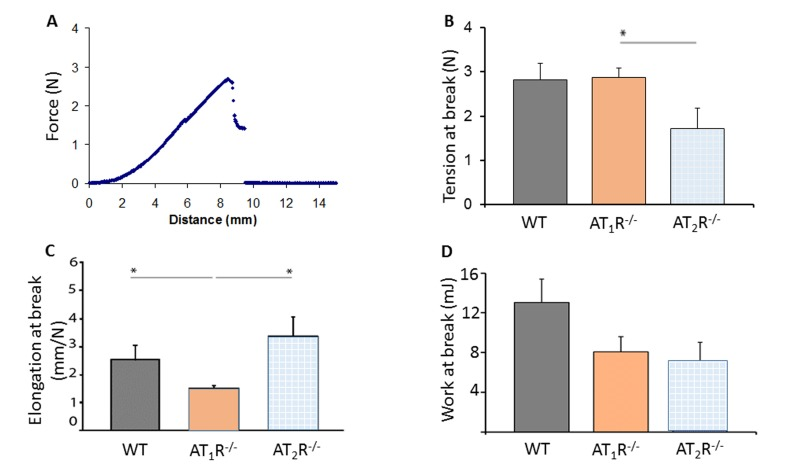
Figure 8. Biomechanical assessment of healed skin in WT, AT1R−/− and AT2R−/− mouse cohorts. (A) Sample representation of tension–elongation curve. (B) Comparison of the average tension at the breaking point of mice groups (mean ± SEM, n = 10; *P < 0.05, Mann–Whitney analysis). (C) Average elongation at the breaking point of both groups (*P < 0.05, t-test). (D) Average work at the breaking point of both groups (calculated from the integral of the curve; *P < 0.05, Mann–Whitney analysis).
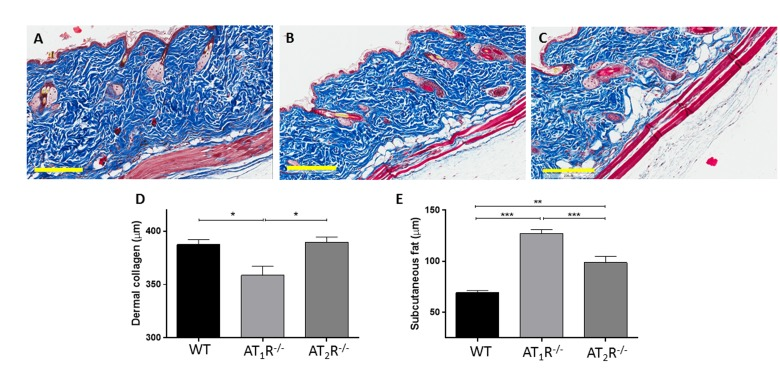
Figure 9. Masson's trichrome staining of skin sections from WT (A), AT1R−/− (B) and AT2R−/− (C) mouse cohorts shows an expanded zone of subcutaneous fat in the angiotensin knockout mice. Quantification of the thickness of the zones of dermal collagen and subcutaneous fat in wild-type and mutant mice is shown. Scale bar 200 μm. *p<0.05, **p<0.005, ***p<0.0005.
Discussion
Several lines of evidence suggest that increased RAS activity through the AT1R plays a crucial role in wound healing [5–9]. Our results further dissects the impact of angiotensin receptors on wound healing. AT2R antagonizes inflammatory signaling, a necessary activating function that leads to the proliferation phase. The lack of AT2R was associated with a slower closure rate during the early stages. This may have resulted from an unopposed pro-inflammatory AT1R, causing delayed resolution of the inflammatory phase and impairing the transition to the proliferative and remodeling phases [1–4;35].
