



Introduction
Replicative senescence of cells, a hallmark of aging, is a state of indefinite growth cessation triggered by the replicative exhaustion of cells and sometimes by cellular damage and oncogenic signaling [1]. Expression of p27KIP1, a pivotal CDK inhibitor and a tight modulator of CDK-dependent phenotypes, increases with cellular senescence and critically promotes cell growth arrest in replicative senescence [2–4].
Although the role of p27KIP1 in replicative senescence has been well established, its regulation is complex and is elicited at multiple levels. The transcriptional factors FoxM1, FOXO1, and FOXO3a are responsible for the transcriptional control of p27 expression [5–6]. However, the fact that p27 mRNA levels are largely unaltered during replicative senescence suggests that the rise in p27 expression in senescent cells may be achieved mainly by regulation at the levels of translation or protein degradation [7]. Indeed, regulation of p27 by ubiquitin-mediated proteolysis, a result of phosphorylation at serine 10 (S10) and threonine 198 (T198), has been intensively reported [8–11]. Other studies have also linked the ubiquitin-mediated proteolysis of p27 to the accumulation of p27 protein in cellular senescence [12–14]. In addition to protein degradation, regulation at the level of mRNA translation is also critical for the expression of p27. For example, RNA-binding proteins HuR and CUGBP1 have shown to represses the translation of p27 by interacting with the internal ribosome entry site (IRES) element located at the p27 5′UTR [15–16]. Finally, a variety of microRNAs are also involved in the silencing of p27 translation in cancer [17–18].
RNA methylation critically modulates the efficiency and accuracy of translation [19–20], RNA stability [21–22], and the biogenesis of small RNAs [21, 23–24]. NSun2 (NOP2/Sun domain family, member 2; MYC-induced SUN domain–containing protein, Misu) mediates MYC-induced cell proliferation. The expression of NSun2 varies throughout the cell cycle, displaying lowest levels during G1 and highest during S phase [25]. Our previous studies revealed that NSun2 methylates the 3′UTRs of mRNAs encoding p16, p53, E2F3, and ErbB2, thereby enhancing the expression of these proteins in cells exposed to oxidative stress [26–27]. NSun2-mediated mRNA methylation could also promote cell proliferation by elevating the translation of CDK1 [28]. However, the role of NSun2 in replicative senescence and the mechanisms underlying this process have not been studied.
In the present study, we report our findings on the role of NSun2-mediated mRNA methylation in replicative senescence. We describe evidence that NSun2 methylates p27 mRNA at the 5′UTR. Residue C64 located in the p27 5′UTR was identified as a major methylation site. Methylation by NSun2 repressed the translation of p27 and CDK1 mRNAs, thereby delaying the process of replicative senescence. Our results high-light the profound impact of NSun2-mediated mRNA methylation on replicative senescence.
Results
NSun2 represses p27 translation by methylating the 5′UTR of p27 mRNA
The present study was prompted by our earlier findings that high levels of NSun2 lowered p27 abundance. As shown in Fig. 1A, overexpression of NSun2 in HeLa cells by transfection of plasmid pNSun2 reduced p27 protein levels by ~70% while silencing of NSun2 by transfection of small interfering (si) RNA increased p27 protein levels by ~4.2 fold. However, neither overexpression nor knockdown of NSun2 influenced p27 mRNA levels (Fig. 1B). These results suggested that the regulation of p27 by NSun2 may not involve altered p27 mRNA transcription or turnover. Because NSun2 had been shown to regulate the expression of p53, p16, E2F3, Bak1, ErbB2, and CDK1 by methylating the mRNAs encoding these proteins [26–28], we tested if NSun2 was capable of methylating p27 mRNA. To this end, the p27 mRNA fragments described in Fig. 2A were used for in vitro methylation assays (Materials and Methods). The p27 cDNA and p16-CR (coding region of p16 mRNA) were included as negative controls, while bacterial tRNA served as a positive control. As shown in Fig. 2B (left and right), tRNA, 5′UTR, 5′UTRa, and 5′UTRa1 were methylated, while p27 cDNA, p16-CR, p27-CR, p27-3′UTR, p27-5′UTRa2, and p27-5′UTRb were not methylated. Accordingly, the methylation site was located at the p27 5′UTR (positions 1-140).
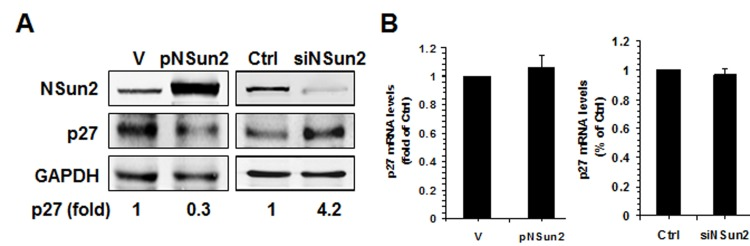
Figure 1. NSun2 regulates p27 expression (A) HeLa cells were transfected with a vector expressing NSun2 (pNSun2) or with a siRNA targeting NSun2. Forty-eight hours later, cell lysates were prepared and subjected to Western blot analysis to assess the levels of proteins NSun2, p27, and GAPDH. (B) RNA prepared from cells described in Fig. 1A was used for RT-qPCR analysis to assess the levels of p27 mRNA. Data represent the means ± SD from 3 independent experiments.
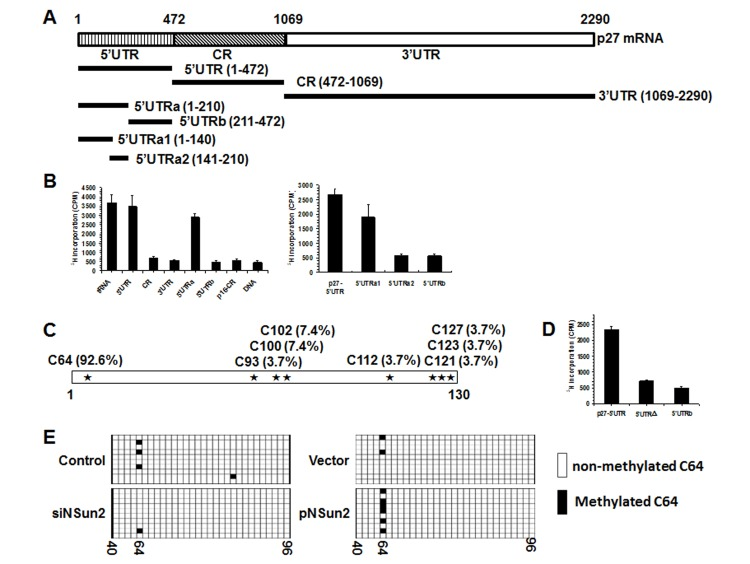
Figure 2. NSun2 methylates p27 5′UTR in vitro and in cells (A) Schematic representation depicting the p27 mRNA fragments used for in vitro methylation assays. (B) Incorporation of 3H-labeled SAM into p27 5′UTR, CR, 3′UTR, 5′UTRa, and 5′UTRb fragments (left) as well as 5′UTR, 5′UTRa1, 5′UTRa2, and 5′UTRb fragments (right). The incorporation of 3H-labeled SAM into p27 cDNA (DNA) and p16-CR (coding region) served as negative controls. The incorporation of 3H-labeled SAM into bacteria tRNA served as a positive control. (C)In vitro methylated 5′UTRa1 fragment was subjected to bisulfate RNA sequencing analysis to identify the methylation sites, as described in “Materials and Methods”. The percent of methylation at different sites is indicated. (D) Incorporation of 3H-labeled SAM into the 5′UTR variant mutating C64 (5′UTRΔ). 5′UTR and 5′UTRb served as negative and positive controls, respectively. (E) HeLa cells were transfected with a pGL3-derived reporter bearing the p27 5′UTR; 24 h later, cells were further transfected with a vector expressing NSun2 or with NSun2 siRNA and cultured for an additional 48 h. RNA was isolated and subjected to bisulfate sequencing analysis to assess the rate of C64 methylation. Open boxes indicate cytosine-to-uracil conversion, read as thymidine in the cDNA (unmethylated), and filled boxes indicate a retained cytosine (methylated). The numbers below the columns refer to cytosine positions in the p27 5′UTR.
