



Introduction
A year ago, I wrote a perspective “Cell cycle arrest is not senescence”, intended to clarify a new meaning of cellular senescence [1]. The perspective was not completely understood in part due to its title. The title was missing the word “yet”, which is now included. As discussed in the article, cell cycle arrest is not yet senescence and senescence is not just arrest: senescence can be driven by growth-promoting pathways such as mTOR, when actual growth is impossible. (This mechanism connects cellular senescence, organismal aging and age-related diseases, predicting anti-aging agents [2-6]). In brief, senescence can be caused by growth stimulation, when the cell cycle is arrested [7, 8]. As one hallmark, senescent cells loose proliferative potential (PP) - the potential to resume proliferation. Importantly, inhibitors of mTOR suppress hallmarks of senescence during cell cycle arrest so cells stay quiescent but not senescent [9-13]. Such quiescent cells, with inhibited mTOR, retain PP. Once again, cells may be arrested but retain PP, the ability to restart proliferation, when allowed. In certain conditions, p53 causes arrest but can preserve PP by inhibiting the mTOR pathway [14-16]. However, this phenomenon should not be misunderstood to indicate that “p53 induces proliferation or prevents arrest or keeps cells proliferating” or “arrested cells retain proliferation”; rather, p53 instead regulates proliferative potential. Although we tried to explain what p53 does exactly (causes arrest, while preserving PP) misunderstanding nonetheless ensued. One solution is not to use the term PP altogether, substituting for it the term RP (regenerative potential). In the organism, stem cells and wound-healing cells, while quiescent, are capable to regenerate tissues after cell loss. Unlike non-senescent cells, senescent cells cannot divide in response to cell loss and therefore lose the potential to regenerate tissues. In cell culture, quiescent cells preserve RP. If the cell cycle is blocked, activation of mTOR causes loss of RP [17]. New concepts need new terminology. Instead of squeezing novel meaning into the old terms, here we present new terms for a new meaning of the aging process. And a central term is gerogenic conversion or geroconversion.
Geroconversion
Mitogens and growth factors activate growth-promoting pathways, which stimulate (a) growth in size and (b) cell cycle progression. When cells proliferate, an increase in cell mass is balanced by division. Withdrawal of growth factors causes quiescence: the quiescent cell neither grows, nor cycles, and its functions and metabolism are low. In contrast, cell cycle blockage, in the presence of growth-stimulation leads to senescence (Figure 1). Hallmarks of senescence include a large flat morphology, senescence-associated beta-galactosidase (SA-beta-gal) staining, cellular over-activation and hyper-function, feedback signal resistance and loss of RP (that is, the inability to restart proliferation when the cell cycle inhibitor is removed). For example, in one well-studied cellular model, inducible ectopic expression of p21 causes cycle arrest (day 1) and senescence (after 3 days) [18, 19]. At first, the arrested cells are quiescent-like: they are not hypertrophic, and they are SA-beta-gal negative and retain RP. Thus, they can restart proliferation when p21 expression is switched off. After 3 days, however, the cells acquire a senescent morphology and, if p21 is then switched off, the cells cannot restart proliferation or die in mitosis [19]. Importantly, while inhibiting the cell cycle, p21 does not inhibit the mTOR pathway [8-17, 20, 21]. mTOR and perhaps some other growth-promoting pathways convert quiescence (day 1) into senescence (day 3). Inhibition of mTOR by rapamycin decelerates this ‘geroconversion’ [8-17, 20, 21].
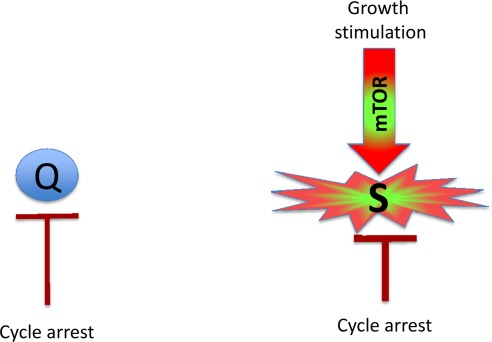
Figure 1. Quiescence versus senescence Q: quiescent cell. In the absence of the growth factors, normal cells undergo cell cycle arrest. S: senescent cell. When cell cycle is arrested, the growth signal (if it cannot reactivate cycling) drives senescence.