We have observed a delayed healing pattern in AT1R−/− throughout all phases of wound healing, which is consistent with the pro-inflammatory and pro-proliferative characteristics of AT1R [12], and is in agreement with previous reports [11]. This is also is supported with the significant reduction in both PCNA and phospho-Histone H3 in healing skin of the AT1R−/− mice. In contrast, by day 8 in AT2R−/− mice, as the healing wounds were transitioning to the proliferative phase we observed a significant upregulation of wound AT1R along with an accelerated rate of healing. Our combined results of accelerated healing with the upregulation of AT1R (in AT2R−/− mice), contrasted with delayed healing in AT1R−/− may suggest phase-dependent role for increased AT1R signaling during the proliferative phase through alterations in TGF-β signaling and alterations in the extracellular matrix [30;36;37].
The relationship between the TGFβ family and angiotensin receptors is not entirely mapped out and remains mechanistically vague. Previous studies have reported that RAS activation through AT1R increases TGFβ signaling [38;39]. Our results demonstrate a significant down regulation of all the three isoforms of TGFβ and the downstream targeting proteins Smad3, Smad4, as well as the phospho-Smad2 and phospho-Smad3 in the AT1R−/− mice. The impact of TGFβ on cutaneous wound healing has been well established. The release of TGFβ1 during early stages of healing prompts the expression of key components such as fibronectin, collagen types I and III, and VEGF [32]. Additionally, TGFβ1 improves angiogenesis to facilitate blood supply to the injured site [40] which then stimulates fibroblasts to allow for wound closure [41]. Whether the decrease in TGFβ1 is causal of the impaired wound healing in the AT1R−/− mice is not known. However, in AT2R−/− mice there was a strong, positive correlation between dermal TGFβ1 and AT1R expression in later stages of wound healing that corresponded to an accelerated wound closure. This is in agreement with previous reports linking AT1R stimulation to increased TGFβ1 expression and collagen maturation [42] and may potentially explain the decreased compliance seen in healed skin in AT1R−/− mice.
Similarly, the second isoform TGFβ2, is involved in granulation tissue formation, angiogenesis and collagen synthesis [43;44]. Impaired wound healing has been demonstrated in TGFβ2 transgenic mice [45]. In AT2R−/− mice, we observed a decrease in the expression of TGFβ2 by day 3 of wound healing. This initial drop corresponded to the impaired healing seen in early phases in AT2R−/− mice. Furthermore, we have observed a significant increase in TGFβ2 by day 7 that was matched with a faster healing rate.
The lack of change in TGFβ3 and blood flow in AT2R−/− mice may suggest that these two factors do not play a significant role in modulating wound healing in response to the knocking out of the AT2R.
Changes in the TGFβdownstream signaling proteins (Smads) have been linked to the rate of wound healing. The down regulation of Smad3 have been linked to acceleration of wound closure [46]. In contrast, the knockdown of Smad4 was associated with aberrant wound healing [47]. Consistent with previous reports on the role of Smad3, we have noted the lowest level of Smad3 in AT2R−/− along with the fastest wound closure rate. The down regulation of Smad4 in AT1R−/− may have played a role in the delayed healing rate.
In summary, the silencing of AT1R delayed wound healing, while the interruption of AT2R accelerated wound healing. Furthermore, this effect in AT2R−/− mice, was at least partially mediated by AT1R, TGFβ1and TGFβ2. This data supports the notion of the antagonistic interaction between AT1R and AT2R.
Mitochondria provide energy and produce reactive oxygen species to drive the increased mitotic and synthetic activity necessary for wound healing. Several groups demonstrated a link between age-related mitochondrial dysfunction and impaired wound healing [48]. The identification of a functional intra-mitochondrial angiotensin system (MAS) [49] may provide additional insight into the RAS interface with wound healing. Activation of the intra-mitochondrial AT2R is coupled to increased nitric oxide generation and inhibition of mitochondrial energy production [49]. Further work is needed to determine the impact of MAS on wound healing.
There are several limitations to our current study. Structurally, mice skin differs from human skin in that mice have much thinner epidermal and dermal layers than humans. Furthermore, mice also have a large subcutaneous muscle layer, which augments wound repair by contraction making further studies in a second animal model (pigs) or humans necessary before extrapolating results to humans. Also, given that we are studying mice that are homozygote knockouts for either the AT1R or the AT2R, partial effects of the genes and the compensatory effects of one angiotensin receptor on the absence of the other receptor are still not clear.
