



Introduction
DNA methylation is widespread across the genomes of different organisms and in mammals usually consists in the enzymatic addition of a methyl group to the carbon-5 of cytosine ring of a CpG dinucleotide. Through the recruitment of methyl-binding proteins, this modification induces an inactive chromatin structure that represses transcription. While the bulk of human genome is generally methylated, the promoters of around 40% of genes contain CpG-rich regions, termed CpG islands, whose methylation status is strictly regulated during development and cellular differentiation [1-3]. Deregulation in methylation patterns can lead to disease onset. More recently it has been shown that tissue- and disease-specific differentially methylated regions (DMR) are more frequent in CpG island shores rather than in CpG islands [4,5].
Although DNA methylation is generally considered a stable modification, it is well established that methylation profiles vary in pathological conditions, and it is also accepted that external factors, such as diet, age, toxins and lifestyle, can induce quantitative hypo- or hyper- DNA methylation [6-13]. Only few studies have focused the effect of such factors on inter-individual variability of DNA methylation patterns among human populations and subjects [14-17]. Regarding time/age, variations in methylation levels of the IGF2/H19 locus were observed in a cohort of newborn twins, both between individuals and within twin pairs [17]. In this study, dizygotic twins (DZ) resulted more discordant than monozygotic twins (MZ), suggesting that a heritable component can affects the epigenetic status of this locus. Moreover, Heijmans and colleagues [16] analyzed the variations in DNA methylation of the same IGF2/H19 locus in a cohort of adolescent and middle-aged twins (13-62 years of age) and demonstrated that a substantial part of the variation observed across individuals was ascribable to heritable factors and single nucleotide polymorphisms (SNPs) in cis, rather than to the cumulative effect of environmental and stochastic factors occurring with age. Although this study involved a high number of twins (N=372), it did not include old twin pairs, and therefore could not appreciate the possible epigenetic variability of the locus occurring later in life (after sixties).
Regarding space/population, only few studies addressed the DNA methylation variability at the population level (population epigenetics) [18], despite its great potential interest owing to the interaction of both environmental and genetic variables in determining the DNA methylation architecture. To our knowledge, the only two available studies have investigated DNA methylation in multiethnic cohorts of healthy women [19] and of men affected by prostate cancer [20].
To better understand the intricate relationship between spatial (geography/ancestry) and temporal (age of the individuals) dimensions on DNA methylation variability, we took advantage of two ad hoc models: i) Cohort 1, constituted of 376 individuals homogeneous for gender (males) and age (age range 30-51), but differing for ancestral geographical origin and place of living, i.e four Italian regions (Northern, Central and Southern Italy, and Sardinia); ii) Cohort 2, constituted of 31 monozygotic (MZ) and 16 dizygotic (DZ) twin couples of different ages, spanning from 22 to 97 years, homogeneous for geographic origin (Northern Italy, Bologna area). Both the models were analyzed for variations in DNA methylation status of 4 target regions in the imprinted IGF2/H19 locus, previously analyzed by other groups [16,17].
Results
Characterization of the IGF2/H19 target regions
Four target regions (amplicons) in the IGF2/H19 locus were analyzed: IGF2AS, 3 kb from a CpG island, in exon 3 of IGF2AS transcript, within the IGF2 DMR 0; H19, upstream the transcription start site of H19 gene, partially overlapping a CpG island (H19 DMR); IGF2_island, within a CpG island in the last exon of IGF2 gene (DMR 2); IGF2_shore, in the shore upstream the island targeted by the IGF2_island amplicon (Figure 1A). IGF2AS and H19 have been previously analyzed by Heijmans et al. [16] and Ollikainen et al. [17], while IGF2_island and IGF2_shore were analyzed here for the first time.
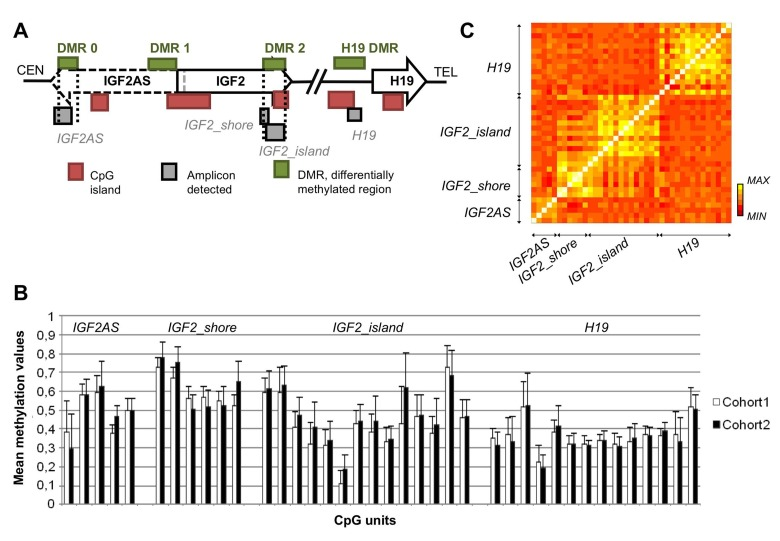
Figure 1. CpGs spatial localization and their methylation patterns (A) Spatial disposition of the amplicon analyzed in relation to functional elements. (B) Mean methylation level with the corresponding standard deviation for each CpGs considering the whole dataset. (C) Correlation matrix of CpG sites of IGF2_shore, IGF2AS, H19 and IGF2_island amplicons.
