



Reactive oxygen species
ROS are a group of compounds derived from the incomplete reduction of molecular oxygen. They include—but are not limited to—the superoxide anion (O2*−), hydrogen peroxide (H2O2), and the hydroxyl radical (OH*). While ROS are generated through a variety of mechanisms, their primary physiological source is cellular respiration. During respiration, electrons are passed through four protein complexes (Complex I, II, III, and IV) that reside in the mitochondrial inner membrane. The majority of electrons proceed through each complex and ultimately to molecular oxygen, which is reduced to water. However, a small percentage of electrons can escape the electron transport chain prematurely, leading to incomplete reduction of molecular oxygen and formation of the superoxide anion O2*−[1]. The anionic nature of O2*− restricts both its ability to diffuse throughout the cell and its reactivity toward electron-rich substrates. O2*− primarily reacts with and inactivates enzymes that contain Fe-S clusters, such as dehydratases and aconitase [2]. O2*− is detoxified by dismutation into H2O2 and O2 in a process that occurs spontaneously but is rapidly accelerated by superoxide dismutase (SOD) [3].
Other ROS include hydrogen peroxide (H2O2) and the hydroxyl radical (OH*). While H2O2 can rapidly diffuse throughout the cell, its reactivity is restricted to proteins containing transition metals (such as Fe-S clusters) or low pKa thiols [4]. The transition metal-catalyzed reduction of H2O2 to OH*, however, is highly reactive. OH* displays the broadest reactivity and indiscriminately oxidizes lipids, nucleic acids, and amino acids (Figure 1).
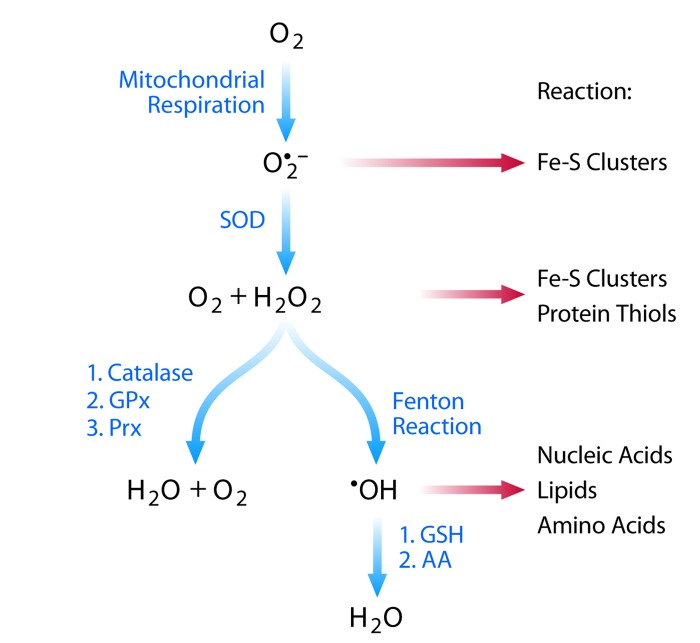
Figure 1. Reactive oxygen species: production and protection Schematic showing the major reactive oxygen species associated with cellular respiration. Blue arrows indicate detoxification mechanisms while red arrows indicate reactivity for each ROS.
Cells have numerous enzymatic mechanisms to detoxify H2O2, including catalase, glutathione peroxidase, and the peroxiredoxins, which reduce H2O2 to H2O and O2. OH* is primarily detoxified by small-molecule reductants, such as glutathione and ascorbate. When these detoxification mechanisms are insufficient to neutralize the ROS, cellular macromolecules may become oxidatively damaged, a state defined as oxidative stress.
It has long been appreciated that oxidative damage increases during aging and that caloric restriction—which has been postulated to mitigate such damage—can increase the lifespan of several model organisms [5]. While the mechanisms governing caloric restriction-mediated lifespan extension and oxidative stress resistance remain incompletely understood, the sirtuin family of proteins has attracted considerable attention for regulating these phenotypes. Here, we discuss data linking the sirtuins to the oxidative stress response, caloric restriction, and longevity.