To determine the formation of m5C or m6A in the methylated fragments, p27-5′UTRa1 methylated in vitro by using nonisotopic S-Adenosyl methionine (SAM) and NSun2, and unmethylated (same reaction without adding NSun2) p27-5′UTRa1 were subjected to MS-HPLC analysis. As shown, m5C was detected in the methylated p27-5′UTRa1 fragment (Supplemental Fig. S1). Identification of m6A from the methylated p27-5′UTRa1 did not show any positive results (not shown). To further identify the methylation site by NSun2, in vitro methylated p27-5′UTRa1 was subjected to bisulfate sequencing analysis. As shown in Fig. 2C, among all of the clones sequenced, C64 was detected in ~92.6% of positive clones. Mutation of C64 (p27 5′UTRΔ) nearly abolished the effect of NSun2 on methylating the p27 5′UTR (Fig. 2D), indicating that C64 is a major methylation site.
To test whether NSun2 was capable of methylating p27 5′UTR in cells, HeLa cells were transfected with a pGL3-derived reporter bearing the p27 5′UTR. Twenty-four hours later, cells were further transfected with a vector expressing NSun2 or with NSun2 siRNA and cultured for an additional 48 h. RNA was isolated and subjected to bisulfate sequencing analysis to assess the rate of C64 methylation. As shown, knockdown of NSun2 reduced the methylation of C64, whereas overexpression of NSun2 increased the rate of C64 methylation (Fig. 2E). These results indicate that NSun2 was able to methylate p27 mRNA in cells.
To further test whether methylation of p27 5′UTR by NSun2 influenced p27 expression levels, pGL3-derived reporters bearing fragments of p27 mRNA were constructed (Fig. 3A, schematic). HeLa cells were transfected with each of these reporters and 24 h later, cells were transfected with control or NSun2-directed siRNAs and cultured for an additional 48 h. Cellular lysates then were collected and subjected to Western blot analysis to assess NSun2, Firefly luciferase, Renilla luciferase, and GAPDH protein levels. As shown in Fig. 3B, knockdown of NSun2 elevated the levels of reporter protein expressed from pGL3-5′UTR, but not that expressed from pGL3, pGL3-CR, pGL3-3′UTR, or pGL3-5′UTRΔ, indicating that methylation by NSun2 at C64 selectively repressed the expression of p27. Because NSun2 regulates the expression of p27 without influencing the levels of p27 mRNA (Fig. 1), we further asked if methylation by NSun2 regulated the translation of p27. To this end, in vitro-transcribed reporter transcripts Luciferase (Luc), Luc-5′UTR, and Luc-5′UTRΔ were methylated in vitro by NSun2 or kept unmethylated. These transcripts then were used for in vitro translation assays and reporter activity was used as readout of the efficiency of translation. As shown in Fig. 3C (left), methylation by NSun2 reduced the reporter activity of Luc-5′UTR (by ~59%), but not that of Luc and Luc-5′UTRΔ. Therefore, methylation of p27 5′UTR by NSun2 represses the expression of p27 at the level of translation.
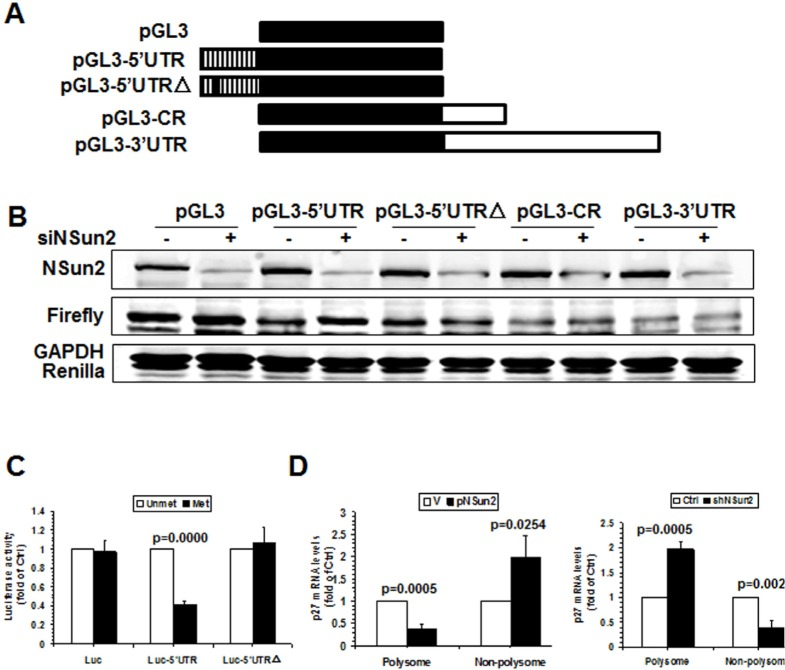
Figure 3. Methylation by NSun2 is functional for regulating the translation of p27 (A) Schematic representation of the pGL3-derived reporter vectors used for reporter gene assays. (B) HeLa cells were transfected with each of the reporter vectors described in Fig. 3A together with a pRL-CMV control reporter. Twenty-four hours later, cells were further transfected with NSun2 siRNA and cultured for an additional 48 h. Firefly luciferase activity against Renilla luciferase activity was analyzed. Data represent the means ± SD from 3 independent experiments; significance was analyzed by Student's t test. (C)In vitro methylated (Met) or unmethylated (Unmet) Luciferase (Luc), luc-5′UTR, and luc-5′UTRΔ reporter transcripts were used for in vitro translation assays. Firefly luciferase activity was measured to determine the translation efficiency. Data represent the means ± SD from 3 independent experiments; significance was analyzed by Student's t test. (D) Cells described in Fig. 1A were used for isolating the polysomal and non-polysomal fractions. RNA prepared from the fractions was subjected to RT-qPCR analysis to assess the presence of p27 mRNA in the polysomal and non-polysomal fractions. Data represent the means ± SD from 3 independent experiments; significance was analyzed by Student's t test.