Mean methylation levels and inter-individual variability of IGF2/H19 locus
Figure 1B reports mean methylation values and their standard deviations for the 4 target regions considering cohort 1 and 2 separately. For cohort 1 the mean DNA methylation levels observed in IGF2AS, IGF2_shore, IGF2_island and H19 were 49%, 60%, 43% and 36% respectively. These values are similar for cohort 2, where the mean methylation values in IGF2AS, IGF2_shore, IGF2_island and H19 were 49%, 62%, 47% and 36%. Notably IGF2AS and H19 DNA methylation values were comparable to those previously reported in literature [16,17].
Considerable inter-individual variation in DNA methylation values was observed within each CpG unit. Within a single amplicon, DNA methylation values of the CpG units were strongly correlated with each others, while correlation values were lower between CpGs belonging to different amplicons (Figure 1C). Intermediate correlation levels were observed only between IGF2_island and IGF2_shore, as expected because of their adjacent chromosomal position.
Space/ancestry: geographical dimension
The Italian cohort comprises 376 middle-aged males (age range: 30-51 years) representative of the four main Italian geographical areas: Northern Italy (141 samples), Central Italy (94 samples), Southern Italy (121 samples) and Sardinia (20 samples) (Figure 2A). We evaluated if the observed variability in methylation levels of the IGF2/H19locus could be explained by ancestry and place of living of the four populations studied.
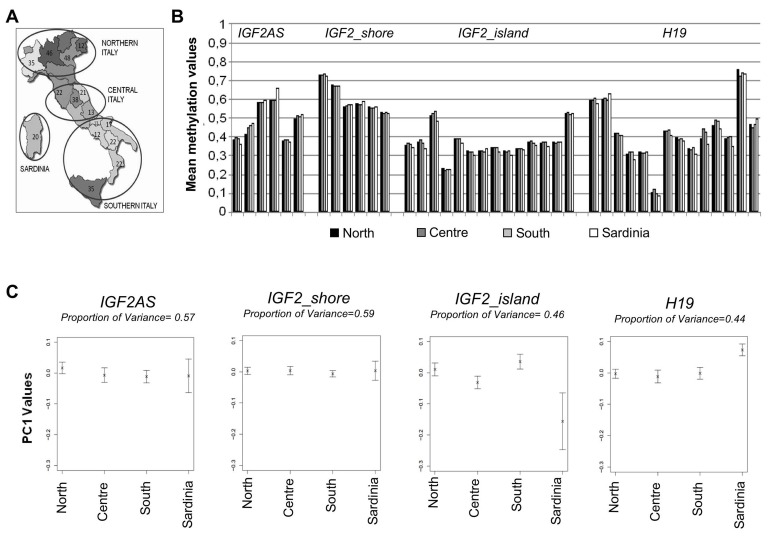
Figure 2. Methylation variation in cohort 1 (A) Number of samples from different Italian districts and corresponding groups made for the statistical analysis. (B) Mean methylation levels of each CpGs unit in the 4 macroarea. (C) PC1 values plotted for each macro-area and the corresponding percentage of variance.
Mean methylation levels of each CpG unit were calculated for the 4 geographical areas (Figure 2B). The results showed that the methylation patterns were comparable throughout all the samples. No evident differences were detectable according to sample ancestry/geography, except for few CpGs (i.e. CpG 20 of IGF2_island amplicon).
Principal component analysis (PCA) was used to capture the most salient patterns of variation in each amplicon for the 4 geographical areas. The percentages of variance explained by principal component 1 (PC1) values are reported in Figure 2C. No statistically significant difference was observed between PC1 values, as confirmed by analysis of variance (ANOVA) (Table 1).