The sirtuins
Sir2 (silent information regulator 2) from Sacchromyces cerevisiae and the conserved mammalian orthologs, collectively called sirtuins, are NAD+-dependent histone/protein deacetylases. Sirtuins catalyze the removal of acetyl groups from the side chain amino group of lysine residues. This reaction consumes NAD+ and generates nicotinamide (NAM) and 2’-O-acetyl-ADP-ribose [6]. Seven sirtuin members have been identified in mammals, named numerically as SIRT1-7. They have distinct subcellular localizations: SIRT1, 6, and 7 are found in the nucleus, SIRT2 is cytosolic, and SIRT3, 4, and 5 are primarily located in the mitochondria [7]. While SIRT1, 2, and 3 have robust deacetylase activities, the other sirtuins exhibit weak or no deacetylase activity. Recently, SIRT5 was found to catalyze the removal of malonyl and succinyl groups from lysines suggesting that the remaining sirtuins may target additional modes of lysine acylation [8, 9].
Sir2 and oxidative stress
Much of the early evidence linking the sirtuins to oxidative stress was obtained by studying the effect of genetic manipulation of Sir2 on aging in eukaryotic model organisms such as S. cerevisiae, C. elegans, and D. melanogaster. Disruption of the SIR2 gene severely shortens lifespan in Saccharomyces cerevisiae, while its overexpression increases lifespan beyond that for wild-type cells [10]. Likewise, overexpression of Sir-2.1, the homolog of yeast Sir2, increases lifespan in C. elegans [11] and Drosophila [12]. Intriguingly, the lifespan extension observed during caloric restriction in yeast is abrogated when Sir2 is deleted begging the question that Sir2 may influence lifespan by diminishing oxidative stress [13]. In support of this notion, overexpression of Sir2 rescues the shortened lifespan phenotype observed when S. cerevisiae are treated with hydrogen peroxide [14]. Moreover, Sir2 regulates the asymmetric segregation of oxidatively damaged proteins from daughter cells to mother cells during cell division in yeast providing a putative mechanistic basis for Sir2's role in oxidative stress resistance and lifespan extension [15]. The role of Sir2 in mediating lifespan extension, however, has been recently called into question with new experiments demonstrating that Sir2 overexpression does not enhance longevity in C. elegans or Drosophila[16]. Clearly, more work is required to resolve these conflicting data and to determine the relative importance of sirtuins for longevity in these metazoans.
The mammalian sirtuins and oxidative stress
The two closest mammalian homologs of yeast Sir2 are the nuclear-localized SIRT1 and the mitochondrion-localized SIRT3 (see Figure 2 for an overview). Strong evidence supports a role for SIRT1 mediating an oxidative stress response by directly deacetylating several transcription factors that regulate antioxidant genes. Notably, SIRT1 activates several members of the FOXO family of transcription factors which promote the expression of stress response genes including SOD2 [17-19]. For example, SIRT1 functions in an autoregulatory loop along with the early growth response protein ERG1 to regulate SOD2 to protect contracting muscle cells from oxidative stress [20, 21].