Similarly other inhibitors of mTOR also suppress geroconversion [10, 11]. For example, in some cell lines, induction of p53 inhibits mTOR [22-26] and other anabolic pathways [27-32], thus suppressing geroconversion in cells arrested by ectopic p21 [14]. By itself, p53 causes cell cycle arrest but can suppress geroconversion [14-17, 33-35]. In cell culture, cell cycle arrest and geroconversion are initiated simultaneously. In proliferating cultured cells (especially cancer cells) mTOR is activated. Many agents cause cell cycle arrest without inhibiting mTOR (or other growth factor-sensing pathways). Once arrested, such cells are rapidly converted to senescent cells. This is accelerated geroconversion. So it may seem that senescence is “caused” by cell cycle arrest. The above examples, however, suggest that senescence is caused by growth-stimulation when the cell cycle is arrested.
In the organism, most cells are arrested and geroconversion can be slow. When chronically stimulated (but still arrested) they can become senescent. This physiological geroconversion can be imitated in cell culture [17].
The terms gerogenic conversion and oncogenic transformation sound alike.This is not a coincidence for choosing the term geroconversion. Gerogenic conversion and oncogenic transformation are two sides of the same process.
Gerogenic oncogenes and gerogenes
Activation of growth factor receptors, Ras and Raf family members and members of the MAPK and PI3K/Akt pathways are universal in cancer [36-38]. All these oncogenes activate the mTOR pathway [39-47]. They are gerogenic oncogenes, which drive the geroconversion of arrested cells. Because strong growth-promoting (mitogenic) signals induce cell cycle arrest [48-54], strong mitogenic signaling causes both conditions of senescence: arrest and mTOR/growth signal (Figure 2A). To avoid senescence, cancer cells must disable cell cycle control (Figure 2B) by either loss of p16, p53 and Rb or activation of c-myc, for example [36-38, 48, 55, 56]. In proliferating cells, gerogenic oncogenes render cells malignant and pro-gerogenic (see below). The same gerogenic oncogenes or their analogs accelerate aging and shorten life span in diverse species from worm to mammals. Therefore, these genes can be termed gerogenes [57]. Thus, the mTOR pathway shortens life span, whereas rapamycin extends life span [58-75]. Not coincidentally, Mutations that increase the life span of C. elegans inhibit tumor growth [76]. Finally, metabolic self-destruction, known as chronological senescence in yeast [60, 61, 77] is also stimulated by gerogenes and is inhibited by rapamycin [78].
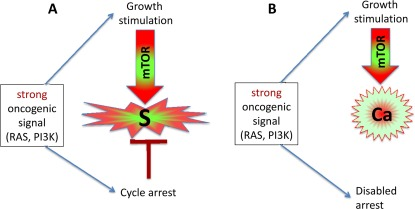
Figure 2. Strong oncogenic signaling, senescence and cancer (A) Strong mitogenic/oncogenic signal can simultaneously cause arrest and activate mTOR. Cells senesce. (B) Disabling of cell cycle control (loss of p16, Rb, p53) can convert senescence to cancer.
Gerosuppressors
Gerosuppressors are genes (and their products) that suppress geroconversion. Gerosuppressors (for example, PTEN, AMPK, sirtuins, TSC2, NF-1 and p53) antagonize the mTOR pathway (see for ref. [57]). Their inactivation shortens life span in model organisms. Gerosuppressors are also tumor suppressors. So gero-suppressors suppress both geroconversion and cancer.
Gerosuppressants
Gerosuppressants are small molecules (such as rapamycin) that suppress geroconversion. Not co-incidentally, rapamycin also extends life span in diverse species from yeast to mammals. They can, in theory, be used to treat age-related diseases by slowing down aging, thus extending both maximal and healthy lifespan.
Gero promoters
Small molecules or drugs that can accelerate or promote geroconversion. One potential candidate is phorbol esters, which can activate mTOR in some cells. Not surprisingly, it is also a tumor-promoter.
Gerogenic pathways
Gerogenic signaling pathways promote geroconversion. Whether gerogenic pathways cause or abrogate cell cycle arrest is irrelevant. For example, strong mitogenic/growth signals can induce cell cycle arrest, instead of proliferation [48-54]. Simultaneously, in arrested cells, growth signals cause geroconversion, leading to senescence (Figure 2A). As another example, the effects of p53 on cell cycle and geroconverion can be dissociated [14].