Given the effects of aging on angiotensin receptors [12;49;50], and that many aged, frail individuals are already on angiotensin receptor blockers, this research highlights the crosstalk between AT1R and AT2R and that pinpointing the exact molecular changes in angiotensin receptors and the impact of angiotensin receptor blockers on wound healing in aged individuals is important for the progression of the field of wound healing.
Methods
Mouse models
This study was approved by the Johns Hopkins Animal Care and Use Committee (ACUC). To ascertain the influence of angiotensin receptors on wound healing, downstream effectors and healed skin quality, we compared 28-week old male C57BL/6J wild type (WT) mice (Jackson Laboratories, Bar Harbor, Maine) to age and gender matched AT1R knockout (AT1R−/−) (Jackson Laboratories, Bar Harbor, Maine) [13] and AT2R knockout (AT2R −/− mice (supplied by our collaborator Dr. Tedashi Inagami, Vanderbilt University, TN) [14;15]. Male mice were employed to avoid the effects of hormonal changes on wound blood flow and healing.
Wounding procedure and area calculation
Mice (N=10 in each group) were anesthetized by a mobile RC2 non-rebreathing anesthesia machine (Vet Equip, Inc. Pleasanton, CA). Buprenorphine (1 mg/kg) was administered by a subcutaneous injection during the first 24 hours. A full thickness 8 mm wound was created by punch biopsy. On days 0, 3, 5, 7, 9, 11 and 13, the wound borders were traced in situ onto clear acetate paper. Images were digitized at 600 dpi (Hewlett Packard Company, Laser Jet 3390, Paolo Alto). Wound areas (in pixels) were calculated using Adobe Photoshop CS3 Image software (Adobe System Inc. San Jose, CA). Wound area on day 0 was taken as a 100% and a wound size ratio obtained with that measurement in each time point. The wounds were checked daily after day 10 until complete closure.
Laser Doppler Perfusion Imaging (LDPI)
Blood flow in the wound areas was measured at days 7 and 11 using a 633 nm, He–Ne scanning laser Doppler imaging device (Moor Instruments, Devon, UK], which utilizes a near-infrared laser diode to measure subcutaneous blood flow as a function of light scattering by moving red blood cells (Doppler shift), as described previously [16].
Physical measurements of tissue strength
Peak force, work to rupture and flexibility of healed skin were calculated at day 21 using a FGV-10XY tensiometer (Checkline by Electromatic, Cedarhurst, NY) to record the force generated as the skin was elongated until rupture as described previously [17].
Histology/Immunofluorescence
Healing skin tissues were embedded in Tissue-Tek O.C.T. Compound (Sakura) and multiple thin sections (5 μm) were cut using a cryostat (Microm). Subsequently, the sections were stained with Masson's Trichrome (Polysciences, Inc.) or using immunofluorescence techniques. Masson's Trichrome staining was carried out according to the manufacturer protocol with the addition of an one hour 10% formalin fix at room temperature (RT) prior to the fixation in Bouin's solution (Meinen 2011). Slides were then digitally scanned and under high-power fields both dermal and subcutaneous fat thicknesses were measured [18] using Aperio ImageScope software (Leica Biosystems, Germany). For immunostaining, the sections were fixed with 4% paraformaldehyde for 15 minutes at RT then blocked with 5% BSA/0.3% TritonX-100/PBS for one hour at RT, incubated with the primary antibodies, overnight at 4°C. For the TGFβsignaling cascade, the following antibodies were purchased commercially: anti- Smad2 (D43B4) rabbit mAb (1:100 dilution, Cat#5339) [19], anti- Smad2/3 (D7G7) Rabbit mAb (1:100 dilution, Cat# 8685) [20], anti- Smad3 (C67H9) rabbit mAb (1:100 dilution, Cat# 9523) [21], anti- Smad4 (cat#9515) [22], anti-Phospho-Smad2 (Ser465/467) (138D4) rabbit mAb (1:100 dilution, cat#3108) [23], and anti-phospho-Smad3 (Ser423/425) (C25A9) rabbit mAb (1:100 dilution, Cat#9520) [24] from Cell Signaling Technology, Beverly, MA. For cell proliferation activity, the following antibodies