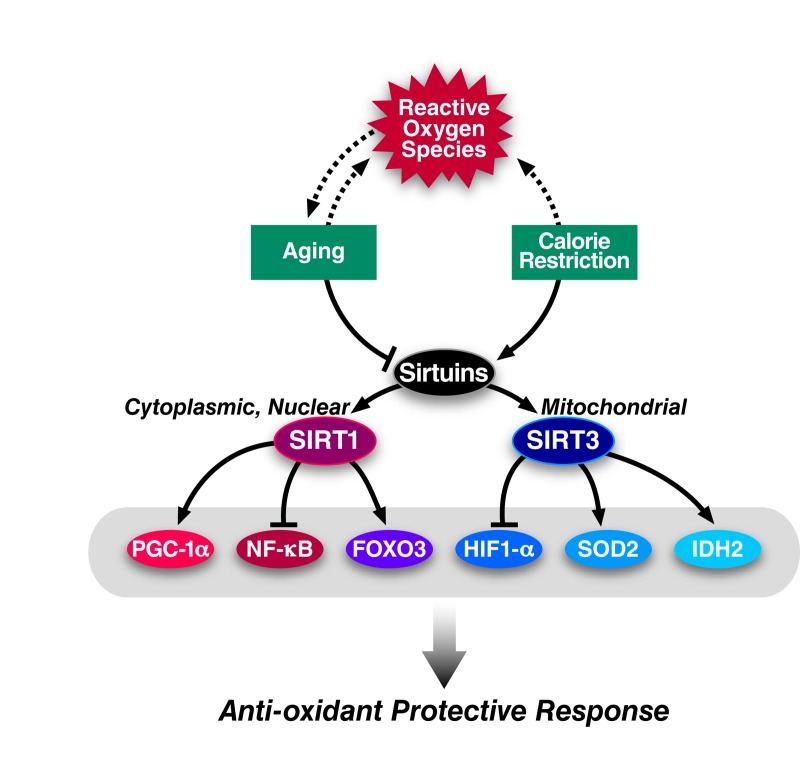
Figure 2. Regulation of ROS by sirtuins Diagram showing how ROS, aging, and caloric restriction interact to influence the activity of the cytoplasmic/nuclear sirtuin, SIRT1, and the mitochondrial sirtuin, SIRT3. Upon activation, SIRT1 and SIRT3 deacetylate several proteins that promote resistance to oxidative stress. Arrows indicate positive regulation while hash-marks indicate negative regulation. Solid lines reflect robust experiment evidence for an interaction while dashed lines indicate putative interactions.
SIRT1 also promotes mitochondrial biogenesis by activating peroxisome proliferator-activated receptor co-activator 1-α (PGC-1α) [22]. PGC-1α increases mitochondrial mass and upregulates the expression of oxidative stress genes including glutathione peroxidase (GPx1), catalase, and manganese SOD (MnSOD) [23]. Finally, SIRT1 inactivates the p65 subunit of NF-ĸB through direct deacetylation. NF-ĸB inhibition suppresses the inducible nitric oxide synthase (iNOS) and nitrous oxide production and thus may lower the cellular ROS load [24]. Given its role in antioxidant response, whether SIRT1 activation contributes to CR mediated lifespan extension has been extensively studied. CR fails to increase the lifespan of SIRT1 knock-out mice, and these mice do not increase their physical activity, a phenotype typically associated with calorically-restricted mice [25, 26]. Similarly, SIRT1 overexpression mimics a caloric restriction phenotype [27]. Precisely how SIRT1 functions during CR remains an open question, but emerging evidence suggests that p53 plays an important role in modulating SIRT1 during CR [28].
Mitochondria account for the majority of cellular ROS production. Mitochondrial SIRT3 deacetylates and activates several enzymes that are critical in maintaining cellular ROS levels. SIRT3 deacetylates SOD2 at two important lysine residues to boost its catalytic activity and the catalytic activity of SOD2 is diminished when SIRT3 is deleted [29]. SIRT3 knock-out mice fail to reduce their levels of lipid peroxidation and protein carbonylation that are typically observed during caloric restriction indicating that SIRT3 is necessary for caloric restriction to mitigate oxidative stress. Additionally, SIRT3 stimulates the activity of mitochondrial isocitrate dehydrogenase, IDH2, during caloric restriction through direct deacetylation [30]. IDH2 promotes the conversation of NADP+ to NADPH which in turn provides the reducing equivalents for conversion of oxidized to reduced glutathione. In support of this biochemical data, SIRT3 is required to protect calorically-restricted mice from age-associated hearing loss [30, 31].