Pro-gerogenic conversion
In proliferating cells, overactivation of the mTOR pathway renders them pro-gerogenic. Cancer cells are proliferating pro-gerogenic cells. When such cells are forcefully arrested, they become senescent. Also, stimulation of mTOR in normal stem cells causes hyper-proliferation, pro-gerogenic conversion and cell exhaustion [79-84], contributing to aging.
Gerogenic cell
Although loss of RP is very useful marker of senescence in cell culture, this marker may not play a key role in age-related pathologies in the organism, because most post-mitotic cells should not be able to restart proliferation anyway. (Notable exceptions are stem, wound-healing and satellite cells). I suggest that active mTOR_in arrested cells is a crucial marker of gerogenic cells and early senescence. Gerogenic (senescent) and quiescent cells can be distinguished by the levels of phosphorylated S6 (pS6), the ribosomal protein that is phosphorylated in response to mTOR activation: high in senescent cells and low in quiescent cells. Levels of pS6 in senescent cells may remain similar to the levels of pS6 in proliferating cells. So senescent/gerogenic cells have many features of proliferating cells. Interestingly, basal (fasting) levels pS6 were elevated in old mice [85]. Gerogenic cells could be defined as arrested cell with activated mTOR. The most physiologically relevant features are hypertrophy, hyperfunction and feedback resistance.
Hypertrophy
Growth signals during cell cycle arrest lead to an enlarged cell morphology. From theoretical perspective, hypertrophy will eventually be limited by activation of lysosomes/autophagy [7]. This phenomenon may explain the activity of SA-beta-Gal, which is lysosomal enzyme [86-88] and active autophagy despite active mTOR [89, 90].
Hyperfunction
Due to over-stimulation, senescent cells are hyperfunctional. For example, For example, senescent fibroblasts secrete many cytokines, growth factors and proteases (the hypersecretory senescence-associated secretory phenotype or SASP), senescent osteoclasts resorb bones, smooth muscle cells contract, platelets aggregate, neutrophils generate ROS, neurons charge, endocrine cells produce hormones. Noteworthy, SASP as a marker of senescence [91-99] is an example of hyperfunction.
Feedback resistance
Overactivation of signaling pathways causes signal resistance due to feedback inactivation of the signaling pathway. As an example, mTOR/S6K overactivation causes insulin and GF resistance [100-104].
Secondary atrophy
Hyperfunctions are associated with hypertrophy and hyperplasia. Yet, at the end, cells may fail either to function or to survive, leading to secondary atrophy. When cells fail, conditions become TOR-independent and terminal. This conceals hyperfunction as an initial cause, misleadingly presenting aging as a decline.
From gerogenic cells to organismal aging
You might notice that an accumulation of molecular damage was never mentioned in this article. It was unneeded. Cellular aging and geroconversion is not caused by accumulation of random molecular damage. Although damage accumulates, I suggest that the organism does not live long enough to suffer from this accumulation with one special exception that illuminates the rule [105]. (The weakness of free radical damage theory was discussed in detail [106-114]).
One definition of organismal aging is an increase in the probability of death. Gerogenic cells (due to their hyper-activity and signal-resistance) may slowly cause atherosclerosis, hypertension, insulin-resistance, obesity, cancer, neurodegeneration, age-related macular degeneration, prostate enlargement, menopause, hair loss, osteoporosis, osteoarthritis, benign tumors and skin alterations. These conditions lead to damage -- not molecular damage but organ and system damage. Examples include beta-cell failure [115], ovarian failure (menopause) [116], myocardial infarction, stroke, renal failure, broken hips, cancer metastases and so on [6, 117]. These are acute catastrophes, which cause death. I suggest that by suppressing geroconversion, gerosuppressants will prevent diseases and extend healthy life span.
Acknowledgments
I thank Judith Campisi (Buck Institute for Age Research, Novato, CA, USA) and Manuel Serrano (Spanish National Cancer Research Center, Madrid, Spain) for reading or editing this manuscript and for excellent suggestions.
Conflicts of Interest
The author of this manuscript has no conflict of interest to declare.