were purchased commercially: anti- Proliferating cell nuclear antigen (PCNA) PCNA (1:2400 dilution, cat# #8607) [25], anti- Histone H3 (D1H2) rabbit mAb (1:100 dilution, Cat#4499) [26], anti-phospho-Histone H3 (Thr3) (cat#9714) [27]; anti- phospho-Histone H3 (Thr11) (cat#9764) [28], anti- phospho-Histone H3 (Ser28) (1:100 dilution, cat# 9713) [29] from Cell Signaling Technology, Beverly, MA. Sections were then incubated with secondary IgG (H+L), F(ab')2 Fragment (Alexa Fluor® 555 Conjugate, (1:1000 dilution, Cat #44091) at RT for 1 hour. Slides were mounted with Vectashield Hard Set with Dapi (Vector Laboratories). All images were taken at 10x, 0.3 NA, PlanNeoflaur on the Zeiss LSM 700. Imaging and quantification were done by the JHU microscope facility by blinded examiners. Automated quantification of mean intensity of fluorescent signal were done using Volocity image analysis software (v6.3, Perkin Elmer, Waltham, Massachusetts).
Quantitative real-time reverse transcription PCR
Total RNA was extracted from half of each wound sample using TRIzol (Invitrogen, Frederick, MD, USA), RNeasy Kit (QIAGEN, Redwood City, CA, USA), based on the manufacturer's protocol. Single-stranded cDNA is synthesized using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). The fold changes in TGF-β 1 (primer sequence: Fwd: 5′GAGCCCGAAGCGGACTACTA 3′; Rev: 5′ CCCGAATGTCTGACGTATTGAAG 3′), TGF-β 2 (primer sequence: Fwd: 5′ AGAATCGTCCGCTTTGATGTC 3′; Rev: 5′ TCTGGTTTTCACAACCTTGCT 3′), TGF-β 3 (primer sequence: Fwd: 5′ CAGGCCAGGGTAGTCAGAG 3′; Rev: 5′ ATTTCCAGCCTAGATCCTGCC 3′.) gene expression of healed tissue samples was normalized to Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the threshold cycle for amplification as
Statistical analysis
Data were presented as mean ± SEM. Differences in means between groups were analyzed for significance using two-way analysis of variance, followed by Holm-Sidak post hoc analysis when appropriate. Complete closure rate as a final outcome, was assessed with the Kaplan-Meier method, using Gehan-Breslow test to determine differences. Student's t-test and Mann–Whitney test were also used to analyze the biomechanical tensiometry data. A probability value of <0.05 was considered statistically significant.
Author Contributions
Mahya Faghih- PCR measurement of angiotensin receptors and TGFβ; drafting of the manuscript; critical revision of the manuscript for important intellectual content.
Sayed Mohammad Hosseini- performance of animal experiments, acquisition of data, critical revision of the manuscript for important intellectual content.
Amir Mehdi Ansari- acquisition of data, critical revision of the manuscript for important intellectual content.
Barbara Smith- IHC staining and quantification of SMADs and wound healing markers. Drafting of the manuscript; critical revision of the manuscript for important intellectual content.
Frank Lay- acquisition of data, critical revision of the manuscript for important intellectual content.
Tedashi Inagami: provided the AT2R−/− mice. Study concept and design; critical revision of the manuscript for important intellectual content.
Guy Marti- provided expertise regarding the animal model and the methods for assessing wound healing including planimetry, laser Doppler perfusion imaging of blood flow and assessment of biodynamic characteristics of the skin with tensiometry. Critical revision of the manuscript for important intellectual content.
John W. Harmon- provided expertise regarding the animal model and the methods for assessing wound healing including planimetry, laser Doppler perfusion imaging of blood flow and assessment of biodynamic characteristics of the skin with tensiometry. Critical revision of the manuscript for important intellectual content.
Jeremy D. Walston - study concept and design; critical revision of the manuscript for important intellectual content.
Peter Abadir- study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; administrative, technical, and material support; study supervision.