Another link between SIRT3 and oxidative stress comes from the field of oncology. Since ROS can severely damage nucleic acids, it is not surprising that oxidative stress can promote tumorigenesis. SIRT3 knock-out mouse embryonic fibroblasts (MEFs) exhibit higher ROS levels, greater genomic instability, and increased sensitivity to oncogenic transformation compared to wild-type fibroblasts (Kim et al, 2010). Intriguingly, overexpression of SOD2 suppresses oncogenic transformation in SIRT3 knock-out MEFs suggesting that SIRT3 may protect against tumorigenesis through an oxidative stress mechanism. In support of the above in vitro data, mice deficient for SIRT3 are more susceptible to cancer and many human tumors display reduced SIRT3 levels compared to healthy tissues [32]. In addition to suppressing the formation of cancer, SIRT3 can also combat established tumors. Overexpression of SIRT3 suppresses tumor proliferation via inhibiting the activity of the hypoxia inducible factor-1α (HIF-1α) [33, 34]. Mechanistically, the HIF-1α protein is stabilized by the presence of ROS and activates a gene expression program that enhances survival and growth in hypoxic environments, as typically found in solid tumors. Given HIF-1α is activated via a ROS-mediated mechanism it is likely that SIRT3 decreases HIF-1α activity by suppression of ROS levels.
Other sirtuins
Besides SIRT1 and SIRT3, other sirtuins also contribute to the cellular response to oxidative stress. SIRT2 deacetylates FOXO3a and promotes cellular resistance to H2O2[35], similar to SIRT1 regulation of oxidative stress via FOXO family members. SIRT6 deficient cells display sensitivity to oxidative stress and a reduced capacity for DNA repair, while SIRT6 knockout mice show many hallmarks of premature aging [36]. In support of the anti-aging effects of SIRT6, male mice overexpressing SIRT6 have a significantly longer lifespan than their wild-type counterparts [37]. Mechanistically, SIRT6 mono-ADP ribosylates poly (ADP-ribose) polymerase 1 (PARP1) to stimulate DNA double-strand break (DSB) repair in response to oxidative stress [38, 39]. Mutations in several DSB repair genes are often associated with premature aging which could explain in part why SIRT6 mutants also have many characteristics of premature aging [40]. Lastly, SIRT7 has been implicated in oxidative stress resistance through an investigation of primary cardiomyocytes from SIRT7 knock-out mice. These cells are increasingly sensitive to both genotoxic and oxidative insults such as doxorubicin and H2O2 compared to wild-type. [41]. Together these studies reflect the importance of the sirtuin family in oxidative stress and will hopefully spur future studies to carefully decipher their mechanisms.
Mitohormesis and mitochondrial hyperacetylation
In 1956, Harman proposed that oxidative stress may cause the observed physiological decline in cellular and organismal functions that occur during aging [42]. This theory has since been supported by studies showing that cellular oxidative damage increases with aging and that overexpression of some anti-oxidant genes increase lifespan in Drosophila[5]. However, transgenic mice over-expressing related anti-oxidant genes do not live longer than their wild-type counterparts raising some doubts on the universal relevance of this theory as a mechanism of aging[43]. These conflicting data suggest a more complex mode of regulation.
Mitohormesis may reconcile many of the seemingly conflicting data relating to the role of oxidative stress and health. First coined by Ristow and colleagues, mitohormesis is an application of the theory of hormesis in which a stressor may have beneficial effects at relatively low doses and deleterious effects at high doses [44]. Conceptually, small and/or transient amounts of reactive oxygen species elicit a protective stress response that may improve lifespan. Relatively large and/or chronic amounts of the same species, however, cause cellular damage or death because they exceed the capacity of the oxidative stress response to maintain homeostasis (Figure 3). In support of this notion, low-doses of paraquat, which induce O2-* formation, increase lifespan in Caenorhabditis elegans [45], while high-doses of paraquat reduce lifespan [46]. Mitohormesis may also explain how caloric restriction extends lifespan. Caloric restriction was originally thought to decrease the production of ROS by dampening mitochondrial respiration. However, mitochondrial respiration actually increases during caloric restriction in yeast, worms, and mice which may in turn increase ROS associated with respiration [47-49]. A moderate ROS increase during caloric restriction can stimulate oxidative stress resistance mechanisms which can minimize oxidative cellular damage over time, delay age-associated phenotypes, and extend lifespan [44, 50]. While attractive, the mechanistic link between metabolic stressors such as caloric restriction and mitohormesis remain unresolved.