References
- 1. Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY). 2011; 3: 94 -101. [PubMed] .
- 2. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087 -2102. [PubMed] .
- 3. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Disc Today. 2007; 12: 218 -224. .
- 4. Blagosklonny MV. Paradoxes of aging. Cell Cycle. 2007; 6: 2997 -3003. [PubMed] .
- 5. Blagosklonny MV and Hall MN. Growth and aging: a common molecular mechanism. Aging. 2009; 1: 357 -362. [PubMed] .
- 6. Blagosklonny MV. mTOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8: 4055 -4059. [PubMed] .
- 7. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358 -362. [PubMed] .
- 8. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 -3361. [PubMed] .
- 9. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8: 1888 -1895. [PubMed] .
- 10. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009; 8: 1896 -1900. [PubMed] .
- 11. Demidenko ZN and Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009; 8: 1901 -1904. [PubMed] .
- 12. Demidenko ZN and Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging (Albany NY). 2009; 1: 1008 -1016. [PubMed] .
- 13. Pospelova TV, Demidenk ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009; 8: 4112 -4118. [PubMed] .
- 14. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 -9664. [PubMed] .
- 15. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2: 344 -352. [PubMed] .
- 16. Leontieva O, Gudkov A, Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9: 4323 -4327. [PubMed] .
- 17. Leontieva OV and Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2: 924 -935. [PubMed] .
- 18. Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, Roninson IB. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999; 18: 4808 -4818. [PubMed] .
- 19. Chang BD, Broude EV, Fang J, Kalinichenko TV, Abdryashitov R, Poole JC, Roninson IB. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000; 19: 2165 -2170. [PubMed] .
- 20. Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, Fornace AJ Jr., Pospelova TV, Pospelov VA. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. 2010; 9: 3945 -3955. [PubMed] .
- 21. Leontieva OV, Demidenko ZN, Gudkov AV, Blagosklonny MV. Elimination of proliferating cells unmasks the shift from senescence to quiescence caused by rapamycin. PLoS One. 2011; 6: e26126 [PubMed] .
- 22. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN transcription by p53. Mol Cell. 2001; 8: 317 -325. [PubMed] .
- 23. Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006; 20: 267 -275. [PubMed] .
- 24. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005; 102: 8204 -8209. [PubMed] .
- 25. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007; 67: 3043 -3053. [PubMed] .
- 26. Feng Z and Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010; 20: 427 -434. [PubMed] .
- 27. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, Sugano S, Sato E, Nagao T, Yokote K, Tatsuno I, Prives C. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010; 107: 7461 -7466. [PubMed] .
- 28. Poyurovsky MV and Prives C. P53 and aging: A fresh look at an old paradigm. Aging (Albany NY). 2010; .
- 29. Vousden KH and Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009; 9: 691 -700. [PubMed] .
- 30. Vigneron A and Vousden KH. p53, ROS and senescence in the control of aging. Aging (Albany NY). 2010; 2: 471 -474. [PubMed] .
- 31. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: Emerging new roles for the tumor suppressor. Oncotarget. 2011; 2: 948 -957. [PubMed] .
- 32. Zawacka-Pankau J, Grinkevich VV, Hunten S, Nikulenkov F, Gluch A, Li H, Enge M, Kel A, Selivanova G. Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: targeting Warburg effect to fight cancer. J Biol Chem. 2011; 286: 41600 -41615. [PubMed] .
- 33. Long JS and Ryan KM. p53 and senescence: a little goes a long way. Cell Cycle. 2010; 9: 4050 -4051. .
- 34. Santoro R and Blandino G. p53: The pivot between cell cycle arrest and senescence. Cell Cycle. 2010; 9: 4262 -4263. [PubMed] .
- 35. Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010; 9: 4256 -4257. [PubMed] .
- 36. Vogelstein B and Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004; 10: 789 -799. [PubMed] .
- 37. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006; 6: 184 -192. [PubMed] .
- 38. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646 -674. [PubMed] .
- 39. Hay N and Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004; 18: 1926 -1945. [PubMed] .
- 40. Sarbassov dos D, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005; 17: 596 -603. [PubMed] .
- 41. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005; 37: 19 -24. [PubMed] .
- 42. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124: 471 -484. [PubMed] .
- 43. Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007; 13: 252 -259. [PubMed] .