Funding
This study was supported by the Johns Hopkins Older Americans Independence Center National Institute on Aging Grant P30 AG021334, National Institute on Aging Grants 1R01AG046441 and K23 AG035005, Nathan Shock in Aging Scholarship Award (PMA), R21AG043284 (JW and PMA), the Wound healing society foundation 3M scholarship(PMA) , NIH research grant HL58205 (TI) and NIH grant S10 OD016374 (microscope Facility).
Conflicts of Interest
The authors declare no conflict of interests.
References
- 1. Scimeca CL, Bharara M, Fisher TK, Kimbriel H, Mills JL, Armstrong DG. An update on pharmacological interventions for diabetic foot ulcers. Foot Ankle Spec. 2010; 3: 285 -302. [PubMed] .
- 2. Pradhan L, Nabzdyk C, Andersen ND, LoGerfo FW, Veves A. Inflammation and neuropeptides: the connection in diabetic wound healing. Expert Rev Mol Med. 2009; 11: e2 [PubMed] .
- 3. Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005; 366: 1736 -1743. [PubMed] .
- 4. Stadelmann WK, Digenis AG, Tobin GR. Physiology and healing dynamics of chronic cutaneous wounds. Am J Surg. 1998; 176: 26S -38S. [PubMed] .
- 5. Hao SY, Ren M, Yang C, Lin DZ, Chen LH, Zhu P, Cheng H, Yan L. Activation of skin renin-angiotensin system in diabetic rats. Endocrine. 2011; 39: 242 -250. [PubMed] .
- 6. Steckelings UM, Wollschlager T, Peters J, Henz BM, Hermes B, Artuc M. Human skin: source of and target organ for angiotensin II. Exp Dermatol. 2004; 13: 148 -154. [PubMed] .
- 7. Yevdokimova N and Podpryatov S. The up-regulation of angiotensin II receptor type 1 and connective tissue growth factor are involved in high-glucose-induced fibronectin production by cultured human dermal fibroblasts. J Dermatol Sci. 2007; 47: 127 -139. [PubMed] .
- 8. Cooper ME. The role of the renin-angiotensin-aldosterone system in diabetes and its vascular complications. Am J Hypertens. 2004; 17: 16S -20S. [PubMed] .
- 9. Rodgers K, Verco S, Bolton L, diZerega G. Accelerated healing of diabetic wounds by NorLeu(3)-angiotensin (1-7). Expert Opin Investig Drugs. 2011; 20: 1575 -1581. .
- 10. Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O'Rourke B, Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. 2011; 108: 14849 -14854. [PubMed] .
- 11. Yahata Y, Shirakata Y, Tokumaru S, Yang L, Dai X, Tohyama M, Tsuda T, Sayama K, Iwai M, Horiuchi M, Hashimoto K. A novel function of angiotensin II in skin wound healing. Induction of fibroblast and keratinocyte migration by angiotensin II via heparin-binding epidermal growth factor (EGF)-like growth factor-mediated EGF receptor transactivation. J Biol Chem. 2006; 281: 13209 -13216. [PubMed] .
- 12. Vajapey R, Rini D, Walston J, Abadir P. The impact of age-related dysregulation of the angiotensin system on mitochondrial redox balance. Front Physiol. 2014; 5: 439 [PubMed] .
- 13. Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci U S A. 1995; 92: 3521 -3525. [PubMed] .
- 14. Ichiki T, Labosky PA, Shiota C, Okuyama S, Imagawa Y, Fogo A, Niimura F, Ichikawa I, Hogan BL, Inagami T. Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature. 1995; 377: 748 -750. [PubMed] .
- 15. Tanaka M, Tsuchida S, Imai T, Fujii N, Miyazaki H, Ichiki T, Naruse M, Inagami T. Vascular response to angiotensin II is exaggerated through an upregulation of AT1 receptor in AT2 knockout mice. Biochem Biophys Res Commun. 1999; 258: 194 -198. [PubMed] .