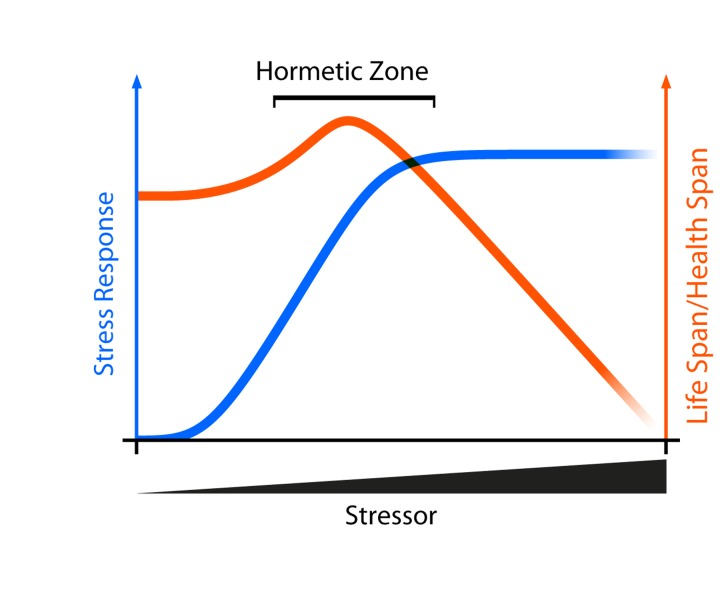
Figure 3. Aging and Mitohormesis Theoretical curve showing how low doses of a stressor may have beneficial effects by activating intracellular stress response pathways. If the stressor exceeds the capacity of the stress response system to maintain homeostasis, then deleterious phenotypes are observed.
It is noteworthy that during caloric restriction, global mitochondrial protein acetylation increases in metabolically active tissue such as liver [51]. In fact, several physiological stresses including fasting and chronic high-fat feeding are associated with mitochondrial hyperacetylation [52, 53]. We speculate here that these stresses may dampen the activity of enzymes that protect against oxidative stress, such as SOD2 and IDH2, via direct acetylation [29, 54, 55]. If true, the subsequent ROS increase would promote gene expression to boost oxidative stress defense thus achieving a new hormetic steady state. In support of this notion, caloric restriction, fasting, and high-fat feeding initially spur an increase in SIRT3 transcription. Increased levels of SIRT3 could then deacetylate SOD2, IDH2, and several other mitochondrial proteins involved in redox regulation to minimize chronic ROS production. Interestingly, this response, while initially appropriate on a short-term high fat diet, is lost during chronic high fat diet feeding (>10 weeks) and associated with a downregulation of PGC-1α[53, 56]. As discussed above, the transcriptional increase of genes involved in mitochondrial energy metabolism and ROS detoxification during calorie restriction is strongly correlated with elevated expression of the transcriptional coactivator PGC-1α. This stress response may be sufficient to counter the mild stress of caloric restriction, but incapable of combating chronic high-fat feeding. When PGC-1α expression decreases, SIRT3 expression ultimately declines, oxidative stress worsens, and the fitness of the organism is diminished (Figure 4).