Table 1. p_values from ANOVA on PC1 values calculated based on DNA methylation levels in samples belonging to different Italian districts
| ANOVA – p_values | |
|---|---|
| IGF2_shore | 0.93 |
| IGF2_island | 0.45 |
| IGF2AS | 0.80 |
| H19 | 0.44 |
Time/age: temporal dimension
The twins cohort (cohort 2) includes 94 twins (62 MZ and 32 DZ) with age ranging from 22 to 97 years all recruited in the area of Bologna (Emilia Romagna, Italy). Cohort 2 was divided into four age classes (20-45, 46-60, 61-75, 76-97) and for each amplicon, mean methylation value and mean standard deviation were calculated (Figure 3). No significant variations in mean methylation levels were observed. In the first 2 age classes IGF2_shore was characterized by a minor range of variation if compared to the other amplicons. Moreover, while mean standard deviations in IGF2_island, IGF2AS and H19 were stable over the time, IGF2_shore showed a doubling in their values in the older age classes.
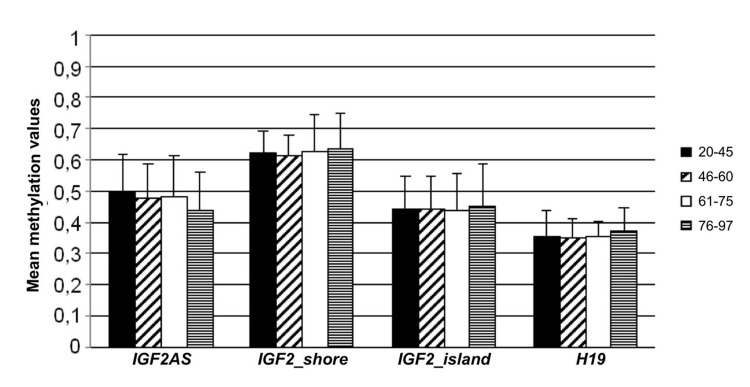
Figure 3. Methylation variation in cohort 2 The twins cohort was divided in 4 age classes (20-45, 46-60, 61-75, 76-97). Mean methylation values and the corresponding standard deviation for the 4 analyzed amplicons (IGF2AS, IGF2_shore, IGF2_island and H19) are reported for each age-class.
In order to estimate DNA methylation variability within the MZ twins couples, Pearson's correlation and Euclidean distance values for each twins pair were calculated. Pearson's correlation was calculated considering methylation values of each CpG from the same amplicon. The correlation values were then plotted against the age of the subjects (Figure 4). H19 was characterized by a high degree of within-twin pairs correlation (Figure 4A) which was maintained up to old ages (Spearman's correlation with age ρ= −0.094). IGF2_island also did not display variations of correlation levels with age (Spearman's correlation with age ρ = −0.105). However, in this case the intra-couple correlation scores were lower, indicating that high levels of variability characterize the CpG island of IGF2 (Figure 4B). Interestingly, both IGF2AS and IGF2_shore showed an age dependent trend (Spearman's correlation ρ = −0.613 and r = −0.651 respectively), with high correlation levels till the age of 50, followed by a progressive decrease of correlation in the couples of older age (Figure 4C-4D). A possible threshold between the age of 56 and 60 years can be envisaged, and after such age limit the values of intra-couple correlation levels are much more scattered.
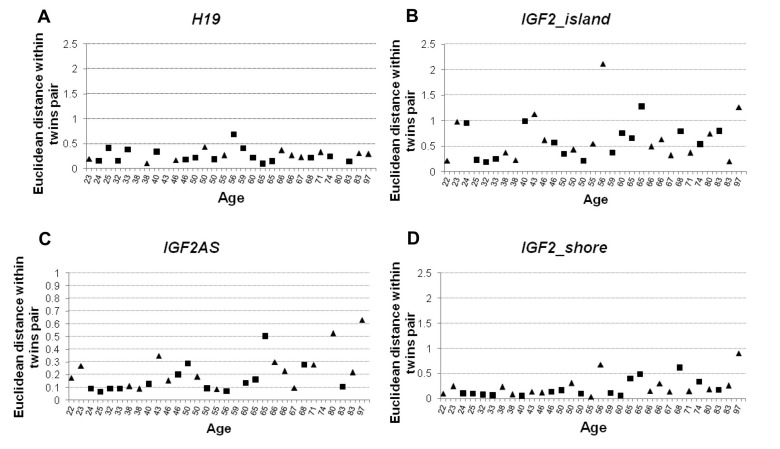
Figure 4. Pearson correlation values calculated within twin pairs in the 4 analyzed amplicons (A) Correlation values within MZ twin couple in H19 amplicon. (B) Correlation values within MZ twin couple in IGF2_island. (C) Correlation values within MZ twin couple in IGF2AS. (D) Correlation values within MZ twin couple in IGF2_shore amplicon. In each figure male individuals are indicated with squares and female using a triangle.