- 44. Dazert E and Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011; .
- 45. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2010; 12: 21 -35. [PubMed] .
- 46. Conn CS and Qian SB. mTOR signaling in protein homeostasis: less is more? Cell Cycle. 2011; 10: 1940 -1947. [PubMed] .
- 47. Ma XM and Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009; 10: 307 -318. [PubMed] .
- 48. Serrano M, Lim AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK1A. Cell. 1997; 88: 593 -602. [PubMed] .
- 49. Lloyd AC, Obermuller F, Staddon S, Barth CF, McMahon M, Land H. Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev. 1997; 11: 663 -677. [PubMed] .
- 50. Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol. 1997; 17: 5588 -5597. [PubMed] .
- 51. Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997; 17: 5598 -5611. [PubMed] .
- 52. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998; 12: 3008 -3019. [PubMed] .
- 53. Zhu JY, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998; 12: 2997 -3007. [PubMed] .
- 54. Kerkhoff E and Rapp UR. High-intensity Raf signals convert mitotic cell cycling into cellular growth. Cancer Res. 1998; 58: 1636 -1640. [PubMed] .
- 55. Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003; 22: 4212 -4222. [PubMed] .
- 56. Hueber AO and Evan GI. Traps to catch unwary oncogenes. Trends Genet. 1998; 14: 364 -367. [PubMed] .
- 57. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010; 9: 3151 -3156. [PubMed] .
- 58. Stipp D. A new path to longevity. Sci Am. 2012; 306: 32 -39. [PubMed] .
- 59. Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003; 426: 620 [PubMed] .
- 60. Powers RWr, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006; 20: 174 -184. [PubMed] .
- 61. Kaeberlein M, Powers RWr, Kristan K. Steffen, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005; 310: 1193 -1196. [PubMed] .
- 62. Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004; 131: 3897 -3906. [PubMed] .
- 63. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004; 14: 885 -890. [PubMed] .
- 64. Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007; 6: 95 -110. [PubMed] .
- 65. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandezr E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogenous mice. Nature. 2009; 460: 392 -396. [PubMed] .
- 66. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326: 140 -144. [PubMed] .
- 67. Moskalev AA and Shaposhnikov MV. Pharmacological Inhibition of Phosphoinositide 3 and TOR Kinases Improves Survival of Drosophila melanogaster. Rejuvenation Res. 2010; 13: 246 -247. [PubMed] .
- 68. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010; 11: 35 -46. [PubMed] .
- 69. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, et al. Rapamycin, But Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. J Gerontol A Biol Sci Med Sci. 2011; 66: 191 -201. [PubMed] .
- 70. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092 -2097. [PubMed] .
- 71. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10: 4230 -4236. [PubMed] .
- 72. Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010; 11: 453 -465. [PubMed] .
- 73. Blagosklonny MV. Rapamycin and quasi-programmed aging: Four years later. Cell Cycle. 2010; 9: 1859 -1862. [PubMed] .
- 74. Bjedov I and Partridge L. A longer and healthier life with TOR down-regulation: genetics and drugs. Biochem Soc Trans. 2011; 39: 460 -465. [PubMed] .
- 75. Katewa SD and Kapahi P. Role of TOR signaling in aging and related biological processes in Drosophila melanogaster. Exp Gerontol. 2011; 46: 382 -390. [PubMed] .
- 76. Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science. 2006; 313: 971 -975. [PubMed] .
- 77. Burtner CR, Murakami CJ, Kennedy BK, Kaeberlein M. A molecular mechanism of chronological aging in yeast. Cell Cycle. 2009; 8: 1256 -1270. [PubMed] .
- 78. Leontieva OV and Blagosklonny MV. Yeast-like chronological senescence in mammalian cells: phenomenon, mechanism and pharmacological suppression. Aging (Albany NY). 2011; 3: 1078 -1091. [PubMed] .
- 79. Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008; 11: 801 -808. [PubMed] .
- 80. Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009; 2: ra75 [PubMed] .
- 81. Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, Williams DA, Kwiatkowski DJ, DePinho RA. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008; 105: 19384 -19389. [PubMed] .
- 82. Gan B and DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009; 8: 1003 -1006. [PubMed] .
- 83. Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009; 5: 279 -289. [PubMed] .