To further investigate the mechanisms underlying the translational regulation of p27 by NSun2, the polysomal presence of p27 mRNA in cells with overexpressed or silenced NSun2 was examined. As shown in Fig. 3D, overexpression of NSun2 decreased the presence of p27 mRNA in the polysomal fraction (by ~66%, p=0.0005) but increased it in the non-polysomal fraction (by ~2.0 fold, p=0.0254). On the other hand, knockdown of NSun2 increased the presence of p27 mRNA in the polysomal fraction (by ~60%, p=0.0005) but decreased it in the non-polysomal fraction (by ~2.0 fold, p=0.0024). Taken together, these findings indicate that the assembly of p27 mRNA in the polysome was repressed by NSun2-mediated methylation, in turn repressing the translation of p27.
By binding to the p27 5′UTR, RNA-binding proteins HuR and CUGBP1 repress p27 translation [15–16]. These findings raised the question that HuR or CUGBP1 might influence the function of NSun2 in repressing the translation of p27. As shown in Supplemental Fig. S2A by RNA pull-down assays, HuR associated with the p27 5′UTR, 5′UTRb, and 3′UTR, while CUGBP1 only associated with the p27 5′UTR and 5′UTRb, in agreement with previous findings [15, 16]. NSun2 interacted with the p27 5′UTR and 5′UTRa, consistent with the results that NSun2 methylated p27 5′UTR and 5′UTRa (Fig. 2B). Although NSun2 was capable of interacting with the p27 3′UTR, this interaction did not lead to the methylation of p27 3′UTR (Fig. 2B). Furthermore, methylation by NSun2 did not influence the association of HuR or CUGBP1 with the p27 mRNA (Supplemental Fig. S2B), and the association of NSun2 with p27 mRNA did not influence the association of HuR and CUGBP1 with p27 mRNA or vice versa (Supplemental Fig. S2C-E). Moreover, knockdown of NSun2 increased the activities of reporters pGL3-5′UTR and pGL3-5′UTRa, but not those of pGL3-5′UTRb or pGL3-5′UTRΔ (Supplemental Fig. S2F). In contrast, knockdown of HuR or CUGBP1 increased the activities of reporters pGL3-5′UTR, pGL3-5′UTRb, and pGL3-5′UTRΔ, but not that of pGL3-5′UTRa (Supplemental Fig. S2F). These results suggest that the translational repression of p27 by NSun2-mediated mRNA methylation is independent of the effects elicited by HuR or CUGBP1.
NSun2 regulation of p27 and CDK1 impacts upon replicative senescence
Apart from p27, NSun2 was also found to regulate the expression of senescence-associated proteins p53, p16, CDK1, and CDC25C [26–28]. To further investigate whether NSun2 regulated the process of replicative senescence, we assessed the levels of proteins NSun2, p53, p16, CDK1, p27, CDC25C, and GAPDH in early-passage [proliferating, ‘Young’ (Y), ~PDL 28], middle-passage [middle (M), ~PDL 40], and late-passage [senescent (S), ~PDL 55] human diploid fibroblasts (2BS) by Western blot analysis. As shown in Fig. 4A (left), the levels of p16 and p27 increased with replicative replicative senescence, while the levels of p53 remained unchanged. However, the levels of proteins NSun2, CDK1, and CDC25C were reduced with replicative senescence. The inverse correlation between NSun2 and p27 levels was also observed in another model of senescence, proliferating and senescent IDH4 cells (Fig. 4A, right). In addition, the levels of CDK1 mRNA decreased (by ~45% in 2BS cells, p=0.0000; by ~63% in IDH4 cells, p=0.0000) but p27 mRNA levels remained unchanged during senescence in both cell types (Fig. 4B). Both endogenous p53 and p16 are constitutively expressed in IDH4 cells, regardless of whether they are maintained in a proliferative state or they are induced to undergo senescence (Fig. S3). Methylated RNA-specific PCR analysis revealed that the levels of methylated CDK1 and p27 mRNAs decreased in the process of replicative senescence (by ~76% for p27 mRNA; by ~46% for CDK1 mRNA) (Fig. 4C), while the levels of unmethylated CDK1 and p27 mRNAs increased in replicative senescence (by ~2.7 fold for p27 mRNA, p=0.0023; by ~5.9 fold for CDK1 mRNA). These results indicate that NSun2-mediated RNA methylation may be able to regulate the expression levels of CDK1, p27, and CDC25C, but not those of p53 and p16, as cells progress to senescence.
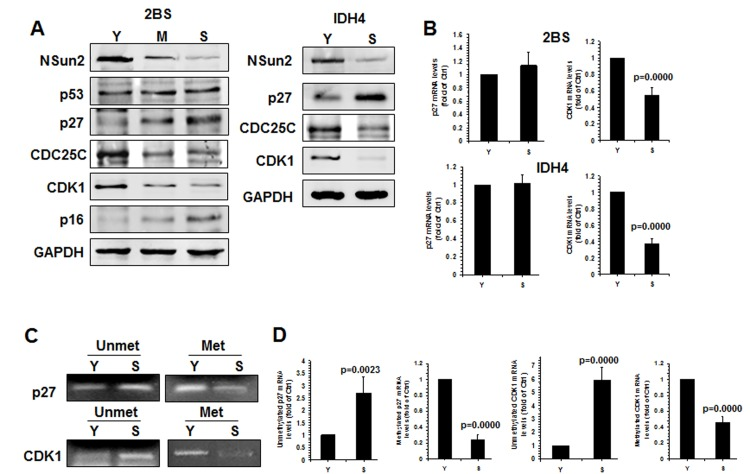
Figure 4. Inverse correlation between p27 levels and NSun2 levels in replicative senescence (A)Left, the levels of proteins NSun2, p53, p16, p27, CDC25C, CDK1, and GAPDH in early-passage (Proliferating ‘Young’, ‘Y’, ~PDL 28), middle-passage (middle, ~PDL 40), and late-passage (Senescent, ‘S’, ~PDL 55) human diploid fibroblasts (2BS) were assessed by Western blotting. Right, the levels of proteins NSun2, p27, and GAPDH in Proliferating (Y) and Senescent (S) IDH4 cells were analyzed by Western blot analysis. (B) RNA was prepared from young (Y, ~PDL 28) and senescent (S, ~PDL 55) 2BS cells as well as young (Y) and senescent (S) IDH4 cells and RT-qPCR analysis was performed to assess the levels of p27 and CDK1 mRNAs. (C) RNA described in Fig. 4B was subjected to methylation-specific PCR analysis to assess the methylation of p27 and CDK1 mRNA. Data are representative from 3 independent experiments. (D) The density of the methylation-specific PCR (% of Ctrl) in Fig. 4C relative to that of the mRNA levels (% of Ctrl) shown in Fig. 4B is shown. Data represent the means ± SD from 3 independent experiments; significance was analyzed by Student's t test.