Then, the Euclidean distance between the CpG units of each amplicon. was calculated for each MZ twins pair.
Regarding H19 and IGF2_island the distance values did not show any age-dependent trend (Figure 5A and 5B, Spearman's correlation r = 0.099 r = 0.216 respective-ly). On the contrary, IGF2AS and IGF2_shore (Figure 5C and 5D) showed an age-dependent increase in the intra-couple Euclidean distances (Spearman's correlation ρ = 0.462 and r= 0.553 respectively), with a scatter of the distance values after the age threshold of 60, as reported for Pearson's correlation analysis.
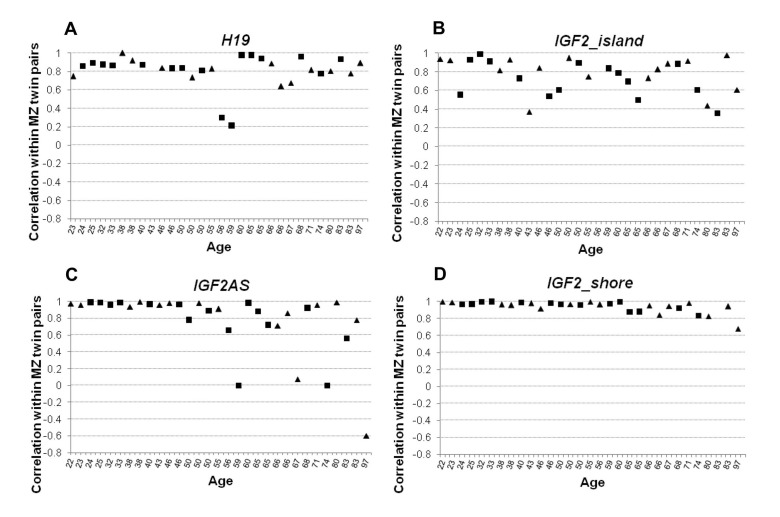
Figure 5. Euclidean distance calculated using DNA methylation values within MZ twin pairs considering the 4 considered amplicons (A) Distance within twin pairs in H19 amplicon. (B) Distance values within twin pairs in IGF2_island amplicon. (C) Distance values within twin pairs in IGF2AS amplicon. (D) Distance values within twin pairs in IGF2_shore amplicon. In each figure male individuals are indicated with squares and female using a triangle.
The data were also subdivided according to sex to exclude an influence on the patterns of correlation and distance values (Figure 4 and Figure 5).
To confirm the previous observations, samples from cohort 2 were divided into 2 groups according to age (below and over 60 years old) and Student t-test on correlation and Euclidean distance values was performed. Significant differences between the 2 groups were found only for IGF2_shore and IGF2AS amplicons (Table 2).
Table 2. Student t-test on correlation and Euclidean distance scores using 60 years old as age threshold
| Amplicon | Age limit | Values considered | p_values |
|---|---|---|---|
| IGF2_shore | 60 | Correlation | 0.00047*** |
| IGF2AS | 60 | Correlation | 0.022* |
| H19 | 60 | Correlation | 0.27 |
| IGF2_island | 60 | Correlation | 0.77 |
| IGF2_shore | 60 | Euclidean distance | 0.0075** |
| IGF2AS | 60 | Euclidean distance | 0.023* |
| H19 | 60 | Euclidean distance | 0.48 |
| IGF2_island | 60 | Euclidean distance | 0.57 |
Evaluation of heritability and environmental influences on DNA methylation
MZ and DZ twins were used to estimate the genetic and environmental influences on DNA methylation of the IGF2/H19 locus.
Firstly for each amplicon intra-couple levels of correlation between MZ and DZ twins were compared. Like MZ, also DZ twins showed high values of correlation in the 4 amplicons (Figure 6), suggesting a strong epigenetic control on this locus. In IGF2AS, H19 and IGF2_island, the intra-couple correlation in MZ was higher compared with that of DZ. Only inIGF2_shore median correlation values were similar among MZ (r=0.94) and DZ (r=0.90) twins. Falconer method confirmed this observation, showing that IGF2_shore is more influenced by environmental factors (h2=0.07) when compared with IGF2AS (h2=0.37), H19 (h2=0.34) and IGF2_island (h2=0.20). As IGF2_shore methylation seems to be more affected by environmental factors, the total lifetime in which the twins did not shared the same environment (i.e. the total lifetime in which they did not live in the same house) was considered. As expected, an increase in percentage of life in which the twins lived separated (S = (years lived in a different house / age)*100) was observed for older couples (Figure 7A).