- 16. Zhang X, Wei X, Liu L, Marti GP, Ghanamah MS, Arshad MJ, Strom L, Spence R, Jeng J, Milner S, Harmon JW, Semenza GL. Association of increasing burn severity in mice with delayed mobilization of circulating angiogenic cells. Arch Surg. 2010; 145: 259 -266. [PubMed] .
- 17. Dou C, Lay F, Ansari AM, Rees DJ, Ahmed AK, Kovbasnjuk O, Matsangos AE, Du J, Hosseini SM, Steenbergen C, Fox-Talbot K, Tabor AT, Williams JA, Liu L, Marti GP, Harmon JW. Strengthening the skin with topical delivery of keratinocyte growth factor-1 using a novel DNA plasmid. Mol Ther. 2014; 22: 752 -761. [PubMed] .
- 18. Castelino FV, Seiders J, Bain G, Brooks SF, King CD, Swaney JS, Lorrain DS, Chun J, Luster AD, Tager AM. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011; 63: 1405 -1415. [PubMed] .
- 19. Tamiya T, Ichiyama K, Kotani H, Fukaya T, Sekiya T, Shichita T, Honma K, Yui K, Matsuyama T, Nakao T, Fukuyama S, Inoue H, Nomura M, Yoshimura A. Smad2/3 and IRF4 play a cooperative role in IL-9-producing T cell induction. J Immunol. 2013; 191: 2360 -2371. [PubMed] .
- 20. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De BJ, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005; 37: 275 -281. [PubMed] .
- 21. Louafi F, Martinez-Nunez RT, Sanchez-Elsner T. MicroRNA-155 targets SMAD2 and modulates the response of macrophages to transforming growth factor-{beta}. J Biol Chem. 2010; 285: 41328 -41336. [PubMed] .
- 22. Wang J, Wang Y, Wang Y, Ma Y, Lan Y, Yang X. Transforming growth factor beta-regulated microRNA-29a promotes angiogenesis through targeting the phosphatase and tensin homolog in endothelium. J Biol Chem. 2013; 288: 10418 -10426. [PubMed] .
- 23. Kong B, Michalski CW, Hong X, Valkovskaya N, Rieder S, Abiatari I, Streit S, Erkan M, Esposito I, Friess H, Kleeff J. AZGP1 is a tumor suppressor in pancreatic cancer inducing mesenchymal-to-epithelial transdifferentiation by inhibiting TGF-beta-mediated ERK signaling. Oncogene. 2010; 29: 5146 -5158. [PubMed] .
- 24. Bergstrom R, Savary K, Moren A, Guibert S, Heldin CH, Ohlsson R, Moustakas A. Transforming growth factor beta promotes complexes between Smad proteins and the CCCTC-binding factor on the H19 imprinting control region chromatin. J Biol Chem. 2010; 285: 19727 -19737. [PubMed] .
- 25. Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, Hung MC. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006; 8: 1359 -1368. [PubMed] .
- 26. Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, Vicente-Suarez I, Pinilla-Ibarz J, Wright KL, Seto E, Bhalla K, Villagra A, Sotomayor EM. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011; 186: 3986 -3996. [PubMed] .
- 27. Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999; 274: 25543 -25549. [PubMed] .
- 28. Sayegh J, Cao J, Zou MR, Morales A, Blair LP, Norcia M, Hoyer D, Tackett AJ, Merkel JS, Yan Q. Identification of small molecule inhibitors of Jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J Biol Chem. 2013; 288: 9408 -9417. .
- 29. Sakabe K and Hart GW. O-GlcNAc transferase regulates mitotic chromatin dynamics. J Biol Chem. 2010; 285: 34460 -34468. [PubMed] .
- 30. Roberts AB, Flanders KC, Kondaiah P, Thompson NL, Van Obberghen-Schilling E, Wakefield L, Rossi P, de CB, Heine U, Sporn MB. Transforming growth factor beta: biochemistry and roles in embryogenesis, tissue repair and remodeling, and carcinogenesis. Recent Prog Horm Res. 1988; 44: 157 -197. [PubMed] .
- 31. Crowe MJ, Doetschman T, Greenhalgh DG. Delayed wound healing in immunodeficient TGF-beta 1 knockout mice. J Invest Dermatol. 2000; 115: 3 -11. [PubMed] .