To test this idea using a different approach, 2BS cells were stably infected with lentiviruses expressing either NSun2 or NSun2 shRNA. As shown in Fig. 5A, infection with the NSun2-expressing lentiviruses reduced the levels of p27 by ~70%, while infection with NSun2 shRNA-expressing lentiviruses increased the levels of p27 by ~5.1 fold. In contrast, the levels of proteins CDK1 and CDC25C increased in cells with overexpressed NSun2 (by ~2.8 fold for CDK1; by ~1.5 fold for CDC25C) but decreased in cells with silenced NSun2 (by ~70% for both CDK1 and CDC25C). However, the levels of proteins p53 and p16 increased moderately in cells with overexpressed NSun2 and decreased only moderately in cells with silenced NSun2 (Fig. 5A). These modest changes may be an indirect reflection of the fact that 2BS cells with overexpressed NSun2 or silenced NSun2 have a constitutively altered senescence phenotype, perhaps as a result of compensatory changes in response to the permanent alterations in NSun2 levels. In agreement with the findings in Fig. 1B and in previous studies [28], neither overexpression of NSun2 nor knockdown of NSun2 altered the levels of p27 and CDK1 mRNAs in a measurable manner (Fig. 5B). In addition, the levels of cellular methylated p27 mRNA increased in cells with overexpressed NSun2 but decreased in cells with silenced NSun2 (Supplemental Fig. S4).
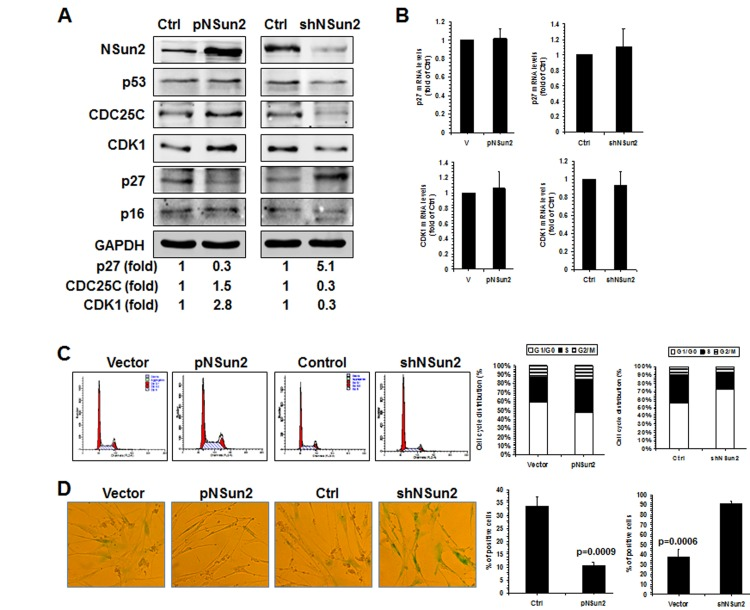
Figure 5. NSun2-p27 regulatory process impacts on the progression of replicative senescence (A) 2BS cells were stably infected with a lentivirus bearing a pHBLV-NSun2 vector (pNSun2) or a pHBLV-shNSun2 vector (shNSun2). Cell lysates were prepared and subjected to Western blot analysis to assess the levels of proteins NSun2, p53, CDC25C, p27, p16, CDK1, and GAPDH. (B) RNA prepared from cells described in Fig. 5A was subjected to RT-qPCR analysis to assess the levels of p27 and CDK1 mRNAs. Data represent the means ± SD from 3 independent experiments. (C) Cells described in Fig. 5A were analyzed for cell cycle distribution (left). The percentage of cells in each cell cycle compartment is presented (right). (D) Cells described in Fig. 5A were analyzed for the activity of SA-β-gal (left). The means ± SD from 3 independent experiments are presented; significance was analyzed by Student's t test (right).
Finally, knockdown of NSun2 inhibited cell growth and increased the proportions of cells displaying the protein marker senescence-associated (SA)-β-galactosidase (~37.5% vs. ~91.0%), while overexpression of NSun2 promoted cell growth and reduced SA-β-galactosidase activity (~33.5% vs. ~10.6%). We next tested if overexpression of the p27 5′UTR fragment could function as a decoy fragment and rescue the effect of NSun2 overexpression in modulating p27, CDK1, and CDC25C levels, in promoting cell growth, and in delaying replicative senescence. As shown in Figure 6, overexpression of the p27 5′UTR fragment in 2BS cells by infecting 2BS cells with a lentivirus expressing luc-5′UTR (which increased p27 5′UTR levels by ~944 fold) rescued the effect of NSun2 overexpression in influencing the expression of p27, CDK1, and CDC25C (Figure 6A), in promoting DNA replication (progression through the S phase; Figure 6B), and in delaying replicative senescence (Control vs. pNSun2, ~46.4% vs. ~20.0%; control+pHBLV-5′UTR vs. pNSun2+pHBLV-5′UTR, ~51.5% vs. ~46.8%) (Figure 6C). Overexpression of the p27 5′UTR modestly increased the SA-β-galactosidase activity in control vector-transfected cells (~46.4% vs. ~51.5%), although the alteration was not statistically significant. In sum, the influence of NSun2 on the expression of p27, CDK1, and CDC25C has a robust impact upon replicative senescence.
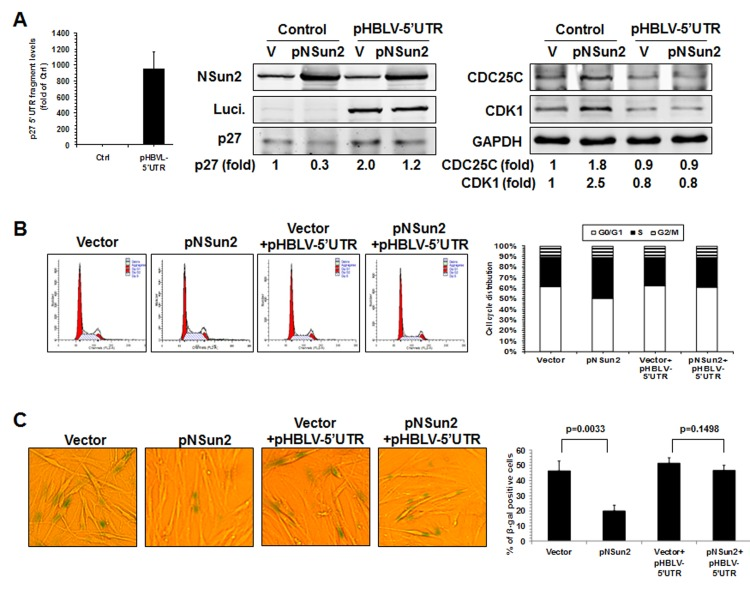
Figure 6. NSun2 delays replicative senescence by regulating p27, CDK1, and CDC25C (A) 2BS cells were stably infected with a lentivirus bearing a pHBLV-NSun2 vector (pNSun2) or with a control lentivirus (Vector), or stably co-infected with a lentivirus bearing a pHBLV-luc-5′UTR vector (Luc-5′UTR) or kept untreated (Control). Cell lysates were prepared and subjected to Western blot analysis to assess the levels of proteins NSun2, luciferase, p27, CDK1, CDC25C, and GAPDH. (B) The cells described in Fig. 6A were analyzed for cell cycle distribution (left) and the percentage of cells in each cell cycle compartment represented (right). (C) Cells described in Fig. 6A were used for analysis of SA-β-gal activity (left). The means ± SD from 3 independent experiments are presented and the significance was assessed by using Student's t test (right).