- 84. Wang CY, Kim HH, Hiroi Y, Sawada N, Salomone S, Benjamin LE, Walsh K, Moskowitz MA, Liao JK. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci Signal. 2009; 2: ra11 [PubMed] .
- 85. Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010; 468: 1100 -1104. [PubMed] .
- 86. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000; 113: Pt 20 3613 -3622. [PubMed] .
- 87. DeJesus V, Rios I, Davis C, Chen Y, Calhoun D, Zakeri Z, Hubbard K. Induction of apoptosis in human replicative senescent fibroblasts. Exp Cell Res. 2002; 274: 92 -99. [PubMed] .
- 88. Hampel B, Malisan F, Niederegger H, Testi R, Jansen-Durr P. Differential regulation of apoptotic cell death in senescent human cells. Exp Gerontol. 2004; 39: 1713 -1721. [PubMed] .
- 89. Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, Tavare S, Inoki K, Shimizu S. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011; 332: 966 -970. [PubMed] .
- 90. Narita M and Young AR. Autophagy facilitates oncogene-induced senescence. Autophagy. 2009; 5: 1046 -1047. [PubMed] .
- 91. CoppŽ JP, Patil CK, Rodier F, Sun Y, Mu-oz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008; 6: 2853 -2868. [PubMed] .
- 92. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001; 98: 12072 -12077. [PubMed] .
- 93. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11: 973 -979. [PubMed] .
- 94. Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppe JP, Campeau E, Beausejour CM, Kim SH, Davalos AR, Campisi J. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011; 124: 68 -81. [PubMed] .
- 95. Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, Nedospasov SA, Hazen SL, Feinstein E, Gudkov AV. p53 is a suppressor of inflammatory response in mice. Faseb J. 2005; 19: 1030 -1032. [PubMed] .
- 96. Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011; 21: 354 -359. [PubMed] .
- 97. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011; 25: 2125 -2136. [PubMed] .
- 98. Pani G. From growing to secreting: new roles for mTOR in aging cells. Cell Cycle. 2011; 10: 2450 -2453. [PubMed] .
- 99. Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, Sotgia F. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011; 10: 2059 -2063. [PubMed] .
- 100. Tremblay F and Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001; 276: 38052 -38060. [PubMed] .
- 101. Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, WaldhŠusl W, Marette A, Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005; 54: 2674 -2684. [PubMed] .
- 102. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004; 431: 200 -205. [PubMed] .
- 103. Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004; 14: 1650 -1656. [PubMed] .
- 104. Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007; 117: 730 -738. [PubMed] .
- 105. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY). 2011; 3: 1130 -1141. [PubMed] .
- 106. Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008; 22: 3236 -3241. [PubMed] .
- 107. Cabreiro F, Ackerman D, Doonan R, Araiz C, Back P, Papp D, Braeckman BP, Gems D. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med. 2011; 51: 1575 -1582. [PubMed] .
- 108. Gems D and Doonan R. Antioxidant defense and aging in C. elegans: Is the oxidative damage theory of aging wrong? Cell Cycle. 2009; 8: 1681 -1687. [PubMed] .
- 109. Lapointe J and Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci. 2009; 67: 1 -8. [PubMed] .
- 110. Van Raamsdonk JM, Meng Y, Camp D, Yang W, Jia X, Benard C, Hekimi S. Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics. 2010; 185: 559 -571. [PubMed] .
- 111. Speakman JR and Selman C. The free-radical damage theory: Accumulating evidence against a simple link of oxidative stress to ageing and lifespan. Bioessays. 2011; 33: 255 -259. [PubMed] .
- 112. Ristow M and Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011; 51: 327 -336. [PubMed] .
- 113. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 114. Blagosklonny MV. Hormesis does not make sense except in the light of TOR-driven aging. Aging (Albany NY). 2011; 3: 1051 -1062. [PubMed] .
- 115. Blagosklonny MV. Rapamycin-induced glucose intolerance: Hunger or starvation diabetes. Cell Cycle. 2011; 10: 4217 -4224. [PubMed] .
- 116. Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging (Albany NY). 2010; 2: 265 -273. [PubMed] .
- 117. Blagosklonny MV. Aging-suppressants: cellular senescence (hyperactivation) and its pharmacologic deceleration. Cell Cycle. 2009; 8: 1883 -1887. [PubMed] .