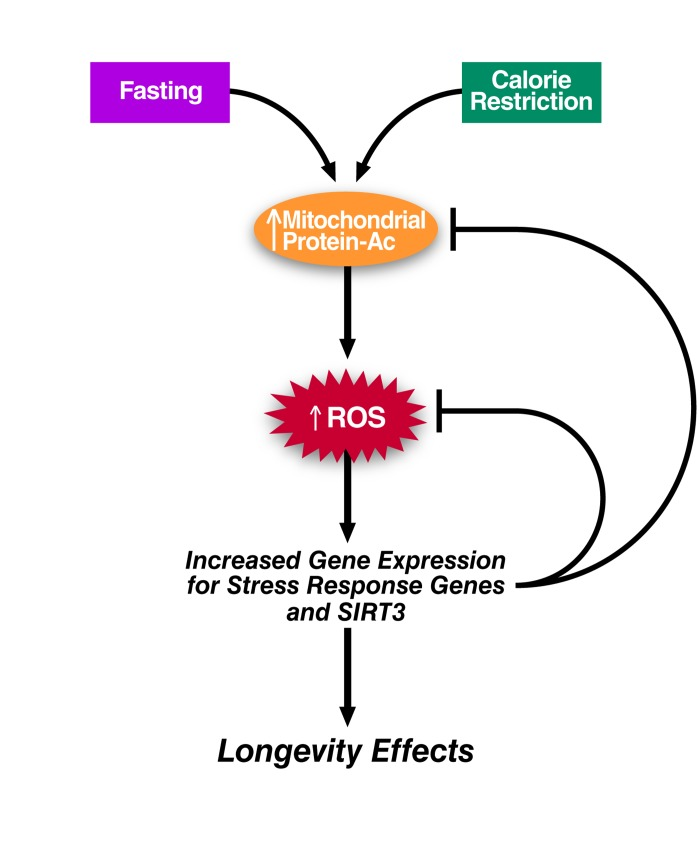
Figure 4. Hyperacetylation and Mitohormesis Theoretical model linking mitochondrial hyperacetylation to the generation of ROS and mitohormesis.
Perspective
While considerable more investigations are required, a causative role for oxidative stress in aging remains one of the most solid aging theories. The studies addressed in this review have demonstrated that sirtuins are intimately linked to the cellular response to oxidative stress. Moving forward, it will be important to develop experimental models in which the levels of oxidative stress and the activities of sirtuins can be precisely modulated to determine if sirtuins have a causative role in lifespan extension. Does mitochondrial sirtuin overexpression in mice extend lifespan? Do long-lived animals exhibit chronic low levels of oxidative stress? From a more practical standpoint, is it possible to rejuvenate tissue function by targeted overexpression of sirtuins to reduce oxidative stress? We look forward to future studies that will undoubtedly address many of these important questions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009; 417: 1 -13. [PubMed] .
- 2. Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008; 4: 278 -286. [PubMed] .
- 3. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003; 552: 335 -344. [PubMed] .
- 4. D'Autreaux B and Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007; 8: 813 -824. [PubMed] .
- 5. Sohal RS and Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996; 273: 59 -63. [PubMed] .
- 6. Blander G and Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004; 73: 417 -435. [PubMed] .
- 7. Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010; 35: 669 -675. [PubMed] .
- 8. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011; 334: 806 -809. [PubMed] .
- 9. Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, Zwaans BM, Tishkoff D, Ho L, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011; 10: M111 -012658. [PubMed] .
- 10. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999; 13: 2570 -2580. [PubMed] .
- 11. Tissenbaum HA and Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001; 410: 227 -230. [PubMed] .
- 12. Rogina B and Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004; 101: 15998 -16003. [PubMed] .
- 13. Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000; 289: 2126 -2128. [PubMed] .
- 14. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, Wright SM, Mills KD, Bonni A, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008; 135: 907 -918. [PubMed] .
- 15. Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003; 299: 1751 -1753. [PubMed] .
- 16. Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvari M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ, Vazquez-Manrique RP, Orfila AM, Ackerman D, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011; 477: 482 -485. [PubMed] .