Discussion
In response to oxidative stress, elevated NSun2 is responsible for the rise in p53 and p16 expression levels [26–27], suggesting that NSun2 is able to promote the process of oxidative stress-induced senescence. However, NSun2 does not appear to regulate replicative senescence by methylating p53 and p16 mRNAs, since reduced NSun2 in senescent HDFs does not result in reduced p53 and p16 levels (Fig. 4). Instead, the evidence obtained in the present study supports the view that NSun2-mediated mRNA methylation represses replicative senescence at least in part by lowering the translation of p27 and elevating the expression of CDK1 (Figs. 1–6). The NSun2-enhanced CDC25C expression, which is mediated by NSun2 regulation of miR125b [25], may also impact upon the process of replicative senescence (Figs. 4–6). It is possible that the role of NSun2 in replicative senescence is different from its role in oxidative stress-induced cellular senescence, since the levels of NSun2 protein decrease in replicative senescence (Fig. 4A) but increase in response to oxidative stress [26–27]. Accordingly, the mechanisms underlying the regulation of expression of certain genes (e.g., CDK1, p27, p16 or p53) during replicative senescence could be different from those operating during oxidative stress-induced senescence.
In previous studies, we demonstrated that methylation of 3′UTRs of different mRNAs by NSun2 increased the expression of p53, p16, E2F3, Bak1, ErbB2, and CDK1 [26–28]. In this study, methylation of p27 5′UTR by NSun2 repressed p27 translation (Figs. 1–3). Therefore, the regulatory effect of NSun2-mediated methylation may depend upon the location of methylation or the mRNAs targeted by NSun2. On the other hand, the association of NSun2 with p27 5′UTR is linked to the methylation of p27 5′UTR (Fig. 2B and Supplemental Fig. S2A). However, the association of NSun2 with p27 3′UTR (Supplemental Fig. S2A) did not result in methylation of p27 3′UTR (Fig. 2B) nor did it affect p27 expression levels (Fig. 3B). Therefore, whether the association of NSun2 with p27 3′UTR regulated the expression of p27 under other specific conditions or processes remains to be studied.
Translational regulation is critical for the elevation of p27, since p27 mRNA levels do not change in the process of replicative senescence (Fig. 4B). Apart from NSun2, HuR and CUGBP1 also repress the translation of p27. Interestingly, HuR and CUGBP1 protein levels declined during replicative senescence [29–31]. Thus, it is plausible to postulate that HuR- and CUGBP1-mediated translational repression may be also involved in the regulation of p27 in replicative senescence, even though they appear to function independently of NSun2.
Proliferation arrest is a hallmark feature of cell senescence [1]. Therefore, genes involved in the regulation of the cell division cycle are extensively involved in the control of cellular senescence. Apart from the elevation in p27 levels, the reduction in CDK1 expression and CDK1 activity also contribute to growth arrest in cellular senescence [13,32]. Therefore, the repression of p27 translation and the elevation of CDK1 translation by NSun2-mediated mRNA methylation may cooperatively promote cell proliferation and delay the process of replicative senescence. In addition, although the role of CDC25C in replicative senescence remains untested, our results suggest that CDC25C may also inhibit growth during senescence (Figs. 4–6).
In general, genes highly expressed in senescent cells tend to show low expression levels in cancer, and vice versa. Indeed, induction of CDK1 has been linked to the growth of human colorectal cancer and acute myeloid leukemia [33–34]; the reduction of p27 has been liked to the genesis and progression of a broad array of human cancers, including cancers of the breast, colon, prostate, ovary, lung, stomach, and others [35–37]. The increase of NSun2 levels observed in a variety of human cancers, including those originating in the colon, esophagus, stomach, liver, oral cavity, pancreas, uterus, cervix, prostate, kidney, bladder, thyroid, breast, and skin [25, 38], may be part of a program of senescence repression involving reduced levels of p27. Besides cancer, p27 has also been implicated in aging-related diseases such as osteoporosis [39], atherosclerosis [40–41], and Alzheimer's disease [38]. It will be particularly important to investigate the possible involvement of the NSun2-p27 regulatory paradigm into these aging-related conditions.
Materials and Methods
Cell culture, transfection, and SA-β-galactosidase activity
Human IDH4 fibroblasts were generously provided by J. W. Shay and cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and dexamethasone (Dex, 1 μg/ml) for constitutive expression of SV40 large T antigen to suppress senescence and stimulate proliferation [29]. Proliferating (‘young’) and senescent IDH4 cells were obtained by incubating cells in charcoal-stripped serum (10%) in the presence (Young) or absence (Senescent) of Dexamethasone (Dex, 1 μg/ml) for 5 days. Early- [Proliferating (P), Young, ~28 population doublings (pdl)], middle- [(M), ~40 pdl], and late-passage [(S) Senescent, ~55 pdl] human diploid 2BS fibroblasts (National Institute of Biological Products, Beijing, China), and HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin, at 37°C in 5% CO2. All plasmids were transfected using lipofectamine 2000 (Invitrogen) and collected 48 to 72 h after transfection for further analysis. SA-β-galactosidase activity (β-gal staining) was performed as described previously [42].