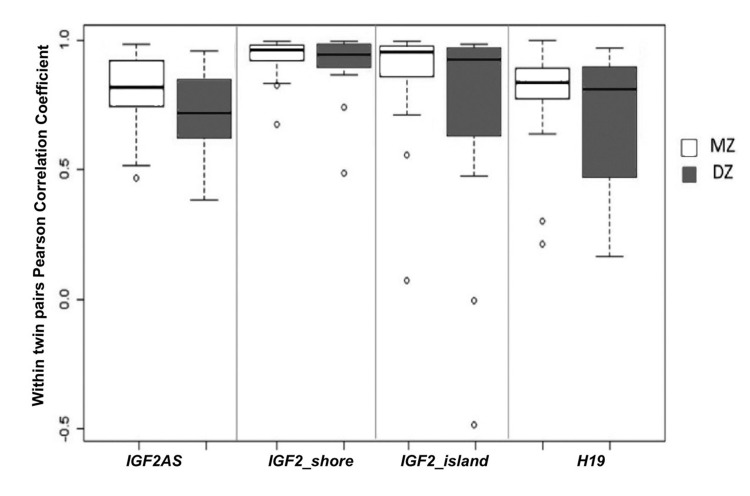
Figure 6. Comparison between intra-couple correlation (ICC) in MZ and DZ twins Boxplot of ICC for each amplicon dived on the basis of zygosity.
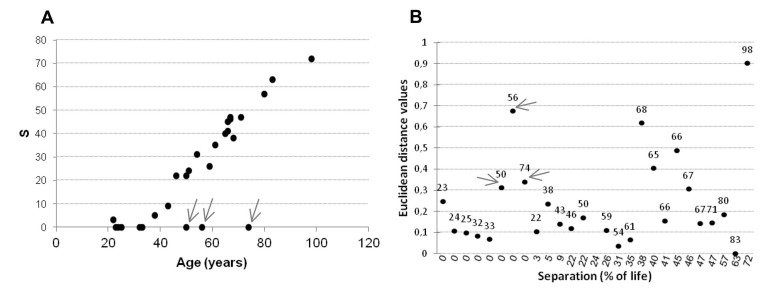
Figure 7. Analysis of twins individual histories (A) Percentage of years lived separately for each twin pairs in relation to their ages. (B) Euclidean distance within twin pairs related to years the twins lived separately in IGF2_shore amplicon. S = (years lived in a different house / age)*100
Low Euclidean distance levels characterized twins with small S values, while a scatter after the threshold ofS=35, which in our cohort corresponds to the age of 60, was observed (Figure 7B). There were 3 main exceptions, indicated with an arrow in Figure 7B. These subjects (aged 50, 56 and 74) were still living in the same house at the time of recruitment (small S values) but were characterized by high Euclidean distance values for IGF2_shore.
Discussion
Our results indicate that the 4 analyzed regions in the IGF2/H19 locus show high inter-individual variability both in cohort 1 (spatial/population dimension) and in cohort 2 (temporal/aging dimension). IGF2AS and H19 have mean methylation values comparable to previous reports [16,17], indicating that methylation profiles are consistent among different populations and that the experimental technique used for methylation analysis is highly reproducible between independent laboratories. On the contrary, IGF2_island and IGF2_shore were analyzed for their methylation values here for the first time.
Previous studies have analyzed methylation variability in multiethnic populations, but the enrolled subjects lived in the same geographical area and were therefore exposed to the same environmental factors [19, 20]. On the contrary, cohort 1 was selected in order to include individuals both belonging to different genetic clines and living in their original geographical area. Our results show that methylation variability in cohort 1 cannot be explained on the basis of the geographic provenience of subjects. This implies that the genetic structure of the Italian population does not influence the methylation pattern of the analyzed loci. At the same time, it means that there are no environmental cues specifically associated with Italian macro-areas able to generate clear population epigenetic signatures in the IGF2/H19 locus.
Methylation of IGF2 promoter has been shown to increase with aging [21]. On the contrary, mean methylation values of the 4 regions that we considered in IGF2/H19 locus do not change according to age in cohort 2. This suggests that methylation levels of different regions in the same locus are differently affected by the aging process. At the same time, in young individuals from cohort 2, DNA methylation variations is smaller in IGF2_shore respect to the other analyzed amplicons indicating a stricter control of its methylation levels. Accordingly previous studies have highlighted that the range of methylation variation depends on the genomic position [14]. Moreover, only in IGF2_shore the range of methylation values increases in the elderly suggesting a clear susceptibility of this region to epimutations that occur with aging.