- 17. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004; 303: 2011 -2015. [PubMed] .
- 18. Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004; 116: 551 -563. [PubMed] .
- 19. van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J Biol Chem. 2004; 279: 28873 -28879. [PubMed] .
- 20. Pardo PS and Boriek AM. An autoregulatory loop reverts the mechanosensitive Sirt1 induction by EGR1 in skeletal muscle cells. Aging (Albany NY). 2012; 4: 456 -461. [PubMed] .
- 21. Pardo PS, Mohamed JS, Lopez MA, Boriek AM. Induction of Sirt1 by mechanical stretch of skeletal muscle through the early response factor EGR1 triggers an antioxidative response. J Biol Chem. 2011; 286: 2559 -2566. [PubMed] .
- 22. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005; 434: 113 -118. [PubMed] .
- 23. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006; 127: 397 -408. [PubMed] .
- 24. Lee JH, Song MY, Song EK, Kim EK, Moon WS, Han MK, Park JW, Kwon KB, Park BH. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes. 2009; 58: 344 -351. [PubMed] .
- 25. Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, Xuan J, Evans M, Harper ME, et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One. 2008; 3: e1759 [PubMed] .
- 26. Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005; 310: 1641 [PubMed] .
- 27. Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007; 6: 759 -767. [PubMed] .
- 28. Tucci P. Caloric restriction: is mammalian life extension linked to p53? Aging (Albany NY). 2012; 4: 525 -534. [PubMed] .
- 29. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010; 12: 662 -667. [PubMed] .
- 30. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010; 143: 802 -812. [PubMed] .
- 31. Zhang D, Liu Y, Chen D. SIRT-ain relief from age-inducing stress. Aging (Albany NY). 2011; 3158 -161. .
- 32. Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010; 17: 41 -52. [PubMed] .
- 33. Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011; 30: 2986 -2996. [PubMed] .
- 34. Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011; 19: 416 -428. [PubMed] .
- 35. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007; 6: 505 -514. [PubMed] .
- 36. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006; 124: 315 -329. [PubMed] .
- 37. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012; 483: 218 -221. [PubMed] .
- 38. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011; 332: 1443 -1446. [PubMed] .
- 39. Van Meter M, Mao Z, Gorbunova V, Seluanov A. Repairing split ends: SIRT6, mono-ADP ribosylation and DNA repair. Aging (Albany NY). 2011; 3: 829 -835. [PubMed] .
- 40. O'Driscoll M and Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006; 7: 45 -54. [PubMed] .
- 41. Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008; 102: 703 -710. [PubMed] .
- 42. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956; 11: 298 -300. [PubMed] .
- 43. Perez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009; 8: 73 -75. [PubMed] .
- 44. Ristow M and Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol. 2010; 45410 -418. .
- 45. Yang W and Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010; 8: e1000556 [PubMed] .
- 46. Ishii N, Takahashi K, Tomita S, Keino T, Honda S, Yoshino K, Suzuki K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat Res. 1990; 237: 165 -171. [PubMed] .
- 47. Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002; 418: 344 -348. [PubMed] .
- 48. Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005; 310: 314 -317. [PubMed] .
- 49. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6: 280 -293. [PubMed] .
- 50. Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005; 126: 987 -1002. [PubMed] .
- 51. Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, Lombard DB. Calorie restriction alters mitochondrial protein acetylation. Aging Cell. 2009; 8: 604 -606. [PubMed] .
- 52. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010; 464: 121 -125. [PubMed] .
- 53. Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011; 44: 177 -190. [PubMed] .
- 54. Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. 2012; 287: 14078 -14086. [PubMed] .
- 55. Ozden O, Park SH, Kim HS, Jiang H, Coleman MC, Spitz DR, Gius D. Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging (Albany NY). 2011; 3: 102 -107. [PubMed] .
- 56. Choudhury M, Jonscher KR, Friedman JE. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging (Albany NY). 2011; 3: 175 -178. [PubMed] .