Transcript preparation
cDNA was used as a template for PCR amplification of the p27 fragments. All 5′ primers contained the T7 promoter sequence (CCAAGCTTCTAATACGACTCACTATAGGGAGA). To prepare templates for the p27 5′UTR (positions 1-472), p27 5′UTR-A (positions 1-210), p27 5′UTR-B (positions 211-472), p27 5′UTRa1 (positions 1-140), p27 5′UTR a2 (positions 141-210), p27 CR (positions 473-1069), and p27 3′UTR (positions 1070-2290), we used the following primer pairs: (T7) CTTCTTCGTCAGCCTCCC and CTTTCTCCCGGGTCTGCA for 5′UTR, (T7) CTTCTTCGTCAGCCTCCC and AGCGGAGAGGGTGGCAAAGCCC for 5′UTR-A, (T7) TGCCTGGTCCCCTCTCCTCT and CTTTCTCCCGGGTCTGCA for 5′UTR-B, (T7) CTTCTTCGTCAGCCTCCC and CTCTCCAAACCTTGCCGGCGTC for 5′UTRa1, (T7) CGGCTGGGTTCGCGGGAC and AGCGGAGAGGGTGGCAAAGCCC for 5′UTRa2, (T7) ATGTCAAACGTGCGAGTGTC and TTACGTTTGACGTCTTCTGAG for CR, (T7) ACAGCTCGAATTAAGAATATG and TAACAAAAGAGGGGAAAACCTATTC for 3′UTR. These PCR products were transcribed in vitro following the manufacturer's instructions (Thermo).
The p27 5′UTR mutants were prepared by overlapping PCR. The p16 3′UTR fragment was described previously [27].
Western blot analysis
Monoclonal anti-CUGBP1, monoclonal anti-p53, monoclonal anti-p16, monoclonal anti-CDC25C, and monoclonal anti-HuR were from Santa Cruz Biotechnology. Polyclonal anti-NSun2 and monoclonal anti-GAPDH were from Abcam. After secondary antibody incubations, signals were detected by Odyssey Imaging System (Gene Company Limited) following the manufacturer's instruction and quantitated by densitometric analysis with Image J software.
Knockdown of NSun2, HuR, and CUGBP1
To silence NSun2, HuR, or CUGBP1, cells were transfected with siRNAs (10 nmol/L each) targeting NSun2 (AGAUGUUAAGAUACUGUUGACCC), HuR (AAGAGGCAAUUACCAGUUUCAUU) or CUGBP1 (GAGCCAACCUGUUCAUCUAUU), or with a control siRNA (UUGUUCGAACGUG UCACGUTT) using RNAiMAX (Invitrogen). Unless otherwise indicated, cells were collected for analysis 48 h after transfection. All knockdown interventions caused less than 1% cell death (by FACS analysis, data not shown).
RNA isolation and real-time qPCR analysis
Total cellular RNA was prepared using the RNeasy Mini Kit (Qiagen). For reverse-transcription (RT) followed by real-time, quantitative (q)PCR analysis of CDK1 and p27, we used following primer pairs: AAATGTGTGTAGGTCTCAC and ATGATTTAAG CCAACTCAAA for CDK1 mRNA, and GGCTCCGGC TAACTCTGA and TCTTCTGTTCTGTTGGCTCTTT for p27 mRNA.
Constructs and reporter gene assays
For construction of the pGL3-derived reporter vectors bearing the p27 5′UTR and 5′UTR mutating C64 (C-G, 5′UTRΔ), the 5′UTR or 5′UTRΔ fragment was amplified by PCR by using primer pairs CCCAAGCTTGGGCTTCTTCGT CAGCCTCCC and CATGCCATGGCATGCTTTCT CCCGGGTCTGCA and inserted between the Hind III and Nco I sites of pGL3-promoter vector (Promega). For constructing the pGL3-derived reporter vectors bearing the p27 5′UTRa, 5′UTRb, CR, and 3′UTR, 5′UTRa, 5′UTRb, CR, and 3′UTR were amplified by PCR using primer pairs CCCAAG CTTCTTCGTCAGCCTCCC and CATGCCAGCGGA GAGGGTGGCAAAGCCC for 5′UTRa, CCCAAGTGCCTGGTCCCCTCTCCTCT and CATGCCCTTTCTCCCGGGTCTGCA for 5′UTRb, CCCAAGCTGTCAAACGTGCGAGTGTC and CATGCCTTACGTTTGACGTCTTCTGAG for CR, and CCCAAGACAGCTCGAATTAAGAATATG and CATGCCTAACAAAAGAGGGGAAAACCTATTC for 3′UTR. Each of these fragments was inserted between HindIII and NcoI sites of the pGL3-promoter vector (Promega). The pcDNA 3.1 vector expressing NSun2 was described previously [27].
For reporter gene assays, each of the pGL3-derived vectors was co-transfected with pRL-CMV vector by Lipofectamine 2000 (Invitrogen). Forty eight hours after transfection, cell lysates were collected and the firefly and renilla luciferase activities were measured with a double luciferase assay system (Promega) following the manufacturer's instructions. All firefly luciferase measurements were normalized to renilla luciferase measurements from the same sample.
Lentiviruses and stable expression of ectopic genes
For the construction of lentivirus vectors expressing NSun2 (pHBLV-NSun2), the coding region of NSun2 was amplified by PCR by using primer pairs CGGAATTCATGGGCGGCGGTCGCGGGGT and CGGGATCCGGTCACCGGGGTGGATGGACC and inserted between EcoRI and BamHI sites of pHBLV-CMVIE-IRES-Puro vector (HANBIO Biotechnology Co., Ltd, Shanghai, China). For construction of the lentivirus vector expressing Luc-5′UTR (pHBLV-luc-5′UTR), the Luc-5′UTR fragment was amplified from the pGL3-5′UTR reporter by PCR by using primers CCGGAATTCTTCTTCGTCAGCCTCCC and CGGGATCCCGTTACACGGCGATCTTTCCGCCCT and inserted between EcoRI and BamHI sites of pHBLV-CMVIE-IRES-Puro vector. For construction of the lentivirus vector expressing NSun2 shRNA (pHBLV-shNSun2), complementary fragments GATCCGCGGCCTCATCATAAGATCTTAGATATTCAAGAGATATCTAAGATCTTATGATGAGGCCGTTTTTTC and AATTGAAAAAACGGCCTCATCATA AGATCTTAGATATCTCTTGAATATCTAAGATCTTATGATGAGGCCGCG were annealed and inserted between the BamHI and EcoRI sites of pHBLV-U6-Zsgreen-puro vector (HANBIO Biotechnology Co., Ltd, Shanghai, China). Viruses were packaged in 293T cells following the instructions from the manufacturer (HANBIO Biotechnology Co., Ltd, Shanghai, China). Stably transfected cells (2BS) were selected in the presence of puromycin (0.5 μg/ml) for 5 days.
Preparation of polysomal fractions
A total of 20 million cells were incubated for 15 min with 100 mg/ml cycloheximide, and total lysates (500 μl) were layered onto a cushion of 30% sucrose in ice-cold buffer containing 20 mM HEPES (pH 7.4), 50 mM potassium acetate, 5 mM magnesium acetate, 1 mM dithiothreitol, 1 unit of RNasin per μl, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, and 0.5 mM phenylmethylsulfonyl fluoride. After centrifugation (Beckman SW40; 100,000 × g for 2 h, 4°C), RNA from the supernatant (nonpolysomal fraction) and the pellet (polysomal fraction) was prepared and used for RT-qPCR analysis.