MZ twins model allowed the investigation of the maintenance of DNA methylation profiles during the lifespan. IGF2AS and IGF2_shore methylation levels were highly concordant within twin pairs, but only until the 60 years age threshold. Homogeneous methylation values in IGF2AS and IGF2_shore until the age of 60 years can be in part due to the genetic identity and the sharing of intrauterine and neonatal environments. It is interesting to observe that the threshold of 60 years coincides with the end or with the significant reduction of the reproductive capacity of individuals. A loss in molecular fidelity after the threshold of 60 years has been described [22]. This can affect also the DNA methylation machinery, impairing its ability to maintain the methylation patterns across cellular divisions [23]. The extent and the rate of this process can be influenced by the genetic background of subjects [24], accordingly in our cohort, a subset of couples maintains the concordance in DNA methylation profiles also at old age.
Comparison between MZ and DZ twins confirms a genetic component in the variability observed in IGF2/H19 locus in our cohort. These analysis highlight also an effect of non-genetic factor, which is stronger for IGF2_shore. This observation is supported by the 3 old twins pairs that had always lived together, in the same house, sharing the same daily habits. They show high distance values in IGF2_shore, similar to those observed in the old couples that lived separately. This data suggests that age-related epimutations are independent from the shared environment and are more likely related to individual histories or stochasticity. A larger sample of old twins that lived together should be considered to confirm this hypothesis.
Previous data show that different imprinted genes have different ability to maintain their methylation status [25]. Our results not only confirm these observations, but also show that within the same imprinted locus there are regions whose methylation maintenance is differently influenced by aging.
It is interesting to highlight that, between the 4 regions that we have analyzed in IGF2/H19 locus, IGF2_shore is the one in which the scatter in methylation variability during aging is more evident. This result confirms recent evidences that indicate CpG shores as the regions most variable in terms of methylation levels across normal and pathological tissues and between different cell types [4].
The ability of imprinted genes to maintain their methylation profile has been proposed as a marker of epigenetic stability [25]. In fact, although imprinted loci are under a strict epigenetic control, a relaxation in their regulation can be observed both in pathological [26] and normal conditions [27]. Further studies are needed to deepen our knowledge on age-related DNA methylation variability of imprinted genes, with specific attention to the shore elements.
In conclusion our data confirm the loss of control over DNA methylation maintenance with age only in specific genomic regions (shore). In particular we observe a sudden increase in intra-couple variability from the age of 60, when the reproductive capacities decline and, as a consequence, the effect of natural selection falls off. On one side this decline could be interpreted as a loss of capacities of transmitting the methylation patterns across cellular divisions. On the other side, as the weaker epigenetic control can result in a reduction of gene expression control, it is possible to speculate that this mechanism could represent a molecular strategy to counteract the physiological fitness decline that occurs during aging by offering a wider range of expression possibilities.
Materials and Methods
Ethics statement
Informed consent was obtained from all partecipants and was approved by the Medical Ethics Committee of the S. Orsola-Malpighi Polyclinic (Bologna, Italy).
Samples
Twins: The twins cohort was recruited between 2004 and 2005 in Bologna and neighboring districts to reduce possible bias associated to different geographical origin of the samples. The cohort comprises 47 twin pairs (31 MZ an 16 DZ pairs) with a mean age of 55 years (age range: 22-97 years; Table 3). Italian population: in order to assemble the most representative and informative Italian sample, with an ancient regional ancestry in the area, and an adequate coverage for both the number of populations and the number of individuals within each population sample, an appropriate and accurate sampling strategy was built based on a preliminary surname-based study (Boattini et al., submitted).
Table 3. Cohort 1 characteristics
| Twins | All | ||
|---|---|---|---|
| Monozygotic | Dizygotic | Total samples | |
| N | 62 (31 pairs) | 32 (16 pairs) | 94 (47 pairs) |
| Mean age (SD; range) | 54.0 (19.4; 22-97) | 57.8 (12.8;36-79) | 55.3 (17.4; 22-97) |
| Male (%) | 32 (51.6%) | 20 (62.5%) | 52 (55.3%) |
Groups of homogeneous provinces (“sampling areas”) were aggregated according to the surname-based clusters. Provinces (“sampling points”) were selected within each sampling area according to the complex geographic and historical Italian backgrounds. Individuals (“samples”) were chosen, within each province, based on two different strategies, the “grandparents criterion” and “founder surnames analysis“, so that were included into the study only those individuals whose four grandparents were born in the same sampling area, taking also into account the presence of founder surnames.
Using this sampling strategy 378 male individuals from 29 provinces scattered in 15 regions of Italy were collected (Figure 2A).