- 32. Finnson KW, McLean S, Di Guglielmo GM, Philip A. Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring. Adv Wound Care (New Rochelle). 2013; 2: 195 -214. [PubMed] .
- 33. Moustakas A and Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009; 136: 3699 -3714. [PubMed] .
- 34. Bergstrom R, Savary K, Moren A, Guibert S, Heldin CH, Ohlsson R, Moustakas A. Transforming growth factor beta promotes complexes between Smad proteins and the CCCTC-binding factor on the H19 imprinting control region chromatin. J Biol Chem. 2010; 285: 19727 -19737. [PubMed] .
- 35. Van de Kerkhof PC, Van BB, Spruijt K, Kuiper JP. Age-related changes in wound healing. Clin Exp Dermatol. 1994; 19: 369 -374. [PubMed] .
- 36. Mauviel A. Transforming growth factor-beta signaling in skin: stromal to epithelial cross-talk. J Invest Dermatol. 2009; 129: 7 -9. [PubMed] .
- 37. Denton CP, Khan K, Hoyles RK, Shiwen X, Leoni P, Chen Y, Eastwood M, Abraham DJ. Inducible lineage-specific deletion of TbetaRII in fibroblasts defines a pivotal regulatory role during adult skin wound healing. J Invest Dermatol. 2009; 129: 194 -204. [PubMed] .
- 38. Burks TN, Andres-Mateos E, Marx R, Mejias R, Van EC, Simmers JL, Walston JD, Ward CW, Cohn RD. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med. 2011; 3: 82ra37 .
- 39. Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, Motomiya Y, Bucala R, Iida S, Tamaki K, Imaizumi T, Cooper ME, Okuda S. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004; 66: 2137 -2147. [PubMed] .
- 40. Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol. 2009; 219: 449 -458. [PubMed] .
- 41. El GH, Elbardisey DM, Eltokhy HM, Teaama D. Effect of transforming growth factor Beta 1 on wound healing in induced diabetic rats. Int J Health Sci (Qassim). 2013; 7: 160 -172. [PubMed] .
- 42. Sakai N, Wada T, Matsushima K, Bucala R, Iwai M, Horiuchi M, Kaneko S. The renin-angiotensin system contributes to renal fibrosis through regulation of fibrocytes. J Hypertens. 2008; 26: 780 -790. [PubMed] .
- 43. Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J Cell Sci. 1995; 108: 985 -1002. [PubMed] .
- 44. Miller MC and Nanchahal J. Advances in the modulation of cutaneous wound healing and scarring. BioDrugs. 2005; 19: 363 -381. [PubMed] .
- 45. Pakyari M, Farrokhi A, Maharlooei MK, Ghahary A. Critical Role of Transforming Growth Factor Beta in Different Phases of Wound Healing. Adv Wound Care (New Rochelle). 2013; 2: 215 -224. [PubMed] .
- 46. Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, Roberts AB. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999; 1: 260 -266. [PubMed] .
- 47. Owens P, Engelking E, Han G, Haeger SM, Wang XJ. Epidermal Smad4 deletion results in aberrant wound healing. Am J Pathol. 2010; 176: 122 -133. [PubMed] .
- 48. Gould L, Abadir P, Brem H, Carter M, Conner-Kerr T, Davidson J, DiPietro L, Falanga V, Fife C, Gardner S, Grice E, Harmon J, Hazzard WR, High KP, Houghton P, Jacobson N, Kirsner RS, Kovacs EJ, Margolis D, McFarland HF, Reed MJ, Sullivan DH, Thom S, Tomic-Canic M, Walston J, Whitney J, Williams J, Zieman S, Schmader K. Chronic Wound Repair and Healing in Older Adults: Current Status and Future Research. Wound Repair Regen. 2014; .
- 49. Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O'Rourke B, Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. 2011; 108: 14849 -14854. [PubMed] .
- 50. Abadir PM. The frail renin-angiotensin system. Clin Geriatr Med. 2011; 27: 53 -65. [PubMed] .