In vitro methylation assays. For in vitro methylation assay, His-tagged NSun2 was prepared as described previously [27]. Briefly, reaction mixtures (50 μl) containing 0.2 nM His-NSun2, 0.01 nM RNA, and 1 μCi of 3H-labeled S-adenosyl-L-methionine (SAM, Amersham Bioscience) in reaction buffer (5 mM Tris HCL[pH 7.5], 5 mM EDTA, 10% glycerol, 1.5 mM dithiothreitol, 5 mM MgCl2) supplemented with inhibitors (leupeptin [1 μg/ml], aprotinin [1 μg/ml], 0.5 mM phenyl-methylsulfonyl fluoride, and RNasin [5 U/μl]), incubated for 45 min at 37°C, as described [27]. E. coli tRNA (0.01 nM, Sigma) and p16 CR (0.01 nM) served as a positive and negative controls, respectively. Unincorporated 3H SAM was removed by using Qiaquick Spin Columns (Qiagen) and incorporated radioactivity was measured by liquid scintillation counting. The non-isotopic methylated RNA fragments were prepared using cold SAM (Sigma) and in vitro-transcribed RNA fragments under similar conditions.
Bisulfate RNA sequencing
This method could identify the methylation site (m5C) within an RNA fragment shorter than 150 nt. Briefly, fragment 5′UTRa1 (positions 1-140), which was methylated by NSun2, was amplified by using primers (T7) CTTCTTCGTCAGCCTCCCTT and CCTCCATTCC CAACAAACCTTGCCGGCGTCGGAGTCGCAGAGCCGTGA.
This fragment (1 μg) was transcribed in vitro and methylated by using non-isotopic SAM. Samples then were dissolved in 10 μl of RNase-free water and mixed with 42.5 μl 5 M sodium bisulfate (Epitect) and 17.5 μl DNA protection buffer (Epitect), incubated in 70°C for 5 minutes and 60°C for 60 min, repeating for 3-5 cycles. After desalting using Micro Bio-spin6 columns, samples were de-sulfonated by 1 M Tris (pH 9.0, 1/1, V/V) at 37°C for 1 h, followed by ethanol precipitation. The bisulfate-converted fragments (0.2 μg) were reverse-transcribed by RevertAid First Strand cDNA Synthesis Kit (Thermo) using primer GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCCTCCATTCCCAACAAAC. The reverse-transcribed products (cDNA) were amplified by PCR by using primers GGGAGATTTTTTTGTTAGT and GCAGGGTCCGAGGTATTC. The PCR products were inserted into the pGEM-T Easy Vector System (Promega). The plasmids purified from single clones were sequenced. The sequences were aligned with the corresponding p27 mRNA sequence and the cytosines retained were considered to be methylated.
Methylation-specific RT-PCR
Two micrograms (2 μg) of cellular RNA or 1 μg of in vitro-methylated RNA fragment was bisulfite-converted as described in ‘bisulfate RNA sequencing’. The converted p27 5′UTR was reverse-transcribed by using primer GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCCAACCACTCTCCAAACCTT. For methylation-specific p27 mRNA RT-PCR analysis, primer pairs TAATAAGGTTAAAAAAGGGGGTTTG TTTGGGC and GCAGGGTCCGAGGTATTC (for methylated fragment) as well as AAAAAAGGGGG TTTGTTTTAAT and GCAGGGTCCGAGGTATTC (for unmethylated fragment) were used.
In vitro translation assays
For in vitro translation assays, a cell-free translation system (Promega) in rabbit reticulocyte lysate (RL) was used. Luciferase (Luc) transcript was in vitro transcribed from pGL3 by using primer pairs (T7) ATGGAAGACGCCAAAAA CAT and TTTTTTTTTTTTTTTTTTTTTTTTTTTTT TACACGGCGATCTTTCCGCCCT. Luc-5′UTR and Luc-5′UTRΔ transcripts were transcribed in vitro from pGL3-5′UTR and pGL3-5′UTRΔ reporters by using primer pairs (T7) CTTCTTCGTCAGCCTCCCTT and TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTACACG CGATCTTTCCGCCCT. Transcripts (0.01 nM) were either methylated by NSun2 or kept untreated. The methylated and non-methylated transcripts were used for in vitro translation assays. The translation efficiency was determined by measuring the activity of firefly luciferase.
Supplementary Materials
Acknowledgments
We thank J. Shay for generously providing us the IDH4 cells.
Funding
This work was supported by Grants 81230008, 81420108016, and 91339114 from the National Science Foundation of China; Grant B07001 (111 project) from the Ministry of Education of People's Republic of China. M.G. was supported by the NIA-IRP, NIH.
Conflicts of Interest
The authors of this manuscript declare no conflict of interests.
References
- 1. Hayflick L and Moorhead PS. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961; 25: 585 -621. [PubMed] .
- 2. Bringold F and Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000; 35: 317 -329. [PubMed] .
- 3. McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998; 8: 351 -354. [PubMed] .
- 4. Alexander K and Hinds PW. Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol Cell Biol. 2001; 21: 3616 -3631. [PubMed] .
- 5. Zeng J, Wang L, Li Q, Li W, Björkholm M, Jia J, Xu D. FoxM1 is up-regulated in gastric cancer and its inhibition leads to cellular senescence, partially dependent on p27 kip1. J Pathol. 2009; 218: 419 -427. [PubMed] .
- 6. Lees SJ, Childs TE, Booth FW. Age-dependent FOXO regulation of p27Kip1 expression via a conserved binding motif in rat muscle precursor cells. Am J Physiol Cell Physiol. 2008; 295: C1238 -1246. .
- 7. Wong H and Riabowol K. Differential CDK-inhibitor gene expression in aging human diploid fibroblasts. Exp Gerontol. 1996; 31: 311 -325. [PubMed] .
- 8. Bencivenga D, Tramontano A, Borgia A, Negri A, Caldarelli I, Oliva A, Perrotta S, Della Ragione F, Borriello A. P27Kip1 serine 10 phosphorylation determines its metabolism and interaction with cyclin-dependent kinases. Cell Cycle. 2014; 13: 3768 -3782. [PubMed] .
- 9. Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006; 20: 47 -64. [PubMed] .
- 10. Kossatz U, Vervoorts J, Nickeleit I, Sundberg HA, Arthur JS, Manns MP, Malek NP. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J. 206 25: 5159 -70. [PubMed] .
- 11. Starostina NG and Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012; 22: 33 -41. [PubMed] .
- 12. Cao X, Xue L, Han L, Ma L, Chen T, Tong T. WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) delays cellular senescence by promoting p27(Kip1) degradation in human diploid fibroblasts. J Biol Chem. 2011; 286: 33447 -33456. [PubMed] .
- 13. Byrne A, McLaren RP, Mason P, Chai L, Dufault MR, Huang Y, Liang B, Gans JD, Zhang M, Carter K, Gladysheva TB, Teicher BA, Biemann HP, Booker M, Goldberg MA, Klinger KW, Lillie J, Madden SL, Jiang Y. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Exp Cell Res. 2010; 316: 258 -271. [PubMed] .