DNA methylation measurement and associated data cleaning
The level of DNA methylation was measured on genomic DNA using a MALDI-TOF mass spectrometry-based method (Epityper, Sequenom, San Diego, CA) as previously described [28]. DNA was extracted from whole blood (QIAamp 96 DNA Blood Kit, Qiagen, Hilden, Germany), quantified using Picogreen (Quant-iT dsDNA Broad-Range Assay Kit Invitrogen, Carlsbad, CA), and 1 μg was treated with sodium bisulfite using the EZ methylation kit (Zymo-Research, Irvine, CA). The treatment converts non-methylated cytosine into uracil, leaving methylated cytosine unchanged. In this way variations in the sequence are produced depending on DNA methylation status of the original DNA molecule. PCR amplification, addition of SAP solution and Transcription/RNase A cocktails were performed according to the protocol provided by Sequenom and the mass spectra were analyzed by EpiTYPER analyzer (Sequenom, San Diego, CA). IGF2_shore amplicon encompassed 276 bp (NCBI build 36, chr11: 2 111 039 - 2 111 314). IGF2AS encompassed 338 bp (NCBI build 36, chr11: 2 126 035 - 2 126 372). H19 encompassed 413 bp (NCBI build 36, chr11: 1 975 948 - 1 976 360). IGF2_island encompassed 453 bp (NCBI build 36, chr11: 2 111 119 - 2 110 666). The primers sequences used to amplify this regions are listed in Table 4.
Table 4. CpG Units considered after data cleaning and primers used to amplify the 4 target regions
| Amplicon Name | CpG sites after data cleaning | Total Cpg Units | Primers sequences |
|---|---|---|---|
| IGF2_shore | CpG 1,2 – CpG 3,4 - CpG 6 - CpG 7 - CpG 8 - CpG 9,10,11 | 6 | Forward: aggaagagagGAAGGGGTTGGTTAGTAGGTGTTTGT Reverse: cagtaatacgactcactatagggagaaggctCCTAAACCCCTTTCCCACTCTCTAA |
| IGF2_island | CpG 1,2 - CpG 4 - CpG 11,12 - CpG 13 - CpG 14 - CpG 15 - CpG 16 - CpG 17 - CpG 18,19 - CpG 20 - CpG 21 - CpG 25 - CpG 26 - CpG 27 | 14 | Forward: aggaagagagTATAGGGGTGGTTTGTTAGGTTAGG Reverse: cagtaatacgactcactatagggagaaggcTAAATCAAAAAAAACCCCAAAAAAAC |
| IGF2AS | CpG 1 - CpG 3 - CpG 4 - CpG 6,7 - CpG 8 | 5 | Forward: aggaagagagTGGATAGGAGATTGAGGAGAAA Reverse: cagtaatacgactcactatagggagaaggctAAACCCCAACAAAAACCACT |
| H19 | CpG 1 - CpG 2 - CpG 6 - CpG 7 - CpG 8 - CpG 9,10 - CpG 12 - CpG 13 - CpG 14,15 - CpG 17 - CpG 18,19 - CpG 20 - CpG 22 - CpG 24 | 14 | Forward: aggaagagagGGGTTTGGGAGAGTTTGTGAGGT Reverse: cagtaatacgactcactatagggagaaggctATACCTACTACTCCCTACCTACCAAC |
In this way, a total of 50 CpG units in the 4 gene regions were interrogated for their methylation level. A rigorous data cleaning process was performed to remove unreliable measurements before statistical analysis. Firstly, DNA samples for which methylation level could be established for <60% of the CpG sites were removed. Secondly, CpG sites with more than 30% missing data points within an amplicon were not analyzed. Finally CpG sites that show a bimodal pattern due to the presence of a SNP were removed. Six CpG sites on four fragments could not be measured independently because the fragments had the same molecular weight and were overlapping in the spectrum. After data cleaning, we were able to analyze a total of 39 CpG units in the 4 genomic regions (Table 4).
Statistical analysis
All analysis were performed using R (http://cran.r-project.org)/). Prior to statistical analysis, methylation data were checked for complete bisulfite conversion using the MassArray package [29]. h2 values were calculated according to Falconer's formula [30].
Acknowledgments
I thank Wei Gu, Arnold Levine and Bert Vogelstein for critical reading of the manuscript and excellent suggestions. The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007-2011) under grant agreement n½ 259679 (“IDEAL”).
Conflicts of Interest
The author of this manuscript has no conflict of interest to declare.
References
- 1. Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science (New York, NY). 2001; 293: 1089 -1093. .
- 2. Morgan HD, Santos Ft, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Human Molecular Genetics. 2005; 14: Spec No 1: R47-58 -R47-58. [PubMed] .
- 3. Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science (New York, NY). 2010; 330: 622 -627. .
- 4. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 2009; 41: 178 -186. [PubMed] .
- 5. Doi A, Park I-H, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature Genetics. 2009; 41: 1350 -1353. [PubMed] .
- 6. Aguilera O, Fernandez AF, Munoz A, Fraga MF. Epigenetics and environment: a complex relationship. J Appl Physiol. 2010; 109: 243 -251. [PubMed] .
- 7. Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Human Molecular Genetics. 2009; 18: 4046 -4053. [PubMed] .
- 8. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105: 17046 -17049. [PubMed] .
- 9. Waterland RA, Lin J-R, Smith CA, Jirtle RL. Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Human Molecular Genetics. 2006; 15: 705 -716. [PubMed] .
- 10. Heijmans BT, Tobi EW, Lumey LH, Slagboom PE. The epigenome: archive of the prenatal environment. Epigenetics: Official Journal of the DNA Methylation Society. 2009; 4: 526 -531. [PubMed] .
- 11. Champagne FA and Curley JP. Epigenetic mechanisms mediating the long-term effects of maternal care on development. Neuroscience and biobehavioral reviews. 2009; 33: 593 -600. [PubMed] .
- 12. Weaver ICG, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S,, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nature neuroscience. 2004; 7: 847 -854. .
- 13. Weaver ICG, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, Szyf M. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005; 25: 11045 -11054. [PubMed] .
- 14. Schneider E, Pliushch G, El Hajj N, Galetzka D, Puhl A, Schorsch M, Frauenknecht K, Riepert T, Tresch A, Müller AM, Coerdt W, Zechner U, Haaf T. Spatial, temporal and interindividual epigenetic variation of functionally important DNA methylation patterns. Nucleic acids research. 2010; 38: 3880 -3890. [PubMed] .
- 15. Bock C, Walter Jr, Paulsen M, Lengauer T. Inter-individual variation of DNA methylation and its implications for large-scale epigenome mapping. Nucleic acids research. 2008; 36: e55 -e55. [PubMed] .
- 16. Heijmans BT, Kremer D, Tobi EW, Boomsma DI, Slagboom PE. Heritable rather than age-related environmental and stochastic factors dominate variation in DNA methylation of the human IGF2/H19 locus. Human Molecular Genetics. 2007; 16: 547 -554. [PubMed] .
- 17. Ollikainen M, Smith KR, Joo EJ-H, Ng HK, Andronikos R, Novakovic B, Abdul Aziz NK, Carlin JB, Morley R, Saffery R, Craig JM. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Human Molecular Genetics. 2010; 19: 4176 -4188. [PubMed] .
- 18. Liu J, Hutchison K, Perrone-Bizzozero N, Morgan M, Sui J, Calhoun V. Identification of genetic and epigenetic marks involved in population structure. PloS One. 2010; 5: e13209 -e13209. [PubMed] .
- 19. Terry MB, Ferris JS, Pilsner R, Flom JD, Tehranifar P, Santella RM, Gamble MV, Susser E. Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008; 17: 2306 -2310. .
- 20. Das PM, Ramachandran K, Vanwert J, Ferdinand L, Gopisetty G, Reis IM, Singal R. Methylation mediated silencing of TMS1/ASC gene in prostate cancer. Molecular cancer. 2006; 5: 28 -28. [PubMed] .
- 21. Issa JP, Vertino PM, Boehm CD, Newsham IF, Baylin SB. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93: 11757 -11762. [PubMed] .
- 22. Hayflick L. Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS genetics. 2007; 3: e220 -e220. [PubMed] .
- 23. Goyal R, Reinhardt R, Jeltsch A. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic acids research. 2006; 34: 1182 -1188. [PubMed] .
- 24. Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekström TJ, Harris TB, Launer LJ, Eiriksdottir G, Leppert MF, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA: The Journal of the American Medical Association. 2008; 299: 2877 -2883. [PubMed] .
- 25. Woodfine K, Huddleston JE, Murrell A. Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenetics & chromatin. 2011; 4: 1 -1. [PubMed] .
- 26. Cui H. Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Disease markers. 2007; 23: 105 -112. [PubMed] .
- 27. Sakatani T, Wei M, Katoh M, Okita C, Wada D, Mitsuya K, Meguro M, Ikeguchi M, Ito H, Tycko B, Oshimura M. Epigenetic heterogeneity at imprinted loci in normal populations. Biochemical and Biophysical Research Communications. 2001; 283: 1124 -1130. [PubMed] .
- 28. Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 15785 -15790. [PubMed] .
- 29. Thompson RF, Suzuki M, Lau KW, Greally JM. A pipeline for the quantitative analysis of CG dinucleotide methylation using mass spectrometry. Bioinformatics (Oxford, England). 2009; 25: 2164 -2170. .
- 30. Falconer DS. Introduction to quantitative genetics. Burnt Mill, Harlow, Essex, England, New York Longman Wiley 1989; .