- 14. Wagner M, Hampel B, Hütter E, Pfister G, Krek W, Zwerschke W, Jansen-Dürr P. Metabolic stabilization of p27 in senescent fibroblasts correlates with reduced expression of the F-box protein Skp2. Exp Gerontol. 2001; 37: 41 -55. [PubMed] .
- 15. Kullmann M, Göpfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 2002; 16: 3087 -3099. [PubMed] .
- 16. Zheng Y and Miskimins WK. CUG-binding protein represses translation of p27Kip1 mRNA through its internal ribosomal entry site. RNA Biol. 2011; 8: 365 -371. [PubMed] .
- 17. Le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafrè SA, Farace MG, Agami R. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007; 26: 3699 -3708. [PubMed] .
- 18. Martínez-Sánchez A and Gebauer F. Regulation of p27(kip1) mRNA expression by microRNAs. Prog Mol Subcell Biol. 2010; 50: 59 -70. [PubMed] .
- 19. Das G, Thotala DK, Kapoor S, Karunanithi S, Thakur SS, Singh NS, Varshney U. Role of 16S ribosomal RNA methylations in translation initiation in Escherichia coli. EMBO J. 2008; 27: 840 -851. [PubMed] .
- 20. Seshadri A, Dubey B, Weber MH, Varshney U. Impact of rRNA methylations on ribosome recycling and fidelity of initiation in Escherichia coli. Mol Microbiol. 2009; 72: 795 -808. [PubMed] .
- 21. Ji L and Chen X. Regulation of small RNA stability: methylation and beyond. Cell Res. 2005; 22: 624 -636. [PubMed] .
- 22. Schaefer M, Pollex T, Hanna K, Tuorto F, Meusburger M, Helm M, Lyko F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010; 24: 1590 -1595. [PubMed] .
- 23. Hussain S, Sajini AA, Blanco S, Dietmann S, Lombard P, Sugimoto Y, Paramor M, Gleeson JG, Odom DT, Ule J, Frye M. NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines Its Processing into Regulatory Small RNAs. Cell Rep. 2013; doi:pii: S2211-1247(13)00321-00325 .
- 24. Kimura S and Suzuki T. Fine-tuning of the ribosomal decoding center by conserved methyl-modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res. 2010; 38: 1341 -1352. [PubMed] .
- 25. Frye M and Watt FM. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr Biol. 2006; 16: 971 -981. [PubMed] .
- 26. Yuan S, Tang H, Xing J, Fan X, Cai X, Li Q, Han P, Luo Y, Zhang Z, Jiang B, Dou Y, Gorospe M, Wang W. Methylation by NSun2 represses the levels and function of miR-125b. Mol Cell Biol. 2014; 34: 3630 -3641. [PubMed] .
- 27. Zhang X, Liu Z, Yi J, Tang H, Xing J, Yu M, Tong T, Shang Y, Gorospe M, Wang W. NSun2 stabilizes p16INK4 mRNA by methylating the p16′UTR. Nature Comms. 2012; 3: 712 https://doi.org/10.1038/ncomms1692 .
- 28. Xing J, Yi J, Cai X, Tang H, Liu Z, Zhang X, Martindale JL, Yang X, Jiang B, Myriam Gorospe M, Wang W. NSun2 Promotes Cell Growth via Elevating CDK1 Translation. Mol Cell Biol. 2015; In press .
- 29. Wang W, Yang X, Cristofalo V, Holbrook N, Gorospe M. Loss of HuR is Linked to Reduced Expression of Proliferative Genes during Replicative Senescence. Mol Cell Biol. 2001; 21: 5889 -5898. [PubMed] .
- 30. Chang N, Yi J, Guo G, Liu X, Shang Y, Tong T, Cui Q, Zhan M, Gorospe M, Wang W. HuR uses AUF1 as a cofactor to promote p16INK4 mRNA decay. Mol. Cell. Biol. 2010; 30: 3875 -86. [PubMed] .
- 31. Iakova P, Wang GL, Timchenko L, Michalak M, Pereira-Smith OM, Smith JR, Timchenko NA. Competition of CUGBP1 and calreticulin for the regulation of p21 translation determines cell fate. EMBO J. 2004; 23: 406 -417. [PubMed] .
- 32. Quadri RA, Arbogast A, Phelouzat MA, Boutet S, Plastre O, Proust JJ. Age-associated decline in cdk1 activity delays cell cycle progression of human T lymphocytes. J Immunol. 1998; 161: 5203 -5209. [PubMed] .
- 33. Fang Y, Yu H, Liang X, Xu J, Cai X. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol Ther. 2014; 15: 1268 -1279. [PubMed] .
- 34. Hedblom A, Laursen KB, Miftakhova R, Sarwar M, Anagnostaki L, Bredberg A, Mongan NP, Gudas LJ, Persson JL. CDK1 interacts with RARgamma and plays an important role in treatment response of acute myeloid leukemia. Cell Cycle. 2013; 12: 1251 -1266. [PubMed] .
- 35. Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008; 14: 159 -169. [PubMed] .
- 36. Blain SW, Scher HI, Cordon-Cardo C, Koff A. p27 as a target for cancer therapeutics. Cancer Cell. 2003; 3: 111 -5. [PubMed] .
- 37. Slingerland J and Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol. 2000; 183: 10 -17. [PubMed] .
- 38. Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, Castellani RJ, Perry G, Smith MA. Increased p27, an essential component of cell cycle control, in Alzheimer's disease. Aging Cell. 2003; 2: 105 -110. [PubMed] .
- 39. Smith E, Redman RA, Logg CR, Coetzee GA, Kasahara N, Frenkel B. Glucocorticoids inhibit developmental stage-specific osteoblast cell cycle. Dissociation of cyclin A-cyclin-dependent kinase 2 from E2F4-p130 complexes. J Biol Chem. 2000; 275: 19992 -20001. [PubMed] .
- 40. Shahzad K, Thati M, Wang H, Kashif M, Wolter J, Ranjan S, He T, Zhou Q, Blessing E, Bierhaus A, Nawroth PP, Isermann B. Minocycline reduces plaque size in diet induced atherosclerosis via p27(Kip1). Atherosclerosis. 2011; 219: 74 -83. [PubMed] .
- 41. Díez-Juan A, Pérez P, Aracil M, Sancho D, Bernad A, Sánchez-Madrid F, Andrés V. Selective inactivation of p27(Kip1) in hematopoietic progenitor cells increases neointimal macrophage proliferation and accelerates atherosclerosis. Blood. 2004; 103: 158 -161. [PubMed] .
- 42. Xu F, Pang L, Cai X, Liu X, Yuan S, Fan X, Jiang B, Zhang X, Dou Y, Gorospe M, Wang W. let-7-repressesed Shc translation delays replicative senescence. Aging Cell. 2014; 13: 185 -192. [PubMed] .